
Abstract
Dimorphic organisms possess the remarkable genome capacity to alternate genetic information between two distinct life forms. In dimorphic Mucorales, lineages can reversibly transition between yeast and mycelium as a function of environmental stimuli. However, how genetic information produces two divergent life evolves in a single genome remains unresolved. Here, we report hundreds of gene families exhibiting convergent evolution for dimorphism. This adaptation involves paralog functionalization and the coordination of expression, with yeast and mycelium-specific paralogs. Though these gene families have distinct functions, all their paralogs are convergently adapted to produce yeast and mycelium morphologies. In addition, dimorphic gene families with related functions form head-to-head structures, coordinating differential expression. This coordinated regulation is controlled by two new genes, dkl and dfl. Loss of function of these genes results in global dysregulation of gene expression and loss of dimorphism. Dimorphic gene families, head-to-head marker loci, and the dfl gene are conserved across various dimorphic species but are absent in closely related monomorphic species. Our findings identify an evolutionary mechanism that integrates and optimizes the genetic information required for two distinct life forms within a single organism.
Similar content being viewed by others


Introduction
The existence of distinct morphological forms within a species is a compelling phenomenon that underscores the diversity of life and the complex interplay between genetics, environment, and evolution. Diverse dimorphic strategies, each with distinct functions, have evolved across species. Sexual dimorphism, for instance, results in morphological differences between sexes, typically affecting traits directly related to reproduction1. In contrast, dimorphism associated with metamorphosis involves a two-stage developmental process characterized by dramatic physical transformations between the larval and adult forms2. Fungal dimorphism differs significantly. The yeast and mycelial forms can exist and reproduce independently, without requiring a transition to the alternate form3,4. While these two forms are commonly observed independently in monomorphic fungal species, dimorphic species exhibiting both forms represent a less frequent occurrence3. Importantly, morphological duality and the ability to transition between forms are key traits of human pathogens, as they facilitate host infection, immune evasion, and dissemination. This is the case for fungi within the order Onygenales5, as well as other Ascomycetes and Basidiomycetes, such as Talaromyces marneffei and the meningitis-causing fungus Cryptococcus spp5,6. These forms do not represent sequential developmental stages within a single program, but rather distinct, independent programs that do not necessitate interconversion. A key distinguishing feature of fungal dimorphism is the reversible and environmentally responsive transition between forms, allowing the fungus to adapt to fluctuating conditions4.
In Mucoromycota fungi, the morphological divergence between yeast and mycelium profoundly influences fungal physiology. The yeast form, characterized as a unicellular microorganism, typically proliferates under anaerobic conditions, employing fermentative metabolism, and reproduces asexually via budding4. Conversely, the mycelial form, a multicellular filamentous organism, thrives in aerobic environments, utilizes oxidative metabolism, and exhibits both sexual and asexual reproductive strategies4. Furthermore, mycelial growth involves complex differentiation, leading to specialized hyphal types and macroscopic structures such as sporangiophores and zygosporangia7. Yeast formation is generally induced by elevated CO2 and glucose concentrations, while mycelial development is stimulated by the presence of oxygen4. Of particular clinical significance is the observed differential virulence between these morphological forms. Certain Mucorales species are etiologic agents of mucormycosis, a severe and often fatal opportunistic fungal infection that predominantly affects immunocompromised individuals, including those with diabetes mellitus, hematological malignancies, and solid organ transplant recipients8. Numerous studies have documented virulence exclusively from the mycelial form, attributing this disparity to enhanced nutrient acquisition and tissue invasion capabilities9,10,11,12.
The current understanding of the dimorphism phenomenon in fungi of the order Mucorales is limited, largely confined to studies in a few organisms within the Mucoraceae family, particularly the model Mucor lusitanicus. These investigations primarily focus on three signaling pathways: cAMP-Dependent Protein Kinase A (PKA)10, ADP-Ribosylation Factors (Arfs)13, and the calcineurin pathway9,14. Studies have demonstrated that exogenous cAMP administration can induce yeast-like growth even under aerobic conditions, thereby modulating PKA activity and influencing subsequent morphological outcomes15. ADP-ribosylation factors (Arfs) are small GTP-binding proteins implicated in vesicular trafficking and cytoskeletal organization. In M. lusitanicus, four Arf-encoding genes (arf1–arf4) have been identified. Arf1 and Arf3 are primarily associated with filamentous growth, whereas Arf2 and Arf4 are linked to yeast-like growth13. Calcineurin is a Ca²⁺/calmodulin-dependent serine/threonine-specific protein phosphatase composed of catalytic A and regulatory B subunits. In M. lusitanicus, three catalytic A subunits (CnaA, CnaB, and CnaC) and one regulatory B subunit (CnbR) have been identified9. Disruption of the cnbR gene results in mutants exhibiting a permanently locked yeast phase, indicating that calcineurin activity is essential for hyphal growth9. Interestingly, protein kinase A (PKA) activity is elevated during yeast growth under conditions of calcineurin inhibition, suggesting a novel connection between PKA and calcineurin pathways in controlling dimorphism14.
Notably, the morphological plasticity of dimorphism in M. lusitanicus is not only crucial for its adaptability and pathogenicity but also plays a significant role in its resistance to antifungal agents mediated by the calcineurin pathway and the RNAi mechanism. Thus, the presence of the antifungal agent FK506 (tacrolimus) inhibits virulent mycelial growth by blocking the calcineurin pathway through binding to FKBP12, resulting in a persistent yeast-like growth. In this state, the fungus can circumvent the blockage through specific silencing of FKBP12, thereby regaining the ability to grow as mycelium in the presence of the antifungal agent16.
In complex species exhibiting sexual dimorphism, genomes have adapted through the evolution of sex chromosomes17, whereas in species with metamorphosis, the maintenance of the larval phase and the transition to the adult phase are tightly controlled by hormonal systems2. However, in Mucorales dimorphism, beyond studies focused on signaling controlling yeast or mycelial form activation, limited information exists regarding how this process has shaped the evolution of genomes that must accommodate information for two distinct life forms. The profound morphological and physiological differences between yeast and mycelium, the independence of these two forms in the fungal life cycle, and the capacity for reversible transition suggest a unique evolutionary trajectory in the genomes of these species.
Here, we show evolutionary adaptations in dimorphic Mucorales species can be elucidated through detailed structural and functional analysis of dimorphic genomes. We identify a novel class of gene families composed of dimorphic paralogs with coordinated expression, reveal the systematic utilization of head-to-head (H2H) structures in coordinating gene functions and differential expression, and discover two novel genes whose loss of function results in general dysregulation and loss of dimorphism. Comparative analyses demonstrate that these mechanisms are conserved across dimorphic Mucorales species but are absent in non-dimorphic relatives, providing critical insights into the evolutionary foundations of fungal dimorphism. These findings establish a framework for understanding the genomic and regulatory innovations that have shaped the dimorphic evolution of Mucorales fungi.
Results
Differential Expression Across Morphological States in Mucor lusitanicus
Mucorales fungi, exemplified by our model organism M. lusitanicus, exhibit a striking cellular dimorphism, enabling them to grow either as yeast or as filamentous mycelia. Furthermore, these fungi can transition between these forms in response to changes in environmental oxygen levels (Fig. 1a, b). To better understand the regulatory pathways involved in dimorphism, we conducted a transcriptomic analysis aimed at capturing the broader repertoire of genes involved in this process, measuring transcript abundance across each growth condition. For that, we generated samples from yeast, primary mycelium, reverse transition, and mature mycelium (Fig. 1a, b). Primary mycelium develops when a yeast culture is transferred to an aerobic medium, where yeast cells germinate to form exponentially growing mycelia with primary metabolism. The reverse transition occurs when the primary mycelium is transferred back to an anaerobic culture, leading to hyphal thickening, septation, and the generation of unicellular yeast cells. Mature mycelium arises when primary mycelium depletes available nutrients, initiating the secondary metabolism and the production of metabolites such as beta-carotene, followed by the development of macroscopic sporangiophores. Transcriptomic data from these four states were analyzed across three distinct comparisons: yeast versus primary mycelium (Y vs PM), yeast versus mature mycelium (Y vs MM), and reverse transition versus primary mycelium (RT vs PM) (Fig. 1b, c, Supplementary Data 1). The distributions of differentially expressed genes (DEGs) across these three comparisons revealed a similar number of genes with higher transcript abundance in the yeast form (hereafter referred to yeast-specific) and genes with higher transcript abundance in the mycelial form (hereafter referred to mycelium-specific genes) in the “Y vs PM” and “RT vs PM” comparisons. In contrast, the “Y vs MM” comparison showed a greater number of mycelium-specific DEGs with higher log2FC values (Fig. 1c, d, Supplementary Data 2). Integration of data from all three comparisons yielded a total of 8181 DEGs, of which 4620 were mycelium-specific and 3561 were yeast-specific (Fig. 1d, Supplementary Data 2). These data indicate that 70% of the M. lusitanicus genome undergoes differential expression during dimorphism, suggesting that most cellular processes respond differentially during this transition (Supplementary Data 3). However, more specific functional enrichment analysis in which we detected significantly overrepresented (FDR < 0.05) functional categories revealed that yeast forms are enriched for functional groups of genes dedicated to metabolism, expression, and synthesis, while mycelia are enriched for genes involved in signaling, as well as the formation and activity of the cytoskeleton (Fig. 1e).

a Images depicting the four dimorphic states analyzed. Scale bars represent 50μm in Yeast, Primary Mycelium, and Reverse Transition images, and 0,25 cm in Mature Mycelium. Representative images from five independent experiments. b Schematic representation of potential transitions between dimorphic states. c Distribution of differential expression values across three comparative analyses. Wald test is integrated in DESeq2 during the differential expression analysis (Padj<0.05). Blue points represent genes upregulated in mycelial states, red points represent genes upregulated in yeast-like states, and black points represent non-significant changes (Log2FC < 1 and p > 0.05). Y: yeast; PM: primary mycelium; MM: mature mycelium; RT: reverse transition. d Venn diagrams illustrating the classification of specific and shared genes among the three comparisons. e Functional enrichment analysis of KOG categories overrepresented in the transcriptomic data.
Coordinated Expression of Dimorphic Gene Families
Previous work described the existence of a family of ferroxidases in M. lusitanicus comprising two paralogs, each specialized for expression in one of the dimorphic forms18. Specifically, the fet3a paralog is expressed in the yeast form, while the fet3b paralog is expressed in the mycelial form. These prior findings were fully validated by the transcriptomic data generated in the present study (Fig. 2a). These ferroxidases are components of the high-affinity iron uptake system (HAIUS), where they function in conjunction with an iron permease, Frt118. Notably, the frt1 permease genes also constitute a two-member gene family with paralogs, frt1a, and frt1b, that also exhibited differential expression across the dimorphic states (Fig. 2a). These results illustrate the dimorphic adaptation of HAIUS, wherein distinct paralogs from two gene families are utilized depending on the dimorphic state of the fungus. To validate that the transcriptional changes detected by RNA-seq correspond to functional differences between the two growth forms, we generated and analyzed two mutant strains: one with deletions of both the mycelium-specific ferroxidase and permease paralogs (∆fet3b∆frt1b), and another with deletions of the yeast-specific paralogs (∆fet3a∆frt1a) (Supplementary Fig. 1). These genes were selected as markers because their activity is essential for growth and other iron-dependent processes. A mutational analysis revealed that disruption of a specific paralog affected only the growth form in which this paralog is predominantly transcribed, thereby demonstrating functional specialization among paralogs (Fig. 2b).

a Dimorphic expression of paralogs from the two gene families comprising the HAIUS system across three comparative transcriptomic analyses described in Fig. 1. Data are presented as mean values ± SD of the Log2FC from three biological replicates that were sequenced from each condition. Data points are represented as yellow dot plots. b Comparison of mycelial and yeast growth (relative units) between the wild-type strain (WT) and mutant strains in the HAIUS system (∆fet3a∆frt1a: mutant in yeast-expressed paralogs; ∆fet3b∆frt1b: mutant in mycelium-expressed paralogs). Growth values (dry weight for mycelium and yeast count for yeast form) were obtained by seeding an identical inoculum of viable spores for the three strains in iron-free liquid YNB medium and were normalized to the values obtained with the same inoculum in rich YPG medium. Boxplots indicate the median, first, and third quartile. Whiskers are drawn down to the 10th percentile and up to the 90th. Significance was tested using unpaired one-sided t-test (P < 0,05); ****: P < 0,0001; *** P < 0.001; **P < 0.01. c Distribution of expression patterns for distinct gene clusters identified in the M. lusitanicus genome. d Percentage distribution of gene groups identified in the M. lusitanicus genome. DEGs: differentially expressed genes; UG: unique genes not forming gene families; GFID: gene families involved in dimorphism; GFDP: gene families with dimorphic paralogs; M: genes expressed in mycelium; Y: genes expressed in yeast. e Distribution of gene family types based on the number of components. TGF: total gene families. f Distribution of differential gene expression across dimorphism-related gene family types. T: GFDP + GFID. Gene expression values from selected gene families were obtained from the same transcriptomic analysis described in Fig. 1, in which three biological replicates were sequenced from each condition. Boxplots indicate the median, first, and third quartile. Whiskers are drawn down to the 10th percentile and up to the 90th. g KEGG functional enrichment analysis comparing gene family types.
The discovery of dimorphic adaptation in HAIUS prompted us to investigate whether this phenomenon represents an isolated case, or a more widespread genomic mechanism employed by M. lusitanicus to coordinate its complex life cycle. To address this, we performed a genome-wide screen for additional dimorphic gene families. First, we conducted a homology-based clustering analysis to group all M. lusitanicus genes sharing ≥40% sequence identity, identifying a total of 1618 gene families encompassing 4195 genes (Supplementary Data 4). Next, we integrated our transcriptomic data to assign each gene to one of three expression classes: mycelium-specific, yeast-specific, or constitutive. Following the expression profiling, we classified each gene family into one of four categories: (1) families with yeast-specific paralogs, (2) families with mycelium-specific paralogs, (3) families with all paralogs expressed constitutively, and (4) dimorphic families containing at least one mycelium-specific paralog and one yeast-specific paralog (Fig. 2c). This analysis uncovered 1196 gene families (3127 genes) associated with dimorphism, including 490 dimorphic families (1341 genes) exhibiting dual paralog expression (Fig. 2c, e, Supplementary Data 5). These findings demonstrate that up to 11.5% of M. lusitanicus genes are organized into dimorphic families that have evolved to utilize paralogs differentially during the dimorphic transition (Fig. 2d). Dimorphic families constituted 32.3% of all gene families identified. Within dimorphic families, the predominant configuration consisted of two paralogs with dimorphic expression, although there were many other families with more than two paralogs (Fig. 2e). Regarding differential gene expression, genes expressed in mycelium generally exhibited higher log2FC values compared to yeast-expressed genes. This difference was even more pronounced in families where all paralogs displayed the same expression pattern (gene families involved in dimorphism but without dimorphic paralogs) (Fig. 2f). Finally, we examined the distribution of general functional categories (KEGG) among the dimorphic families compared to the remaining families (Fig. 2g). This analysis revealed that dimorphic families are distributed across all functional categories like other gene families, suggesting that the evolution of dimorphic families in dimorphic fungi is not contingent on specific cellular functions but instead reflects a more general adaptation of fungal physiology.
Dimorphism uses clusters in a head-to-head orientation to coordinate gene expression
Analysis of paralog expression within HAIUS gene families, coupled with gene localization data, revealed a distinctive locus arrangement (Fig. 3a). The two ferroxidase paralogs are positioned adjacent to the two permease paralogs, forming two functionally linked gene clusters in a head-to-head (H2H) orientation, sharing the same promoter region. This H2H gene configuration has previously been described in other organisms19. However, the novelty found in M. lusitanicus is notable not only for clustering functionally related genes but also for the dimorphic specialization of each locus. One locus encompasses the permease and ferroxidase paralogs expressed specifically in mycelia, while the other contains the paralogs expressed in yeast (Fig. 3a). These two loci thus exhibit a dual functional association: the clustering of genes encoding proteins functioning in the same pathway, and the co-localization of paralogs expressed in the same dimorphic state. To dissect the regulatory mechanism governing these loci, we generated mutant strains with swapped promoter regions (Fig. 3b, Supplementary Fig. 2). Expression analysis of the ferroxidase and permease paralogs in these mutants confirmed that the yeast- and mycelium-specific paralogs exhibit reciprocal expression patterns upon promoter swapping. This demonstrated that the dimorphic regulation of these genes is controlled by these special H2H promoter regions.

a Structure and expression of the two paralogous loci of the HAIUS system. Pm: mycelium promoter; Py: yeast promoter. b Functional analysis (promoter exchange and RT-qPCR) of the two paralogous loci of the HAIUS system. Y2M (promoter of yeast-to-mycelium), mutant in which both loci have a mycelium promoter; M2Y (promoter of mycelium-to-yeast), mutant in which both loci have a yeast promoter; WT (wild type). Data are presented as mean values ± SD of the Log2FC from three biological samples that were tested from each condition. Data points are represented as grey dot plots c KEGG functional enrichment analysis comparing total H2H structures with dimorphic H2H structures. d Identification of enriched sequence motifs in the promoters of H2H structures.
Having established the unique features of the HAIUS loci, we sought to determine whether this arrangement is specific to this system or, analogous to dimorphic gene families, represents a more generalized mechanism for coordinating gene expression across dimorphic states. To address this, we screened the M. lusitanicus genome for loci meeting the following criteria: two genes on the same DNA region in opposite orientations and separated by a distance similar in size to that found between ferroxidase and permease genes. This screening identified 2092 genes (1046 pairs) in an H2H configuration (Supplementary Data 6). These identified genes were then cross-referenced with our expression data to select H2H loci where both genes exhibit similar expression patterns—either both expressed in yeast, both in mycelia, or neither differentially expressed during the yeast-to-mycelium transition. This secondary filtering yielded 918 genes (459 pairs) expressed under the same condition. Focusing specifically on genes involved in dimorphism (i.e., both expressed in either yeast or mycelia), we identified 712 genes (359 pairs) (Supplementary Data 7). Functional category analysis (KEGG) of this substantial number of dimorphism-associated H2H genes, compared to all H2H genes, revealed a similar distribution, again indicating a homogeneous integration of this regulatory mechanism into general fungal physiology (Fig. 3c). Finally, we analyzed the promoter sequences of the dimorphism-associated H2H gene group, searching for sequence enriched motifs (p-val <0.05) that were differentially represented between yeast and mycelium promoters (Fig. 3d). Promoters from yeast genes were found to contain in a higher frequency two heterogeneous motifs encompassing all four nucleotides. In contrast, promoters from mycelium-specific contain two more repetitive motifs: one composed of T and C nucleotides, and the other consisting exclusively of A and G nucleotides (Fig. 3d).
Structure and Gene Distribution of a Dimorphic Genome
Following transcriptomic and functional analyses, we performed a genome-wide analysis to determine the distribution and density of the various gene groups identified across the M. lusitanicus genome (Supplementary Fig. 3). Exclusively mycelial genes (3922) constituted 33.8% of the total, while exclusively yeast genes (2914) represented 25.1% (Fig. 2d, Supplementary Fig. 3, and Supplementary Data 8 and 9). These two groups comprise both single-copy genes and genes belonging to families where all paralogs are expressed in the same dimorphic state. Genes belonging to dimorphic families comprise a third group, including paralogs expressed in yeast (665 genes, 5.7% of total) and paralogs expressed in mycelia (676 genes, 5.8% of total) (Fig. 2d, Supplementary Fig. 3, and Supplementary Data 5). Collectively, all differentially expressed genes (DEGs) during dimorphism represented 71.4% of the total, compared to 29.6% expressed constitutively. Of the 2092 genes comprising H2H structures, 34% (712) were differentially expressed during dimorphism (Supplementary Fig. 3 and Supplementary Data 6 and 7). The distribution and density of all these gene groups were relatively uniform across M. lusitanicus chromosomes (Supplementary Fig. 3).
Identification of Novel Regulators of Dimorphic Gene Expression
Dimorphic gene families must possess a specific regulatory system that dictates the differential expression of their component paralogs. To identify novel regulators implicated in the control of dimorphic families, we employed a DNA affinity chromatography-pulldown approach, using promoters from these families as bait. Specifically, we selected the two promoters from the paralogous loci of HAIUS (Fig. 3a), one from the yeast-expressed locus and the other from the mycelium-expressed locus, and hybridized them with their corresponding nuclear extracts. Complexes immunoprecipitated by the biotin-labeled promoter probes were subjected to mass spectrometry analyses. This approach identified two candidate regulatory proteins, Mucci3 | 1468915 (an F-box domain-containing protein) and Mucci3 | 1471074 (a kinase domain-containing protein). Structural analysis of these proteins revealed that Mucci3 | 1468915 contains 434 amino acids that fold into two well-defined interacting domains (Fig. 4a). Within the second domain, a small conserved region (amino acids 293-329) exhibits similarity to F-box domains (Fig. 4a). Mucci3 | 1471074 is an 845 amino acid protein with a single central domain corresponding to a conserved kinase-like domain (Fig. 4a).

a Predicted structure of the dfl gene product using AlphaFold3. Inset: protein structure highlighting two identified domains in violet and yellow, with the conserved F-box region in cyan. b Predicted structure of the dkl gene product using AlphaFold3. Inset: protein structure highlighting the kinase domain in red. c Comparison of growth on solid medium between the wild-type strain and two independent dkl mutants (left) and dfl mutants (right). d Wild-type and mutant strains grown on solid aerobic medium (4X magnification), liquid aerobic medium (20X magnification), and liquid anaerobic medium (20X magnification). The white arrow indicates hyphal swellings observed in the dkl mutant. e Yeast production during reverse transition in the wild-type and two independent mutants of the dkl and dfl genes. For yeast production, 5 mg of primary mycelium was grown in 2 ml of YPG during 12 hours. Data are presented as mean values ± SD of five biological replicates from each condition. Significance was tested using unpaired one-sided t-test (P < 0,05); *** p < 0.001; **p < 0.01. f Transcriptomic data showing dysregulation of differential expression in dkl and dfl mutants for four different gene families. Blue bars: genes expressed in mycelium; red bars: genes expressed in yeast. Data are presented as mean values ± SD of the Log2FC from the three biological samples (replicates) that were sequenced from each condition. Data points are represented as grey dot plots. Gene expression data from selected gene families were extracted from the transcriptomic analyses made from three biological replicates sequenced from each strain and condition. g Heatmap representing dysregulation of all differentially expressed genes during reverse transition in the wild-type strain compared to ∆dkl and ∆dfl mutants. Three biological replicates were sequenced from each strain and condition.
To analyze the function of these genes, deletion mutants were generated (Supplementary Fig. 4 and 5). Deletion of each of these genes individually altered the normal progression of dimorphism (Fig. 5). At the macroscopic level, mutants deleted for the Mucci3 | 1468915 gene (designated dimorphism f-box like, dfl) showed reduced radial growth and asexual sporulation, while mutants for Mucci3 | 1471074 (designated dimorphism kinase-like, dkl) exhibited a slight increase in pigmentation (Fig. 4c). At the microscopic level, dfl mutants displayed normal hyphae but a reduction in yeast formation, whereas dkl mutants presented thickened hyphae, nodule formation, and a complete absence of yeast cells (Fig. 4d). Quantitative measurement of yeast production in these mutants revealed that the deletion of dfl resulted in a decrease to less than half the level of yeast production compared to the wild-type strain, while deletion of dkl completely abolished yeast production (Fig. 4e).

a Growth under aerobic and anaerobic conditions, and gene structure of the HAIUS system in two dimorphic Mucorales (M. lusitanicus and M. circinelloides) and two non-dimorphic Mucorales (R. microsporus and R. delemar). Representative images from five independent experiments. b Experimental validation by RT-qPCR of the differential expression in paralogs from permease and ferroxidase families in M. circinelloides. Data are presented as mean values ± SD of the Log2FC from three biological samples that were tested from each condition. Data points are represented as grey dot plots. c Phylogenetic clustering of Ftr1 permease orthologs. Pink: orthologs similar to the yeast permease of M. circinelloides (Y). Blue and green: orthologs similar to the mycelium permease of M. circinelloides (M). Green: Mucorales from the Rhizopodaceae family, which only present orthologue genes to the one expressed in mycelium in M. circinelloides. The question mark denotes a prediction derived from phylogenetic analysis that has not yet undergone experimental validation. d Phylogenetic clustering of Fet3 ferroxidase orthologs. Pink: orthologs similar to the yeast ferroxidase of M. circinelloides (Y). Blue and green: orthologs similar to the mycelium ferroxidase of M. circinelloides (M). Green: Mucorales from the Rhizopodaceae family, which only present orthologue genes to the one expressed in mycelium in M. circinelloides. The question mark denotes a prediction derived from phylogenetic analysis that has not yet undergone experimental validation. e Comparison of orthogroups defined in M. lusitanicus with orthogroups in M. circinelloides (Mc, dimorphic, blue) and R. microsporus and R. delemar (Rm and Rd, non-dimorphic, yellow). Morphological and functional categories: Yeast (L), Mycelium (M), Constitutive (C), and Dimorphic (D, genes with at least one paralog that is differentially expressed). Orthogroups: MG (orthogroups where Mucor lusitanicus has a greater number of components than the compared organism), ME (orthogroups where Mucor lusitanicus has the same number of components as the compared organism), and ML (orthogroups where Mucor lusitanicus has fewer components than the compared organism), NNO (genes that were not assigned to any orthogroup).
Next, we investigated the effect of dfl and dkl mutations on the differential expression of genes involved in dimorphism. We performed a transcriptomic analysis of the two mutant strains and the wild-type strain during the reverse mycelium-to-yeast transition. Specific analysis of the expression of the HAIUS genes, under the control of the promoters used as probes in the pull-down assay, showed strong dysregulation of all four genes in both mutants. The mycelium-specific permease, Frt1b, exhibited a strong reduction in expression in mycelia, while the yeast-specific permease, Frt1a, switched expression to mycelia; these changes were evident in both mutants (Fig. 4f). Similarly, the two ferroxidases, Fet3b and Fet3a, altered their expression in coordination with their corresponding permease within the H2H structure (Fig. 4f). We also analyzed four additional genes from two dimorphic families not related to iron metabolism or H2H structures. These genes belong to a family with two manganese/iron superoxide dismutases (misdb and misda) and another family with two NADH-ubiquinone reductases (nurb and nura). All four genes showed dysregulation in both mutants, which in most cases was a switch in the direction of expression. Only misda showed the same direction of expression in the ∆dkl mutant but with a strong reduction compared to the wild-type (Fig. 4f).
These results indicated a significant role for the dfl and dkl genes in the regulation of dimorphic families. To investigate whether this regulation extends to the set of genes involved in dimorphism, we compared the transcript abudance of the total set of differentially expressed genes in the wild-type strain with the two mutant backgrounds (Fig. 4g). The ∆dfl mutant strain showed that 29.3% of the genes maintained the differential expression observed in the wild type, while 11.7% switched the direction of expression, and 58.9% were no longer differentially expressed (Fig. 4g and Supplementary Fig. 6). The ∆dkl mutant strain showed that only 4.4% of genes were expressed similarly to the wild type, 5.8% switched the direction of expression, and 89.8% were no longer differentially expressed (Fig. 4g and Supplementary Fig. 6). This comparison demonstrated a strong dysregulation of the dimorphism process, coinciding with the loss of dimorphic capacity observed in the mutant phenotypes, indicating an important and general role of the dkl and dfl genes in the regulation of dimorphism in M. lusitanicus.
Conservation of Dimorphic Gene Family Structure and Regulatory Elements in Mucorales
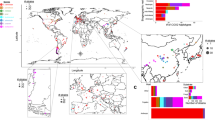
The presence of hundreds of gene families that have coordinately adapted their paralogs for differential function during dimorphism, the use of H2H structures to control the co-expression of different gene pairs, and the identification of new regulatory elements represent novel aspects defining the dimorphic genome of M. lusitanicus. Our next objective was to study the degree of conservation of these novel aspects in other Mucorales. We began by specifically analyzing particular loci across dimorphic and non-dimorphic species. The chosen loci were those involved in HAIUS, as they fulfill the criteria of containing families with paralogs differentiated in yeast and mycelium and arranged in H2H structures. Mucor circinelloides was incorporated into de study as a dimorphic fungus due to its close phylogenetic proximity to M. lusitanicus, while Rhizopus microsporus and Rhizopus delemar were selected as non-dimorphic representatives. The dimorphism of M. circinelloides and the lack thereof in R. microsporus and R. delemar were experimentally validated (Fig. 5a), which also confirmed the curious case of the Rhizopus genus, which not only does not produce yeasts but is also capable of growing as mycelium under anaerobic conditions20. M. circinelloides presents orthologous loci fully equivalent to those identified in M. lusitanicus, showing two H2H loci, each containing a permease and a ferroxidase (Fig. 5a). Experimental validation of gene expression in M. circinelloides showed that one permease and one ferroxidase are expressed in the mycelial form, while their paralogs are expressed in the yeast form, demonstrating the existence of gene families with dimorphic paralogs in M. circinelloides (Fig. 5b). Furthermore, it was confirmed that the permease expressed in mycelium is located in H2H with the mycelial ferroxidase, with a paralogous H2H locus existing for the yeast genes (Fig. 5a, b). However, R. microsporus and R. delemar have lost one ortholog for the permease and another ortholog for the ferroxidase, leaving a single copy of each and losing the H2H structure.
In the subsequent analysis, we searched for new fungi closely related to M. lusitanicus (Mucoraceae and Mycotyphaceae) that fulfilled the synteny observed in M. circinelloides (Ellisomyces anomalus, Thamnidium elegans, Mucor mucedo, Cokeromyces recurvatus, and Mycotypha africana) and others closely related to R. microsporus and R. delemar (Rhizopodaceae) that fulfilled the loss of synteny and families with dimorphic paralogs (Sporondiniella umbellata, Rhizopus stolonifer, and Syzygites megalocarpus) (Supplementary Fig. 7a). Using the amino acid sequences of all the orthologs of the permeases and ferroxidases of M. lusitanicus, a phylogenetic and multiple alignment analysis was performed (Fig. 5c, d). In this analysis, it can be observed that when two paralogs exist, one always groups with the yeast ortholog of M. lusitanicus and M. circinelloides, while the other paralog groups with the mycelial ortholog present in R. microsporus and R. delemar. Fungi that only have one ortholog always group with the ortholog of the non-dimorphic fungi R. microsporus and R. delemar (Fig. 5c, d). This grouping between the yeast and mycelium orthologs was also observed in other gene families that are not involved in iron absorption or do not form H2H structures (Supplementary Fig. 7b and 7c).
Subsequently, we performed a comparative genomic analysis between dimorphic and non-dimorphic species to find evolutionary differences determined by dimorphism (Fig. 5e). The protein sequences of these fungi were collected to perform a comparative analysis based on the generation of orthogroups, which are based on sequence similarity and phylogenetic relationships between the compared organisms. The orthogroups were generated by comparing M. lusitanicus with M. circinelloides to find similar patterns between dimorphic fungi, and by comparing M. lusitanicus with R. microsporus and R. delemar to find differences with non-dimorphic fungi. Once the orthogroups were generated, transcriptomic analyses were used to classify each gene of M. lusitanicus into four morphological and functional categories: Yeast (L), Mycelium (M), Constitutive (C), and Dimorphic (D, genes with at least one paralog that is differentially expressed). For each comparison, three groups of orthogroups were generated: MG (orthogroups where Mucor lusitanicus has a greater number of components than the compared organism), ME (orthogroups where Mucor lusitanicus has the same number of components as the compared organism), and ML (orthogroups where Mucor lusitanicus has fewer components than the compared organism). Additionally, genes that were not assigned to any orthogroup (NNO) were identified. For each orthogroup in the MG, ME, ML, and NNO groups, a count was made of the number of genes classified in the L, M, C, and D categories. Subsequently, the observed proportions of genes in each morphological group were calculated and compared with the expected proportions in the general population of M. lusitanicus genes, analyzing whether the genes of each category were over- or under-represented compared to what was expected in each of the groups. The most interesting results were found in the ML group, where M. lusitanicus has fewer components than the compared organism; dimorphic fungi present a greater proportion of genes associated with the dimorphic group, while in non-dimorphic organisms, a decrease is observed in the number of genes belonging to this group (Fig. 5e). This difference indicates that dimorphic organisms maintain a more robust set of genes specific for the regulation of dimorphism, while non-dimorphic organisms seem to have lost or reduced the presence of these genes. This same analysis was performed with the genomes of the fungi used in the phylogenetic analyses (Fig. 5c, d). As in the previous case, the comparisons were performed in a one-to-one manner, confronting Mucor with another species and defining the groups (MG, ME, ML, and NNO) in the same way as in the comparisons between M. lusitanicus and R. microsporus. Regarding the group ML, the results indicated that T. elegans, C. recurvatus, E. anomalus, and M. africana behaved similarly to the dimorphic fungi M. circinelloides and M. lusitanicus, while R. stolonifer, S. umbellata, and S. megalocarpus behaved like the non-dimorphic fungi R. microsporus and R. delemar Supplementary Fig. 8). Moreover, the gene dfl is conserved in all the fungi that are similar to M. lusitanicus (Mucoraceae and Mycotyphaceae), whereas it is lost in the family of Rhizopodaceae (Supplementary ig. 9). These differences and similarities also correspond to those obtained in the analysis of the iron loci (Fig. 5a, b, Supplementary Fig. 7a and 9) and their phylogenetic conservation (Fig. 5c, d, Supplementary Fig. 9), suggesting their possible use as predictive markers of the dimorphic character in mucoralean fungi.
Comparative Analysis of Dimorphism-Associated Gene Families Across Dimorphic Fungi
To expand our understanding of the conservation of dimorphism-associated genes beyond M. lusitanicus, we analyzed publicly available datasets and performed a comparative genomic analysis across a diverse panel of thermally dimorphic fungi. We first analyzed the transcriptomic dataset of C. neoformans generated by Hu et al.21. Specifically, we evaluated the log2FC values of four gene families used as dimorphism markers in M. lusitanicus: ferroxidases (fet3a, fet3b, fet3c), permeases (ftr1a, ftr1b), dismutases (misda, misdb), and reductases (nura, nurb) (Supplementary Fig. 10a). We also examined the genomic locations of these genes and their potential head-to-head (H2H) configurations (Supplementary Fig. 10b). The results revealed that the dismutase and reductase families of M. lusitanicus are not conserved in C. neoformans, where only a single ortholog of each is present. In contrast, C. neoformans contains expanded ferroxidase and reductase gene families (three and two orthologs, respectively), although only the ferroxidase paralogs showed differential transcription during dimorphism. These results point to both conservation and divergence of dimorphism-associated gene families between M. lusitanicus and C. neoformans.
To place these findings in a broader evolutionary context, we conducted a comprehensive orthogroup analysis across 11 thermally dimorphic fungi: Histoplasma capsulatum, Paracoccidioides brasiliensis, P. lutzii, Sporothrix schenckii, T. marneffei, C. neoformans, C. gattii, Coccidioides posadasii, C. immitis, and Blastomyces dermatitidis, in addition to M. lusitanicus. Orthogroup inference and cross-species comparison revealed extensive variability in gene family conservation, expansion, and loss (Supplementary Fig. 10d). Expansions specific to M. lusitanicus were identified, while several Onygenales species lacked one or more of the studied gene families. In addition to the global overview, we specifically examined the four marker gene families associated with dimorphic development in M. lusitanicus (Supplementary Fig. 10c).
Together, these results highlight both conserved and lineage-specific features of dimorphism-associated gene families across fungal pathogens and emphasize the unique evolutionary trajectory of Mucorales.
Discussion
In this study, we conducted a genomic and transcriptomic analysis of the global physiology of the fungus M. lusitanicus during the dimorphic transition to elucidate the evolutionary strategies underlying this remarkable process. As an initial step, we defined four distinct physiological states of the dimorphic transition for transcriptomic analyses. This classification was crucial, as it provided a comprehensive overview of all genes involved in the process, complementing and expanding upon previous partial studies22.
Our transcriptomic approach revealed that an overwhelming majority of the fungal genes (70%) participate in the dimorphism of Mucorales, a proportion significantly higher than reported in other fungi23,24. These findings indicate that the genomes of dimorphic Mucorales have undergone substantial adaptation to the dimorphic process. This adaptation is also reflected in the pronounced differences between the yeast and mycelial forms, as well as in the unique dimorphic characteristics of these organisms, including the independence of the two morphotypes and the reversible transition between them.
Notably, functional analysis of dimorphic genes unveiled the most striking evolutionary mechanism: the presence of hundreds of gene families with functionalized paralogs exhibiting coordinated expression during dimorphism. The adaptation of paralogs to different conditions (e.g., substrate, cellular or tissue localization, regulatory mechanisms) is a well-documented phenomenon25. In fungi, numerous examples exist of specific gene families whose paralogs have evolved to perform the same function under different conditions26,27,28. However, the observation that a single organism harbors 490 gene families with distinct functions, distributed across fungal physiology, all of which have adapted their paralogs to the same process, dimorphism, represents a novel evolutionary mechanism. This mechanism appears to be designed to manage the dual nature of dimorphism at the genomic level.
A particularly intriguing aspect of this mechanism is the coordinated expression of paralogs within these families. All paralogs adapted to the yeast form are co-expressed under anaerobic conditions, whereas those adapted to the mycelial form are expressed in the presence of oxygen. The biological rationale behind these adaptations becomes evident when considering the substantial environmental differences encountered by the yeast and mycelial morphotypes. Despite their necessity in both morphotypes, dimorphic family paralogs exhibit differential expression, suggesting that their functions cannot be effectively executed unless their protein structures are specifically adapted to the environmental context in which they operate. Our hypothesis posits that the existence of dimorphic families represents an evolutionary solution to the challenge of maintaining essential functions in both yeast and mycelium, a challenge that cannot be resolved by simple constitutive expression.
This hypothesis is exemplified by the HAIUS system, in which a ferroxidase and a permease function together in the cell membrane to facilitate iron uptake29. The yeast-adapted paralogs of these families have evolved to function in an anaerobic liquid environment and within the molecular context of the yeast cell wall, whereas the mycelium-adapted paralogs have specialized for a solid, oxidative environment and a differentially composed cell wall30. Beyond the HAIUS system examined in this study, similar adaptations may exist in other gene families. For example, the cytochrome P450 family includes two paralogs, CYP51 F1 and CYP51 F5, which are conserved in Mucorales species and exhibit structural differences potentially linked to resistance against short-tail azole antifungals31. Our study identified this family as one of the dimorphic families, with CYP51 F1 expressed in mycelium and CYP51 F5 in yeast (Supplementary Data 5). A possible explanation for these findings is that dimorphism led to the emergence of two paralogs with structural differences to adapt ergosterol biosynthesis to the yeast and mycelial membranes, consequently affecting their interactions with antifungal molecules. The fact that dimorphic gene families constitute 11.5% of the total genes and 32.3% of total gene families, along with their homogeneous distribution across cellular functions, underscores the global and genomic nature of this evolutionary process.
In addition to generating dimorphic families, dimorphism in Mucorales has also influenced genome organization, impacting gene regulation and function. This is exemplified by the widespread presence of head-to-head (H2H) gene arrangements associated with dimorphism identified in this study. The H2H configuration, in which two genes are located in opposite DNA strands with divergent transcriptional orientations, often involves a shared promoter region32,33. This bidirectional promoter coordinates the expression of both genes34, thereby enhancing the efficiency of the associated biological process19. Our work on the HAIUS system provides experimental evidence that genes arranged in H2H pairs are functionally related to iron uptake. Furthermore, our results demonstrate not only the coordinated expression of functionally linked gene pairs but also their differential regulation in mycelium and yeast. The dimorphic process has utilized a substantial proportion of H2H structures in the M. lusitanicus genome to enhance gene expression coordination, clustering functionally related genes that require differential expression between morphotypes. The high prevalence of genes in H2H arrangements with differential expression, along with their homogeneous distribution across cellular functions, highlights the adaptive advantage of this genomic organization in dimorphic fungi. Additionally, our results represent a functional analysis of the M. lusitanicus genome, identifying genes grouped into pairs that are potentially involved in the same cellular processes.
A genome predominantly dedicated to dimorphism, the presence of gene families with dimorphic paralogs, and the structural organization of H2H gene pairs linked to dimorphism represent complex adaptations requiring a high degree of coordination in gene expression. These observations suggested the existence of a dedicated regulatory mechanism for this coordination. In this study, we identified two genes, dkl and dfl, whose functions play a pivotal role in coordinating the expression of dimorphism-related genes. The absence of these genes results in a partial or complete block in the transition from mycelium to yeast. The DKL protein contains a central domain with putative kinase activity, suggesting a role in signal transduction35. However, the lack of conservation in the rest of the protein and the absence of known orthologs preclude its classification within any established pathway. Similarly, the DFL protein exhibits two distinct structural domains, with only a small portion of one domain showing similarity to F-box domains, implying potential involvement in protein-protein interactions36. The second domain remains of unknown function. Structural predictions and conservation analyses indicate that neither protein possesses DNA-binding domains, despite their identification through DNA pull-down assays targeting dimorphic promoters. These findings suggest that both proteins may be part of multiprotein complexes that include additional DNA-binding components. The identification of these proteins provides a foundation for further exploration of the pathways in which they participate and the regulatory mechanisms underlying dimorphism in Mucorales.
Dimorphic gene families are conserved in M. circinelloides, a dimorphic species closely related to M. lusitanicus, as are key H2H structures such as those in the HAIUS system. However, both elements are absent in non-dimorphic species. Comparative analyses across additional species lacking expression data indicate that, when a species retains both HAIUS paralogs, one exhibits greater similarity to the yeast ortholog of M. lusitanicus and the other to the mycelial ortholog. In contrast, species retaining only a single gene per HAIUS component consistently show greater similarity to the M. lusitanicus mycelial ortholog. These analyses suggest that the retention of yeast-expressed paralogs and the HAIUS system could serve as molecular markers for dimorphism in other species. Based on these markers, Mucorales families such as Mucoraceae and Mycotyphaceae are predicted to contain a high number of dimorphic species, whereas families such as Rhizopodaceae are expected to predominantly comprise non-dimorphic species. Our study lays the groundwork for future research into how dimorphism has shaped the genomes, lifestyles, and phylogeny of Mucorales fungi.
Our results indicate that most of the dimorphism-associated gene families identified in Mucorales are not conserved in other fungal lineages, suggesting that gene duplication and loss events have played a central role in shaping the diversification of gene families potentially involved in fungal dimorphism. Nevertheless, we identified a reduced core set of orthogroups conserved across multiple groups of dimorphic fungi, including the ferroxidase gene family, for which we confirmed differential expression in C. neoformans. These findings suggest that, while the majority of dimorphism-associated gene families in Mucorales are lineage-specific, other dimorphic fungi may rely on distinct sets of specialized gene families that fulfill functions required for their particular modes of dimorphism. Overall, these global patterns underscore both the conservation of a limited set of core orthogroups and the lineage-specific evolutionary trajectories that have shaped the molecular basis of dimorphism in fungi.
In conclusion, this study demonstrates that dimorphism in M. lusitanicus is driven by a complex and extensive genomic adaptation, including paralog diversification, coordinated gene expression through H2H arrangements, and a dedicated regulatory network. These findings illuminate the evolutionary strategies underlying dimorphism in Mucorales, providing a foundation for future studies on its impact on fungal physiology, pathogenicity, and phylogeny. Given the direct involvement of dimorphism in pathogenesis, alongside its regulatory influence on key virulence-associated processes such as iron uptake and antifungal resistance, the insights gained from this research also provide a critical foundation for understanding virulence in Mucorales.
Methods
Strains, growth, and transformation conditions
The fungal strains utilized in this study were Mucor lusitanicus CBS277.49, Mucor circinelloides 1006PhL1, Rhizopus microsporus ATCC11559, and Rhizopus delemar 99-880Broad. M. lusitanicus CBS277.49 and its leucine auxotrophic derivative, R7B, served as wild-type controls, according to their respective auxotrophic requirements. The uracil and leucine auxotrophic strain MU402, derived from R7B, was employed for gene deletion experiments37. Standard growth medium was supplemented with uridine (200 mg/L) or 1,10-phenanthroline (50 μM) (Sigma-Aldrich) when required18. Transformants of the MU402 strain, selected using the pyrG marker, were cultivated on Minimal Media with Casamino acids (MMC)37. For iron-limited experiments utilizing the HAUIS system, yeast nitrogen base (YNB) medium was prepared using Milli-Q water, and all cultures were maintained in plastic vessels to minimize iron contamination. Anaerobic conditions were achieved by culturing strains in a deoxygenated medium within an anaerobic jar, utilizing Anaerobe Container System Sachets (Becton Dickinson Ireland). All fungal strains were incubated at 26 °C under illuminated conditions unless otherwise specified. Protoplast transformation of MU402 was performed via electroporation. Briefly, germinated spores were digested with15 μg/ml chitosanase RD (US Biologicals, MA) and then electroporated with electrical pulses generated by a Bio-Rad Gene Pulser Xcell using an exponential decay pulse38,39. Escherichia coli DH5α (Thermo Fisher Scientific) was used for all cloning procedures.
Mutant strain generation and validation
Gene deletions and promoter exchanges were generated through double cross-over homologous recombination, employing DNA cassettes synthesized via overlapping PCR. These cassettes were designed to incorporate the pyrG gene, serving as a selectable marker, flanked by 1 kb upstream and downstream DNA sequences homologous to the target gene, facilitating precise deletion through homologous recombination. For protomer exchange, we generated a cassette containing the ferroxidase-permease gene pair with the opposite promoter: the yeast promoter for fet3b and ftr1b (strain M2Y) and the mycelium promoter for fet3a and ftr1a (strain Y2M). This cassette also included the pyrG selectable marker downstream of the ftr1 locus. To ensure full cassette integration, CRISPR-Cas9 technology was used following the protocol published by Lax et al.40,41. A guide RNA was designed to facilitate cassette insertion (gRNAY: GATCAAGAAAAAATGAATAA for Y2M construction, and gRNAM: TGGGAGACATTTAGTAACCA for M2Y). Briefly, ribonucleoprotein complexes were assembled in vitro according to the published protocol and the manufacturer’s instructions (IDT Technologies). Transformation was performed by electroporation of the DNA cassette along with the CRISPR-Cas9 ribonucleoprotein complex Gene deletions were validated by PCR using a primer complementary to the marker gene and a second primer positioned upstream of the recombination region. Homo- or heterokaryotic status was confirmed by PCR using primers flanking the deletion site, enabling amplification of both wild-type and mutant alleles if present. All primers employed for construct amplification and PCR validation are detailed in Supplementary Table 1. All PCR amplifications were performed using Herculase II Fusion DNA polymerase (Agilent), with annealing temperatures optimized for each primer pair according to the manufacturer’s recommendations.
qRT-PCRs
Quantitative real-time PCR (qRT-PCR) was performed to analyze the expression of fet3a, fet3b, ftr1a, and ftr1b genes in M. lusitanicus and M. circinelloides, utilizing gene-specific primers detailed in Supplementary Table 1. Total RNA was extracted using the NYZ total RNA kit (NYZtech) and subjected to on-column DNase I treatment (Sigma). Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using the iScriptTM cDNA synthesis kit (Bio-Rad). cDNA amplification was conducted in triplicate using a reaction mixture containing 2x Power SYBR® Green Master Mix (Applied Biosystems, Waltham, MA, USA), 150 nM of gene-specific primers, and 100 ng of cDNA. Real-time PCR was performed using the QuantStudioTM 1 real-time PCR system (Applied Biosystems) according to the instrument’s established experimental template. Briefly, 10 μL reactions were denatured at 95 °C for 2 minutes and amplified for 40 cycles (95 °C for 15 seconds for denaturation and 60 °C for 1 minute for hybridization, elongation, and fluorescence detection). Melting curve analysis and non-template controls were included to identify non-specific amplification products. Relative gene expression was normalized to the amplification levels of the constitutively expressed tfc1 gene, primers for which are listed in Supplementary Table 1. Given that the amplification efficiencies of the target genes and the tfc1 endogenous control were approximately equivalent, relative gene expression was calculated using the delta-delta CT (ΔΔCT) method.
RNA extraction and RNA-seq analysis
Total RNA was extracted using the NYZ total RNA kit (NYZtech) and subjected to on-column DNase I treatment (Sigma). Three biological replicates were analyzed for each expression experiment. Yeast samples (Y) were obtained from liquid cultures grown in YPG39 medium within 50 ml conical tubes. Each culture was initiated with an inoculum of 1*10^6 spores/ml and incubated under anaerobic conditions for 24 hours. Primary mycelium samples (PM) were harvested from yeast cultures that were transferred to aerated flasks and incubated at 250 rpm for 2 hours. Mature mycelium (MM) samples were obtained from solid YPG medium grown for 24 hours. Reversion transition samples (RT) were generated by transferring primary mycelial cultures to anaerobic conditions and incubating them for 2 hours in MMC medium. RNA sequencing and library preparation were performed by Novogene (Cambridge, UK) using the Illumina platform. The quality of raw mRNA reads was assessed using FASTQC v0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, accessed on 17 September 2024), both before and after the removal of the adapter and contaminant sequences with Trimmomatic v0.3942. Cleaned mRNA reads were then mapped to the M. circinelloides f. lusitanicus v3.0 genome using star v2.7.11a43. To quantify the reads mapped to Mucor genes, the FeatureCounts v2.0.1 tool was utilized44. The resulting count table was then used as input for differential expression (DE) analysis, which was performed with DESeq2 v2.11.40.845. Loci were considered differentially expressed if they had a false discovery rate (FDR)-adjusted p-value ≤ 0.05 and a log2 fold change (log2FC) ≥ |1.0 | . Whenever the general term “gene expression” is used in the manuscript, we refer to the transcript abundance identified by the RNAseq experiments.
Functional enrichment analysis
Gene Ontology (GO) enrichment analysis was performed using the Blast2GO tool46 to identify overrepresented biological processes, molecular functions, and cellular components among the differentially expressed genes (DEGs). GO term enrichment was evaluated using a false discovery rate (FDR)-adjusted p-value threshold of ≤ 0.05 to ensure statistical significance. In addition to the overall enrichment analysis of DEGs, a separate enrichment analysis was conducted for the set of yeast-specific genes (overexpressed genes) and another for mycelium-specific genes (downregulated genes). In the KEGG enrichment analysis, genes assigned to clusters with expression data derived from transcriptomic analysis and identified as differentially expressed were functionally analyzed using KEGG annotation data to determine their associated biological pathways. Similarly, genes within dimorphic clusters, H2H genes, and the dimorphic H2H genes were analyzed using the same approach.
Gene family identification in M. lusitanicus
To identify gene families in M. lusitanicus, protein sequences of the fungus were analyzed using CD-HIT v4.8.1 (Cluster Database at High Identity with Tolerance), a tool designed to reduce sequence redundancy and perform clustering based on sequence identity47. Protein sequences were retrieved from the Joint Genome Institute (JGI) database48 and filtered to ensure clear annotation and the absence of invalid characters in the FASTA headers, facilitating downstream analyses. CD-HIT was executed with a sequence identity threshold (c) of 0.4, allowing sequences with at least 40% identity to be clustered together. The word size parameter (n) was set to 2, determining the length of the k-mer used for sequence comparison, while the global clustering mode parameter (g) was set to 1, enabling a more flexible sequence alignment. All other parameters were kept at their default values. As a result, two output files were generated: a.clstr file, containing information about the formed clusters, including the sequence identifiers assigned to each cluster, and a representative sequence file, which includes only the centroid sequences of each cluster. The cluster file, along with the associated protein identifiers, was used for further analyses.
Classification and expression-based organization of gene clusters
Once the clusters and the gene identifiers composing them were obtained, a preprocessing step was performed. This step included the integration of gene expression data for each cluster, derived from the transcriptomic analysis. Additionally, data on the number of proteins present in each cluster were incorporated. Following data compilation, a series of filtering steps were applied. First, clusters containing at least two genes were selected. Next, only differentially expressed genes from the transcriptomic dataset were retained. A second filtering step was then performed to identify clusters containing at least one gene expressed in the yeast phase and at least one in the mycelial phase. These were distinguished from clusters composed exclusively of yeast-specific genes and those with genes overexpressed in the mycelial phase. The former were classified as dimorphic clusters. Clusters composed of non-differentially expressed genes were classified as constitutive, while those consisting exclusively of yeast-specific or mycelium-specific genes were categorized as yeast clusters and mycelial clusters, respectively.
H2H identification and characterization
A custom script was developed to identify genes arranged in a head-to-head (H2H) configuration. The script utilizes a BED file, generated by processing the GFF3 file containing genomic annotations. This BED file provides information on the genomic locations and strand orientations (forward or reverse) of the genes. To identify H2H genes in the M. lusitanicus genome, the script applies a 500 bp threshold, based on the observation that co-regulated genes in M. lusitanicus are typically found within this distance. Additionally, for genes to be considered H2H, they must be located on opposite strands of DNA. For the identification of H2H gene pairs associated with dimorphism, an additional criterion is applied: the genes must be expressed in the same morphological state (either yeast or mycelium). To accomplish this, the analysis incorporates gene expression data, which is combined with the BED file to identify the relevant dimorphic H2H genes. To perform co-expression analyses, BAM (Binary Alignment Map) files—resulting from the alignment of RNA-seq reads to the reference genome—were used to normalize coverage to bins per million reads (BPM) in 50 bp bins using the bamCoverage function from deepTools v3.2.149 The resulting bigWig files were visualized using the pyGenomeTracks module of deepTools, incorporating the annotations of H2H genes.
Analysis of the promoters of H2H genes
BED files were generated for the intergenic regions of yeast and mycelium H2H genes. For this, the GFF3 file and the corresponding H2H gene files for each morphology were used to obtain BED files for the bidirectional promoters. Subsequently, using Bedtools (v2.31.0) getfasta50, DNA sequences from these regions were extracted from the genome of this organism obtained from the JGI. To identify common motifs in the intergenic regions of yeast and mycelium H2H genes, the sequences obtained in the previous step were analyzed. As a negative control and to rule out the presence of random motifs, sequences from yeast regions were used in the analysis of mycelial bidirectional promoters and vice versa. These sequences serve to compare the specificity of the motifs found in the H2H sequences. The MEME v5.5.8 (Multiple EM for Motif Elicitation) tool in discriminative mode was used to identify these motifs in the regulatory sequences51.
Gene density distribution
To determine the distribution of genes involved in dimorphism across the M. lusitanicus genome, scaffold sizes were extracted from the reference genome to ensure that the analysis was consistent with the genomic structure. Fixed 50 Kb windows were created across the genome, and these windows were aligned with gene annotation data to ensure compatibility for subsequent analyses. Gene density was calculated by mapping gene lengths onto each 50 Kb window, generating a BEDGraph file that visualized gene density across the genome at 50 Kb resolution. This data was then visualized using Circos v0.69.852, a software package that facilitates the creation of high-quality, circular visualizations, ideal for exploring relationships between genomic positions. This procedure was applied to the entire M. lusitanicus genome, as well as specifically to yeast genes, mycelial genes, genes belonging to dimorphic families, total H2H genes, and H2H genes involved in dimorphism.
DNA pull-downs
Nuclear extracts were prepared from Mucor lusitanicus primary mycelium (PM) grown under aerobic conditions and reverse transition (RT) cultures grown under anaerobic conditions53. Briefly, fungal tissues were homogenized in STMDPS buffer (0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 50 mM Tris-HCl, pH 7.4, 1 mM DTT). The homogenate was filtered through a 75-μm mesh, and nuclei were pelleted by centrifugation at 400 g for 10 min. Nuclear pellets were lysed in nuclear lysis buffer (420 mM NaCl, 20 mM HEPES, pH 7.9, 20% v/v glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.1% NP-40, and complete protease inhibitor cocktail without EDTA (Roche)) and clarified by centrifugation at 12,000 g for 10 min. For each DNA pulldown assay, 10 μg of biotin-labeled double-stranded DNA (dsDNA) probes (Supplementary Table 1) were immobilized on 50 μl of streptavidin-conjugated magnetic beads (GE Healthcare) by incubation for 2 h at room temperature in DNA binding buffer (10 mM sodium phosphate, pH 7.0, 140 mM NaCl, 0.05% NP-40). The immobilized probes were then incubated with 500 μg of nuclear extract in a total volume of 700 μl of protein binding buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM DTT, 0.25% NP-40, and complete protease inhibitor cocktail) for 3 h. Following four washes with protein binding buffer, bound proteins were eluted, digested with trypsin (Promega), and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The resulting MS/MS data were searched against the UniProt Mucor lusitanicus proteome database.
Protein conservation and structural predictions
Proteomes of representative species were retrieved from the Joint Genome Institute (JGI) Mycocosm genome portal48. Sequences of M. lusitanicus Dkl and Dfl proteins were queried against the selected proteomes using iterative HMMER jackhmmer searches (E-value \(\le\) 1×10-3) (v3.3.2) (http://hmmer.org). A reciprocal BLASTp search (v2.10.1)54 was conducted, and sequences that failed to produce a hit were discarded. An additional search using Pfam-A database55 using HMMER hmmscan (v3.3.2) (http://hmmer.org) served to remove hits that lacked the f-box domain (Dfl orthologs), and kinase domain (Dkl orthologs). A final list and a matrix including information about the presence or absence of the putative orthologs were generated (Supplementary Fig. 8).
Structures of Dfl and Dkl were predicted using AlphaFold3 by submitting protein sequences to Alphafold3 server56. Structural models were visualized with ChimeraX (v1.8)57 and f-box and kinase domains were colored for visualization.
Comparative transcriptomic analysis
To evaluate the effect of gene deletions in the dfl and dkl genes on the expression of dimorphic genes, a comparative transcriptomic analysis was performed. Differentially expressed genes during the reverse transition (mycelium to yeast) at 2 hours were used as a reference for wild-type (WT) expression, comprising yeast-specific genes (upregulated) and mycelium-specific genes (downregulated). Changes in the expression of these yeast- and mycelium-specific genes were then analyzed in the transcriptomes of the two mutants. To quantitatively assess the expression changes, the percentage of genes that retained their original expression pattern (upregulated or downregulated), as well as those exhibiting opposite expression patterns, was calculated by comparing the transcriptomic data of the WT with those of each mutant.
Comparative genomic analysis between dimorphic and non-dimorphic species of Mucorales
Sequence comparison and phylogenetic analyses of the gene families of the HAIUS system were made with the online version of MAFFT v7 (https://mafft.cbrc.jp/alignment/server/index.html, accessed on 9 January 2025). Protein sequences from M. lusitanicus and other dimorphic and non-dimorphic organisms were retrieved from the JGI database and formatted into FASTA files. Genomic comparative analyses were performed using OrthoFinder58 with default parameters, conducting pairwise comparisons between M. lusitanicus and each selected organism to generate orthogroups based on sequence similarity and phylogenetic relationships. The orthogroups were classified into three categories: MG (where M. lusitanicus had more genes than the compared organism), ME (same number of genes), and ML (fewer genes). Genes from M. lusitanicus were assigned to these groups and further classified into four morphological and functional categories: Yeast (L), Mycelium (M), Constitutive (C), and Dimorphic (D), based on transcriptomic data. The number of genes in each category within the MG, ME, and ML groups was counted, and the observed proportions were compared to the expected proportions in the total M. lusitanicus genome. To normalize these comparisons, the base-2 logarithm of the ratio between observed and expected proportions was calculated, indicating whether genes were overrepresented or underrepresented in each group. Additionally, genes not assigned to any orthogroup were identified, classified into the same morphological categories, and analyzed using the same approach to compare their distribution with genes assigned to orthogroups.
Comparative Analysis of Dimorphism-Related Orthogroups Across Dimorphic Fungi
Proteomes of eleven dimorphic fungi, including major human pathogens (H. capsulatum, P. brasiliensis, P. lutzii, S. schenckii, T. marneffei, C. neoformans, C. gattii, C. posadasii, C. immitis, B. dermatitidis, and M. lusitanicus), were retrieved from the Joint Genome Institute (JGI)48. Orthogroups were inferred with OrthoFinder (2.5.5)59 using complete predicted proteomes as input. All-versus-all similarity searches and clustering were applied to define orthogroups, which were then used to assess conservation, expansion, and contraction of gene families across species.
Dimorphism-associated gene families characterized in M. lusitanicus were mapped to their corresponding orthogroups. Orthogroup composition was compared across species to identify conserved families, expansions, and losses. These data were visualized in a heatmap showing copy number variation. Four families previously linked to dimorphism in M. lusitanicus were analyzed in detail: fet (OG0000056), ftr (OG0005754), nur (OG0011887), and misd (OG0001018). Orthogroup assignments were used to assess presence/absence and copy number variation across fungi. A heatmap was generated to illustrate these patterns, and supplementary tables provide gene identifiers, orthogroup assignments, and pairwise comparisons with M. lusitanicus.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The complete raw data that support the findings of this study have been submitted to the Gene Expression Omnibus (GEO) under the accession number GSE293402. Source data are provided with this paper.
Code availability
The complete data analysis pipeline, including all scripts, datasets, annotations, software versions, and auxiliary files required to reproduce the results of this study, is available on GitHub: (https://doi.org/10.5281/zenodo.17662492). Any future updates to the code and supplementary materials will also be provided there.
References
Slavenko, A. et al. Evolution of sexual size dimorphism in tetrapods is driven by varying patterns of sex-specific selection on size. Nat. Ecol. Evolution 2024, 1–10 (2024).
Gilbert, S. F. Metamorphosis: The Hormonal Reactivation of Development (Sinauer Associates, 2000).
Orlowski, M. Mucor dimorphism. Microbiol Rev. 55, 234–258 (1991).
Gauthier, G. M. Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog. 11, e1004608 (2015).
Van Dyke, M. C. C., Teixeira, M. M. & Barker, B. M. Fantastic yeasts and where to find them: the hidden diversity of dimorphic fungal pathogens. Curr. Opin. Microbiol 52, 55–63 (2019).
Wang, F., Han, R. H. & Chen, S. An Overlooked and Underrated Endemic Mycosis—Talaromycosis and the Pathogenic Fungus Talaromyces marneffei. Clin. Microbiol. Rev. 36, e0005122 (2023).
Lee, S. C. & Heitman, J. Sex in the Mucoralean Fungi. Mycoses 57, 18 (2014).
Cornely, O. A. et al. Global guideline for the diagnosis and management of mucormycosis: an initiative of the European Confederation of Medical Mycology in cooperation with the Mycoses Study Group Education and Research Consortium. Lancet Infect. Dis. 19, e405–e421 (2019).
Lee, S. C., Li, A., Calo, S. & Heitman, J. Calcineurin plays key roles in the dimorphic transition and virulence of the human pathogenic zygomycete Mucor circinelloides. PLoS Pathog. 9, e1003625 (2013).
Valle-Maldonado, M. I. et al. The heterotrimeric G-protein beta subunit Gpb1 controls hyphal growth under low oxygen conditions through the protein kinase A pathway and is essential for virulence in the fungus Mucor circinelloides. Cell Microbiol. 22, e13236 (2020).
Boyce, K. J. & Andrianopoulos, A. Fungal dimorphism: The switch from hyphae to yeast is a specialized morphogenetic adaptation allowing colonization of a host. FEMS Microbiol. Rev. 39, 797–811 (2015).
Lax, C. et al. Genes, pathways, and mechanisms involved in the virulence of mucorales. Genes 11, 317 (2020).
Patiño-Medina, J. A. et al. Control of morphology and virulence by ADP-ribosylation factors (Arf) in Mucor circinelloides. Curr. Genet 64, 853–869 (2018).
Vellanki, S. et al. A novel resistance pathway for calcineurin inhibitors in the human-pathogenic mucorales mucor circinelloides. mBio 11, e02949-19 (2020).
Moriwaki-Takano, M., Iwakura, R. & Hoshino, K. Dimorphic Mechanism on cAMP Mediated Signal Pathway in Mucor circinelloides. Appl Biochem Biotechnol. 193, 1252–1265 (2021).
Calo, S. et al. Antifungal drug resistance evoked via RNAi-dependent epimutations. Nature 513, 555–558 (2014).
Ellegren, H. Sex-chromosome evolution: recent progress and the influence of male and female heterogamety. Nat. Rev. Genet. 2011 12, 157–166 (2011).
Navarro-Mendoza, M. I. et al. Components of a new gene family of ferroxidases involved in virulence are functionally specialized in fungal dimorphism. Sci. Rep. 8, 7660 (2018).
Chang, D. T. H., Wu, C. Y. & Fan, C. Y. A study on promoter characteristics of head-to-head genes in Saccharomyces cerevisiae. BMC Genomics 13, S11 (2012).
Tang, X. et al. Application and analysis of rhizopus oryzae mycelia extending characteristic in solid-state fermentation for producing glucoamylase. J. Microbiol Biotechnol. 28, 1865–1875 (2018).
Hu, P. et al. A unique cell wall synthetic response evoked by glucosamine determines pathogenicity-associated fungal cellular differentiation. PLoS Genet 17, e1009817 (2021).
Homa, M. et al. Differential Gene Expression of Mucor lusitanicus under Aerobic and Anaerobic Conditions. J Fungi (Basel) 8, 404 (2022).
Nigg, M., Laroche, J., Landry, C. R. & Bernier, L. RNAseq Analysis Highlights Specific Transcriptome Signatures of Yeast and Mycelial Growth Phases in the Dutch Elm Disease Fungus Ophiostoma novo-ulmi. G3 (Bethesda) 5, 2487–2495 (2015).
Du, M. et al. Unraveling the dynamic transcriptomic changes during the dimorphic transition of Talaromyces marneffei through time-course analysis. Front Microbiol 15, 1369349 (2024).
Hardison, R. C. Evolution of Hemoglobin and Its Genes. Cold Spring Harb. Perspect. Med 2, a011627 (2012).
Heineike, B. M. & El-Samad, H. Paralogs in the PKA Regulon Traveled Different Evolutionary Routes to Divergent Expression in Budding Yeast. Front. Fungal Biol. 2, 642336 (2021).
Ballou, E. R., Nichols, C. B., Miglia, K. J., Kozubowski, L. & Alspaugh, J. A. Two CDC42 paralogs modulate C. neoformans thermotolerance and morphogenesis under host physiological conditions. Mol. Microbiol 75, 763 (2009).
Smoak, R. A., Snyder, L. F., Fassler, J. S. & He, B. Z. Parallel expansion and divergence of an adhesin family in pathogenic yeasts. Genetics 223, iyad024 (2023).
Philpott, C. C. Iron uptake in fungi: A system for every source. Biochimica et. Biophysica Acta (BBA) - Mol. Cell Res. 1763, 636–645 (2006).
Bartnicki-Garcia, S. & Lippman, E. Fungal morphogenesis: cell wall construction in Mucor rouxii. Science 165, 302–304 (1969).
Caramalho, R. et al. Intrinsic short-tailed azole resistance in mucormycetes is due to an evolutionary conserved aminoacid substitution of the lanosterol 14α-demethylase. Sci. Rep. 7, 15898 (2017).
Rennie, S. et al. Transcription start site analysis reveals widespread divergent transcription in D. melanogaster and core promoter-encoded enhancer activities. Nucleic Acids Res 46, 5455–5469 (2018).
Chen, Y., Li, H., Li, Y. Y. & Li, Y. Pan-Cancer Analysis of Head-to-Head Gene Pairs in Terms of Transcriptional Activity, Co-expression and Regulation. Front Genet 11, https://doi.org/10.3389/fgene.2020.560997 (2021).
Yang, W. et al. Promoter-sharing by different genes in human genome--CPNE1 and RBM12 gene pair as an example. BMC Genomics 9, 456 (2008).
Cornell, T. T. & Shanley, T. P. Signal transduction overview. Crit Care Med 33, S410–S413 (2005).
Skowyra, D., Craig, K. L., Tyers, M., Elledge, S. J. & Harper, J. W. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91, 209–219 (1997).
Nicolás, F. E. et al. Mutants defective in a Mucor circinelloides dicer-like gene are not compromised in siRNA silencing but display developmental defects. Fungal Genet Biol. 44, 504–516 (2007).
Gutiérrez, A., López-García, S. & Garre, V. High reliability transformation of the basal fungus Mucor circinelloides by electroporation. J. Microbiol Methods 84, 442–446 (2011).
Nicolás, F. E. et al. Molecular tools for carotenogenesis analysis in the mucoral Mucor circinelloides. Methods Mol. Biol. 1852, 221–237 (2018). Humana Press Inc.
Carlos Lax, María Isabel Navarro-Mendoza, Carlos Pérez-Arques, Eusebio Navarro, Francisco Esteban Nicolás, V. G. Transformation and CRISPR-Cas9 – mediated homologous recombination in the fungus Rhizopus microsporus. STAR Protoc. https://doi.org/10.1016/j.xpro.2022.101237 (2022).
Lax, C. et al. Stable and reproducible homologous recombination enables CRISPR-based engineering in the fungus Rhizopus microsporus. Cell Rep. Methods 1, 100124 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Grigoriev, I. V. et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699-704 (2014).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic Acids Res 43, W39–W49 (2015).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645 (2009).
He, S. et al. 6mA-DNA-binding factor Jumu controls maternal-to-zygotic transition upstream of Zelda. Nat. Commun. 10, 1–14 (2019).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinforma. 10, 1–9 (2009).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–D419 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 1–14 (2015).
Acknowledgements
This research was funded the MCIN/AEI/ 10.13039/501100011033 by “ERDF A way of making Europe,” by the “European Union” (grant PID2021-124674NB-I00 to F.E.N. and V.G.), Fundación Séneca-Agencia de Ciencia y Tecnología de la Región de Murcia (20897/PI/18 and 21969/PI/22 to V.G.). We would like to thank Joseph Heitman (Duke University School of Medicine) for his critical review of the original draft.
Author information
Authors and Affiliations
Contributions
G.T. and M.I.N.M. contributed equally to this work. G.T. conducted most of the bioinformatic analyses and reviewed the manuscript draft. M.I.N.M. generated all the HAIUS mutants, contributed to the discovery of the dimorphic gene families, and reviewed the manuscript draft. C.L. conducted bioinformatic analyses and reviewed the manuscript draft. C.P.A. helped with the transcriptomic analyses and reviewed the manuscript draft. G.N.D.S. characterized the phenotype and transcriptome of dkl and dfl mutants. E.N. managed the project and provided materials. V.G. analyzed the results and designed, supervised, and coordinated the project. F.E.N. participated in the generation of mutants, phenotype analyses, bionformatic analyses, wrote the original draft, analyzed the results and designed, supervised, and coordinated the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sinem Beyhan and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tahiri, G., Navarro-Mendoza, M.I., Lax, C. et al. Coordinated gene family evolution shapes the genome of dimorphic Mucorales. Nat Commun 17, 2148 (2026). https://doi.org/10.1038/s41467-026-68866-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68866-7
