
Abstract
Obesity impairs metabolic flexibility—the capacity to adapt to fluctuating energy demands. Emerging evidence suggests that dietary interventions, particularly time-restricted feeding (TRF), may help restore this flexibility. In this study, we demonstrate that feeding upregulates PRMT3 and asymmetric dimethylarginine (ADMA)-containing proteins via insulin–pAKT signaling, while fasting reduces their expression. Pharmacological inhibition of PRMT3 attenuates diet-induced obesity (DIO) and enhances adipocyte glycolysis in male mice. Mechanistically, PRMT3 drives the expression of citrate transporter SLC25A1 during feeding through direct arginine methylation. A 16:8 TRF regimen normalizes PRMT3 and ADMA levels while suppressing SLC25A1 expression. Notably, PRMT3 inhibition recapitulates the metabolic benefits of 16:8 TRF and improves metabolic flexibility. Furthermore, adipocyte-specific deletion of Slc25a1 in male mice protects against DIO and enhances insulin sensitivity. Collectively, these findings identify PRMT3-mediated arginine methylation in vWAT as a nutrient-responsive regulatory axis that impairs metabolic flexibility in obesity, which is a potential therapeutic target.
Similar content being viewed by others

Introduction
Recent research has highlighted the health benefits of dietary restriction, including fasting, calorie and nutrient restrictions1,2. These practices induce adaptive metabolic responses across various organs, improving weight management, promoting ageing delay, and contributing to health optimization3. Among these metabolic benefits, dietary restrictions have shown promising effects in restoring fuel switching, known as metabolic flexibility, which is impaired by overnutrition4. Although dietary-restricted rodent studies have consistently demonstrated positive outcomes5,6,7, human trials have shown mixed results, and challenges in compliance further complicate the reliability of these studies8,9,10,11. This controversy has sparked interest in investigating the molecular mechanisms behind the metabolic benefits of dietary restriction. Researchers are now seeking to develop methods that can replicate these beneficial effects, specifically to aid in combating obesity and associated cardiometabolic complications. Several key signaling pathways and molecules—such as insulin-AKT, FOXO, mammalian target of rapamycin (mTOR), nicotinamide adenine dinucleotide (NAD + ), AMP-activated protein kinase (AMPK), fibroblast growth factor 21 (FGF21), and circadian rhythms—have been identified as relevant in mediating the anti-obesity effects of dietary restriction7,12. Reducing the intake of the essential amino acid methionine has been shown to be particularly effective in preventing weight gain and enhancing metabolism in both rodent and human studies13,14,15,16. Furthermore, hepatic levels of S-adenosylmethionine (SAMe), a metabolite of methionine, positively correlate with body mass index (BMI) and contribute to fasting-induced metabolic adaptations17. Moreover, inhibition of methionine adenosyl transferase (Mat1a) in the liver also demonstrates potential in mitigating diet-induced obesity and metabolic syndrome18. However, it remains unclear how downstream SAMe-mediated substrate methylation might influence adiposity and fasting responses, particularly in adipose tissue.
Results
Diurnal Expression Patterns of Arginine Methylation in eWAT
Protein arginine methylation—a post-translational modification catalyzed by protein arginine methyltransferases (PRMTs)—utilizes S-adenosylmethionine (SAMe), derived from methionine, as a methyl donor19. To explore the relationship between arginine methylation and nutritional state, we assessed total arginine methylation levels across metabolically active tissues, including liver, brown adipose tissue (BAT), and epididymal white adipose tissue (eWAT), by collecting samples at 4 h intervals over a 24 h period.
We found that proteins bearing asymmetric dimethylarginine (ADMA)—but not mono-methylarginine (MMA) and symmetric dimethylarginine (SDMA)—displayed distinct diurnal expression patterns in eWAT. Specifically, MMA and ADMA levels decreased during the daytime fasting period and increased during the nocturnal feeding period, paralleling the natural feeding behavior of mice (Fig. 1a and Supplementary Fig. S1b, S1a). In contrast, no significant diurnal changes in MMA, ADMA, or SDMA levels were observed in the liver or BAT (Supplementary Fig. S1c, d).

a Western blot and quantification of proteins carrying different types methylarginine (MMA, ADMA, SDMA) in eWAT of 12 weeks male B6N mice sacrificed at indicated time points in a 12-hour light/dark cycle (n = 4). b Western blot analysis and quantification of proteins carrying MMA and ADMA from eWAT of 12 weeks male mice at ad libitum, fasted for 36 h, or fasted for 36 h and then refed a standard chow diet for 3–6 h (n = 5, 6 and 6). AL, ad libitum; F-36, fasted 36 h. c, d Concentrations of SAMe in serum (c) and eWAT (d) at ad libitum (AL), fasted for 36 h (F-36), or fasted for 36 h and then refed a standard chow diet for 3–6 h (Refed). (n = 6 for serum and 9, 10 and 10 for eWAT). e Relative levels of proteins carrying MMA and ADMA in eWAT from 20-week-old male mice fed with 12 weeks of regular diet (ND) or a high-fat diet (HFD) (n = 4). f Associations of relative levels of MMA-carrying proteins from human visceral fat with indicated body mass index (BMI). g Quantification of relative levels of MMA-carrying proteins in human visceral fat from individuals with BMI < 25 (n = 4) and BMI > 25 (n = 8). h Western blot analysis of ADMA-carrying proteins from human visceral fat with indicated BMI. i Associations of relative levels ADMA-carrying proteins from human visceral fat with indicated BMI as quantified from (h). j Quantification of relative levels ADMA-carrying proteins in human visceral fat from individuals with BMI < 25 (n = 4) and BMI > 25 (n = 8). k Associations of ADMA levels from human serum samples with indicated BMI. Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
To further characterize this regulation, we subjected mice to defined fasting and refeeding cycles. MMA and ADMA levels in eWAT declined during fasting and rapidly rebounded within 3–6 h of refeeding (Fig. 1b and Supplementary Fig. S1e). Notably, serum SAMe concentrations—though not tissue levels in eWAT—also dropped during fasting and recovered upon refeeding (Fig. 1c, d).
Next, we assessed the impact of diet-induced obesity (DIO) on arginine methylation. After 12 weeks of high-fat diet (HFD), eWAT from obese mice showed marked increases in MMA- and ADMA-modified proteins (Fig. 1e), and the normal circadian oscillation of ADMA-containing proteins was abolished (Supplementary Fig. S1f). Consistent with these findings, in vitro differentiation of inguinal WAT (iWAT) preadipocytes revealed a progressive increase in MMA and ADMA levels during adipocyte maturation (Supplementary Fig. S1g, h).
To investigate whether these findings translate to humans, we analyzed visceral adipose tissue from individuals with body mass indexes (BMIs) ranging from 22.5 to 31.4. Western blot analysis revealed significantly higher levels of MMA-modified proteins in individuals with BMI > 25 compared to those with BMI < 25 (Fig. 1f, g and Supplementary Fig. S2a), consistent with patterns seen in rodent DIO models. ADMA-containing proteins also increased with BMI in human adipose tissue (Fig. 1h–j). Notably, ADMA levels in visceral WAT did not differ significantly between normoglycemic and hyperglycemic individuals (Supplementary Fig. S2b, c). However, serum levels of free ADMA positively correlated with BMI in both males and females (Fig. 1k and Supplementary Fig. S2d, e). Together, these results demonstrate that elevated levels of MMA and ADMA in adipose tissue are associated with obesity in both mice and humans, suggesting that they may have potential as metabolic biomarkers.
PRMT3 Regulates adipose ADMA during fasting in an insulin–pAKT-dependent manner
The diet-dependent changes in MMA and ADMA levels suggest that Type I PRMTs—enzymes responsible for catalyzing mono- and asymmetric dimethylarginine modifications on target proteins—likely mediate the fasting- and obesity-associated methylation patterns observed in rodent and human visceral adipose tissue. To identify the specific PRMT involved in regulating arginine methylation during fasting and refeeding in white adipose tissue, we first examined the expression of Prmt genes before and after 8 days of preadipocyte differentiation in vitro. This allowed us to compare Prmt expression profiles between preadipocytes and mature adipocytes. Notably, mRNA levels of Prmt1–6 (except Prmt2) were upregulated by day 8, with Prmt3 and Prmt5 showing the most pronounced increases (Supplementary Fig. S3a). Complementary RNA sequencing analysis of epididymal white adipose tissue (eWAT) from wild-type mice at Zeitgeber Time (ZT) 4 and ZT16 confirmed the expression of all Prmt genes except Prmt8, with Prmt1, Prmt4, and Prmt5 being the most abundantly expressed (Supplementary Fig. S3b).
PRMT1 has previously been identified as the predominant enzyme responsible for catalyzing ADMA formation in various tissues20. To evaluate its role in white adipose tissue, we assessed PRMT1 abundance in eWAT following fasting. No significant change in PRMT1 protein levels was observed in eWAT after fasting (Supplementary Fig. S3c, d). To further investigate, we administered the established PRMT1 inhibitor AMI-1 to mice subjected to a 12 h fast. While AMI-1 treatment did lead to a moderate reduction in ADMA levels in eWAT, inhibition of PRMT3 using SGC707 produced a markedly greater effect (Supplementary Fig. S3e, f).
Given that adipocyte-specific deletion of Prmt1 has been shown to promote insulin resistance in diet-induced obesity (DIO) models21, we shifted focus to Prmt3, Prmt4, and Prmt5 as alternative candidates. In line with mRNA expression data, protein levels of PRMT3, PRMT4, and PRMT5 were all upregulated during adipogenic differentiation (Fig. 2a), suggesting potential functional roles in mature adipocytes. Notably, only PRMT3 and PRMT4—both Type I PRMTs—exhibited dynamic changes in expression that mirrored the patterns of MMA and ADMA during fasting and refeeding (Fig. 2b, c).

a Western blot analysis of PRMT3, 4 and 5 at d0, d2, d4 and d8 during adipogenic differentiation of preadipocytes isolated from iWAT, representative image from 2 independent repeats. b, c Western blot (b) and quantification (c) of PRMT3, 4 and 5 in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 36 h, or fasted for 36 h and then refed a normal chow diet for 3–6 h (n = 5, 4 and 4). AL, ad libitum; F-36, fasted 36 h. d, e Western blot analysis (d) and quantification (e) of PRMT3 protein levels in eWAT of 12 weeks old male B6N mice sacrificed at the indicated time points in a 12 h light/dark cycle (n = 4). f, g Western blot analysis (f) and quantification (g) of PRMT3 protein levels in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 12 h, or fasted for 36 h (n = 4, 5, 5 and 5). AL, ad libitum; F-12, fasted 12 h; F-36, fasted 36 h. (h, i) Western blot analysis (h) and quantification (i) of levels of proteins containing MMA and ADMA in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 12 h, or fasted for 12 h plus 30 mg/kg SGC707 or fasted for 36 h (n = 5, 6, 7 and 7). AL, ad libitum; F-12, fasted 12 h; F12 + SGC: fasted 12 h plus 30 mg/kg SGC707; F-36, fasted 36 h. Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
Further temporal analysis in mouse eWAT revealed that PRMT3 protein levels followed a diurnal rhythm, decreasing during the daytime fasting phase and increasing during nocturnal feeding (Fig. 2d, e). No comparable diurnal pattern was detected in PRMT3 levels from brown adipose tissue (BAT) or liver (Supplementary Fig. S3g), aligning with the tissue-specific ADMA and MMA changes observed in eWAT. These findings support a model in which PRMT3 acts as a key regulator of diurnal arginine methylation in eWAT proteins.
To validate the specificity of the anti-PRMT3 antibody used in our study, we expressed and purified recombinant mouse PRMT3 protein (rPRMT3) in a prokaryotic system. Immunoblotting confirmed that the antibody specifically recognized rPRMT3 (Supplementary Fig. S3h). We also examined PRMT4 protein levels in eWAT, which did not display diurnal variation (Supplementary Fig. S3i), distinguishing it from PRMT3. Further analysis revealed that PRMT3 protein levels in eWAT progressively declined with increasing fasting duration—showing modest reductions after 12 h and reaching a nadir at 36 h (Fig. 2f, g). In contrast, PRMT3 levels remained unchanged in BAT and liver across time points (Supplementary Fig. S3j), reinforcing its role as the primary adipose-specific PRMT responsive to nutritional cues.
To investigate the metabolic role of PRMT3-mediated arginine methylation in eWAT during fasting, we administered SGC707—a selective chemical inhibitor of PRMT3—to mice. Following a 12-hour fast, eWAT from untreated mice showed no significant change in MMA or ADMA levels. However, the combination of SGC707 treatment and 12 h fasting markedly accelerated the decline in MMA and ADMA levels, producing a reduction comparable to that observed after 36 h of fasting (Fig. 2h, i). Notably, PRMT3 inhibition had no effect on ADMA levels in ad libitum–fed mice (Supplementary Fig. S4a, b). These findings indicate that PRMT3 specifically mediates the fasting-induced, diurnal regulation of arginine methylation in visceral adipose tissue.
To identify the upstream signals regulating PRMT3, we investigated the pathways activated by feeding that promote arginine methylation in white adipose tissue. Glucose-induced insulin secretion facilitates energy storage in WAT via activation of AKT signaling. Specifically, PRMT3 protein levels—markedly reduced in mouse eWAT after 36 h of fasting—were rapidly restored within 3 h of either insulin injection or refeeding (Fig. 3a, b). This restoration coincided with increased phosphorylation of AKT at serine 473 and an elevated pAKT/AKT ratio following both insulin administration and refeeding (Fig. 3a, b). Furthermore, insulin treatment after fasting elevated ADMA levels in eWAT, an effect that was abrogated by PRMT3 inhibition with SGC707 (Supplementary Fig. S4c, d). These findings indicate that insulin–pAKT signaling acts upstream of PRMT3, mediating feeding-induced arginine methylation in visceral adipose tissue.

a Western blot analysis of pAKT and PRMT3 in eWAT of 12 weeks male B6N mice at ad libitum, fasted for 36 h, fasted for 36 h and then refed a normal chow diet for 3 h or fasted for 36 h, then treated with insulin via intraperitoneal injection for 3 h. AL, ad libitum; F-36, fasted 36 h; Insulin, fasted 36 h and 3 h after insulin injection; Refed: fasted 36 h and 3 h of chow diet. b Quantification of relative PRMT3 and pAKT/AKT levels from (a) (n = 5, 6, 7 and 7). c Western blot analysis of pAKT, AKT and PRMT3 in eWAT of 12 weeks old male B6N mice at fasted for 36 h, fasted for 36 h, then treated with insulin via intraperitoneal injection for 3 h or fasted for 36 h then treated with insulin plus MK-2206. F-36, fasted 36 h; Insulin, fasted 36 h and 3 h after insulin injection; MK2206: fasted 36 h then treated with insulin plus MK-2206. d Quantification of relative PRMT3 levels from (c) (n = 6). e, f Immunoprecipitation assays using MYC-tagged PRMT3 in 293 T cells and western blot analysis of pAKTs473 (e) and pT/S (f), representative image from 3 independent repeats. g Adenovirus carrying a FLAG-tagged PRMT3 was infected into differentiated adipocytes in vitro, then immunoprecipitation with a FLAG antibody and western blot analysis of pT/S to confirm PRMT3 phosphorylation, a representative image from 3 independent repeats. h Immunoprecipitation assays using MYC-tagged PRMT3 in 293 T cells and western blot analysis of pAKTs473 and pT/S with or without MK-2206, representative image from 3 independent repeats. i Western blot analysis of pAKT, PRMT3 and AMDA in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 36 h, or ad libitum plus MK-2206 or fasted for 12 h plus MK-2206. AL, ad libitum; F-36, fasted 36 h; AL + MK: ad libitum plus MK-2206; F-12 + MK: fasted for 12 h plus MK-2206. j Quantification of relative levels of proteins carrying ADMA from (i) (n = 3). Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
To determine whether PRMT3 is regulated by the insulin–pAKT signaling pathway, we treated mice with MK-2206, a well-characterized AKT phosphorylation inhibitor22. MK-2206 effectively blocked the insulin-induced upregulation of PRMT3 in eWAT, while not affecting PRMT3 levels in brown adipose tissue (BAT) or liver (Fig. 3c, d and Supplementary Fig. S4e, f). In vitro, MK-2206 treatment of differentiated adipocytes similarly reduced PRMT3 protein levels, whereas PRMT5 levels remained unchanged (Supplementary Fig. S4g). Interestingly, PRMT4 levels increased following MK-2206 treatment (Supplementary Fig. S4g), suggesting distinct regulatory mechanisms among PRMT isoforms. Notably, PRMT3 exhibited a band shift upon both insulin and MK-2206 treatment, similar to that observed for AKT, implying a possible direct post-translational modification of PRMT3 in response to insulin signaling and pAKT inhibition (Supplementary Fig. S4h).
To determine whether PRMT3 is a direct phosphorylation target of pAKT, we overexpressed a MYC-tagged PRMT3 plasmid and performed immunoprecipitation assays. These assays demonstrated that PRMT3 could co-immunoprecipitate with pAKT, indicating a physical interaction (Fig. 3e). Using a phospho-threonine/serine (pT/S) antibody, we confirmed that PRMT3 was phosphorylated at threonine/serine residues (Fig. 3f). An in vitro phosphorylation assay further showed that pAKT enhances PRMT3 phosphorylation (Supplementary Fig. S4i).
To validate this finding in adipocytes, we used an adenoviral construct expressing FLAG-tagged PRMT3 to infect differentiated adipocytes in vitro. Immunoprecipitation with a FLAG antibody confirmed phosphorylation of PRMT3 at T/S residues (Fig. 3g). Importantly, MK-2206 treatment reduced PRMT3 phosphorylation following insulin stimulation, further supporting the role of pAKT in this process (Fig. 3h). Moreover, co-treatment with MK-2206 and 12-hour fasting decreased both PRMT3 and ADMA levels in mouse eWAT (Fig. 2i, j) and similarly suppressed MMA and ADMA levels in cultured adipocytes (Supplementary Fig. S4j, k). Together, these results establish that the insulin–pAKT–PRMT3 signaling axis governs fasting-regulated arginine methylation in visceral adipose tissue.
Inhibition of PRMT3 ameliorates diet-induced obesity by enhancing adipocyte glucose metabolism
Given the role of PRMT3 in mediating arginine methylation in response to nutritional state in eWAT, we investigated its physiological relevance in metabolic regulation. Mice were treated with the selective PRMT3 inhibitor SGC70723, at 30 mg/kg. Tissue distribution analysis revealed that SGC707 was primarily localized in the liver and feces, with detectable concentrations in adipose depots, including eWAT (Supplementary Fig. S5a, b).
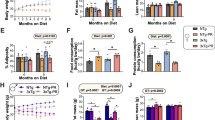
After 8 weeks on a high-fat diet (HFD), mice receiving SGC707 for 10 weeks showed significantly reduced body weight compared to vehicle-treated controls (Fig. 4a, b). Body composition analysis confirmed a marked reduction in fat mass (Fig. 4c, d). Metabolic testing demonstrated improved glucose tolerance and insulin sensitivity, as evidenced by glucose and insulin tolerance tests (GTT and ITT) (Fig. 4e, f). SGC707 treatment also led to significant reductions in the weights of brown adipose tissue (BAT), inguinal WAT (iWAT), and eWAT (Fig. 4g, h). Liver weight was also decreased, accompanied by a reduction in hepatic steatosis associated with diet-induced obesity (DIO) (Fig. 4h–j).

a Body weight of male B6N mice fed with HFD for 8 weeks and then randomly grouped to receive 30 mg/kg SGC707 injection (SGC707) or buffer control (Ctrl) (n = 8). b Representative images of mice from the SGC707 and Ctrl groups after 10 weeks of treatment. c, d Body fat mass and lean mass of mice from SGC707 and Ctrl groups (n = 8 per group). e, f Glucose tolerance (e) and insulin tolerance (f) were performed on mice from the SGC707 and Ctrl groups after 8 weeks of treatment (n = 8). g, h Representative images of various adipose tissues (g) and weights of adipose tissues and liver (h) of mice from SGC707 and Ctrl groups after 10 weeks of SGC707 treated (n = 7). i Representative images of the liver of mice from the SGC707 and Ctrl groups after 10 weeks of SGC707 treatment. j Representative H&E staining image of liver and eWAT from SGC707 and Ctrl groups, Bar=100μm. k–p After 9 weeks of SGC707 treatment, body weight (k), O2 consumption (l) and average VO2 levels (m), O2 consumption over 24 h versus body weight (n), RER (o) and delta RER levels (p) were measured (n = 7 and 8). q–t Isolateion of stromal vascular fraction (SVF) preadipocytes from mouse eWAT and differentiated them into adipocytes. The expression levels of genes related to glucose metabolism were tested by qPCR after 12 h of SGC707 treatment (q) (n = 3). r Seahorse analysis of ECAR in mouse eWAT-derived adipocytes treated with or without SGC707 (n = 5). s Quantification of the glycolic ability of cultured eWAT adipocyte with or without SGC707 treatment from (r). t Targeted-metabolomic analysis to determine intermediates in the glycolysis pathway of cultured eWAT adipocytes with or without SGC707 treatment (n = 3). Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
Histological analysis revealed smaller adipocytes in eWAT following treatment (Fig. 4j). Early intervention with SGC707 prevented HFD-induced weight gain and improved metabolic outcomes without affecting lean muscle mass, indicating selective fat loss (Supplementary Fig. S6a–g). Liver and eWAT histology confirmed reduced steatosis and smaller adipocytes (Supplementary Fig. S6h). GTT and ITT results were similarly improved with early SGC707 administration (Supplementary Fig. S6i, j). Despite the reductions in adiposity and body weight, SGC707-treated mice showed no significant differences in oxygen consumption, carbon dioxide production, or food intake compared to controls (Fig. 4k–n and Supplementary Fig. S7a–d). However, nighttime energy expenditure was specifically elevated (Supplementary Fig. S7e, f), and the delta respiratory exchange ratio (ΔRER) increased significantly, indicating improved metabolic flexibility and a shift toward greater carbohydrate utilization (Fig. 4o, p and Supplementary Fig. S7g, h). In addition, there were no difference in food intake (Supplementary Fig. S7i, j).
The specific inhibition of MMA and ADMA formation in eWAT by SGC707—alongside improvements in glucose tolerance (GTT), insulin sensitivity (ITT), and increased ΔRER—prompted further investigation into the effects of PRMT3 inhibition on adipocyte glucose metabolism. Stromal vascular fraction (SVF) preadipocytes were isolated from mouse eWAT and differentiated in vitro. Following 12 h of SGC707 treatment, the expression of key glucose metabolism genes—including Pkm, Gapdh, Hk2, and Glut4—was significantly upregulated compared to vehicle-treated controls (Fig. 4q). Seahorse metabolic flux analysis further revealed that SGC707 enhanced the glycolytic capacity of these differentiated adipocytes (Fig. 4r, s).
Similarly, in cultured porcine white adipocytes, SGC707 treatment increased the expression of glucose metabolism-related genes, including Pkm, Gapdh, Tpi1, and Pgam1 (Supplementary Fig. S7k). Targeted metabolomic profiling confirmed significant elevations in glycolytic intermediates and end-products, including fructose-6-phosphate, fructose-1,6-bisphosphate, glucose-6-phosphate, and lactic acid (Fig. 4t). In addition, metabolomic analysis of eWAT following SGC707 administration combined with 12 h fasting identified glucose and citrate as the top two significantly altered metabolites (Supplementary Fig. S7l). These findings indicate that the increased ΔRER observed in DIO mice treated with SGC707 reflects enhanced adipocyte glucose metabolism, linking PRMT3 inhibition to improved metabolic flexibility and energy utilization in white adipose tissue.
To further investigate the in vivo function of PRMT3 in epididymal fat, we employed an AAV-mediated adipocyte-specific knockdown (KD) of Prmt3 (Supplementary Fig. S8a). This approach effectively reduced PRMT3 protein levels in eWAT, confirming successful gene silencing (Supplementary Fig. S8b). Eight weeks after local AAV injection into eWAT of DIO mice, no significant differences in body weight or food intake were observed between KD and control groups (Supplementary Fig. S8c, d).
Metabolic chamber analysis revealed that Prmt3 KD led to increased VO₂, heat production, and RER, indicating enhanced energy expenditure and a shift toward carbohydrate utilization (Supplementary Fig. S8e–g). Furthermore, Prmt3 KD improved glucose clearance, as demonstrated by reduced blood glucose levels and area under the curve (AUC) during glucose tolerance testing (Supplementary Fig. S8h). These findings suggest that adipocyte-specific inhibition of PRMT3 in eWAT enhances systemic glucose metabolism and promotes metabolic flexibility in obese mice.
SLC25A1 Is a Direct Target of PRMT3 in a Feeding-Dependent Manner
Metabolic flux analysis using 13C-labeled glucose revealed an increased incorporation of isotopic carbon into citrate in eWAT (Supplementary Fig. S7m). Given the observed reduction in citrate levels following PRMT3 inhibition (Supplementary Fig. S7l, Supplementary Data 1), we hypothesized that PRMT3-mediated arginine methylation may regulate citrate metabolism. Citrate exports from mitochondria to the cytosol—crucial for de novo lipogenesis—is mediated by the citrate transporter SLC25A124,25. To assess whether SLC25A1 was a feeding-dependent and tissue-specific substrate of PRMT3, we examined its expression across metabolic tissues. SLC25A1 protein levels were specifically reduced in eWAT, but not in liver or brown adipose tissue (BAT), following 12 hours of fasting combined with SGC707 treatment (Fig. 5a, b). Although previous bioinformatic analyses had predicted potential interactions between SLC25A1 and either PRMT3 or PRMT526, our co-immunoprecipitation (Co-IP) experiments in 293 T cells overexpressing PRMT3 and SLC25A1 confirmed a specific interaction: SLC25A1 co-precipitated with PRMT3, but not with PRMT5 (Fig. 5c, S9a). These findings identify SLC25A1 as a direct and specific target of PRMT3 in a nutritionally regulated, tissue-selective manner. Further evidence of arginine methylation on SLC25A1 was obtained using anti-ADMA and anti-MMA antibodies, which successfully pulled down SLC25A1, indicating the presence of both mono-methylated and asymmetrically di-methylated arginine residues on the protein (Fig. 5d and Supplementary Fig. S9b). Treatment with the PRMT3 inhibitor SGC707 led to a reduction in methylated SLC25A1 levels, as detected by ADMA-specific immunoblotting (Fig. 5e and Supplementary Fig. S9c).

a, b Western blot (a) and quantification (b) of SLC25A1 in eWAT of 12-week-old male B6N mice at ad libitum and fasted for 12 h with or without 30 mg/kg SGC707 (n = 3). AL, ad libitum; F-12, fasted 12 h. c Immunoprecipitation assays using Anti-FLAG antibody in 293 T cells transfected with FLAG-tagged SLC25A1 and HA-tagged PRMT3, and western blot analysis of HA and FLAG. d Immunoprecipitation assays using Anti-ADMA antibody and western blot analysis of FLAG in 293 T cells transfected with FLAG-tagged SLC25A1 and HA-tagged PRMT3, representative image from 3 independent repeats. e Immunoprecipitation assays using Anti-FLAG antibody in 293 T cells transfected with FLAG-tagged SLC25A1 and HA-tagged PRMT3 with or without SGC707 treatment, and western blot analysis of Anti-ADMA antibody, representative image from 3 independent repeats. f Protein sequence alignment of SLC25A1 from Drosophila melanogaster to Homo sapiens, indicating the conserved R282 and R285 sites. g Structural prediction of WT SLC25A1 (SLC25A1WT) and R282G/R285C (SLC25A1Mut) by AlphaFold 3. h Immunoprecipitation assays using Anti-FLAG antibody in 293 T cells transfected with PRMT3 and SLC25A1WT/SLC25A1Mut and WB analysis of ADMA confirmed that SLC25A1 mutation reduced its methylation, representative image from 3 independent repeats. (i) Immunoprecipitation assays using Anti-ADMA antibody in 293 T cells transfected with PRMT3 and SLC25A1WT/SLC25A1Mut and WB analysis of FLAG confirmed that SLC25A1 mutation reduced its methylation, representative image from 3 independent repeats. j Immunoprecipitation assays using Anti-FLAG antibody in 293 T cells transfected with PRMT3 and SLC25A1WT/SLC25A1Mut and WB analysis of MYC confirmed that SLC25A1 mutation reduced its interaction with PRMT3, a representative image from 3 independent repeats. (k, l) WB analysis (k) and quantification (l) of protein stability of SLC25A1WT and SLC25A1Mut with or without PRMT3 by CHX assay (n = 3). Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
Sequence alignment revealed that arginine residues R282 and R285—located within the citrate-binding domain of SLC25A1—are conserved across species and represent potential methylation sites (Fig. 5f, g). Structural modeling indicated that mutation of these residues (SLC25A1Mut) reduced the protein’s interfacial contact area, potentially altering its conformation (Fig. 5g). Co-IP experiment in 293 T cells expressing PRMT3 with either SLC25A1WT/SLC25A1Mut confirmed that methylation was significantly reduced in the mutant form (Fig. 5h). Consistently, anti-ADMA antibodies pulled down SLC25A1WT but not SLC25A1Mut, indicating that R282 and R285 are the primary sites of asymmetric dimethylation (Fig. 5i). Moreover, the interaction between PRMT3 and SLC25A1 was weakened by mutation of these residues (Fig. 5j).
To assess the functional consequence of arginine methylation on protein stability, we performed a cycloheximide (CHX) chase assay. The results demonstrated that PRMT3 stabilized SLC25A1 protein in a methylation-dependent manner (Fig. 5k, l), supporting a model in which arginine methylation enhanced SLC25A1 protein stability and function.
The expression of SLC25A1 in eWAT displayed a diurnal pattern that closely mirrored that of PRMT3 (Fig. 6a, b). In line with the fasting-induced reduction in PRMT3, SLC25A1 protein levels were slightly decreased after 12 hours of fasting and markedly reduced after 36 hours (Fig. 6c, d). Refeeding for just 3 hours after fasting restored SLC25A1 levels in eWAT (Fig. 6e, f), further supporting its feeding responsiveness.

a, b Western blot (a) and quantification (b) of SLC25A1 in eWAT of 12 weeks old male B6N mice sacrificed at the indicated time points in a 12-hour light/dark cycle (n = 4). c, d Western blot analysis (c) and quantification (d) of SLC25A1 protein levels in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 12 h, or fasted for 36 h (n = 4, 5, 5 and 5). AL, ad libitum; F-12, fasted 12 h; F-36, fasted 36 h. (e, f) Western blot (e) and quantification (f) of SLC25A1 in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 36 h, or fasted for 36 h and then refed a normal chow diet for 3–6 h (n = 7, 7 and 9). g, h Western blot (g) and quantification (h) of SLC25A1 in eWAT of 8 weeks old male mice restricted to the daytime (ZT0-12; DRF, “daytime-fed”) or restricted to the nighttime (ZT12-24; NRF, “nighttime-fed”) for 30 days (n = 3). i, j Western blot (i) and quantification (j) of SLC25A1 at ZT0 and ZT12 in eWAT of mice after NRF or DRF (n = 3). k, l Western blot analysis (k) and quantification (l) of pAKT, AKT, SLC25A1 and PRMT3 in eWAT of 12 weeks old male B6N mice at ad libitum, fasted for 36 h, fasted for 36 h, then treated with insulin via intraperitoneal injection for 3 h or fasted for 36 h then treated with insulin plus MK-2206 (n = 6, 6, 6 and 5). AL, ad libitum; F-36, fasted 36 h; Insulin, fasted 36 h and 3 h after insulin injection; MK2206: fasted 36 h then treated with insulin plus MK-2206. (m, n) Western blot analysis (m) and quantification (n) of pAKT, PRMT3 and SLC25A1 in cultured adipocytes with or without MK-2206 treatment (n = 6). o, p Western blot analysis (o) and quantification (p) of SLC25A1 protein levels in eWAT at fasted for 36 h (Ctrl), fasted for 36 h, then treated with insulin via intraperitoneal injection for 3 h (Insulin) or fasted for 36 h then treated with insulin plus SGC707 (Insulin + SGC707) (n = 4 for SLC25A1 protein levels), the results were calculated from 4 independent repeats as shown in (o). Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
To evaluate the dependence of SLC25A1 expression on feeding time, we subjected mice to night-restricted feeding (NRF) or daytime-restricted feeding (DRF) for one month (Supplementary Fig. S9d). These experiments revealed a strong dietary association: SLC25A1 protein levels increased with feeding and decreased with fasting (Fig. 6g, h). Under DRF and NRF conditions, SLC25A1 expression at Zeitgeber Time 0 (ZT0) and ZT12 showed an anti-phasic pattern (Fig. 6i, j), consistent with feeding status. Notably, Slc25a1 mRNA levels in eWAT remained unchanged across ad libitum (AL), NRF, and DRF conditions (Supplementary Fig. S9e, f), indicating that regulation occurs primarily at the post-translational level.
Insulin injection restored SLC25A1 protein levels in fasted mice, mimicking the effects seen with PRMT3 (Fig. 6k, l). Conversely, treatment with MK-2206, an inhibitor of AKT phosphorylation, blocked the insulin-induced increase in SLC25A1 expression (Fig. 6k, l). These effects were specific to eWAT and were not observed in BAT or liver (Supplementary Fig. S9g). Similarly, MK-2206 treatment reduced SLC25A1 protein levels in cultured adipocytes (Fig. 6m, n). Furthermore, insulin increased SLC25A1 levels under fasting conditions, but this effect was abolished when PRMT3 was inhibited with SGC707 (Fig. 6o, p), demonstrating that insulin–pAKT signaling regulates SLC25A1 expression via PRMT3. Together, these findings establish that SLC25A1 is a direct methylation target of PRMT3 and is upregulated in response to feeding through the insulin–pAKT–PRMT3 signaling axis.
Time-restricted feeding reduces PRMT3/SLC25A1 levels and ameliorates diet-induced obesity
Recent studies have underscored the metabolic benefits of time-restricted feeding (TRF) and intermittent fasting, particularly their anti-obesity and anti-diabetic effects6,27. Given the sensitivity of PRMT3-mediated protein methylation and SLC25A1 expression to fasting and insulin–pAKT signaling, we examined whether TRF modulates the PRMT3–SLC25A1 axis in epididymal white adipose tissue (eWAT).
Using a 16:8 TRF regimen (feeding from 9 p.m. to 5 a.m.) under a regular chow diet, we observed a marked reduction in MMA, ADMA, PRMT3, and SLC25A1 protein levels in eWAT, with no corresponding changes in the liver (Fig. 7a, b). When the same TRF protocol was applied to mice on a high-fat diet (HFD) (Fig. 7c), it led to a significant reduction in body weight without affecting total food intake (Fig. 7d and Supplementary Fig. S10a–c). Metabolic chamber analysis revealed that TRF did not significantly alter overall oxygen consumption or carbon dioxide production (Supplementary Fig. S10d–i). However, nighttime energy expenditure and the ΔRER were significantly increased (Fig. 7e, f and Supplementary Fig. S10j–l), suggesting enhanced metabolic flexibility. In addition, mice under the 16:8 TRF protocol exhibited increased physical activity during the nighttime feeding window (Supplementary Fig. S10m, n). Further analysis revealed that TRF significantly improved glucose homeostasis. Mice subjected to TRF exhibited lower blood glucose levels during glucose tolerance tests (GTT) and insulin tolerance tests (ITT), along with reduced AUC and increased area above the curve (AAC), indicating enhanced glucose clearance and insulin sensitivity (Supplementary Fig. S10o–r).

a, b Western blot analysis (a) and quantification (b) of proteins carrying MMA and ADMA, PRMT3 and SLC25A1 from eWAT and liver of 12 weeks old male mice at ad libitum or fasted for 16 h (n = 5). AL, ad libitum; F-16, fasted 16 h. c Experimental design depicting the timing of HFD access as ad libitum or 16:8 (ZT13 to ZT21) time-restricted feeding (TRF) in wild-type male mice, diagram Created in BioRender. Jia, Z. (2025) https://BioRender.com/rmpiiac. d Body weight of mice after 7 weeks of HFD in FA (High-fat diet, ad libitum) or FT (High-fat diet, TRF) group (n = 16). e, f 48 h RER rhythms and delta RER of mice from the FA or FT group after 7 weeks of treatment (n = 16). g Experimental design depicting the timing of 30 mg/kg SGC707 injection at ZT0 or ZT12 and solvent control (Ctrl). h Body weight of mice from ZT0, ZT12 and Ctrl groups after 5 weeks of treatments. i, j 48 h RER rhythms and delta RER of mice from ZT0, ZT12 and Ctrl groups (n = 5). k–m Western blot analysis (k, l) and quantifications (m) of relative protein levels from eWAT of FA or FT group collected at ZT1 and ZT13 (n = 3). n Blood glucose concentration of mice from ZT0, ZT12 and Ctrl groups, fasted from ZT12 to ZT1 (n = 7). o Relative expression levels of key genes involved in lipid and glucose metabolism from eWAT of ZT0, ZT12 and Ctrl groups (n = 5). Data are mean ± s.e.m. Statistical significance was determined using an unpaired two-tailed Student’s t test. Source data are provided as a Source Data file. Elements in (c) and (g) were obtained from Biorender with a publication license.
After both 5 and 12 weeks of TRF, significant reductions in liver and adipose tissue weights were observed compared to ad libitum-fed controls, while skeletal muscle mass remained unchanged, highlighting the tissue specificity of TRF-induced fat loss (Supplementary Fig. S10s, t). TRF also effectively prevented high-fat diet (HFD)-induced hepatic steatosis, as shown by histological analysis (Supplementary Fig. S10u, v). Notably, elevated serum S-adenosylmethionine (SAMe) levels in diet-induced obese (DIO) mice were significantly reduced following 16:8 TRF (Supplementary Fig. S10w), further supporting the role of TRF in modulating methylation-related metabolic pathways.
To explore the time-dependent metabolic effects of PRMT3 inhibition, we assessed the impact of a single SGC707 injection in combination with 12 hours of fasting. This treatment effectively reduced total ADMA levels in eWAT at ZT12 but not at ZT0 (Supplementary Fig. S11a, b), indicating a temporal dependency in PRMT3 activity. To determine whether the timing of PRMT3 inhibition influenced metabolic outcomes, SGC707 was administered to HFD-fed mice at either ZT0 or ZT12. In both cases, SGC707 significantly reduced body weight, weight gain, and fat mass without affecting lean mass (Fig. 7g, h and Supplementary Fig. S11c–f). Glucose tolerance was also improved in treated mice (Supplementary Fig. S11g, h). Consistent with findings from the 16:8 TRF protocol, SGC707 treatment did not alter overall oxygen consumption or carbon dioxide production (Supplementary Fig. S11i, j). However, injections at either ZT0 or ZT12 led to a significant increase in the ΔRER, suggesting enhanced metabolic flexibility. These effects occurred without changes in food intake and closely mirrored those induced by TRF (Fig. 7i, j and Supplementary Fig. S11k).
Significant volume reductions in white adipose tissues and SLC25A1 protein levels were observed in both ZT0 and ZT12 SGC707-treated groups (Supplementary Fig. S11l–n). Following 12 weeks of 16:8 TRF, marked reductions in MMA, ADMA, PRMT3, and SLC25A1 protein levels were detected specifically in eWAT at ZT13 (fasted state) (Fig. 7k–m and Supplementary Fig. S12) but not at ZT1 (fed state) or in the liver (Supplementary Fig. S13). Interestingly, while SLC25A1 levels decreased at both ZT13 and ZT1 in eWAT, the reduction was more pronounced in the fasted state (Fig. 7l, m and Supplementary Fig. S12).
Although both ZT0 and ZT12 SGC707 treatment groups exhibited comparable anti-diet-induced obesity (anti-DIO) effects, fasting glucose levels were significantly lower in mice treated at ZT12 compared to those treated at ZT0 (Fig. 7n). In addition, key genes involved in lipid and glucose metabolism—including Cpt2, Mcad, Fasn, Acaca, Gapdh, Hk2, and Glut4—were more strongly upregulated in eWAT of ZT12-treated mice (Fig. 7o). These findings suggest that pharmacological inhibition of PRMT3 by SGC707 mimics the beneficial metabolic effects of 16:8 TRF by promoting glucose metabolism and suppressing SLC25A1 expression in a time-of-day–dependent manner.
Adipocyte-specific Slc25a1 deletion enhances glucose metabolism in a time-dependent manner
Given the association between reduced SLC25A1 levels and weight loss, we investigated the functional role of SLC25A1 in adipocytes in vivo. Protein levels of SLC25A1 were significantly upregulated during adipogenesis in white adipocytes and further increased in eWAT following diet-induced obesity (DIO) induced by a high-fat diet (HFD) (Fig. 8a–c). To assess the adipocyte-specific function of SLC25A1, we generated Slc25a1 adipocyte-specific knockout (Slc25a1-AKO) mice by crossing Adiponectin-Cre mice with Slc25a1-floxed mice. In Slc25a1-AKO mice, SLC25A1 protein levels were markedly reduced in eWAT, while liver expression remained unaffected, confirming tissue specificity (Fig. 8d). At 13 weeks of age, there were no significant differences in body weight, fat mass, or lean mass between Slc25a1-AKO and wild-type (WT) littermates under baseline conditions (Supplementary Fig. S14a–c). However, Slc25a1-AKO mice exhibited improved glucose tolerance, as evidenced by lower blood glucose levels during glucose tolerance testing (GTT) (Supplementary Fig. S14d). In addition, there were no differences in the weights of ATs and liver (Supplementary Fig. S14e).

a, b Western blot analysis (a) and quantification (b) of SLC25A1 at d0, d2, d4 and d8 during adipogenic differentiation of preadipocytes isolated from iWAT (n = 3). c Relative levels of SLC25A1 in eWAT of 20 weeks old male mice fed with 12 weeks of normal diet (ND) or high-fat diet (HFD) (n = 4). d Western blot results show the specific deletion of SLC25A1 in eWAT but not liver from Slc25a1AKO mice, representative image from 2 independent repeats. e Body weight of male WT (Slc25a1-flox/flox) and Slc25a1AKO (Adiponectin-Cre; Slc25a1 flox/flox) mice during 10 weeks with HFD (n = 10 and 11). f Body composition of WT and Slc25a1AKO mice after 10 weeks of HFD (n = 5 and 7). g, h Blood glucose levels and area under the curve (AUC) or area above the curve (AAC) of male WT and Slc25a1AKO mice during glucose tolerance test or insulin tolerance test after 10 weeks of HFD, respectively (n = 10 and 11). i Tissue weights of various adipose tissues, liver and tibial anterior muscle (TA) after 10 weeks of HFD (n = 6 and 8). j Representative H&E staining image of liver and eWAT from WT and Slc25a1AKO mice after HFD from 4 pairs of animals, Bar = 250 μm. k Levels of cholesterol, HDL, LDL, and TG from the serum of WT and Slc25a1AKO mice after HFD (n = 5). l Blood glucose of WT and Slc25a1AKO mice after HFD, fasted from zt1 to zt7 (n = 10 and 11). m Seahorse analysis of ECAR in mouse eWAT-derived adipocytes treated with or without CTPI-2 (n = 5). n Quantification of the glycolic ability of cultured eWAT adipocyte with or without CTPI-2 treatment from (m). o Levels of compensatory glycolysis from cultured adipocytes from Ctrl, CTPI-2 treated, and Slc25a1AKO mice (n = 6, 6 and 7). p Gene Ontology (GO) annotation to identify the key up-regulated pathways in eWAT of Slc25a1AKO mice compared to WT. q Heatmap of DEGs enriched in glucose metabolism. r, s Western blot analysis (r) and quantification (s) of SLC25A1, GAPDH, PC and PKM1/2 in eWAT of WT and Slc25a1AKO mice (n = 3). t 48 h VO2 rhythms and average VO2 levels at indicated time frames (n = 7 and 10). Data are mean ± s.e.m. Statistical significance was determined using unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
Upon HFD feeding, Slc25a1-AKO mice exhibited reduced body weight beginning at 5 weeks post-treatment, primarily attributable to a decrease in lean mass, while fat mass remained unchanged (Fig. 8e, f). Deletion of Slc25a1 significantly enhanced glucose metabolism, as shown by improved glucose clearance and insulin sensitivity in GTT and ITT) (Fig. 8g, h). Fasting blood glucose levels were also significantly lower in Slc25a1-AKO mice compared to WT controls (Fig. 8l). Despite these metabolic benefits, no differences were observed in adipose tissue mass or adipocyte size between the two groups (Fig. 8i, j), suggesting that changes in adiposity did not drive the improvements. Interestingly, liver weight was reduced, and histological analysis revealed amelioration of HFD-induced hepatic steatosis in Slc25a1-AKO mice (Fig. 8j). In addition, serum biochemical analysis showed significant reductions in total cholesterol and HDL cholesterol levels (Fig. 8k). Treatment of cultured mouse eWAT-derived adipocytes with CTPI-2, a specific inhibitor of SLC25A128, recapitulated the enhanced glycolytic capacity observed with PRMT3 inhibition (Fig. 8m, n). Seahorse metabolic flux analysis further confirmed that both genetic deletion and pharmacological inhibition of SLC25A1 promoted compensatory glycolysis in adipocytes (Fig. 8o).
RNA sequencing of eWAT from Slc25a1-AKO mice revealed that loss of Slc25a1 predominantly impacted genes involved in lipid and carbohydrate metabolism (Fig. 8p). Notably, glycolysis-related genes such as Gapdh, Pdk1, Hk2, Gpam, Pkm, and Acss2 were significantly upregulated (Fig. 8q and Supplementary Data 2). These transcriptomic changes were accompanied by increased protein levels of pyruvate carboxylase (PC) and GAPDH in eWAT (Fig. 8r, s), further supporting enhanced glycolytic flux.
Metabolic chamber analyses showed that Slc25a1-AKO mice exhibited elevated oxygen consumption, carbon dioxide production, and energy expenditure (EE) during the early dark phase (ZT12–ZT18), coinciding with the mice’s active/feeding period (Fig. 8t and Supplementary Fig. S14f–i). Together, these findings suggest that lowering SLC25A1 levels in eWAT—whether through Slc25a1 deletion, ZT12-targeted SGC707 injection, or 16:8 time-restricted feeding—enhances adipocyte glucose metabolism and energy expenditure, thereby preventing diet-induced obesity and improving insulin sensitivity.
We then locally injected AAV9-Fabp4-Slc25a1 into the eWAT of high-fat diet–induced obese mice and subsequently treated them with the PRMT3 inhibitor SGC707 for four weeks. Two weeks post-injection, Slc25a1 overexpression markedly impaired glucose tolerance following intraperitoneal glucose administration (Supplementary Fig. S15a, b). Consistently, SGC707 treatment failed to improve glucose tolerance in mice overexpressing Slc25a1 (Supplementary Fig. S15a, b). Furthermore, metabolic chamber analyses revealed that Slc25a1 overexpression significantly decreased ΔRER (Supplementary Fig. S15c, d) and heat production (Supplementary Fig. S15e, f), and SGC707 treatment did not reverse these effects. Similarly, SGC707 treatment failed to increase PC protein levels or Hk2 mRNA expression in eWAT from Slc25a1-overexpressing mice (Supplementary Fig. S15g, h). Together, these results demonstrate that Slc25a1 overexpression counteracts the beneficial metabolic effects of PRMT3 inhibition, supporting the notion that reduced SLC25A1 mediates, at least in part, the phenotype observed with PRMT3 loss of function.
Discussion
Our study advances the understanding of time-restricted feeding (TRF) by elucidating a molecular pathway in adipose tissue that promotes metabolic health and supports weight loss. We demonstrate that TRF—particularly the 16:8 regimen6, modulates protein arginine methylation dynamics in visceral adipocytes through the activity of PRMT3. This modulation is closely linked to insulin–pAKT signaling, which regulates levels of asymmetric dimethylarginine (ADMA)-modified proteins. ADMA levels rise postprandially and fall during fasting, aligning with diurnal nutrient cycles. Importantly, ADMA-enriched proteins accumulate specifically in the visceral fat of obese individuals, highlighting their potential as therapeutic targets. Our findings further reveal that pharmacological inhibition of PRMT3 with SGC707 recapitulates the metabolic benefits of TRF, including enhanced glucose metabolism and improved metabolic flexibility. Mechanistically, PRMT3 exerts its effects in part by regulating the mitochondrial citrate transporter SLC25A128,29.
linking arginine methylation to glycolytic reprogramming in adipocytes. This citrate-driven metabolic shift is critical for the beneficial outcomes observed during TRF, underscoring PRMT3’s role as a nutrient-responsive mediator that integrates feeding cues with adipocyte metabolic remodeling. Importantly, our findings show that pharmacological inhibition of PRMT3 using SGC707 restores impaired metabolic flexibility in obese mice, as evidenced by a significant increase in ΔRER—a hallmark of improved fuel switching capacity. This effect closely mirrors the metabolic benefits observed with TRF. Metabolic flexibility refers to the body’s ability to adapt fuel utilization in response to changing metabolic demands, notably the transition from lipid oxidation during fasting to glucose utilization during feeding. This process is tightly regulated by insulin and is a key determinant of insulin sensitivity30,31. A normal, healthy transition from fasting to feeding, which involves shifts in fuel selection, is primarily driven by insulin to store energy for future use. Consistent with this, we demonstrate that insulin–pAKT signaling activates PRMT3-mediated arginine methylation in epididymal white adipose tissue (eWAT) specifically during the fed state, linking hormonal cues to post-translational modification events that regulate nutrient handling. While visceral white adipose tissue (vWAT) is well known for buffering free fatty acids and coordinating systemic energy flux32, its role in glucose uptake and metabolism—particularly in the context of obesity and insulin resistance—remains underexplored. Our data suggests that glucose metabolism in vWAT, regulated through PRMT3 and SLC25A1, plays a previously unrecognized role in maintaining metabolic flexibility under nutritional stress.
Our observation that enhanced adipocyte glycolysis—induced either by SGC707-mediated PRMT3 inhibition or adipocyte-specific Slc25a1 deletion—attenuates both obesity and hepatic steatosis suggests that impaired adipocyte glucose metabolism contributes directly to weight gain, particularly when metabolic flexibility is mistimed or dysregulated. These findings support the emerging concept that disruptions in nutrient-sensing mechanisms within adipose tissue play a causal role in the pathogenesis of diet-induced obesity.
Recent studies have suggested that time-restricted feeding (TRF) reshapes the transcriptome of multiple organs, including adipose tissue7,33,34, in part through circadian rhythm–driven regulation. However, accompanying proteomic studies—particularly in the liver—have highlighted that many metabolic adaptations occur at the level of protein modification rather than gene expression35,36. This raises important questions about the extent to which post-translational modifications contribute to TRF-driven metabolic improvements in peripheral tissues, such as white adipose tissue (WAT). Our results emphasize the importance of protein arginine methylation in the metabolic adaptation of white adipose tissue (WAT).
Our data highlights a critical role for protein arginine methylation in the metabolic reprogramming of WAT. Prior work has demonstrated that inhibiting hepatic SAMe synthesis through MAT1A blockade reduces adiposity17,18, suggesting a broader link between methylation pathways and systemic energy balance. Building on this, we propose that PRMT3-mediated asymmetric dimethylarginine (ADMA) modification serves as a nutrient-responsive metabolic sensor in visceral WAT, translating feeding cues into metabolic adaptations. Interestingly, TRF has been shown to prevent obesity even in circadian-deficient mice, consistent with our findings that Prmt3 and Slc25a1 mRNA levels remain stable across time points—supporting their regulation at the post-transcriptional level and suggesting circadian independence. Given that rhythmic feeding patterns are preserved in mice lacking functional circadian clocks37, our results reinforce the concept that nutrient timing, rather than intrinsic circadian oscillations, is a dominant driver of adipose tissue metabolic adaptation.
Elevated circulating levels of asymmetric dimethylarginine (ADMA) are well-established risk factors for cardiovascular disease, primarily due to ADMA’s inhibitory effect on nitric oxide synthase—an enzyme critical for vascular homeostasis and anti-atherogenic function38,39. In our study, increased ADMA levels in both visceral adipose tissue and plasma in the context of obesity suggest that localized PRMT3 activity may contribute to broader systemic metabolic disturbances. This implicates visceral PRMT3 as a potential upstream driver of obesity-associated cardiovascular risk. The role of dimethylarginine dimethylaminohydrolase (DDAH), which degrades ADMA, merits further investigation. Enhancing DDAH activity could represent a complementary strategy to reduce ADMA accumulation and mitigate the cardiovascular complications of obesity40,41,42. Furthermore, genetic studies linking PRMT3 to increased stroke risk among African Americans43, point to possible ethnic or genetic modifiers of PRMT3 function, emphasizing the importance of personalized approaches in the development of PRMT3-targeted therapies. Finally, the context-dependent effects of PRMT3 inhibition with SGC707—varying by dosage, timing, and metabolic state—highlight the necessity for precision medicine strategies that account for individual circadian rhythms and metabolic profiles in the treatment of obesity and its complications.
Our study opens several avenues for future research. First, the differential responsiveness of adipose depots to metabolic cues, such as fasting and time-restricted feeding (TRF), warrants further investigation. Understanding these depot-specific dynamics could lead to more targeted interventions for abdominal obesity and its associated metabolic complications. Second, while both SGC707 treatment and adipocyte-specific Slc25a1 deletion partially reduced obesity and hepatic steatosis, the incomplete nature of these effects suggests that other tissues contribute to the observed phenotypes. PRMT3 has previously been shown to facilitate liver X receptor (LXR)-mediated lipogenesis in hepatocytes, and SGC707 protects against non-alcoholic fatty liver disease (NAFLD) in models where T0901317 and palm oil activate LXR44,45. In addition, SGC707 treatment has been reported to reduce hepatic steatosis, plasma triglyceride levels, and atherosclerosis in LDL receptor-deficient mice fed a Western-type diet46. Furthermore, our finding that Prmt3 knockdown in eWAT improves glucose metabolism without significantly affecting body weight suggests a dissociation between local metabolic benefits and systemic energy balance. This highlights the importance of organ–organ communication in shaping whole-body metabolic outcomes. To dissect these inter-tissue interactions, developing tissue-specific Prmt3 knockout models—particularly in adipose tissue, liver, and potentially the vasculature—would offer valuable insights into the coordinated roles of PRMT3 across metabolic organs and help inform strategies for tissue-targeted therapies.
Our findings establish a clear link between PRMT3-mediated arginine methylation in adipose tissue and the regulation of systemic metabolic health. Specifically, we identify the PRMT3–SLC25A1 axis as a key modulator of adipocyte glucose metabolism, with implications for the prevention and treatment of obesity, type 2 diabetes, and related metabolic disorders. By elucidating how time-restricted feeding (TRF) and PRMT3 inhibition influence adipocyte function and systemic energy homeostasis, our study reveals therapeutic strategies aimed at restoring metabolic flexibility. These results underscore the importance of a comprehensive, tissue-specific approach to identifying nutrient-responsive sensors and effectors in adipose tissue, which are critical determinants of whole-body metabolic health. However, in the absence of an isotope-labeled SAMe–based in vitro assay, it remains unclear whether methylation of the identified arginine residues (R282 and R285), which are critical for citrate binding, directly modulates citrate metabolism or indirectly affects SLC25A1 protein stability.
Methods
Animal care and mouse strains
Animal studies were conducted in the accredited SPF animal facility in the CAM-SU Genomic Resource Center and approved by the Animal Care and Use Committee of CAM-SU under a protocol of ZJ-2021-1 approved on 12/24/2021. Mice were maintained at room temperature (23–25 °C) when indicated with a 12 h light/dark cycle (illuminated from 8:00) and free access to acidified water and radiated and autoclaved food unless specified. All mice were maintained on a C57BL/6 N background. C57BL/6N-Slc25a1tm1c(EUCOMM)Wtsi/Cmsu were obtained by injecting mutant mouse ES cells into mouse blastocysts CAM-SU Genomic Resource Center. The Adipocyte-specific deletion of Slc25a1 was generated by breeding Adiponectin-Cre (stock #010803) and Slc25a1-flox/flox mice. The pups were identified by PCR followed by sequence analysis using primers (Cre-F: ACGGACAGAAGCATTTTCCA; Cre-R: GGATGTGCCATGTGAGTCTG; flox-F: GGAAGGCAATCCTAGCAGGTGGGTC; flox-R: CAGCTGCGTCTTCACGTATTCGGTC).
Wildtype male C57BL/6 N were obtained from CAM-SU Genomic Resource Center. For fasting experiments, male C57BL/6 N (10–12 weeks of age) were divided into three groups: ad libitum and fasted (12 h or 36 h). The fasted group fasted for 12 h or 36 h, began at ZT12, and were sacrificed at ZT0 with the ad libitum group. For fasting and refeeding experiments, male C57BL/6 N (10–12 weeks of age) were divided into two groups: fasted and refed. The fasted group fasted for 36 h, and the refed group fasted for 36 h and then refed the chow diet for 3–6 h before the study.
Human samples
Visceral adipose tissues from individuals with different BMIs were surgically collected from the Second Affiliated Hospital of Soochow University under a protocol of JD-LK-2020-038-01 approved by the Ethics Community of the Second Affiliated Hospital of Soochow University. Human serum was isolated from venous blood collection under standard procedures from the Second Affiliated Hospital of Soochow University. The signed informed consent has been collected from all participants.
Quantitation of ADMA in human plasma samples
Human plasma samples were extracted with an extraction solvent containing internal standards (40 ng/mL d7-ADMA). The whole extraction process was the same as for drug analysis. The ADMA standard curve was made by dissolving the standard to a series concentration using 5% BSA as a surrogate matrix. Standards d7-ADMA (purity: 98%) were purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA), and ADMA (purity: 98%) from targetMOl (Boston, MA, USA).
UPLC separation was performed using a Shimadzu Nexera X2 liquid chromatography system with a binary gradient pump, autosampler (4 °C), and column oven (40 °C) for chromatographic separation. A hydrophilic interaction liquid chromatographic column (HILIC, 150 × 2.1 mm, 1.7 µm, Atlantis, Waters) was applied for separation. The injection volume was 2 µL. A binary eluent system was used at a flow rate of 0.3 mL/min. Mobile phase A was an aqueous buffer containing 10 mM ammonium formate, and 0.1% formic acid, and mobile phase B was acetonitrile with 0.1% formic acid. The programmed gradient was 0-0.5 min: 90% B, 0.5–1 min: 90% to 70% B, 1–6 min: 70% to 65% B, 6–6.5 min: 65%-40% B, 6.5-7.5 min: 40% B, 7.5–8 min: 40%-90% B, 8-9 min: 90% B. Multiple reaction monitoring (MRM) data acquisition mode (for ADMA: 203.1–46.3) were performed on an AB Sciex 6500 QTrap® hybrid triple quadrupole/linear ion trap mass spectrometer with IonDrive™ TurboV ion source and ESI probe. Spectra were obtained in ESI+ under the following conditions: curtain gas 35 psi, ion spray voltage (IS) 5500 V, the pressure of ion source gas 1 55 psi, ion source gas 2 55 psi, declustering potential 60 V, entrance potential 10 V, and collision cell exit potential 10 V. Data collection and quantification were carried out by SCIEX Analyst and SCIEX OS, respectively.
Feeding schedule and diets
For all high-fat diets (HFD, XTHF60, Xietong Pharmaceutical Bio-engineering) feeding experiments, mice began dietary intervention at 6–8 weeks old with a body weight of ~ 25 g. Under 16:8 TRF, mice were allowed access to food between ZT13 (9 p.m.) and ZT21 (5 a.m.). Food access was regulated by transferring mice daily between cages with food and water and cages with water only. As a control, ad-lib-fed mice were also transferred between feeding cages at the same time.
For NRF and DRF, mice were allowed access to food between nighttime (8 p.m. to 8 a.m.) or daytime (8 a.m. to 8 p.m.) for one month, separately.
Drug treatments
SGC707 (S7832, PRMT3 inhibitor), CTPI-2 (S2968, SLC25A1 inhibitor) and MK-2206 2HCL (S1078, AKT1/2/3 kinase inhibitor) were purchased from MaoKang Biotechnology. For prevention or reversion phenotypes of mice fed an HFD, SGC707 was administered at 10 mg/kg or 30 mg/kg via intraperitoneal injection on alternate days, respectively. SGC707 was diluted in DMSO (at 5% final concentration), and DMSO served as the vehicle control.
For MK-2206 treated experiments, MK-2206 was diluted in DMSO (at 5% final concentration) and administered at 50 mg/kg via intraperitoneal injection, with DMSO serving as a vehicle control.
Drug analysis by using HPLC-MS/MS
The drug was fully dissolved in methanol at 1 µg/mL for HPLC-MSMS analysis. The drug compounds in mice tissue samples were first extracted with methanol and acetonitrile (1:9, v/v). After thorough vortex and ultrasonication, the samples were centrifuged (×18,000 g, 4 °C for 15 min) to extract the supernatants. Following vacuum dryness (Savant SPD2010, Thermo scientific), the residues were redissolved in acetonitrile and H2O (1:1, v/v) for further analysis. All analytical chemicals, including formic acid (Honeywell), methanol (Macklin), acetonitrile (Fisher) and H2O (ThermoFisher) used in the analysis were MS-grade.
The extracts were analyzed using HPLC-QTOF MS/MS (1290II-6530C, Agilent) equipped with a Thermo Hypersil GOLD aQ C18 column (2.1 × 100 mm,1.9 μm) with a 24 min gradient. Flow rate 0.4 ml/min, Column temperature 40 °C. MS-grade water with 0.1% formic acid (v/v) was used as mobile phase A, and MS-grade ACN with 0.1% formic acid (v/v) as mobile phase B. The gradient started at 5% B, keep 1 min, gradually increased to 55% B at 12 min, and to 100% B at 17 min and maintained until 20 min, then reverted to the initial condition of 5% at 22 min and then held at 5% to 24 min. Sample analysis was conducted in positive ionization mode (ESI + ) and collected in full-scan and targeted MS/MS modes, respectively. The nitrogen temperature of the ion source was set to 250 °C, the dry gas 10 L/min, the nebulizer 45 psi, the sheath gas temperature 350 °C, the sheath gas flow rate 11 L/min, the nozzle voltage 1000 V, the Vcap 4000 V, the fragmentor 100 V, the skimmer 45 V, and the detection window 20ppm. For targeted MS/MS analysis, the collision energy was set to 20 eV.
Data was collected by Agilent data acquisition software in centroid mode. The Masshunter quantitative analysis software from the vendor was further used to extract the targeted ion chromatogram and MSMS spectra and to calculate the retention time, peak areas, signal-to-noise ratio, etc.
HEK293T, primary adipocyte and porcine adipocyte culture
HEK293T cells (CRL-3216, ATCC) were grown in Dulbecco’s modified eagle medium (DMEM)/high glucose with 10% fetal bovine serum (FBS) as well as 100 μ/mL penicillin and 100 μg/mL streptomycin (15140-122, Gibco). All cell lines were maintained under standard conditions (37 °C,5% CO2).
For primary adipocyte culture, subcutaneous and epididymal adipose tissues from 6- to 8-week-old mice were minced into small pieces, followed by incubation with 1.25 mg/mL Collagenase from Clostridium histolyticum (C0130-5G, Sigma-Aldrich) in DMEM containing Penicillin/ Streptomycin for 40–50 min in a 37 °C shaker water bath. Cell suspension buffer was filtered through a 100 μm cell strainer and centrifugated for 10 min at 700 × g to get the stromal vascular fraction pellet. For white adipocyte differentiation, the stromal vascular fraction was cultured in DMEM complete medium to 90–95% confluence and then differentiated in DMEM complete medium containing (0.64 μM dexamethasone, 0.625 mM IBMX, 1.6 μg/ml insulin and 1 μM rosiglitazone) for 4 days, change medium every 2 days. Cells were then changed to DMEM complete medium containing 1.6 μg/ml insulin every 2 days until more than 90% of cells were fully differentiated to mature adipocytes before the following experiment. Insulin (PB180432) was obtained from Pricella Biotechnology; Dexamethasone (D4902), 3-isobutyl-methylxanthine (410957, IBMX), Rosiglitazone (557366-M) and 3,3’,5-Triiodo-L-thyronine (T2877, T3) were purchased from Sigma-Aldrich.
Immortalized porcine white pre-adipocytes were cultured in growth medium containing Dulbecco’s modification of Eagle’s medium (DMEM), 15% FBS (Gibco), 1% penicillin/streptomycin (Invitrogen) at 37 °C with 5% CO2. For porcine white adipocytes, adipogenic differentiation, the cells were induced with induction medium (IM) containing DMEM, 10% FBS (Gibco), 500 μM 3-isobutylmethylxanthine (Yeasen Biotechnology Co., Ltd.), 5 μg/mL insulin (WISENT Inc.), 1 μg/mL rosiglitazone (Yeasen Biotechnology Co., Ltd.), and 1 μM dexamethasone (Yeasen Biotechnology Co., Ltd.) for 3 days and then differentiated in differentiation medium (DM) contains DMEM, 10% FBS and 5 μg/mL insulin for 2 days.
Histological analysis
Mice were anesthetised with isoflurane and then euthanasia via cervical dislocation, and adipose and liver tissues were harvested and fixed with 4% paraformaldehyde. Paraffin embedding, tissue sectioning, and hematoxylin and eosin staining were performed.
Total RNA extraction and real-time PCR
Mice were anesthetized with isoflurane, and then euthanasia via cervical dislocation and tissues samples were collected and frozen with liquid nitrogen. Total RNA was extracted from cells or tissues by using Trizol Reagent (15596026, Invitrogen) according to the manufacturer’s instructions. Synthesis of cDNA was performed using 1 μg total RNA from each sample using HiScript IV Reverse Transcriptase (R423-01, vazyme). Quantitative PCR (qPCR) was carried out using the ChamQ Universal SYBR qPCR Master Mix (Q711, vazyme) and analyzed on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific). Primer sequences were obtained from PrimerBank.
Transcriptome sequencing
Total RNA was extracted according to the instructions manual of the TRlzol Reagent (Life Technologies, California, USA). RNA concentration and purity were measured using NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA), following the manufacturer’s recommendations. Index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X. First-strand cDNA was synthesized using a random hexamer primer and M-MuLV Reverse Transcriptase. Second-strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adapter with hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 240 bp in length, the library fragments were purified with the AMPure XP system (Beckman Colter, Beverly, USA). Then 3 μl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. Then, PCR was performed using Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. Finally, PCR products were purified (AMPure XP system), and library quality was assessed using the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v4-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina platform and paired-end reads were generated.
Immunoprecipitation
The cells were collected and suspended in 1000 μL lysis buffer (1% Nonidet P-40, 1 mM EDTA, 150 mM NaCl in 50 mM Tris-HCL, pH 7.5) containing protease inhibitors cocktail (B14001, Bimake) and phosphatase inhibitors cocktail (B15001, Bimake), then passed 12 times through a 22 G needle followed by centrifugation at 12,000 × g for 10 min at 4 °C. Eighty microlitres of supernatants mixed with membrane solubilization buffer plus the 5 × loading buffer as the input sample. 800 μL of supernatants were immunoprecipitated with beads conjugated with the indicated antibodies at 4 °C for 6–8 h. After incubation, the beads were washed four times with a lysis buffer, and the precipitates were eluted with 5 × loading buffer. Elutes and whole-cell extracts were resolved on SDS-PAGE, followed by a western blot with the indicated antibodies. The following plasmids were constructed by standard molecular cloning technics: pcDNA3.1-PRMT3-MYC encodes full-length human PRMT3 followed by MYC epitope tag, pCMV-PRMT3-HA encodes full-length mouse PRMT3 followed by HA epitope tag, pcDNA3.1-PRMT5-MYC encodes full-length human PRMT5 followed by MYC epitope tag, pCGN-SLC25A1-Flag encodes full-length mouse SLC25A1 followed by Flag epitope tag.
Immunoblotting
Mice were anesthetized with isoflurane, and then euthanasia via cervical dislocation and tissues samples were collected and frozen with liquid nitrogen. Cells or tissues were collected and homogenized in the RIPA buffer (1% Nonidet P-40, 0.1% SDS, 1 mM EDTA, 150 mM NaCl in 50 mM Tris-HCL, pH 7.5) supplemented with protease inhibitor cocktail and phosphatase inhibitors cocktail. The protein concentrations of lysates were determined using the BCA method (A55864, Thermo Fisher Scientific). The equal amounts of total proteins were resolved by SDS–PAGE and transferred to PVDF membranes (IPVH00010, Merck Millipore). Immunoblots were blocked with 5% non-fat milk (1172GR500, BioFroxx) in Tris-buffered saline (TBS) containing 0.01% Tween 20 (TBST), probed with indicated primary antibodies overnight at 4 °C. After washing in TBST three times, blots were incubated with horseradish peroxidase-conjugated secondary antibodies diluted in TBST supplemented with 5% skim milk for 1 h at room temperature and followed by at least 3 washes with TBST. Immunoreactivity was developed with enzymatic detection using SuperPico ECL Chemiluminescence Kit (E422, E423, Vazyme).
Primary antibodies used for immunoblots were as follows: anti-Mono-Methyl Arginine (8015, MMA, diluted 1:1000), Asymmetric Di-Methyl Arginine Motif (13522, ADMA, diluted 1:1000), Symmetric Di-Methyl Arginine Motif (13222, SDMA, diluted 1:1000), Normal anti-rabbit lgG (2729), Normal anti-mouse lgG (68860), Phospho-Akt (Ser473) (4060, diluted 1:1000), AKT (9272, diluted 1:1000), PRMT4 (3379, diluted 1:1000), PRMT5 (79998, diluted 1:1000), PKM1/2 (3106, diluted 1:1000), Pyruvate Carboxylase (49381, PC, diluted 1:1,000), LDHA (2012, diluted 1:1000), CPT2 (52552, diluted 1:1000), PGK1 (63536, diluted 1:1000), PCK1 (12940, diluted 1:1000), Phospho-HSL (Ser660) (45804, diluted 1:1000) antibodies were purchased from Cell Signaling technology. Anti-ATGL (55190-1-AP, diluted 1:1000), HSL (17333-1-AP, diluted 1:1000), HSP90 (13171-1-AP, diluted 1:1000), PRMT3 (17628-1-AP, diluted 1:1000), Beta Actin (66009-1-Ig, diluted 1:1000), GAPDH (60004-1-Ig, diluted 1:1000), Alpha Tubulin (66031-1-Ig, diluted 1:5000), SLC25A1 (15235-1-AP, diluted 1:1000) antibodies were obtained from Proteintech. Anti-FLAG-M2 (F1804, diluted 1:5000), FLAG (SAB4301135, diluted 1:5000), HA (H6908, diluted 1:5000), FLAG (H3663, diluted 1:5000), MYC (C3956, diluted 1:5000), MYC (M4439, diluted 1:5000) antibodies were purchased from Sigma. Horseradish peroxidase-conjugated goat anti-mouse (115-035-003, diluted 1:5000) and anti-rabbit (711-035-152, diluted 1:5000) secondary antibodies were from Jackson ImmunoResearch Laboratories.
Glucose and insulin tolerance test
Glucose and insulin tolerance tests (GTT, ITT) were performed by an intraperitoneal injection of glucose (2 g/kg body weight), insulin (0.03 μg/kg body weight) in mice following 14 hours fasting (for GTT) or 6 hours fasting (for ITT). Blood was collected from a small incision from the tip of the tail before injection (Time 0), then 15, 30, 60, 90 and 120 min after injection of glucose or insulin. Blood glucose levels were measured by a blood glucose meter (Accu-Chek® Active, Roche).
Indirect calorimetry and body composition measurement
The oxygen consumption (VO2) and carbon dioxide production (VCO2) of the mice were measured using an indirect calorimetry system (Oxymax, Columbus Instruments). The system was kept at a stable environmental temperature (24 °C) that also had a 12 h light (8 AM-8 PM) and a 12 h dark cycle (8 PM-8 AM). Mice were individually placed in each chamber with free access to food and water. Mice were adapted to the chamber for 24 h before the measurements. The data were presented as uncorrected energy expenditure levels. Average energy expenditure of day (8 AM-8 PM) and night (8 PM-8 AM) values were the average mean value of all points measured during the 12 h. Total body fat and lean mass in live animals without anesthesia were measured by using a Minispec LF50 body composition analyzer located in the Small Animal Facility of CAM-SU. Animals were placed in a specially sized, clear plastic holder without sedation or anesthesia. The holder was then inserted into a tubular space on the side of the Minispec LF50 system. The animals were forced to not move in the holder to guarantee the accuracy of the result. Each scan took about 2 minutes.
Blood biochemistry
Blood biochemistry was performed by using a clinical chemistry analyzer (Hitachi 7100). An appropriate volume of blood that was required (160–200 μl of plasma) for the test was collected from each mouse (mice were anesthetized with isoflurane) and transferred to a gel tube containing lithium Heparin and centrifuge for 15 minutes at 5000 × g in a refrigerated centrifuge set at 4 °C. If plasma samples cannot be analyzed immediately, keep them at − 80 °C before analysis. Use plasma samples undiluted or diluted to a ratio of 1:2 with deionised water if the volume was insufficient.
Targeted metabolomics
Collected samples use a liquid mass platform (UPLC-TQ-MS) to detect energy metabolism-related metabolites in biological (Metabo- Profile, Shanghai, P. R. China). The solvents used were chromatographic grade methanol, acetonitrile, and formic acid, which were purchased from Merck Chemicals, Germany. The experimental ultrapure water was prepared by the Millipore Reference ultrapure water system (Billerica, MA, USA) equipped with a 0.22 μm filter head for LC/MS. Before processing the sample, the sample needs to be thawed in an ice bath to reduce sample degradation. Weigh 10 mg tissue into a 1.5 mL EP tube, add 20 μL ultrapure water and 10 magnetic beads, homogenize for 3 min (BB24, Next Advance, Inc., Averill Park, NY, USA), and then add 120 μL pre-cooling. The methanol solution containing internal standard was homogenized again for 3 min. Centrifuge at 18000 × g for 20 min at 4 °C (Microfuge 20 R, Beckman Colter, Inc., Indianapolis, IN, USA), take 100 μL the supernatant in a 1.5 mL EP tube, and concentrate at 4 °C (Labconco, Kansas City, MO, USA), add 100 μL 80% methanol solution for reconstitution, centrifuge and transfer to 96-well plate, waiting for sample injection analysis. An ultra-high pressure liquid chromatography-triple quadrupole mass spectrometer (ACQUITY-I UPLC / Xevo TQ-S) from Waters Corporation was used to detect energy metabolism-related metabolites. The raw data generated by UPLC-TQ-MS will use Waters MassLynx software (v4.1, Waters, Milford, MA, USA) for peak extraction, integration, identification and quantitative analysis of each metabolite. Self-developed iMAP software (V1.0, Metabo-Profile, Shanghai, China) is used for subsequent statistical analysis, such as PCA, OPLS-DA and one-dimensional statistical testing.
Measurement of ECAR
ECAR of CTPI-2 (S2968, SLC25A1 inhibitor) and SGC707 treated adipocytes were analyzed using an XF96 Analyzer (Seahorse Bioscience). In brief, 2 × 104 cells per well were plated in Seahorse XF96 plates in 200 μl of DMEM supplemented with 2 mM glutamine. The plates were incubated for 1 h at 37 °C in a humidified incubator without CO2. After assessing the ECAR at baseline, the following compounds were sequentially added to the analyzer, and the change in ECAR was measured: (1) after the addition of 10 mM glucose; (2) after the sequential addition of 1 μM oligomycin and (3) after the sequential addition of 50 mM 2-DG. In this assay, maximum ECAR occurs after the addition of oligomycin. All ECAR measures were performed three times in a mix–wait–measure cycle of 3, 2, and 3 min, respectively. Rotenone (S2348), 2-Deoxy-D-glucose (S4701,2-DG), Oligomycin A (S1478) and Indomethacin (S1723) from Selleck Chemicals; Antimycin A (MS0070) were obtained from Shanghai MaoKang Biotechnology, China.
Statistical analysis
Data analysis was conducted mainly in Prism 8.0.1. The rhythmicity of gene expression was evaluated using JTK analysis, with a p-value of <0.05 indicative of rhythmicity. All analyses were conducted using the Student’s t test (two-tail). All experimental data were presented as mean ± SEM. Comparisons with P-values < 0.05, < 0.01 or< 0.001 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the supplementary materials. Source data are provided with this paper. RNA-seq and Metabolomics data has been updated as Supplementary Data. Source data are provided in this paper.
Code availability
No new code has been generated in this study.
References
Mihaylova, M. M. et al. When a calorie is not just a calorie: Diet quality and timing as mediators of metabolism and healthy aging. Cell Metab. 35, 1114–1131 (2023).
Longo, V. D. & Mattson, M. P. Fasting: molecular mechanisms and clinical applications. Cell Metab. 19, 181–192 (2014).
Green, C. L., Lamming, D. W. & Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 23, 56–73 (2022).
Smith, R. L., Soeters, M. R., Wüst, R. C. & Houtkooper, R. H. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr. Rev. 39, 489–517 (2018).
Chaix, A., Lin, T., Le, H. D., Chang, M. W. & Panda, S. Time-restricted feeding prevents obesity and metabolic syndrome in mice lacking a circadian clock. Cell Metab. 29, 303–319. e304 (2019).
Hatori, M. et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 15, 848–860 (2012).
Hepler, C. et al. Time-restricted feeding mitigates obesity through adipocyte thermogenesis. Sci. Signal. 378, 276–284 (2022).
Bao, R. et al. Effects of time-restricted feeding on energy balance: a cross-over trial in healthy subjects. Front. Endocrinol. 13, 870054 (2022).
Liu, D. et al. Calorie restriction with or without time-restricted eating in weight loss. N. Engl. J. Med. 386, 1495–1504 (2022).
Quist, J. S. et al. Effects of 3 months of 10-h per-day time-restricted eating and 3 months of follow-up on bodyweight and cardiometabolic health in Danish individuals at high risk of type 2 diabetes: the RESET single-centre, parallel, superiority, open-label, randomised controlled trial. Lancet Healthy Longev. 5,e314–e325 (2024).
Teong, X. T. et al. Intermittent fasting plus early time-restricted eating versus calorie restriction and standard care in adults at risk of type 2 diabetes: a randomized controlled trial. Nat. Med. 29, 963–972 (2023).
Weir, H. J. et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26, 884–896 (2017).
Hasek, B. E. et al. Dietary methionine restriction enhances metabolic flexibility and increases uncoupled respiration in both fed and fasted states. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R728–R739 (2010).
Malloy, V. L. et al. Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging Cell 5, 305–314 (2006).
Malloy, V. L. et al. Methionine restriction prevents the progression of hepatic steatosis in leptin-deficient obese mice. Metabolism 62, 1651–1661 (2013).
Plaisance, E. P. et al. Dietary methionine restriction increases fat oxidation in obese adults with metabolic syndrome. J. Clin. Endocrinol. Metab. 96, E836–E840 (2011).
Capelo-Diz, A. et al. Hepatic levels of S-adenosylmethionine regulate the adaptive response to fasting. Cell Metab 35, 1373–1389 (2023).
Sáenz de Urturi, D. et al. Methionine adenosyltransferase 1a antisense oligonucleotides activate the liver-brown adipose tissue axis preventing obesity and associated hepatosteatosis. Nat. Commun. 13, 1096 (2022).
Blanc, R. S. & Richard, S. J. M. C. Arginine methylation: the coming of age. Mol. Cell 65, 8–24 (2017).
Tang, J. et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275, 7723–7730 (2000).
Choi, S. et al. Depletion of Prmt1 in adipocytes impairs glucose homeostasis in diet-induced obesity. Diabetes 70, 1664–1678 (2021).
Hirai, H. et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967 (2010).
Kaniskan, H. Ü et al. A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew. Chem.127, 5255–5259 (2015).
Liu, W. et al. IL-1R-IRAKM-Slc25a1 signaling axis reprograms lipogenesis in adipocytes to promote diet-induced obesity in mice. Nat. Commun. 13, 2748 (2022).
Mosaoa, R., Kasprzyk-Pawelec, A., Fernandez, H. R. & Avantaggiati, M. L. The mitochondrial citrate carrier SLC25A1/CIC and the fundamental role of citrate in cancer, inflammation and beyond. Biomolecules 11, 141 (2021).
Wei, H.-H. et al. A systematic survey of PRMT interactomes reveals the key roles of arginine methylation in the global control of RNA splicing and translation. Sci. Bull. 66, 1342–1357 (2021).
Cienfuegos, S. et al. Effects of 4-and 6-h time-restricted feeding on weight and cardiometabolic health: a randomized controlled trial in adults with obesity. Cell Metab. 32, 366–378. e363 (2020).
Tan, M. et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 27, 2143–2157 (2020).
Infantino, V., Iacobazzi, V., Menga, A., Avantaggiati, M. L. & Palmieri, F. J. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα-and IFNγ-triggered inflammation. Biochim. et Biophys. Acta Gene Regul. Mech. 1839, 1217–1225 (2014).
Goodpaster, B. H. & Sparks, L. M. Metabolic flexibility in health and disease. Cell Metab. 25, 1027–1036 (2017).
Galgani, J. E., Moro, C. & Ravussin, E. Metabolic flexibility and insulin resistance. Am. Physiol. Endocrinol. Metab. 295, https://doi.org/10.1152/ajpendo.90558.2008 (2008).
Reshef, L. et al. Glyceroneogenesis and the triglyceride/fatty acid cycle. J. Biol. Chem. 278, 30413–30416 (2003).
Deota, S. et al. Diurnal transcriptome landscape of a multi-tissue response to time-restricted feeding in mammals. Cell Metab. 35, 150–165. e154 (2023).
Xin, H. et al. A multi-tissue multi-omics analysis reveals distinct kineztics in entrainment of diurnal transcriptomes by inverted feeding. Iscience 24, 102335 (2021).
Huang, R. et al. Multi-omics profiling reveals rhythmic liver function shaped by meal timing. Nat. Commun. 14, 6086 (2023).
Mauvoisin, D. et al. Circadian and feeding rhythms orchestrate the diurnal liver acetylome. Cell Rep. 20, 1729–1743 (2017).
Xin, H. et al. Daytime-restricted feeding enhances running endurance without prior exercise in mice. Nat. Metab. 5, 1236–1251 (2023).
Leone, A., Moncada, S., Vallance, P., Calver, A. & Collier, J. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339, 572–575 (1992).
Antoniades, C. et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur. Heart J. 30, 1142–1150 (2009).
Teerlink, T., Luo, Z., Palm, F. & Wilcox, C. S. Cellular ADMA: regulation and action. Pharmacol. Res. 60, 448–460 (2009).
Jacobi, J. et al. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E-deficient mice by lowering asymmetric dimethylarginine. Am. J. Pathol. 176, 2559–2570 (2010).
Xiao, H.-B. et al. Effect of asymmetric dimethylarginine on atherogenesis and erythrocyte deformability in apolipoprotein E deficient mice. Life Sci. 81, 1–7 (2007).
Shendre, A. et al. Admixture mapping of subclinical atherosclerosis and subsequent clinical events among African Americans in 2 large cohort studies. Circ. Cardiovasc. Genet. 10, e001569 (2017).
Kim, D. -i et al. PRMT3 regulates hepatic lipogenesis through direct interaction with LXRα. Diabetes 64, 60–71 (2015).
Nahon, J. E., Groeneveldt, C., Geerling, J. J., van Eck, M. & Hoekstra, M. Inhibition of protein arginine methyltransferase 3 activity selectively impairs liver X receptor-driven transcription of hepatic lipogenic genes in vivo. British J. Pharmacol. 175, 3175–3183 (2018).
de Jong, L. M. et al. PRMT3 inhibitor SGC707 reduces triglyceride levels and induces pruritus in Western-type diet-fed LDL receptor knockout mice. Sci. Rep. 12, 483 (2022).
Acknowledgements
We thank all members of the Jia and Zhang laboratories for helpful discussions. We thank CAM-SU for excellent guidance and assistance with experiments performed in this study. Research support was provided by STI2030-Major Projects (2021ZD0203400 to Y.Z.), National Natural Science Foundation of China (32100944 to Z.J. and 32271206 to Y.Z.), Natural Science Foundation of Jiangsu Province (BK20210715 to Z.J.), Gusu Innovation and Entrepreneur Leading Talents project (ZXL2023200 to Z.J.), Natural Science Foundation of Jiangsu Province (No. BK20255001), Ministry of Science and Technology (2018YFA0801102 to Y.D.). YZ’s lab was supported by the Suzhou International Joint Laboratory for Diagnosis and Treatment of Brain Diseases.
Author information
Authors and Affiliations
Contributions
Z.J. and Y.Z. conceived the study, Z.J. and A.V. wrote the manuscript. Z.J., Y.Z., A.V., and Z.H. designed experiments. Z.H., X.L., X.C., You, Q.C., H.Z., M.D., and S.L. performed experiments. Z.J., Z.H., and K.C. visualized and curated the data. Y.L., K.C., Y.F., L.S., L.W., T.S., S.K., and Y.D. contributed reagents/materials/analysis tools. Z.J., Y.Z., and A.V. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rui Beleza and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Z., Liu, X., Chen, X. et al. PRMT3-mediated post-translational adaptation to fasting regulates metabolic flexibility. Nat Commun 17, 2264 (2026). https://doi.org/10.1038/s41467-026-68883-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68883-6
