
Abstract
Protein C-termini can vary due to errors or programmed regulation, contributing to proteome diversity, yet their impact on the proteome remains poorly understood. Although aberrant C-termini are often linked to protein degradation, it is unclear if this holds true universally. In this study, we examine how C-terminal variations—arising from disease-associated nonstop mutations, alternative splicing, and translational readthrough—affect protein half-lives. Our findings indicate that, contrary to previous studies, erroneous C-termini can either stabilize or destabilize proteins. We have identified multiple oncoproteins and tumor suppressors whose protein stability is altered by disease-relevant nonstop mutations. Notably, we have found that C-terminal variations commonly influence the stability of canonical proteins, extending beyond their role in protein quality control. Furthermore, we have uncovered C-terminal features that distinguish erroneous from wild-type proteins and reveal that hydrophobic C-termini are targeted by a complex ubiquitin ligase network. Overall, our work broadens the understanding of C-terminal-dependent protein degradation and supports that C-terminal variation is a widespread strategy for generating protein forms with distinct half-lives to exert diverse biological functions.
Similar content being viewed by others

Introduction
Diversity in a given protein’s C-terminal tail is a common feature of proteomes. This diversity can arise from various errors, such as genetic mutations, spontaneous transcriptional or translational mistakes, erroneous splicing, and posttranslational damage. For instance, nonsense and frame-shifting indel (insertion-deletion) mutations produce truncated proteins that terminate with internal peptides or frameshift C-termini, respectively. Nonstop mutations (those occurring within the stop codon of a gene) lead to the expression of full-length proteins displaying C-terminal extensions encoded by the 3’ untranslated region (UTR). Translational misreading, such as ribosomal frameshifting and stop codon readthrough, also generates proteins with abnormal C-termini. It has been estimated that basal stop codon readthrough occurs in mammalian cells at levels ranging from 0.01 to 0.1%, depending on the type of stop codon, and spontaneous frameshift errors can occur at a rate of approximately one in 105 codons1,2. These anomalous C-termini were shown to exhibit elevated hydrophobicity, which marks the resulting defective proteins for elimination via Bag6 chaperone-associated E3 ligases—namely RNF126 and RNF1153,4. However, it remains unclear if Bag6-E3 complexes alone are responsible for recognizing the entire spectrum of hydrophobic C-termini in human cells. Moreover, apart from a few documented examples5,6, the effects of disease-associated nonstop C-terminal extensions on the respective mutated proteins remain largely unknown. A recent study systematically investigated the impact of disease-relevant nonstop mutations7. However, that study overlooked the potential influence of canonical C-termini on protein half-life, which may affect the validity of its conclusions.
In addition to unintended errors, programmed regulatory mechanisms such as mRNA alternative splicing and programmed translational recoding events also expand the repertoire of protein C-terminal variants. Alternative splicing occurs in up to 98% of human multi-exon coding genes and often leads to the expression of nearly identical protein isoforms with subtle differences at their C-termini. Programmed stop codon readthrough (context-dependent readthrough) is a well-known form of translational reprogramming, whereby stop codons are interpreted as sense codons that incorporate either a canonical amino acid or a specialized one, such as selenocysteine (Sec) or pyrrolysine (Pyl), resulting in the expression of proteins with extended C-termini8,9,10,11. Ribosome profiling and mass spectrometry analyses have revealed that programmed readthrough is much more widespread than previously appreciated12,13,14,15. Although the frequency of occasional stop codon readthrough is low, it increases dramatically in programmed readthrough, reaching up to 10-80%, depending on the sequence context of the mRNA and trans factors1,10,16. It has been shown previously that isoform-specific C-termini regulate the stability of SRY, MAX, and SMN2 proteins, with these variations contributing to sex determination, the hypoxia response, and the severity of spinal muscular atrophy, respectively17,18,19. Our lab has shown that C-terminal extensions in selenoproteins inhibit C-degrons and subsequently prevent selenoprotein degradation20. However, current attention on C-terminal-dependent protein degradation is primarily focused on its role in protein quality control3,21,22,23,24. How this pathway influences the canonical proteome and affects diverse biological processes has yet to gain global attention.
In this work, we conduct a comprehensive analysis of the impacts on protein half-life of C-terminal variants resulting from disease-relevant nonstop mutations, alternative mRNA splicing, and programmed translational readthrough. We also investigate how C-terminal amino acid composition and patterns influence protein stability and apply this information to compare canonical and deviant C-termini. Moreover, we perform CRISPR screens to identify additional ubiquitin ligases that target hydrophobic C-termini. Our work not only expands the list of protein species subjected to C-terminal-dependent protein degradation but also indicates that, beyond maintaining proteome fidelity, C-terminal alterations serve as a universal mechanism for modulating the abundance of active proteins to meet various biological needs.
Results
Disease-associated nonstop mutations can either prolong or impair protein half-life
Compared to frameshift and nonsense mutations, nonstop mutations have received less clinical attention because they produce proteins of near-normal length and presumed biological function. Multiple previous reports have suggested that readthrough (RT) C-termini resulting from nonstop mutations and translational readthrough can promote protein degradation5,6,7,21. For example, Ghosh et al. examined the effects of cancer-associated nonstop mutations by fusing the corresponding RT C-termini to the C-terminus of GFP7. They found that these aberrant C-termini generally reduced GFP expression, and accordingly classified them as being primarily suppressive. However, one limitation of the approach adopted by Ghosh et al. is that it overlooks the potential influence of the wild-type (WT) C-termini on protein half-life. Rather than performing a direct comparison between the effects of RT and WT C-termini on GFP stability, they compared the stability of GFP fused with RT C-termini to GFP alone (Supplementary Fig. 1A, top panel). Here, we found that, similar to RT C-termini, tagging GFP with WT protein C-termini can also lead to GFP degradation (Fig. 1A). However, surprisingly, the degronic potency of the WT C-termini can be either stronger (e.g., MDC, MYH2) or weaker (e.g., TOPORS, PRDM1) than that of their RT counterparts (Fig. 1B). This outcome suggests that nonstop mutations and translational readthroughs may not only be suppressive, but could also be supportive or stimulative, thereby increasing protein abundance.

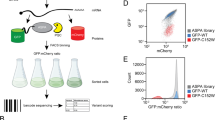
A Comparison of protein stability between GFP alone and GFP fused to the C-terminal 20 residues of the indicated wild-type (WT) proteins, as measured by GPS assays. B Analysis of GFP stability when tagged with WT or readthrough (RT) C-termini, as indicated on the left. C Schematic of the high-throughput platform used to assess the impact of WT and disease-relevant RT C-termini on protein stability. Oligonucleotides encoding 58-residue WT and RT C-termini were cloned into the GPS reporter and introduced into HEK293T cells to generate stable GPS reporter lines. Cells were sorted into eight bins based on GFP/RFP ratios, and oligonucleotides from each bin were sequenced to calculate a Protein Stability Index (PSI) for each peptide, with lower PSI values indicative of stronger degradation signals. D Correlation of PSI values across two independent biological replicates. E Violin plots showing the distributions of mean PSI for WT and RT peptides across two biological replicate screens. Solid and dashed lines indicate the median and the first/third quartiles, respectively. Asterisks indicate statistical significance (****, p = 3.9 × 10−44; unpaired two-tailed Students’ t test). F Ranked distribution of mean ∆PSI (PSIRT-PSIWT) values for genes carrying nonstop mutations across two biological replicate screens. G Proportions of RT extensions resulting in stabilization or degradation of protein stability, or no effect (neutral). H Pie charts comparing the results from Ghosh et al.7 with findings from the current study. Source data are provided as a Source Data file.
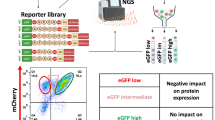
To test this hypothesis, we performed a GPS (global protein stability)-based peptidomic screen to directly compare the degradation effects of disease-relevant nonstop RT C-termini and their WT counterparts (Fig. 1C and Supplementary Fig. 1A, bottom panel). The GPS approach utilizes a lentiviral-based reporter that co-expresses two fluorescent proteins—GFP fused to the peptide of interest and RFP as an internal control—via an internal ribosome entry site (IRES). Since both the GFP-peptide fusion construct and RFP are translated from the same mRNA, the GFP/RFP ratio represents a measure of the effect of the peptide sequence on GFP stability25. We assessed 2227 nonstop mutations from the pan-cancer NonStopDB dataset6, as well as an additional 799 nonstop mutations and 415 readthrough single-nucleotide polymorphisms (SNPs), some of which are associated with human hereditary diseases5. Specifically, we cloned oligonucleotides encoding the last 58 residues of WT and RT proteins into the GPS reporter, introduced these constructs into HEK293T cells via single-copy viral transduction, and sorted the GPS reporter cells into eight bins based on GFP/RFP ratios, with these latter indicatives of the stability of the expressed GFP-peptide fusion construct in each cell. We retrieved oligonucleotides from each bin for sequencing, calculated the Protein Stability Index (PSI) of each peptide, and then determined the difference in PSI between the RT and WT peptides (∆PSI) (Fig. 1C). A positive ∆PSI value indicates that the nonstop mutation increases the stability of the corresponding protein, and vice versa. We conducted two biological replicates of the screen, which were highly correlated (Fig. 1D).
Consistent with the notion that canonical protein C-termini have evolved to evade C-terminal-mediated degradation26,27, we found that, on average, RT C-termini are more effective at promoting protein degradation than WT C-termini (Fig. 1E). However, each individual nonstop mutation may either enhance (9.51%) or reduce (28.4%) the stability of the corresponding proteins (Fig. 1F, G), using a threshold of |ΔPSI | ≥ 1. Next, we compared our screening results with those of Ghosh et al., who reported that most nonstop mutations reduce protein expression and that the remainder could be considered neutral7. Among the 1908 mutations common to both studies, only 42.89% of those previously annotated as destabilizing actually reduced host protein stability, and in fact, 6.01% were found to increase it. Furthermore, more than 27% of mutations previously considered neutral had measurable effects on protein stability: 16.54% led to stabilization, whereas 11.08% caused degradation (Fig. 1H). The complete dataset is available in Supplementary Data 1.
The length of RT tails resulting from each nonstop mutation varies depending on the location of the downstream stop codon within the 3’ UTR. We examined if changes in protein stability are correlated with RT tail length. Although we did detect a positive correlation, our data also indicate that in many cases even small extensions can significantly influence protein stability (Fig. 2A). To identify short degron motifs, we focused on RT extensions of 20 amino acids or fewer (Fig. 2B). Notably, many of the observed degradation or stabilization events can be attributed to known C-degrons, such as diGlu, diGly and other Gly-based degrons26,28 (Fig. 2C and Supplementary Fig. 1B). Introducing these motifs led to protein destabilization, whereas capping existing C-degrons with RT C-terminal extensions resulted in protein stabilization (Fig. 2D). In addition to known motifs, we identified previously unreported C-terminal sequences that influence protein stability (Fig. 2E and Supplementary Fig. 1C). We validated their effects in full-length proteins using GPS assays (Fig. 2F) and cycloheximide (CHX)-chase experiments with FLAG-tagged constructs (Supplementary Fig. 1D).

A Correlation between the length of RT extensions and the magnitude of changes in protein stability (∆PSI). Box plots indicate medians (central line), first and third quartiles (box edges), and 10th and 90th percentiles (whiskers). B Comparison of PSI values between WT and RT peptides for nonstop mutations resulting in ≤ 20-residue extensions. Blue triangles denote degradation or stabilization caused by the introduction or disruption of known C-degrons. C C-terminal sequences corresponding to blue triangles in (B), with known degron motifs highlighted in blue. D Validation of protein stability changes using full-length WT and RT proteins. The type of nonstop mutation is labeled on the blots. E C-terminal sequences of proteins analyzed in (F). F Validation of stability changes caused by nonstop mutations using full-length constructs. G Impact of nonstop mutations on the stability of oncoproteins and tumor suppressors identified in the screen, based on TSGene and ONGene annotations54,55. H, I Validation of changes in nonstop mutation-induced protein stability in full-length tumor suppressors, oncoproteins, and proteins associated with genetic diseases. Source data are provided as a Source Data file.
Next, we explored the potential pathogenic relevance of protein stability changes caused by nonstop mutations, given that alterations in protein abundance are a key mechanism in cancer and other diseases. Our data revealed a modest bias toward destabilization in tumor suppressor proteins compared to oncoproteins (Fig. 2G). We validated experimentally the nonstop mutation-induced stabilization of several oncoproteins (CCND2, SNCG, FOXA1, HEY1, CPNE9) and the degradation of several tumor suppressor proteins (MAX, PTEN, RAD23B, LIMD1, CTCF, AHNAK, DAB2, RBM4, BCL2L11, DUSP9) by GPS and CHX-chase assays (Fig. 2H, Supplementary Fig. 2A–C). In addition, we confirmed degradation of the proteins MOCS2, NHP2, and FHL1 caused by genetic disease-associated nonstop mutations (Fig. 2I, Supplementary Fig. 2B, C). The differential degradation of wild-type or nonstop mutation-induced readthrough proteins is mainly proteasome-dependent (Supplementary Fig. 2D). Although nonstop mutations account for ~ 0.2% of all codon-changing mutations, they remain among the least studied due to their presumed mild effects29. Our work provides the most comprehensive functional annotation of disease-relevant nonstop mutations to date, demonstrating that—like nonsense and missense mutations—nonstop mutations may substantially affect cellular processes by altering the abundance of the respective proteins.
Alternative splicing or programmed readthrough diversifies protein C-termini to impact protein stability
In addition to mutations and translational errors, mRNA alternative splicing and programmed translational readthrough contribute to the diversity of the protein C-terminome. Though most studies on C-terminal-mediated degradation have focused on its role in protein quality control, the effects of physiological C-terminal diversification on the proteome remain largely unexplored.
We conducted a sequence analysis of protein isoforms derived from alternative mRNA splicing. Our results reveal that isoforms with distinct C-termini are remarkably widespread. Up to 62.5% of human genes produce isoforms with more than one type of C-terminus, and 22.4% generate isoforms with five or more distinct C-terminal variations (Fig. 3A). Focusing specifically on isoforms that differ only at their C-termini (i.e., with the remainder of the protein sequence being identical), we found that more than 3000 genes encode isoforms with variations of fewer than 20 amino acids, and more than 1000 genes produce isoforms differing by fewer than five residues. Prior research on SMN2 protein has demonstrated that isoform-specific C-terminal variation affects its stability19. To determine if this represents a general phenomenon, we examined 11 additional genes and observed that isoforms of the majority (7 out of 11) exhibited distinct protein half-lives, indicating that C-terminal diversification is a common mechanism for regulating isoform stability (Fig. 3B, C and Supplementary Fig. 3A).

A Analysis of isoform-specific C-terminal variants (last 20 residues) generated by alternative splicing across 21,784 human genes. B Comparison of protein stability between full-length isoforms of the indicated genes. C C-terminal sequences of the isoforms analyzed in (B). Differences between isoforms are highlighted in red. D, E Comparison of stability between full-length canonical proteins and their corresponding programmed RT variants. Source data are provided as a Source Data file.
Programmed translational readthrough has been identified in more than 100 human genes through techniques such as ribosome profiling, mass spectrometry, and reporter assays (Supplementary Fig. 3B and Supplementary Data 2). There is already evidence that the extended C-termini generated by programmed readthrough can influence protein stability. For instance, our previous study uncovered that recoding the UGA codon into Sec increases selenoprotein stability20. We have also verified that the double-readthrough form of MTCH2 (MTCH2xx) is less stable than its shorter variants (MTCH2, MTCH2x) 30 (Fig. 3D). To further assess the impact of programmed readthrough, we examined ten selected genes and found that readthrough events affected protein stability in six of them, including CGGBP1, BTG1, TIMP1, SQSTM1, PRDX4, and VDR (Fig. 3E and Supplementary Fig. 3C). In these cases, the readthrough isoforms were either more or less stable than their canonical forms. Together, these findings underscore the universal and functional significance of protein C-terminal diversification—both canonical and aberrant—in regulating protein half-lives across the proteome.
Amino acid composition and arrangement in protein C-termini influence protein half-lives
Given the widespread influence of protein C-termini on protein stability, we investigated the specific features of the C-terminus that affect protein half-lives. Previous studies, including our own, have defined several C-degron motifs that promote protein degradation26,28. However, our research has indicated that most C-terminal sequences influencing stability do not adhere to well-defined consensus motifs. This sequence degeneracy likely accounts for the broad impact of C-termini on the proteome. Supporting this idea, we observed that attaching commonly used epitope tags to protein C-termini also alters their half-lives (Fig. 4A). Traditionally, the identification of functional motifs relies on multiple sequence alignment to detect conserved residues, followed by site-directed mutagenesis for validation. However, this method has limitations; principally, in that it struggles to detect ambiguous, complex, or dropout features. Furthermore, conclusions drawn from mutagenesis are often template- and amino acid-specific, and extending such analyses across multiple sequences or residues is labor-intensive. To address these challenges, we employed a context-independent add-in approach. To do so, specific amino acid features were introduced into randomized peptide sequences, enabling assessment in a pooled manner of their effects across diverse sequence contexts. Previously, we applied this random peptide platform effectively to study how sequences surrounding known C-degrons influence degradation efficiency27.

A Comparison of GFP stability with or without C-terminal fusion to various epitope tags. B Schematic of peptide templates designed to examine the influence of amino acid identity and position on protein stability. C Random peptide assays conducted in biological triplicate to assess degron activity across libraries with varying amino acid compositions and arrangements. Data are shown as mean \(\pm \)SD. Amino acids are color-coded by physicochemical properties. The gray bar represents the control template, with fully randomized 12-mer (terminal) or 15-mer (composition) peptides. Asterisks indicate statistical significance compared to control (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; unpaired two-tailed Students’ t test). D Relative degradation levels of peptide templates with dispersed versus clustered arrangement of the specified amino acids were quantified using the biological triplicate experiments. Percent degradation was normalized and compared to the reference template: x#x#xxx#xx#xxxx. Data are shown as mean \(\pm \)SD. Asterisks indicate statistical significance compared to control (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; unpaired two-tailed Students’ t test). All of the p-values in (C) and (D) are provided in Source Data file. Source Data are provided as a Source Data file.
To systematically explore the influence of amino acid identity, position, and arrangement on protein stability, we designed five peptide templates that varied either in terms of their terminal residue or overall composition (Fig. 4B). The results revealed several key trends. The presence of small terminal amino acids (Gly, Ala), or increased overall frequencies of Arg, Cys, or hydrophobic residues (Val, Ile, Leu, Met, Phe, Tyr, Trp), tended to promote degradation. In contrast, enrichment of acidic residues (Asp, Glu) was associated with increased stability (Fig. 4C). The positional context of these residues proved critical. For instance, although hydrophobic residue enrichment generally promoted degradation, their presence at the terminal position did not elicit the same effect. Similarly, Gly triggered degradation only when positioned at the extreme C-terminus. Most notably, the arrangement of amino acids also played a crucial role in determining protein half-life. Even when the total number of specific residues was held constant, clustering (rather than dispersing) them prompted a pronounced effect. In particular, consecutive Cys residues exerted the strongest synergistic degradative impact (Fig. 4D). These findings highlight that not only the identity of C-terminal residues, but also their positioning and arrangement, are key determinants of protein stability.
Enrichment of Cys and Trp, along with depletion of Asp and Glu, drives protein degradation arising from readthrough and frameshift events
Next, we compared the C-termini of canonical and aberrant proteins using insights from our random peptide assay as reference points (Figs. 5, 6). We focused our analysis on canonical cytosolic proteins, as proteins localized to different compartments or organelles are likely subject to distinct evolutionary pressures23,27. For example, secreted proteins may not encounter cytosol-localized E3 ligases. Specifically, we examined the C-termini of human cytosolic proteins using principal component analysis (PCA) of the final 150 amino acid residues. We detected clear signals of selective pressure on protein stability, evident in both amino acid composition and terminal residue usage. Specifically, we observed a gradual decrease in hydrophobic amino acids toward the C-terminus (PC1), along with a marked depletion of Gly and Ala at the extreme C-terminus (PC2) (Fig. 5A, B and Supplementary Fig. 4A, B). The percentages next to PC1 and PC2 represent the proportion of total variance in the dataset explained by each principal component. In addition to amino acid composition, amino acid arrangement also appears to be under evolutionary constraints. The frequency of consecutive hydrophobic or acidic residues at the C-termini of human cytosolic proteins deviates significantly from random expectations, being lower for hydrophobic clusters and higher for acidic clusters (Fig. 5C). We extended this analysis to canonical cytosolic proteins across multiple species, recognizing that global amino acid usage differs substantially among the kingdoms of life. Interestingly, although C-terminal compositional biases were evident across species, they were always relative to the overall proteomic amino acid usage of each species (Fig. 5D, E). Notably, these trends are not uniformly conserved. For example, a reduction in hydrophobic amino acids was observed in mouse, nematode, yeast, and bacterial proteomes, but it was either absent or less pronounced in those of flies (Fig. 5F, left panel). Moreover, unlike cytosolic proteins in human, mouse, nematode and yeast, the terminal residues of cytosolic proteins in fly, plant, and bacterial proteomes do not appear to deviate from the remainder of the C-terminal region along PC2 (Fig. 5E). These differences may reflect species-specific biology, such as the high prevalence of stop codon readthroughs in flies and plants12,15, or the absence of E3 ligases targeting Gly/Ala C-degrons in bacteria.

A PCA of the last 150 residues of canonical human cytosolic proteins. B Principal component loading plots corresponding to (A), with amino acids color-coded according to physicochemical properties. C Analysis of consecutive hydrophobic or acidic residues in the last 50 residues of human cytosolic proteins. Frequencies of contiguous stretches of varying lengths (x-axis) are normalized against a background generated from 10,000 random iterations. Central line in box plots indicates medians; box edges indicate first and third quartiles; whiskers represent min/max values; asterisks indicate statistical significance (****, p < 0.0001; two-sided permutation test). All of the p-values are provided in the Source Data file. D PCA of amino acid usage in the last 150 residues of canonical cytosolic proteins across species, without (left) and with (right) normalization to each species’ overall proteomic amino acid usage. Data points from different species are represented by different shapes, and amino acid positions are color-coded. E PCA of C-terminal amino acid usage across canonical cytosolic proteins from the indicated species. F Loading plots corresponding to (E), with amino acids color-coded as in (B). Source Data are provided as a Source Data file.

A PCA of the C-terminal regions of canonical and aberrant proteins. Analyzed regions include the last 20 residues of canonical and aberrant proteins. B Fold changes in relative frequencies of the 20 amino acids at the C-termini of canonical, RT and frameshift proteins, grouped into four categories based on similarity. C Fold changes in Cys, Trp, Asp and Glu frequencies at the C-termini of canonical and aberrant proteins across multiple species. D Stability comparison between full-length canonical proteins and their RT variants containing C-terminal Cys/Trp-enriched extensions. C-terminal sequences are shown above, with Cys and Trp highlighted in blue. E Stability comparison between canonical proteins and RT variants with or without Cys/Trp-to-Ala substitutions. F PSI comparison for RT peptides before and after substituting Cys/Trp (top) or Asp/Glu (bottom) with Ala. Source data are provided as a Source Data file.
Our PCA revealed distinct compositional differences between canonical and aberrant C-termini arising from readthrough and frameshift events (Fig. 6A and Supplementary Fig. 4C, D). Contrary to previous reports that have suggested a general increase in hydrophobicity in aberrant proteins, our analysis showed that only Cys, Trp, Arg, and Pro are consistently enriched in all types of erroneous C-termini. Among these residues, Cys and Trp—two of the rarest amino acids—exhibited the most significant increases (Fig. 6B). In parallel, we observed a consistent depletion of the acidic, stabilizing residues Asp and Glu in aberrant C-termini. This opposing trend, i.e., enrichment of Cys and Trp, yet depletion of Asp and Glu, was conserved across species (Fig. 6C). To assess the functional impact of Cys and Trp enrichment, we compiled a list of proteins displaying readthrough extensions that were enriched for these residues and compared their stability to their canonical counterparts. We found that these readthrough variants were consistently less stable (Fig. 6D), and this destabilization was largely attributable to Cys and Trp, as substituting them with Ala significantly improved protein stability (Fig. 6E). Further evidencing the roles of Cys, Trp, Asp, and Glu in determining protein stability, we introduced Ala substitutions into the readthrough extensions of the disease-associated nonstop mutation genes presented in Fig. 1. This set of experiments confirmed that Cys and Trp contribute to degradation, whereas Asp and Glu confer stability (Fig. 6F).
Thus, our findings indicate that canonical C-termini have evolved under selective pressure to minimize degradation, whereas aberrant proteins resulting from frameshift or stop codon readthrough can be recognized by the atypical amino acid composition of their C-terminal tails. This compositional signal provides a broad, sequence-independent cue for identifying deviant proteins. In addition to the increased hydrophobicity observed for some readthrough products (see below), enrichment for Cys and Trp, coupled with depletion of Asp and Glu, have emerged from our data as key determinants driving the selective clearance of abnormal proteins, thereby preserving proteome integrity.
Multiple ubiquitin ligases collaborate to target erroneous proteins with hydrophobic C-termini for degradation
Previous studies have suggested that elevated C-terminal hydrophobicity is a hallmark of erroneous proteins, with such hydrophobic tails being recognized by the Bag6 chaperone-RNF126 ubiquitin ligase complex to trigger proteasome-mediated degradation3,4. However, it remains unclear if the Bag6-RNF126 complex alone is sufficient to recognize the full spectrum of hydrophobic C-termini in human cells. In addition, the influence of hydrophobic residue arrangement and surrounding sequence context on this degradation mechanism remains enigmatic.
To address these questions, we examined the degradation pathways of seven readthrough proteins with hydrophobic extensions, identified from the disease-associated nonstop mutation screen described above (Figs. 1, 7A). As indicated by their negative ∆PSI values, all seven hydrophobic C-termini promote degradation of their respective host proteins. Surprisingly, only one of these—TMEM70-RT—proved to be a target of the Bag6-RNF126 complex (Fig. 7B), challenging the notion that this pathway is solely responsible for recognizing hydrophobic C-termini. To explore alternative degradative mechanisms, we performed CRISPR/Cas9 screens for three hydrophobic variants (RGS18-RT, UBA5-RT, RAB18-RT), and identified MARCH6 and RNF139 as additional E3 ligases involved in recognizing hydrophobic C-termini (Fig. 7C). Notably, knockout of Bag6/RNF126, RNF139, or MARCH6 failed to prevent degradation of all hydrophobic variants, such as LAMTOR5-RT and ARHGEF35-RT, implying either the involvement of yet unidentified E3 ligases or functional redundancy among known ones (Fig. 7D).

(A) Amino acid sequences and ∆PSI values of the RT regions analyzed in (B). Hydrophobic residues are highlighted by orange boxes. B Stability comparison of GFP fused to the indicated RT peptides, with or without Bag6 or RNF126 knockout via sgRNAs. Knockdown of APPBP2 serves as a control. C CRISPR screens identifying proteolytic machinery components involved in degradation driven by the indicated RT peptides. Each dot in the plot represents a gene identified from the screening results, and it is positioned according to its confidence and effect size, as quantified by the casTLE score (see “Methods”). D Stability comparison for GFP fused to the indicated RT peptides, with or without sgRNA-mediated knockout of APPBP2, Bag6, RNF126, RNF139 or MARCH6. E Amino acid sequences of RGS18-RT and its mutant variants. First and second hydrophobic stretches are highlighted by orange or red boxes, respectively; the scrambled forms of the stretches are marked by dashed boxes; the TN motif is marked in blue. F Stability comparison between GFP alone and GFP fused to the peptides indicated on the right. G Stability comparison of GFP fused to the indicated RT peptides, with or without sgRNA-mediated knockout of the proteins labeled on the right. Source data are provided as a Source Data file.
Since multiple E3 ligases target hydrophobic C-termini, we adopted RGS18-RT as a model to dissect how the arrangement of hydrophobic residues and local sequence context influence degradation. RGS18-RT harbors two hydrophobic segments, H1 (LILLIFM) and H2 (LYICF), separated by the two residues TN. We generated a series of RGS18-RT mutants, i.e., constructs retaining only one hydrophobic segment (with or without TN), scrambled variants, and constructs with altered TN positioning (Fig. 7E). Although all of the resulting variants promoted degradation when fused to the C-terminus of GFP, the extent of degradation varied widely, even with minor sequence alterations (Fig. 7F). For example, the H1-TN variant (LRKLILLIFMTN) promoted stronger degradation than TN-H1 (LRKTNLILLIFMTN), despite both having the same amino acid compositions. Similarly, TN-H2 (LRKTNLYICF) was more destabilizing than its scrambled counterpart, TN-S2 (LRKTNFCIYL). Crucially, these subtle sequence differences not only modulated degradation levels but also dictated which E3 ligase mediated recognition (Fig. 7G). For instance, the H2 variant was specifically targeted by MARCH6, whereas inclusion of the TN motif upstream (TN-H2) shifted recognition to RNF139. Neither ligase recognized the scrambled version of the TN-H2 variant, i.e., TN-S2, highlighting the sequence specificity of substrate recognition. Similar context-dependent shifts in ligase specificity were also observed among H1-based constructs (Fig. 7G).
Together, these findings uncover the unexpected complexity in the ubiquitin ligase network targeting the hydrophobic C-termini arising from diverse erroneous events. They also demonstrate that degradation is not dictated solely by the presence of hydrophobic residues, but also their precise arrangement and the surrounding sequence context, representing factors that determine both degradation efficiency and the identity of the responsible degradation machinery.
Discussion
Protein C-termini have been implicated in various biological processes, including protein targeting, subcellular anchoring, and the formation of protein complexes31,32. In this study, we further explore their extensive role in regulating protein stability across both canonical and defective proteomes.
Contrary to a previous study7, our findings demonstrate that disease-associated nonstop mutations can either enhance or reduce protein stability. Specifically, we have uncovered stabilization of oncoproteins such as FOXA133, CCND234, SNCG35, HEYL36 and CPNE937 due to cancer-associated nonstop mutations. Conversely, we have validated the degradation of multiple tumor suppressor and disease-causing proteins driven by nonstop mutations. Furthermore, we highlight the critical role of C-terminal variation in regulating the stability of protein isoforms. Given that many alternative splicing and programmed translation events are tissue-specific and influenced by environmental cues, these differences in stability may endow key regulatory advantages. For example, while the stable PAX3b isoform is broadly expressed, the unstable PAX3a isoform is restricted to the cerebellum, esophagus, and skeletal muscle38. PAX3b can reduce melanocyte migration and promote apoptosis, whereas PAX3a cannot39. Similarly, the stable EDF1α isoform is expressed in most tissues, whereas the labile EDF1β isoform exhibits tissue-specific expression40. These findings indicate that alternative splicing offers an alternative mechanism for achieving tissue-specific protein abundances, complementing conventional transcriptional regulatory mechanisms.
Ribosome profiling and mass spectrometry studies have revealed that translational readthrough is far more frequent than previously recognized10,12,13,14,15. Transcriptome and translatome data have further indicated that stop codon readthrough occurs at an elevated frequency in tumor tissues41. The widespread nature of these readthrough events has sparked debate over whether they represent mere translational errors or instead serve functional biological roles12,42,43,44. Given that environmental stress can increase readthrough rates45,46, and that the resulting C-terminal extensions can substantially influence protein stability (as documented herein), our findings support the notion that leaky stop codons enhance proteomic plasticity, enabling cells to dynamically adapt to changing conditions. Notably, readthrough therapy, also known as stop codon suppression, is a key therapeutic strategy for treating nonsense mutations, which account for ~ 10% of all pathogenic variants. This approach involves the use of small molecules to promote ribosomal readthrough of premature stop codons, thereby restoring full-length protein production47,48. However, our results raise critical concerns about the safety of such therapies, as these compounds may also enhance readthrough at natural stop codons, potentially causing unintended, proteome-wide side effects.
We also examined both cis and trans factors that contribute to C-terminal sequence-driven protein degradation. Although several specific C-terminal motifs, known as C-degrons, have been identified by ourselves and others24,26,28, our research indicates that most degradation-promoting C-terminal sequences do not conform to clear motif patterns. Instead, protein stability is largely governed by the overall amino acid composition of the C-terminus, with clusters of similar or identical residues playing a particularly prominent role. This compositional bias provides a broad, sequence-independent marker for recognizing aberrant proteins. Although previous studies have proposed that increased hydrophobicity is a hallmark of aberrant protein C-termini3,4,21, we report here that enrichment for two rare amino acids, i.e., cysteine and tryptophan, along with a reduction in acidic amino acids, are more reliable features distinguishing aberrant from canonical C-termini. These compositional cues facilitate selective elimination of erroneous proteins, thereby safeguarding proteome fidelity.
Although stretches of hydrophobic residues are known to promote protein degradation, the mechanisms underlying how these stretches are recognized appear to be more complex than previously appreciated. In addition to the well-characterized Bag6-RNF126 complex, we have identified at least two additional E3 ligases that target hydrophobic C-termini. Strikingly, we demonstrate that permuting hydrophobic residues or shuffling adjacent hydrophilic residues not only alters the degree of degradation but also shifts E3 ligase specificity. This outcome indicates that recognition of hydrophobic protein regions is more selective and dependent on sequence context than previously appreciated. Since such hydrophobic clusters are also present within the intrinsically disordered regions of cytosolic proteins, we speculate that evolutionary pressure has shaped their sequence composition to minimize the risk of inadvertently targeting folded, functional proteins as degradation targets.
Finally, our findings identify potential therapeutic avenues. Dysregulated protein abundance underlies numerous diseases, and restoring proteins to their proper levels is a key therapeutic objective. The protein C-terminus offers an attractive platform for modulation, as it is relatively unconstrained by structural demands, and subtle sequence alterations can dramatically influence protein half-life. We propose an innovative approach to restore protein levels by inducing gene-specific stop codon readthrough or alternative splicing to activate latent C-terminal degrons or stabilizing elements encoded in 3’UTRs or alternative reading frames. This concept mirrors the mechanism of the FDA-approved drug Evrysdi-Risdiplam49, which treats spinal muscular atrophy by modulating SMN2 splicing to favor expression of the stable isoform SMN2d while suppressing the unstable SMN2a isoform, which is otherwise identical but contains a C-terminal degron19,50.
In summary, our study provides as a solid foundation for a systems-wide understanding of C-terminal-mediated protein degradation and its biological functions. It also offers valuable insights into how protein stability can be predicted and engineered, with applications in biotechnology and therapeutic development.
Method
Cell culture, lentivirus production, and gene knockout
HEK293T cells (ATCC® CRL-3216) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Hyclone), 100 μg/ml streptomycin, and 100 μ/ml penicillin (Gibco). Cells were maintained at 37 °C in a humidified incubator with 6% CO₂. To block proteasome dependent protein degradation, cells were treated with 1 nM bortezomib (BioVision) for 6 hr.
For lentivirus production, HEK293T cells were co-transfected with packaging plasmids pRev, pTat, pHIV gag/pol, pVSVG, and the lentiviral construct of interest using the TransIT-293 transfection reagent (Mirus Bio). Viral supernatants were harvested 48 hours post-transfection. For transduction, cells were incubated with viral supernatants in the presence of 8 μg/mL polybrene (Sigma-Aldrich) for 24 h.
To achieve gene knockout, stable Cas9-expressing HEK293T cells were transduced with lentiviruses encoding gene-specific sgRNAs. Cells were analyzed five days post-infection. Target sequences and catalog information are listed in Supplementary Table 1.
GPS assay and random peptide platform
To generate GPS reporter constructs, peptides or genes of interest were cloned into pLenti-GPS RFP-IRES-GFP or BFP-IRES-GFP vectors using Gibson assembly (New England Biolabs) or Gateway recombination (Invitrogen). HEK293T cells were transduced with lentiviruses expressing reporter constructs at low multiplicity of infection (MOI ~ 0.1) and selected with 1 μg/ml puromycin (Clontech) for 7 days to establish stable GPS reporter lines.
GFP/RFP or GFP/BFP fluorescence ratios were quantified by flow cytometry using a BD LSR Fortessa system (BD Biosciences) with lasers at 561 nm, 488 nm, and 405 nm for RFP, GFP, and BFP excitation, respectively. For standard GPS assays, a minimum of 10,000 cells was analyzed using FlowJo software. We analyzed the results by first gating single cells based on FSC and SSC signals, and then selecting RFP-positive cells (Supplementary Fig. 5). All GPS analysis were done in biological triplicate.
For the random peptide assay, oligonucleotides encoding 12- or 15-residue random peptides were synthesized (Life Technologies) and amplified by polymerase chain reaction (PCR) using 5’ and 3’ linker sequences. Random residues were encoded using NNK degenerate codons (N = A/T/G/C; K = G/T). PCR products were cloned into the pDONR223 vector via Gibson assembly and transferred into the pLenti-GPS reporter vector using Gateway LR recombination. To reduce synthesis bias, three independent libraries were constructed for each design using separately synthesized oligonucleotides. Each library had an estimated complexity of ~ 1010 unique variants. Due to the large complexity of random peptide libraries (~ 1010), 100,000 cells per library were analyzed in triplicate. The percentage of cells with reduced GFP/RFP ratios (termed “% degradation”) was used as an index of peptide-mediated degradation27.
GPS-peptidomic screen for disease-associated nonstop mutations
Disease-associated nonstop mutations were collected in September 2021 from TCGA (https://portal.gdc.cancer.gov/) and the NonStopDB database (http://NonStopDB.dkfz.de). Readthrough-associated SNPs were retrieved from dbSNP (https://www.ncbi.nlm.nih.gov/snp/). All coding and 3’UTR sequences were verified using Ensembl and NCBI databases. DNA sequences encoding the final 58 amino acids of canonical and mutated proteins were synthesized (Twist Biosciences), PCR-amplified using a KAPA HiFi HotStart kit (Roche), and cloned into the GPS vector via Gateway recombination. A library complexity of > 103-fold was maintained throughout cloning.
The GPS-peptidomic reporter cell library was generated by lentiviral transfection at an MOI of ~ 0.05 to minimize double-integration events. The library was generated at ~ 1000-fold representation and sorted into eight bins based on GFP/RFP ratios using a BD FACSAria II SORP sorter (BD Biosciences). Genomic DNA from each bin was extracted and used for PCR amplification of integrated oligos using TaKaRa Ex Taq® DNA Polymerase Hot-Start Version (Takara Bio) with the following primers:
-
Forward primer (5’-GAGCTGAAGGGCATCGACTTCAAGG-3’)
-
Reverse primer (5’-GCGTCAGATGTGTATAAGAGACAG-3’)
PCR conditions: 21 cycles of 98 °C for 10 s, 60 °C for 30 s, 72 °C for 30 s, with a final extension at 72 °C for 5 min. Products were purified using a MinElute PCR purification kit (QIAGEN) and quantified using QubitTM fluorometric quantification (Thermo). A second PCR (8 cycles, same conditions) was performed to add Illumina indices using the primers listed in Supplementary Table 2. Indexed products were purified with AMPure XP beads (Beckman Colter) and quantified via a Bioanalyzer 2100 system (Agilent). Sequencing was performed using the MiSeq Reagent Kit v3 (Illumina) with the primer 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’. Only reads that perfectly matched the designed sequences were retained. Peptides with ≥ 60 reads in both replicates were included in downstream analysis. PSI values were calculated to evaluate degradation-promoting activity.
Cycloheximide-chase assay
Genes of interest were cloned into a Lenti-based vector via Gateway recombination to generate FLAG-tagged reporter constructs. Stable cell lines were established by lentiviral infection as described above. Cells were treated with 100 µg/mL cycloheximide (Calbiochem), and samples were collected at multiple time points. Protein abundance was analyzed by immunoblotting using primary antibodies against the Flag-tag (Sigma-Aldrich, M2 at 1:1000) and GAPDH (GeneTex, 100118 at 1:1000).
CRISPR/Cas9 screen
Stable Cas9-expressing GPS reporter cells were transduced with lentiviruses from the Proteostasis sublibrary of the Human CRISPR Deletion Library (a gift from Michael Bassik, Addgene #101930)51 at an MOI of 1. After 10 days, mCherry + cells with the top 5% highest GFP/BFP ratios were sorted, while the total population was used as a control. Genomic DNA was extracted, and sgRNA sequences were PCR-amplified as described previously52. Sequencing was performed using a MiSeq Reagent Kit V3 (Illumina), and sgRNA enrichment was analyzed using the casTLE algorithm53. According to the analysis pipeline, casTLE applies an Empirical Bayesian framework to estimate the functional impact of gene knockouts in CRISPR-Cas9 pooled screens. It accounts for variability in guide RNA (gRNA) efficacy and potential off-target effects by modeling the observed data across multiple gRNAs per gene. The analysis begins by mapping each gRNA to the corresponding target gene and extracting read counts from both treatment and control conditions. These counts are then normalized using median-based scaling, and log2 fold-changes are computed relative to the reference samples to quantify the effect of each gRNA. casTLE then integrates the fold-change distributions across all gRNAs to infer a gene-level effect size, confidence score, and false discovery rate (FDR). The final output is a ranked list of genes, enabling identification of biologically meaningful hits.
Bioinformatics of canonical and aberrant protein C-termini
To examine human protein isoforms, sequences were downloaded from the Ensembl database (Homo_sapiens.GRCh38.pep.all.fa, December 2022). C-terminal variants were defined as the last 20 amino acids differing among isoforms from the same gene. Isoforms differing only at the C-terminus were identified by clustering those with identical N-terminal sequences, yet varied within the final 20 residues.
Cross-species canonical protein sequences were obtained from NCBI for:
-
Human (Homo sapiens; GCF_000001405.39_GRCh38.p13)
-
Mouse (Mus musculus; GCF_000001635.27_GRCm39)
-
Fly (Drosophila melanogaster; GCF_000001215.4_Release_6_plus_ISO1_MT)
-
Plant (Arabidopsis thaliana; GCF_000001735.4_TAIR10.1)
-
Nematode (Caenorhabditis elegans; GCF_000002985.6_WBcel)
-
Yeast (Saccharomyces cerevisiae; GCF_000146045.2_R64)
-
Bacteria (Escherichia coli; GCF_000005845.2_ASM584v2)
To analyze the C-termini of canonical cytosolic proteins, we annotated protein subcellular localizations using the PANTHER Knowledgebase (http://www.pantherdb.org/). Only proteins annotated as cytosolic were included in the analysis.
Aberrant readthrough C-termini were simulated by in-frame translation of the 3’UTR (excluding yeast and bacteria due to an absence of available data). Frameshifts arise when the reading frame shifts by + 1 or – 1 nucleotide, resulting in entirely altered downstream amino acid sequences. To model these events and generate frameshifted C-terminal sequences in silico, we removed the first nucleotide (for +1 frameshift) or the first two nucleotides (for – 1 frameshift) from all protein-coding genes. These modified sequences were then translated to produce all possible + 1 and – 1 frameshift peptides. Peptides shorter than 20 residues were discarded, and redundant sequences were removed prior to amino acid composition analysis.
Principal Component Analysis and amino acid enrichment analysis
To investigate positional amino acid preferences within the terminal 150 residues of proteins, we developed a normalization method for amino acid and position that helps to account for variations in amino acid frequencies and sequence lengths. We calculated the normalized ratio (\({r}_{{ij}}\)) of each of the 20 standard amino acids at every position within the last 150 residues and then divided the frequency across all positions in the protein sequences:
The numerator represents the observed frequency of the i-th amino acid (\({c}_{{ij}}\)) at the j-th position, normalized by the total number of amino acids (\({t}_{j}\)) at that same position (j-th). The denominator represents the expected frequency of the i-th amino acid (\({C}_{i}\)) across all sequences, normalized by the total number of all amino acids (T). After dividing the observed frequency by the expected frequency, the equation yields a normalized value that indicates whether a particular amino acid is more or less likely to appear at a specific position than would be expected by chance. This approach resulted in a 20 × 150 matrix representing the normalized relative amino acid frequencies at each position. This matrix of ratios was then utilized to perform Principal Component Analysis (PCA) for each position using the prcomp function in R. In the PCA, the percentages shown next to each principal component indicate the proportion of total variance in the dataset explained by that component. The first principal component (PC1) captures the largest possible variance, whereas the second principal component (PC2) captures the second-largest variance, subject to being orthogonal to PC1.
To assess the continuity of hydrophobic and acidic amino acids in the C-termini of human proteins, we analyzed the last 50 residues of each canonical human protein. For each peptide, we identified and counted the consecutive stretches of hydrophobic (I, L, V, M, Y, F, W) and acidic (D, E) residues of length 2, 3, and ≥ 4, respectively. The total number of each stretch type was then summed across the whole peptide set to obtain the observed frequencies. To establish baseline frequencies of the stretches that are amino-acid-composition-dependent, we randomized the amino acid order within each peptide—preserving the original composition—and repeated the same stretch-counting procedure. This process was performed 10,000 times to produce distributions of baseline frequencies for each stretch type, minimizing bias from individual randomizations. Observed frequencies were then normalized against the baseline frequencies to calculate observed-to-expected ratios. Statistical significance of enrichment or depletion for each stretch type was assessed by testing whether these ratios deviated significantly from 1 using a two-sided permutation test with 100,000 permutations.
Statistical analyses
To justify the statistical significance for the PSI threshold (|ΔPSI | ≥ 1) used in our GPS screen of disease-associated nonstop mutations, we generated a background distribution of ΔPSI values by calculating PSI differences between two biological replicates. We then assessed the significance of the ± 1 thresholds by evaluating the right and left tail probabilities of this distribution. The corresponding probabilities are 1.45 × 10–4 and 1.4 × 10–4, respectively.
All experiments using the random peptide library were performed as three independent biological replicates. The statistical significance of the GPS screening results and random peptide assays were determined by an unpaired two-tailed Students’ t test. Enrichment for hydrophobic and acidic stretches was evaluated using a two-sided permutation test based on deviations from the expected observed-to-expected ratio of 1. Statistical significance was defined as a P-value less than 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequencing data from this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE298390 (Protein C-Terminal Variations Broadly Proteostasis) Source data are provided in this paper.
References
Dabrowski, M., Bukowy-Bieryllo, Z. & Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes-The impact of RNA sequence. RNA Biol. 12, 950–958 (2015).
Choi, J., O’Loughlin, S., Atkins, J. F. & Puglisi, J. D. The energy landscape of -1 ribosomal frameshifting. Sci. Adv. 6, eaax6969 (2020).
Kesner, J. S. et al. Noncoding translation mitigation. Nature 617, 395–402 (2023).
Müller, M. B. D., Kasturi, P., Jayaraj, G. G. & Hartl, F. U. Mechanisms of readthrough mitigation reveal principles of GCN1-mediated translational quality control. Cell 186, 3227–3244.e20 (2023).
Shibata, N. et al. Degradation of stop codon read-through mutant proteins via the ubiquitin-proteasome system causes hereditary disorders. J. Biol. Chem. 290, 28428–28437 (2015).
Dhamija, S. et al. A pan-cancer analysis reveals nonstop extension mutations causing SMAD4 tumour suppressor degradation. Nat. Cell Biol. 22, 999–1010 (2020).
Ghosh, A. et al. Suppressive cancer nonstop extension mutations increase C-terminal hydrophobicity and disrupt evolutionarily conserved amino acid patterns. Nat. Commun. 15, 9209 (2024).
Palma, M. & Lejeune, F. Deciphering the molecular mechanism of stop codon readthrough. Biol. Rev. Camb Philos. Soc. 96, 310–329 (2021).
Namy, O., Rousset, J. P., Napthine, S. & Brierley, I. Reprogrammed genetic decoding in cellular gene expression. Mol. Cell 13, 157–168 (2004).
Manjunath, L. E., Singh, A., Som, S. & Eswarappa, S. M. Mammalian proteome expansion by stop codon readthrough. Wiley Interdiscip Rev. RNA 14, e1739 (2023).
Labunskyy, V. M., Hatfield, D. L. & Gladyshev, V. N. Selenoproteins: molecular pathways and physiological roles. Physiol. Rev. 94, 739–777 (2014).
Dunn, J. G., Foo, C. K., Belletier, N. G., Gavis, E. R. & Weissman, J. S. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife 2, e01179 (2013).
Jungreis, I. et al. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 21, 2096–2113 (2011).
Mangkalaphiban, K. et al. Transcriptome-wide investigation of stop codon readthrough in Saccharomyces cerevisiae. PLoS Genet. 17, e1009538 (2021).
Zhang, Y. et al. Readthrough events in plants reveal plasticity of stop codons. Cell Rep. 43, 113723 (2024).
Zhang, Z., Khanal, N., Dykstra, A. B. & Daris, K. Stop-Codon readthrough in therapeutic protein candidates expressed from mammalian cells. J. Pharm. Sci. 113, 1498–1505 (2024).
Miyawaki, S. et al. The mouse Sry locus harbors a cryptic exon that is essential for male sex determination. Science 370, 121–124 (2020).
Peter, S. A., Isaac, J. S., Narberhaus, F. & Weigand, J. E. A novel, universally active C-terminal protein degradation signal generated by alternative splicing. J. Mol. Biol. 433, 166890 (2021).
Cho, S. & Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–442 (2010).
Lin, H. C. et al. SELENOPROTEINS. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/Sec decoding. Science 349, 91–95 (2015).
Arribere, J. A. et al. Translation readthrough mitigation. Nature 534, 719–723 (2016).
Thrun, A. et al. Convergence of mammalian RQC and C-end rule proteolytic pathways via alanine tailing. Mol. Cell 81, 2112–2122 (2021).
Hasenjäger, S., Bologna, A., Essen, L. O., Spadaccini, R. & Taxis, C. C-terminal sequence stability profiling in Saccharomyces cerevisiae reveals protective protein quality control pathways. J. Biol. Chem. 299, 105166 (2023).
Kong, K. E., Shankar, S., Rühle, F. & Khmelinskii, A. Orphan quality control by an SCF ubiquitin ligase directed to pervasive C-degrons. Nat. Commun. 14, 8363 (2023).
Yen, H. C., Xu, Q., Chou, D. M., Zhao, Z. & Elledge, S. J. Global protein stability profiling in mammalian cells. Science 322, 918–923 (2008).
Koren, I. et al. The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell 173, 1622–1635 (2018).
Yeh, C. W. et al. The C-degron pathway eliminates mislocalized proteins and products of deubiquitinating enzymes. EMBO J. 40, e105846 (2021).
Lin, H. C. et al. C-Terminal end-directed protein elimination by CRL2 ubiquitin ligases. Mol. Cell 70, 602–613 (2018).
Hamby, S. E., Thomas, N. S., Cooper, D. N. & Chuzhanova, N. A meta-analysis of single base-pair substitutions in translational termination codons (‘nonstop’ mutations) that cause human inherited disease. Hum. Genomics 5, 241–264 (2011).
Manjunath, L. E. et al. Stop codon read-through of mammalian MTCH2 leading to an unstable isoform regulates mitochondrial membrane potential. J. Biol. Chem. 295, 17009–17026 (2020).
Chung, J. J., Shikano, S., Hanyu, Y. & Li, M. Functional diversity of protein C-termini: more than zipcoding? Trends Cell Biol. 12, 146–150 (2002).
Sharma, S. & Schiller, M. R. The carboxy-terminus, a key regulator of protein function. Crit. Rev. Biochem. Mol. Biol. 54, 85–102 (2019).
Augello, M. A., Hickey, T. E. & Knudsen, K. E. FOXA1: master of steroid receptor function in cancer. EMBO J. 30, 3885–3894 (2011).
Büschges, R. et al. Amplification and expression of cyclin D genes (CCND1, CCND2 and CCND3) in human malignant gliomas. Brain Pathol. 9, 435–442 (1999).
Kang, S. M. et al. Modulation of dendritic cell function by the radiation-mediated secretory protein γ-synuclein. Cell Death Discov. 1, 15011 (2015).
Han, L. et al. HEYL Regulates neoangiogenesis through overexpression in both breast tumor epithelium and endothelium. Front. Oncol. 10, 581459 (2020).
Tang, H. et al. The CPNE family and their role in cancers. Front. Genet. 12, 689097 (2021).
Tsukamoto, K., Nakamura, Y. & Niikawa, N. Isolation of two isoforms of the PAX3 gene transcripts and their tissue-specific alternative expression in human adult tissues. Hum. Genet. 93, 270–274 (1994).
Wang, Q., Kumar, S., Slevin, M. & Kumar, P. Functional analysis of alternative isoforms of the transcription factor PAX3 in melanocytes in vitro. Cancer Res. 66, 8574–8580 (2006).
Kabe, Y. et al. The role of human MBF1 as a transcriptional coactivator. J. Biol. Chem. 274, 34196–34202 (1999).
Wang, N. & Wang, D. Genome-wide transcriptome and translatome analyses reveal the role of protein extension and domestication in liver cancer oncogenesis. Mol. Genet. Genomics 296, 561–569 (2021).
Fan, Y. et al. Heterogeneity of Stop Codon Readthrough in Single Bacterial Cells and Implications for Population Fitness. Mol. Cell 67, 826–836 (2017).
Li, C. & Zhang, J. Stop-codon read-through arises largely from molecular errors and is generally nonadaptive. PLoS Genet. 15, e1008141 (2019).
von der Haar, T. & Tuite, M. F. Regulated translational bypass of stop codons in yeast. Trends Microbiol. 15, 78–86 (2007).
Zhang, H. et al. Metabolic stress promotes stop-codon readthrough and phenotypic heterogeneity. Proc. Natl. Acad. Sci. USA 117, 22167–22172 (2020).
Romero Romero, M. L. et al. Environment modulates protein heterogeneity through transcriptional and translational stop codon readthrough. Nat. Commun. 15, 4446 (2024).
Keeling, K. M., Xue, X., Gunn, G. & Bedwell, D. M. Therapeutics based on stop codon readthrough. Annu. Rev. Genomics Hum. Genet. 15, 371–394 (2014).
Bidou, L., Allamand, V., Rousset, J. P. & Namy, O. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol. Med. 18, 679–688 (2012).
Ratni, H., Scalco, R. S. & Stephan, A. H. Risdiplam, the first approved small molecule splicing modifier drug as a blueprint for future transformative medicines. ACS Med. Chem. Lett. 12, 874–877 (2021).
Zhou, J., Zheng, X. & Shen, H. Targeting RNA-splicing for SMA treatment. Mol. Cells 33, 223–228 (2012).
Morgens, D. W. et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 8, 15178 (2017).
Mathiowetz, A. J., Roberts, M. A., Morgens, D. W., Olzmann, J. A. & Li, Z. Protocol for performing pooled CRISPR-Cas9 loss-of-function screens. STAR Protoc. 4, 102201 (2023).
Morgens, D. W., Deans, R. M., Li, A. & Bassik, M. C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 34, 634 (2016).
Zhao, M., Kim, P., Mitra, R., Zhao, J. & Zhao, Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 44, D1023–D1031 (2016).
Liu, Y., Sun, J. & Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genomics 44, 119–121 (2017).
Acknowledgements
We thank Dr. M. Bassik (Stanford University School of Medicine, Stanford, CA) for sharing the sgRNA-CRISPR knockout library; Y.M. Lin and N.C. Hsu of the Flow Cytometry Core, and S.Y. Tung of the Genomic Core, Institute of Molecular Biology, Academia Sinica, for technical assistance; K.L. Hsu for organizing disease-associated nonstop mutations; and J. O’Brien for English editing. This work was supported by Investigator Award AS-IA-108-L02 from Academia Sinica, and by grants 111-2311-B-001-014-MY3, 111-2326-B-001-008, 112-2326-B-001-005, and 113-2326-B-001-005 from the National Science Council of Taiwan.
Author information
Authors and Affiliations
Contributions
C.Y. Chu, S.Y. Hsu, C.W. Yeh, and S.C. Chen carried out the GPS-peptidomic and CRISPR/Cas9 screens. C.Y. Chu, S.Y. Hsu, and L.T. Lee performed the validation experiments. C.Y. Chu and L.C. Wang conducted random peptide assays and mutagenesis experiments to characterize protein C-termini. K.H. Yeh, S.Y. Hsu, and C.H. Yu performed the bioinformatic analyses. H.C. Yen supervised the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Domnita-Valeria Rusnac and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chu, CY., Hsu, SY., Yeh, CW. et al. Protein C-terminal variations impact proteostasis. Nat Commun 17, 2288 (2026). https://doi.org/10.1038/s41467-026-68979-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68979-z
