
Abstract
The valorization of plastic waste represents a major challenge of the 21st century due to its severe environmental impact. Here, we report a stepwise hydrogen spillover-constructed CuCo/CoOx catalyst that enables near-quantitative conversion of waste polyethylene terephthalate to p-xylene (>99.9%), significantly outperforming the performance of various Cu- and Co-based catalysts as well as previously reported noble metal catalysts. The stepwise hydrogen spillover induces the formation of partially phase-transformed Co0 species and abundant oxygen-vacancy-rich Co0/CoOx interfaces. The former enhances H2 dissociation efficiency, while the latter facilitates C-O bond activation in polyethylene terephthalate and regulates substrate-product adsorption equilibria, synergistically contributing to the exceptional catalytic performance. The catalyst demonstrates broad applicability to more than 30 real-world polyester plastics. Furthermore, techno-economic analysis reveals significant reductions in CO2 emissions and competitive processing costs. This breakthrough in near-quantitative polyethylene terephthalate conversion and stepwise hydrogen spillover-enabled active site construction offers valuable insights into plastic waste upcycling and the design of advanced heterogeneous catalysts.
Similar content being viewed by others

Introduction
The continuous consumption of plastic materials has led to the accumulation of over 4.9 billion metric tons of plastic waste in landfills and natural ecosystems, a figure projected to rise to 12 billion metric tons by 20501,2. The environmental burden associated with plastic waste has intensified markedly3,4,5,6,7,8,9, with widespread pollution across terrestrial, marine, and atmospheric environments. Such widespread contamination facilitates the bioaccumulation of plastic particles within living organisms, thereby disrupting ecological systems and posing potential threats to human health10,11,12,13,14. Among various plastic types, polyethylene terephthalate (PET) represents the most extensively utilized polyester worldwide15, owing to its low density, high mechanical strength, excellent abrasion and chemical resistance, and cost-effectiveness. The global annual production of PET exceeds 82 million tons, primarily utilized in the fabrication of textiles and fibers, rigid packaging materials, disposable beverage bottles, and plastic films. PET accounts for approximately 8% of global plastic production and contributes around 12% to total solid plastic waste16,17,18,19.
The chemical upcycling of PET into high-value-added chemicals represents a critical pathway in advanced PET waste valorization. Conventional approaches such as hydrolysis20,21 and alcoholysis22,23 have been widely explored, while emerging methods such as hydrogenolysis and coupled alcoholysis-hydrogenolysis24 offer promising alternatives. Current studies on PET hydrogenolysis have revealed a wide distribution of products25,26,27,28,29, yet achieving near-quantitative yields of specific targeted compounds remains challenging, owing to the complex hydrogenation behavior of multiple functional groups. Among the potential products, p-xylene (PX) is of particular interest due to its relevance in three important industrial domains: high-end chemical manufacturing (e.g., polyester fibers), general-purpose solvents, and specialized applications in the pharmaceutical and laboratory sectors. Notably, these applications demand ultra-high purity PX, with rigorous control over trace impurities. This underscores the urgent need to develop highly selective catalytic systems for the PX production from PET. However, current reports on metal-based catalysts (e.g., Ru-, Pt-based systems) have shown limited success in achieving such selectivity, primarily due to undesired side reactions such as decarboxylation and aromatic ring hydrogenation27,30,31,32,33.
The C–O cleavage in hydrogenolysis requires Lewis acid-supported metal catalysts (Ma/MbOx), where synergistic catalysis at the interface between hydrogen dissociation at metallic (Ma0) sites and Lewis acid (MbOx)-facilitated C–O bond activation enables efficient transformation34,35,36. To maximize this synergistic catalysis, the construction of M/MOx systems composed of homologous transition metals is considered advantageous, owing to their favorable electronic interactions and band structure compatibility. Non-precious Co/CoOx has emerged as a promising candidate, leveraging the moderate hydrogen activation ability of metallic Co species and the strong oxygen affinity of CoOx for the activation of oxygen-containing substrates37,38. Co-based catalysts, such as Co/TiO238, Co–Fe–Al39, and Co-Cu-Al39, have demonstrated effective degradation of PET. Among these catalysts, Co/TiO2 and Co-Cu-Al exhibit relatively poor selectivity towards PX, and Co-Fe-Al exhibits high PX selectivity. However, there have been no reports yet on achieving highly selective conversion of PET into PX using Co-based catalysts within the single-metal category. Conventional high-temperature reduction techniques commonly employed in Co/CoOx preparation often lead to active site agglomeration and phase crystallization, thereby reducing the density of accessible active sites and weakening interfacial synergy40. Consequently, the rational design of structurally tailored Co/CoOx catalysts with abundant synergistic sites for the near-quantitative conversion of PET to PX remains highly desirable but has yet to be realized.
Hence, we report a unique stepwise hydrogen spillover-constructed CuCo/CoOx catalyst, enabling efficient PET conversion into PX with a near-quantitative yield (>99.9%) (Fig. 1). The stepwise hydrogen spillover is as follows: CuO is first reduced to Cu0, which initiates hydrogen spillover to reduce CoOx; the resulting Co0 with superior hydrogen activation ability drives a secondary spillover that accelerates CoOx reduction. Compared with monometallic Co systems, the stepwise hydrogen spillover allows the generation of partially phase-transformed Co0 species at lower temperatures, while simultaneously creating a high density of vacancy-rich Co0/CoOx interfaces. Partially phase-transformed Co achieves an optimal balance between hydrogen dissociation ability and active site density, markedly outperforming both fully transformed and untransformed states. The vacancy-rich Co0/CoOx interfaces exhibit enhanced C–O bond activation capacity and afford favorable adsorption/desorption of substrates and products. Additionally, we directly capture the signals of ester bond activation in PET, leading to the formation of an aldehyde-like intermediate, using in-situ FT-IR, providing unambiguous molecular-level evidence for ester bond activation and offering distinctive insights into the hydrogenolysis mechanism of PET. This unique catalyst demonstrates broad applicability across 28 real-world PET plastics, including single-component PET, PET with additives and PET-based mixed plastics, achieving near-quantitative PX in most cases. Furthermore, this catalyst demonstrates significant potential for the conversion of 5 other aromatic polyesters (e.g., PBT, PBAT) as well as aliphatic polyesters (e.g., PLA, PCL and PBS). Techno-economic analysis further confirms substantial CO2 emission reduction alongside cost competitiveness, highlighting its potential to reconcile environmental sustainability with industrial viability.

PET refers to polyethylene terephthalate.
Results
Catalytic performance in hydrogenolysis of PET to PX
The catalytic performance of various Co-based catalysts, including Co/CoOx, CuCo/CoOx, NiCo/CoOx, RuCo/CoOx, and FeCo/CoOx, for the hydrogenolysis of PET was initially evaluated (Fig. 2a). The catalytic performance results follow this trend: CuCo/CoOx > Co/CoOx ≈ NiCo/CoOx > RuCo/CoOx > FeCo/CoOx. These results indicate that a significant enhancement in the hydrogenolysis activity of PET is observed only when Cu species are incorporated into the CoOx-based catalyst. For individual Co- or Cu-based catalysts supported on conventional supports (TiO2, SiO2 and Al2O3), the PX yield remained below 25%. Thermogravimetric analysis (TGA) (Figs. S1–4) and GC results (Figs. S5–7) suggest that the catalytic activity of Co/TiO2, Co/Al2O3, and Cu/SiO2 catalysts is exceptionally low, resulting in only limited depolymerization of PET, which largely retains its polymeric structure. These results indicate that catalysts primarily composed of metallic Co or Cu species exhibit limited catalytic activity, and these conventional supports have no obvious contribution. In contrast, the Co/CoOx catalyst achieved a significantly higher PX yield of 52.5%, suggesting a potential synergistic effect between metallic Co and CoOx species. Upon incorporation of Cu, the CuCo/CoOx catalyst achieved a near-quantitative PX yield exceeding 99.9%, almost two-fold improvement compared to Co/CoOx. To exclude the influence of CuOx species, systematic evaluations were conducted on both Cu/CuOx and Co/CuOx. The results demonstrated that neither the Cu–CuOx synergy nor the Co–CuOx interaction exhibited appreciable catalytic activity. In summary, the superior performance observed in the CuCo/CoOx system can be unequivocally attributed to the synergistic cooperation between Co and CoOx species. The introduction of Cu may enhance the synergistic interaction between metallic Co and CoOx species by facilitating the formation of unique active sites, thereby contributing to the observed improvement in catalytic performance. Importantly, the catalytic performance for the PX production surpasses that of a series of noble metal catalysts as reported in the literature27,30,31,32,33.

a Catalytic performance of Co or Cu-based catalysts, Reaction conditions: 0.1 g catalyst, 0.1 g PET, 10 mL of 1,4-dioxane, 600 rpm, 230 °C, 2 MPa H2, 10 h; Effect of H2 pressure and temperature. b The stability of CuCo/CoOx. Reaction conditions: 0.1 g catalyst, 0.1 g PET, 10 mL 1,4-dioxane, 600 rpm, 230 °C, 2 MPa H2, 10 h. c 1H NMR spectra of the products in 1,4-dioxane and the pure 1,4-dioxane after reaction. a Products in 1,4-dioxane with higher purity (GC grade, 99.9%). d TGA curve of CuCo/CoOx after reaction. e GC spectra of reaction results. f Possible reaction pathway for the conversion of PET to PX over CuCo/CoOx. Reaction conditions: 0.1 g catalyst, 0.1 g substrate, 10 mL 1,4-dioxane, 600 rpm, 230 °C, 2 MPa H2, 2 h. PX refers to p-xylene.
Subsequent optimization of reaction conditions was conducted to maximize catalytic performance. The results demonstrated that low temperature hindered the reaction progress, while excessively high temperature also adversely affected the PX yield. Thermogravimetric analysis (TGA) (Fig. S8) indicated that the weight loss profile of the solid residues obtained at 250 °C did not correspond to typical PET degradation patterns (Fig. S4), which was attributed to undesired polymerization side reactions triggered by thermal overstress. Consequently, 230 °C was identified as the optimal reaction temperature. In addition, the hydrogen pressure exhibited a strong correlation with catalytic activity. Under low H2 pressures, the reaction mixture remained turbid and could contain partially degraded PET fragments and oligomeric intermediates. The highest PX yield (>99.9%) was achieved at 2 MPa H2. However, further increasing the hydrogen pressure resulted in decreased PX yield. This decrease is likely associated with the over-hydrogenation of aromatic rings, resulting in the formation of cycloalkyl intermediates, which show non-planar configurations. Such configurations render the cleavage of the chemically robust C–O linkages more challenging, as previously reported31. Based on these findings, the optimal reaction conditions were determined to be 230 °C and 2 MPa H2. Additionally, we optimized the solvent selection by evaluating several common organic solvents (tetrahydrofuran, cyclohexane, and 1,4-dioxane) as well as water. The results confirmed that 1,4-dioxane is the most promising solvent (for a detailed discussion, see Supporting Information below Table S1). Moreover, the catalyst demonstrated excellent reusability across four consecutive reaction cycles, maintaining consistently high PX yields (Fig. 2b), thereby highlighting its potential for practical applications.
To evaluate the purity of the product PX, a series of characterization techniques were performed. 1H nuclear magnetic resonance (1H NMR) analysis was performed on both the pure solvent and the reaction raw solution (Fig. 2c). The results demonstrated that, apart from the peaks corresponding to deuterated chloroform (CDCl3), solvent, and trace inherent impurities in solvent (Fig. 2c), only the distinct chemical shifts corresponding to PX were observed. Additionally, the H signals of PX at the α- and β-positions were maintained at 4:6 in solvents of varying purities, further confirming the high purity of the obtained PX and the absence of any by-products (Fig. S9). Consistently, gas chromatography (GC) analysis (Fig. 2e) showed a single product peak attributable to PX, after excluding the signals from the solvent (including residual peaks from needle washing) and the internal standard. TGA of the spent catalyst (Fig. 2d) showed no measurable mass loss, implying complete conversion of PET and the absence of undecomposed polymer residues. In addition, XRD, Raman spectroscopy, XPS, TEM, and FTIR spectroscopy, conducted on the catalyst before and after the reaction (Figs. S11–S14, S10), revealed no significant structural changes and detectable accumulation of organic residues (for a detailed discussion, see Supporting Information below Figs. S11–S14, S10). In short, these results unambiguously confirm that the near-quantitative production of PX was successfully achieved.
To elucidate the degradation pathway of PET over CuCo/CoOx, we conducted the hydrogenolysis of PET under mild conditions (210 °C, 2 MPa, 1 h), which successfully captured 2-hydroxyethyl 4-methylbenzoate (Fig. S15). Furthermore, the conversion of dimethyl terephthalate as a model substrate under milder conditions (190 °C, 1 MPa, 1 h) yielded intermediates such as 4-methylbenzyl alcohol and methyl p-methylbenzoate (Fig. S16). These results confirm that the reaction initially involves the cleavage of ester bonds, generating p-methylbenzene intermediates (e.g., 2-hydroxyethyl 4-methylbenzoate and ethyl p-methylbenzoate). These intermediates are subsequently hydrogenated to form 4-methylbenzyl alcohol, which undergoes deoxygenation to produce PX. To further validate this pathway, we investigated the conversion of model intermediates, including bis (2-hydroxyethyl) terephthalate, terephthalic acid, 4-methylbenzyl alcohol and ethyl p-methylbenzoate (Fig. 2f). The results showed that bis (2-hydroxyethyl) terephthalate, 4-methylbenzyl alcohol and ethyl p-methylbenzoate were efficiently converted to PX, with the highest yield (99.9%) achieved using 4-methylbenzyl alcohol as the substrate. In contrast, terephthalic acid exhibited negligible conversion. These findings support a reaction pathway in which PET hydrogenolysis proceeds through ester bond hydrogenation to form 4-methylbenzyl alcohol, followed by deoxygenation, rather than via a terephthalic acid intermediate. Based on these observations, a three-step reaction pathway is proposed for PET hydrogenolysis as Fig. 2f: (i) initial depolymerization, (ii) asymmetric hydrogenolysis, and (iii) terminal dehydroxylation. Furthermore, GC analysis of the gaseous products revealed the formation of both methane and ethane (Fig. S17), indicating that the ethylene glycol segments of PET undergo hydrogenolysis and C–C bond cleavage in parallel with the main depolymerization pathway.
Catalyst characterizations
To elucidate the morphological and electronic structures of the designed catalysts, comprehensive characterization was performed. Inductively coupled plasma optical emission spectroscopy (ICP-OES) verified that the actual Cu loadings were in close agreement with the nominal values (Table S2, Fig. S18). TEM images of Co/CoOx (Fig. 3a, b) and CuCo/CoOx (Fig. 3d, e) at varying magnifications reveal the formation of smaller nanoparticles in the CuCo/CoOx sample, indicative of reduced crystallinity and increased structural disorder. High-resolution TEM (HR-TEM) image of Co/CoOx (Fig. 3c) displays well-resolved lattice fringes at 0.203 nm and 0.246 nm, corresponding to the (002) plane of hexagonal close-packed (hcp) Co and the (111) plane of CoO, respectively. An interfacial structure (Interface I) between metallic Co and CoOx is clearly observed. The HR-TEM image of CuCo/CoOx (Fig. 3f) demonstrates the simultaneous presence of the (111) plane of face-centered cubic (fcc) Co (0.205 nm) alongside the (002) and (101) planes of hcp-Co (0.203 nm and 0.191 nm, respectively). This coexistence provides direct evidence of a phase transformation within the CuCo/CoOx catalyst from hcp-Co to fcc-Co. Noteworthy, in addition to Interface I (hcp-Co/CoOx), two additional types of interfaces, namely abundant Interface II between fcc-Co and CoOx and Interface III between hcp-Co and fcc-Co, are observed. The appearance of Interface III (hcp-Co/fcc-Co) further suggests the partial phase transformation from hcp-Co to fcc-Co. Overall, HR-TEM analysis reveals that the incorporation of Cu induces partial phase transformation and lattice distortions, thereby promoting the formation of numerous interfaces, with Interface II (fcc-Co/CoOx) being particularly prevalent. Moreover, energy-dispersive X-ray spectroscopy (EDS) elemental mapping (Fig. 3g–j) confirms the homogeneous spatial distribution of Co, Cu, and O elements within CuCo/CoOx, indicating high elemental dispersion and structural uniformity.

a TEM image of Co/CoOx. b, c HR-TEM images of Co/CoOx. d TEM image of CuCo/CoOx. e, f HR-TEM images of CuCo/CoOx. g–j EDS spectroscopy mapping profiles of CuCo/CoOx. k XRD patterns. l Raman spectra. m EPR spectra. n The IR spectra of adsorbed pyridine on CuCo/CoOx and Co/CoOx catalysts. o, p XPS spectra of (o) O1s and (p) Co 2p3/2 in CuCo/CoOx and Co/CoOx catalysts.
To further understand the phase transformation, comparative XRD analyses were performed. The diffraction patterns of unreduced catalysts (Fig. S19) reveal negligible differences in the characteristic peaks of Co3O4 between the two samples, indicating that the phase transformation primarily occurs during the reduction process. As shown in Fig. 3k, the XRD patterns of the reduced catalysts exhibit distinct differences. The Co/CoOx sample displays characteristic diffraction peaks of Co (2θ = 41.7, 44.8, 47.6, 75.9°) (PDF#05-0727). Notably, a discernible redshift of the metallic Co peak at 44.8°, corresponding to the (111) plane, is observed in CuCo/CoOx. This shift is attributed to a partial phase transformation from hcp-Co to fcc-Co during the reduction process41, consistent with the HR-TEM observations. To elucidate the influence of phase transformation on catalytic performance and to assess the implications of complete phase transition at elevated temperatures, the pre-reduced CuCo/CoOx catalyst was subjected to thermal treatment at 500 °C under an inert Ar atmosphere. The structural characterizations of CuCo/CoOx-500 (Figs. S20–S24) revealed the complete disappearance of hcp-Co diffraction features, accompanied by a pronounced increase in the crystallinity of the fcc-Co phase (for a detailed discussion, see Supporting Information below Figs. S20–S24). Catalytic evaluations under optimized conditions showed a decrease in PX yield to 94.6% compared with the untreated CuCo/CoOx catalyst (Fig. S25). This decrease in activity is likely attributed to the competing effects of enhanced intrinsic activity associated with the fcc-Co phase and a reduced number of accessible active sites resulting from particle agglomeration. Importantly, it is evident that the introduction of Cu significantly decreases the crystallinity of the catalyst, likely due to the occurrence of lattice distortions and the formation of oxygen vacancies42. To identify the occurrence of lattice distortions and the formation of oxygen vacancies, several characterization techniques were systematically employed. Raman spectra (Fig. 3l) show that the bands at 186, 501 and 592 cm−1 are assigned to the F2g vibrational mode, corresponding to Co-O stretching vibrations associated with Co2+ species. The band at 461 cm−1 is attributed to the Eg mode of the Co3O4 spinel structure, arising from lattice vibrations induced by the synergistic interaction between Co3+ and Co2+ ions43. For the Co/CoOx catalyst, the A1g mode at 661 cm−1 is characteristic of octahedrally coordinated cobalt species (CoO6). Notably, in the case of the CuCo/CoOx catalyst, the A1g mode exhibits a red shift from 661 to 651 cm−1, indicating the presence of lattice distortion. This shift suggests that the incorporation of Cu species induces structural defects and intensifies lattice distortion44, thereby facilitating a partial phase transformation from hcp-Co to fcc-Co and promoting the generation of a greater number of oxygen-vacancy-rich Co0/CoOx interfacial sites. The Electron Paramagnetic Resonance (EPR) results (Fig. 3m) exhibit signals at g = 2.003, which are attributed to unpaired electrons associated with oxygen vacancies45. Notably, the CuCo/CoOx catalyst displays a significantly stronger signal intensity compared to Co/CoOx, indicating a higher concentration of oxygen vacancies. The FTIR of pyridine adsorption show the three peaks that centered at 1596, 1581 and 1438 cm−1, which can be assigned to the Lewis acid sites on the surface46 (Fig. 3n). Notably, the peaks corresponding to Lewis acid sites exhibit substantially larger integrated areas on the CuCo/CoOx catalyst compared to Co/CoOx, clearly indicating a higher density of Lewis acid sites. This pronounced increase suggests the presence of a greater number of coordinatively unsaturated CoOx sites, which are closely associated with enhanced oxygen vacancy concentrations. Ultraviolet-visible (UV-Vis) diffuse reflectance spectroscopy (Fig. S26) shows that CuCo/CoOx exhibits significantly stronger light absorption than Co/CoOx, suggesting that Cu incorporation modulates the electronic band structure and promotes the formation of additional defect states associated with oxygen vacancies. Furthermore, XPS was performed to systematically investigate the surface electronic states of the catalysts. O 1 s spectrum was deconvoluted into three distinct components: lattice oxygen (OL) at 529.6 ± 0.1 eV, oxygen vacancies (Ov) at 531.3 ± 0.1 eV, and adsorbed oxygen species (Oa) at 533.4 eV (Fig. 3o). Quantitative analysis indicated a notable increase (Figure. S27) in the relative concentration of oxygen vacancies from 27.9% in Co/CoOx to 35.4% in CuCo/CoOx. In addition, Co 2p3/2 XPS (Fig. 3p) analysis revealed that the Co0/Co2+ ratio increased significantly from 0.19 to 0.60, indicating a more balanced distribution between Co0 and Co2+ in CuCo/CoOx47. Consequently, the population of interfacial active sites experienced a substantial increase. The Cu 2p XPS spectra (Figs. S28, S29) revealed a surface ratio of (Cu0+ Cu⁺)/Cu2+ = 1.73 for CuCo/CoOx47,48. The Cu LMM Auger spectra (Fig. S30) further showed a Cu0/Cu⁺ ratio of 1.14, confirming the substantial presence of Cu0 and CuOx species on the surface of CuCo/CoOx48. The specific surface area and pore size of Co/CoOx and CuCo/CoOx were characterized using the Brunauer-Emmett-Teller method (Table S3). Compared to Co/CoOx, CuCo/CoOx exhibited a larger specific surface area, pore volume, and average pore size, favoring greater exposure of active sites and enhancing mass transfer during the reaction process. In summary, the above multi-technique characterizations provide compelling evidence that Cu incorporation induces lattice distortion, facilitates partial hcp-to-fcc phase transformation, and enhances the concentration of oxygen vacancy-rich interfacial Co-CoOx sites, all of which could contribute to the improved catalytic performance of the CuCo/CoOx system. Furthermore, the CuCo/CoOx catalyst still retained its structure after the reaction (for a detailed discussion, see Supporting Information below Figs. S10–13).
Based on the aforementioned characterization results, it is evident that the incorporation of Cu induces a substantial decrease in overall crystallinity, resulting in a more amorphous structural nature that may facilitate the exposure of additional surface active sites. More notably, two critical and distinctive structural features have been identified: 1) the partial phase transformation of hcp-Co to fcc-Co; 2) the introduction of lattice distortion that generates oxygen vacancy-rich Co0/CoOx interfaces, with the fcc-Co0/CoOx interfaces being predominant. Since the influence of the distinctive structural characteristics on catalytic performance has been exhibited, these findings naturally lead to two fundamental scientific questions that will be systematically addressed in subsequent investigations: First, what are the formation mechanisms underlying these unique structural features? Second, what are the molecular-level structure-activity relationships governing this catalytic system?
Identification of stepwise hydrogen spillover
To elucidate the formation mechanism of the unique active sites structure, a comprehensive investigation was conducted by integrating H2-temperature programmed reduction (H2-TPR) (Fig. 4a), hydrogen spillover-controlled experiments (Fig. 4b) and density functional theory (DFT) calculations (Fig. 4c). The H2-TPR profile of Co/CoOx exhibits a minor reduction peak at 200–300 °C, corresponding to the reduction of Co3+ to Co2+, and a dominant peak at 300–400 °C, assigned with the complete reduction of Co2+ to Co0. Upon Cu incorporation, the entire reduction peaks shift downward by approximately 100 °C. This shift is primarily attributed to hydrogen spillover, which may proceed via two possible pathways: 1) Cu0-mediated spillover: CuO is first reduced to metallic Cu0, followed by the migration of dissociated hydrogen species (H*) from Cu0 to adjacent CoOx, thereby accelerating its reduction; 2) Co0-mediated spillover: CoOx undergoes initial partial reduction to Co0, and the resulting metallic Co0, which subsequently promotes further hydrogen dissociation and then spillover to surrounding CoOx domains. To elucidate the difference in reducibility between CuO and Co3O4, both oxides were individually supported on inert (SiO2) and reducible (TiO2) supports, and subjected to H2-TPR. In all cases, CuO exhibited a consistently lower reduction temperature than Co3O4, likely due to its lower Gibbs free energy of formation. Therefore, it can be concluded that the Cu-mediated spillover pathway is thermodynamically more favorable. Further insight was gained from the H2-TPR analysis of a physically mixed Co/CoOx and Cu/TiO2 (Cu/SiO2) sample in a 1:1 mass ratio, which showed only a modest downward shift in reduction temperature, suggesting weaker interactions and diminished spillover efficiency compared to the integrated CuCo/CoOx system.

a H2-TPR profiles of Co/CoOx, CuCo/CoOx, Co/TiO2, Cu/TiO2, Co/SiO2, Cu/SiO2 and physically mixed Co/CoOx and Cu/TiO2 (Cu/SiO2) samples in a 1:1 mass ratio. b Hydrogen spillover experiments of unreduced Co/CoOx, CuCo/CoOx, Cu/SiO2, physically mixed Co/CoOx and Cu/SiO2 samples in a 1:1 mass ratio and reduced Co/CoOx, CuCo/CoOx. c Calculated adsorption energies of H2 and dissociation energy of H2 on Cu (111), hcp Co (101) and fcc Co (111). d The proposed mechanism of stepwise hydrogen spillover on CuCo/CoOx.
Then, the WO3 coloration experiments show that pronounced changes in color upon exposure to unreduced Co/CoOx and CuCo/CoOx catalysts, with WO3 transitioning to yellow-green and deep blue, respectively (Fig. 4b), reflecting markedly enhanced hydrogen spillover after Cu incorporation. In contrast, unreduced Cu/SiO2 exhibits negligible chromatic change, whereas a physical mixture of unreduced Cu/SiO2 and Co/CoOx in a 1:1 mass ratio induced only limited color variation, indicating the reduced hydrogen spillover efficiency due to limited contact between Cu and CoOx. These stark contrasts provide compelling evidence for intensive hydrogen spillover phenomena during the reduction of CuCo/CoOx, in agreement with H2-TPR results. Furthermore, the reduced catalysts exhibit discernible hydrogen spillover effects, although the associated coloration is markedly diminished compared to their pre-reduced state. This decrease in chromatic intensity provides further evidence for substantial hydrogen spillover occurring during the reduction process.
To further understand the hydrogen spillover phenomena, DFT calculations were carried out to systematically evaluate both the hydrogen adsorption energies and dissociation energy barriers on metallic Cu0 and Co0 surfaces with distinct crystallographic structures (Fig. 4c, Figs. S31–33). Based on lattice parameter variations identified by XRD and predominant surface orientations observed via TEM, atomistic models were constructed for hcp-Co(101), fcc-Co(111), and Cu(111) surfaces to simulate representative hydrogen activation sites. The calculations revealed that hydrogen exhibits stronger chemisorption on Co0 surfaces compared to Cu0. More importantly, the energy barrier for H2 dissociation on fcc-Co was found to be as low as 0.05 eV, significantly lower than that on hcp-Co (0.18 eV) and Cu (0.48 eV), indicating facile hydrogen activation on fcc-Co. Furthermore, the H2 dissociation process on fcc-Co is more exothermic (−0.87 eV) compared to that on hcp-Co (−0.83 eV) and Cu (−0.55 eV), indicating a thermodynamically more favorable activation on the fcc-Co surface. These results provide a mechanistic rationale for the experimentally observed hydrogen spillover process: although Cu0 acts as the initial site for H2 dissociation and primary spillover, the emergence of Co0 species, especially fcc-Co species, enables a more efficient secondary spillover due to its superior capability of H2 dissociation.
Building upon these findings, a stepwise hydrogen spillover mechanism is proposed (Fig. 4d). Initially, CuOx species are preferentially reduced to metallic Cu0 sites capable of dissociating H2 into active H* species. These H* species subsequently migrate to adjacent CoOx regions, promoting the reduction of a small amount of CoOx to Co0. The formed Co0 sites, particularly those with fcc-structure, then act as secondary spillover centers, further facilitating hydrogen activation and migration for the reduction of more CoOx species. In summary, the unique stepwise hydrogen spillover process not only facilitates the partial phase transformation of hcp-Co into fcc-Co, but also promotes the formation of oxygen vacancy-rich Co0/CoOx interfaces, among which the fcc-Co0/CoOx interfaces are dominant.
Unique roles of CuCo/CoOx
Subsequently, the focus lies in explaining why the active sites constructed via the aforementioned strategies exhibit such distinctive catalytic efficiency. Due to the inherent difficulty in precisely quantifying the number of active sites on the catalyst surface, kinetic orders for the substrate and H2 could not be determined. Nevertheless, within the range of kinetic testing conditions, a systematic investigation was conducted to examine the relationship between product yield and both substrate concentration and hydrogen pressure. The results indicate that both factors exhibit comparable levels of influence, as demonstrated by the similar slopes obtained from fitting the data in Fig. 5a. This convergence suggests that hydrogen partial pressure and substrate concentration exert equally significant and synergistic effects on catalytic efficiency. Considering the critical role of reactant adsorption and activation in heterogeneous catalysis, particular focus was placed on investigating the adsorption and activation behaviors of H2 and PET on the CuCo/CoOx catalyst via temperature-programmed desorption (TPD) analysis.

a PX production efficiency under different H2 pressure and p-methylbenzoate concentration. Reaction conditions: 0.05 g catalyst, 0.2 g ethyl p-methylbenzoate, 10 mL of 1,4-dioxane, 600 rpm, 190 °C, 1 h, H2 pressure ranging from 0.5 MPa to 2 MPa; 0.03 g catalyst, 10 mL of 1,4-dioxane, 600 rpm, 190 °C, 1 h, 2 MPaH2, weight of ethyl p-methylbenzoate ranging from 0.5 g to 2.0 g; b H2-TPD profiles of CuCo/CoOx and Co/CoOx. c Time profile of hydrogenolysis on CuCo/CoOx and Co/CoOx. d Ethyl p-methylbenzoate-TPD profiles of CuCo/CoOx and Co/CoOx. e PX-TPD profiles of CuCo/CoOx and Co/CoOx. f–h In situ methyl benzoate-absorbed FTIR spectra in the presence of Ar on CuCo/CoOx and Co/CoOx. i In situ methyl benzoate-conversion FTIR spectra in the presence of 10% H2-Ar on CuCo/CoOx and Co/CoOx. j Proposed catalytic mechanism on CuCo/CoOx. PX refers to p-xylene.
First, H2-TPD analysis (Fig. 5b) reveals two distinct desorption peaks for both Co/CoOx and CuCo/CoOx catalysts. The low-temperature peak is attributed to the adsorption of hydrogen on metallic species, while the high-temperature peak is likely associated with hydrogen spillover processes. Notably, the CuCo/CoOx catalyst exhibits a significantly larger total desorption area compared to Co/CoOx, suggesting a higher density of active sites available for H2 adsorption and activation. This observation is consistent with previous DFT calculations (Fig. 4c), which demonstrate that the fcc-Co phase present in CuCo/CoOx exhibits superior hydrogen adsorption and dissociation capabilities compared to the hcp-Co phase in Co/CoOx. In addition, XRD analysis (Fig. 3k) reveals reduced overall crystallinity in the CuCo/CoOx catalyst, implying a greater abundance of exposed active sites. Moreover, the high-temperature desorption peak for CuCo/CoOx shifts to a substantially higher temperature, indicating stronger binding or stabilization of spillover-formed hydrogen species. This shift is corroborated by WO3 coloration experiments using both Co/CoOx and CuCo/CoOx catalysts (Fig.4b), which confirm enhanced hydrogen spillover capability on CuCo/CoOx. The resulting Co–OH species formed via spillover exhibit improved thermal stability, likely due to the presence of abundant oxygen vacancies. These vacancies may facilitate hydrogen retention and stabilization, thereby accounting for the elevated desorption temperature observed49,50.
The time course of ethyl p-methylbenzoate conversion revealed that CuCo/CoOx exhibits higher activity consistently. In contrast, when Co/CoOx achieved a yield of 50% at 6 h, the reaction progressed minimally thereafter with the increased yield of PX, implying the existence of competitive adsorption between PX and the substrate (Fig. 5c). The TPD of ethyl p-methylbenzoate was conducted after in situ reduction of Co/SiO2, Cu/SiO2 at 500 °C, and CuO at 145 °C to obtain Co0, Cu0, or CuOx sites (Fig. 5d). The results show negligible adsorption of ethyl p-methylbenzoate on all catalysts. In contrast, both Co/CoOx and CuCo/CoOx exhibit significant adsorption, with the desorption peak area for CuCo/CoOx being 3.5-fold larger than that for Co/CoOx. This suggests a notably higher density of substrate adsorption sites, primarily located on CoOx, rather than Cu0 or CuOx. Cu species (whether Cu0 or CuOx) do not act as the primary active sites in the catalytic process, but instead function as components that significantly contribute to the formation of the oxygen vacancy-rich Co0/CoOx interface active sites through stepwise hydrogen spillover. The enriched coordinatively unsaturated CoOx sites in CuCo/CoOx account for its abundant active sites.
To investigate the role of unsaturated CoOx at the Co/CoOx interfaces facilitating substrate activation and validate the relationship between unsaturated CoOx species and substrate adsorption capacity, Co3O4 was reduced at 250 °C and 350 °C to obtain CoOx species with varying degrees of coordination saturation (denoted as Co/CoOx-250 and Co/CoOx-350, respectively), distinct from the Co/CoOx sample. XPS (Fig. S34), EPR (Fig. S35), Raman (Fig. S36), and pyridine-IR analyses (Fig. S37) confirm that Co/CoOx reduced at 300 °C possesses the most abundant unsaturated CoOx species, which are closely associated with enhanced oxygen vacancy concentrations51. Subsequently, in-situ FTIR adsorption-desorption experiments (Fig. S38) and TPD experiments (Fig. S39) further reveal a strong positive correlation between substrate adsorption-activation and the presence of unsaturated CoOx species.
In order to investigate the role of different metals in active site construction and their structure-activity relationships, we systematically characterized a series of MCo/CoOx (M = Ru, Fe, Ni, Cu, Co) catalysts (Figs. S40–S43) (for a detailed discussion, see Supporting Information below Figs. S38–S43). The findings are as follows: Fe and Ni are incapable of inducing hydrogen spillover. Although Ru can initiate hydrogen spillover to promote the reduction of CoOx, excessive reduction inhibits the formation of low-crystallinity fcc-Co and low-coordinated, oxygen vacancy-rich CoOx species. In contrast to other MCo/CoOx catalysts, the introduction of Cu plays a critical role in initiating the stepwise hydrogen spillover process. This process not only promotes the partial phase transition from hcp-Co to fcc-Co but also facilitates the formation of oxygen vacancy-rich Co0/CoOx interfaces, with fcc-Co0/CoOx interfaces being the predominant species. As we have robustly demonstrated, the synergy between Co0 (particularly fcc-Co) and oxygen vacancy-rich CoOx species at the abundant Co0/CoOx interfacial sites significantly enhances the catalytic activity.
In addition, comparative TPD studies of PX revealed distinct differences in product adsorption strengths (Fig. 5e). PX exhibited notably weaker adsorption on CuCo/CoOx, which facilitates its rapid desorption and promotes continuous catalytic turnover. In contrast, on Co/CoOx, PX displayed much stronger adsorption, with its desorption temperature nearly coinciding with that of ethyl p-toluate. This overlap implies significant competitive adsorption between the substrate and the product. Accumulation of PX significantly suppresses the reaction progress, consistent with the observed reaction profiles. These findings, together with prior characterizations and TPD analyses of H2 and substrate adsorption-desorption, support a synergistic mechanistic model. The lattice distortion within CuCo/CoOx reduces crystallinity and promotes the formation of fcc-Co domains, which exhibit enhanced hydrogen dissociation capabilities. Concurrently, interfacial CoOx sites with abundant vacancies induced by lattice distortion promote substrate adsorption and product desorption. These processes are further promoted by interfacial hydrogen spillover, collectively contributing to the superior catalytic performance of CuCo/CoOx.
To gain deeper insight into the catalytic mechanism at the molecular level, a series of in situ FTIR spectroscopy experiments was conducted using methyl benzoate as a model compound (Fig. 5f–i). The characteristic band observed at 1729 cm−1 is assigned to the C=O stretching vibration, while the peaks at 1278 cm−1 and 1110 cm−1 correspond to the asymmetric and symmetric C–O stretching vibrations, respectively52. Notably, all three bands exhibited enhanced intensity on the CuCo/CoOx catalyst compared to Co/CoOx, indicating a higher density of active adsorption sites. Temperature-programmed in situ FTIR adsorption-desorption experiments on both catalysts revealed contrasting vibrational shifts: with increasing temperature, the C–O bands exhibited red shifts, whereas the C = O band underwent distinct blue shifts. This inverse shift behavior strongly suggests that the C–O bond, rather than the C = O bond, is preferentially adsorbed and activated. The preferential activation of the C–O bond is proposed to alter the electronic environment of adjacent functional groups, resulting in strengthened (blue-shifted) C = O vibrations. Importantly, on the CuCo/CoOx catalyst, additional IR bands emerged at 1694 cm−1, closely matching those of aldehyde species53, and their intensities increased progressively with temperature. In contrast, no such bands were observed on Co/CoOx, indicating a superior ability of CuCo/CoOx to activate and cleave the C–O bond in ester groups. This provides compelling mechanistic evidence that on CuCo/CoOx, the initial activation of the C–O bond leads to the formation of aldehyde-like intermediates. To investigate the differences in adsorption behavior between the substrate and the solvent 1,4-dioxane on CuCo/CoOx, we performed TPD and in-situ FT-IR experiments using 1,4-dioxane as a probe molecule (Fig. S44). The results showed an absence of desorption peaks or infrared signals corresponding to 1,4-dioxane, indicating negligible adsorption on the catalyst surface. These findings suggest that the catalyst can selectively adsorb and activate the substrate, and 1,4-dioxane remains chemically inert and stable as the solvent throughout the reaction process. Subsequently, in situ hydrogenolysis was initiated by introducing hydrogen gas over the CuCo/CoOx catalyst. As the reaction proceeded, the characteristic vibrational signals of ester groups gradually diminished, suggesting smooth and continuous conversion. However, no detectable signals corresponding to aldehyde intermediates were detected during this process, implying their rapid conversion into subsequent products. Their highly transient nature renders them difficult to capture by in situ spectroscopic techniques.
Based on the aforementioned results, the catalytic mechanism over CuCo/CoOx is proposed (Fig. 5j). The process initiates with the adsorption and activation of the C–O bond in PET on the oxygen-vacancy-rich CoOx interface. Subsequently, H2 is dissociated on metallic sites, predominantly fcc-Co species, generating active H* species that spill over to adjacent CoOx surfaces. These H* species attack the activated C–O bonds, resulting in their cleavage and the formation of aldehyde intermediates. The C = O group within the aldehyde is further activated and hydrogenated on CoOx to form a hydroxyl intermediate, which then undergoes C–O bond cleavage to yield the final product PX. Finally, PX desorbs from the catalyst surface, completing the catalytic cycle. In short, the distinctive catalytic role of CuCo/CoOx includes that compared to Co/CoOx, CuCo/CoOx exhibits superior H2 dissociation due to the presence of fcc-Co phases. The oxygen-vacancy-rich interfacial CoOx species not only facilitates the activation of C–O bonds in PET but also modulates the adsorption equilibrium between the substrate and product, thereby continuously driving the reaction forward.
CuCo/CoOₓ for real-world plastic waste conversion and techno-economic assessment
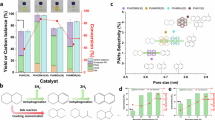
To evaluate the practical applicability of the CuCo/CoOx catalyst, its performance was systematically assessed for the conversion of 28 real-world PET plastics, including single-component PET, PET with additives and PET-based mixed plastics involving various feed ratios (Fig. 6a–c, Figs. S45–S49 and Tables S4, S5). Following simply shearing, all tested feedstocks can be efficiently converted into PX over CuCo/CoOx (Fig. 6a). To investigate the impact of typical additives present in waste plastics, a range of representative compounds was examined54 (Fig. 6c). The majority of these additives (1–4 and 7 in Fig. 6c) exhibited negligible influence on catalytic activity, maintaining PX yields above 99.9%. Some additives (5 in Fig. 6c) induced only mild inhibition, with yields retained at ~ 95%, whereas a few specific species (6, 8 in Fig. 6c) exerted more pronounced poisoning effects. Furthermore, the CuCo/CoOx catalyst demonstrates remarkable potential for the selective conversion of PET in mixed plastics, enabling its high-selectivity conversion to PX without interference from other plastics (Fig. 6b). Furthermore, this catalyst demonstrates significant potential for the conversion of 5 other aromatic polyesters (e.g., PBT, PBAT) as well as aliphatic polyesters (e.g., PLA, PCL, PBS) (for a detailed discussion, see Supporting Information below Tables S4, S5). These results highlight the intrinsic robustness and tolerance of the CuCo/CoOx system toward real-world impurities.

a The direct hydrogenolysis of single-component PET plastics. Reaction conditions: 0.1 g CuCo/CoOx, 0.1 g plastic, 10 mL 1,4-dioxane, 250 °C, 2 MPa H2, 600 rpm, 12 h. b The direct hydrogenolysis of PET-based mixed plastics. Reaction conditions: 0.1 g CuCo/CoOx, 0.1 g plastics, 10 mL 1,4-dioxane, 250 °C, 2 MPa H2, 600 rpm, 12 h. c The direct hydrogenolysis of PET with additives. Reaction conditions: 0.1 g CuCo/CoOx, 0.1 g of PET plastics, 5 wt% additives, 10 mL 1,4-dioxane, 230 °C, 2 MPa H2, 600 rpm, 10 h. d Comparison of environmental and economic benefits between naphtha-to-PX and PET-to-PX routes. PET refers to polyethylene terephthalate, PX refers to p-xylene.
To benchmark the techno-economic and environmental advantages of the PET-to-PX process, a comprehensive comparison was conducted against conventional naphtha-to-PX production, with both routes maintaining elemental mass balance (Figs. S50, S51). In terms of economic efficiency, the PET-to-PX pathway exhibits a lower production cost of ¥5.90 per kg PX and a higher profit margin of ¥3.30 per kg PX, representing a 106.3% increase compared to the conventional naphtha-based routes. A life cycle assessment (LCA) performed using Simpro and Aspen simulations further indicates a significantly reduced global warming potential (GWP) of 0.45 kg CO2 eq/kg PX, reflecting a 30.7% decrease in emissions (Fig. 6d). A comprehensive life-cycle carbon footprint analysis, including feedstock acquisition, transportation, and production, further confirms the considerable carbon reduction potential of this upcycling strategy (Fig. 6d). Additional environmental advantages include the valorization of by-product gases (methane and ethane) for natural gas generation, high-purity PX output, and exceptional catalyst durability, which allows for multiple recycling cycles. Detailed parameter settings, economic comparisons, environmental evaluation results and in-depth discussion are provided in Supporting Information (Figs. S50–S53 and Tables S6–S88). Collectively, these findings establish the CuCo/CoOx-catalyzed PET-to-PX conversion as a viable, economically attractive, and environmentally superior alternative to traditional petroleum-based PX production, offering a sustainable route for plastic waste valorization and fossil resource mitigation.
Discussion
In summary, we have developed a CuCo/CoOx catalyst that harnesses a unique stepwise hydrogen spillover mechanism to realize unprecedentedly high selectivity in PET hydrogenolysis to p-xylene (>99.9%), markedly surpassing the catalytic performance of a wide range of Cu- and Co-based catalysts as well as state-of-the-art noble metal systems. This distinctive stepwise hydrogen spillover pathway, initiated by Cu0-facilitated hydrogen activation and amplified through Co0-driven secondary spillover, promotes the formation of partially phase-transformed Co0 species alongside abundant oxygen-vacancy-rich Co0/CoOx interfaces. The former substantially enhances H2 dissociation, while the latter critically enables efficient C–O bond activation and finely tunes the adsorption-desorption equilibria between substrates and products, collectively underpinning the exceptional catalytic efficacy. Especially, we directly observed the activation signals of ester bonds in PET, leading to the formation of an aldehyde-like intermediate, through in situ FT-IR spectroscopy. This provides clear molecular-level evidence of ester bond activation and offers deeper insights into the hydrogenolysis mechanism of PET. The catalyst’s demonstrated versatility across diverse post-consumer plastic feedstocks underscores its practical and scalable applicability. Combined with favorable techno-economic metrics, including reduced carbon emissions and cost competitiveness, the system demonstrates significant application potential. The achievement of near-quantitative conversion of waste PET plastics into PX, coupled with the innovative construction of active sites through a stepwise hydrogen spillover, represents a significant advancement for plastic waste upcycling and the rational design of advanced catalysts.
Methods
Synthesis of catalysts
The Co3O4 supports were synthesized with a precipitation method using cobalt nitrate as the synthetic precursor37. Typically, 17.46 g Co(NO3)2·6H2O and 6.63 g (NH4)2CO3 were dissolved in 200 ml deionized water, respectively. Then the solution of (NH4)2CO3 was added slowly into the solution of Co(NO3)2·6H2O with vigorous stirring to form a precipitate until the pH ≈ 9. After that, the resulting suspension was stirred at 65 °C for 1 h and then allowed to maintain at room temperature for 16 h. After filtration and washing with deionized water multiple times, ensuring the pH reached 7, the solid phase was dried under vacuum at 100 °C for 12 h and finally calcined in air at 500 °C for 4 h to obtain Co3O4 supports. The CuO supports were synthesized in the same way.
CuCo/CoOx was prepared by a simple impregnation method. Detailly, the Co3O4 support was added to a 5 mL aqueous solution containing 0.3802 g Cu(NO3)3·3H2O. After stirring at room temperature overnight to ensure the uniform dispersion of Cu, the obtained sample was dried at 80 °C for 16 h, and then calcined at 500 °C in the air for 3 h.
Cu/TiO2, Co/TiO2, Cu/SiO2 and Co/SiO2 were synthesized in the same way as CuCo/CoOx, with supports changed. All catalysts should be reduced at 300 °C for 3 h under flowing H2 (10% H2-Ar mixed gas) before reaction.
Catalytic test and product analysis
The detailed reaction conditions are described in the figure captions and table footnotes. In a typical reaction with PET conversion as an example, 0.1 g of PET balls, 0.1 g of catalyst, and a solvent mixture of 10 mL of 1,4-dioxane were loaded into a stainless-steel autoclave reactor in a 50 mL stainless steel batch reactor equipped with a Polytetrafluoroethylene (PTFE) liner and magnetic rotor. After the reactor was purged with H2 three times and charged to the target H2 pressure (2 MPa), the reaction was conducted at the target temperature (230 °C) with a magnetic stirring speed of 600 rpm. Upon completion, the reactor was cooled naturally. The quantitative analysis of reacting products was performed using gas chromatography (GC-2060) with a flame ionization detector equipped with HP-5 capillary columns. The column temperature began at 50 °C (held for 5 min) and was then increased to 280 °C at a heating speed of 10 °C min−1 (held for 10 min); the total running time was 50 min. The liquid solution was separated from the solid catalyst by centrifugation and was directly analyzed when 1,4-dioxane was used as a solvent. Biphenyl was used as an internal standard for the quantification of the liquid products. The PX yield was calculated following the Eq. (1):
Characterizations and in situ reaction studies
The crystal structure of the catalyst was analyzed via X-ray diffraction with Cu Kα (λ = 0.15406 nm) radiation (XRD, D8 ADVANCE, Bruker Co., Germany) at 40 kV and 40 mA. The morphology of catalysts was examined by transmission electron microscopy (TEM) with an FEI Talos 200X (USA) field emission electron microscope. The actual Cu loading in the sample was detected by a Thermo ScientificTM iCAPTM PRO XP ICP-OES. Raman spectra were recorded using a HORIBA XploRATM PLUS equipped with an Ar⁺ laser (532 nm wavelength). Surface composition and chemical states of the products were characterized by X-ray photoelectron spectroscopy (XPS, PHI 5000 Versaprobe III, Japan) equipped with monochromatic Al Kα radiation, with binding energies corrected using the C 1 s peak at 284.8 eV as a reference. Defects in catalysts were determined using electron paramagnetic resonance (EPR, MEX-nano, Bruker, Germany). Optical properties were obtained using a UV-vis spectrophotometer (UV-2600, Shimadzu, Japan). The 1H nuclear magnetic resonance (1H NMR) spectrum was recorded on an AV 500 MHz Fourier-transform superconducting nuclear magnetic resonance spectrometer. Thermal gravimetric analysis (TGA) of the catalyst was conducted on a HS-TGA-101 Thermogravimetric Analyzer. Fourier-transform infrared (FTIR) spectroscopy on the catalyst was performed on a FOLI10-R Fourier-transform infrared spectrometer.
FTIR of pyridine adsorption was recorded on the FOLI10-R Fourier-transform infrared spectrometer with an in-situ cell and MCT detector. Following the dissolution of a 5 mg sample in cyclohexane, the solution was applied dropwise onto a 160 mg KBr pellet and uniformly spread. The sample was in situ reduced at a H2 (10% H2-Ar mixed gas) atmosphere at 300 °C for 30 min, and then heated at 400 °C for 30 min under vacuum in order to remove the physically adsorbed water. After cooling to 100 °C, a background scan was performed. Pyridine vapor was then introduced into the cell at room temperature for 20 min. Subsequent evacuation was performed at 100 °C for 30 min followed by spectral acquisitions. The spectra presented were obtained by subtracting the spectra recorded before and after pyridine adsorption.
In-situ FTIR measurements of methyl benzoate adsorption were collected with the FOLI10-R Fourier-transform infrared spectrometer with an in-situ cell and MCT detector. Firstly, the catalysts were pre-treated in the in-situ cell in a H2 (10% H2-Ar mixed gas) atmosphere at 300 °C for 30 min and then switched to an Ar atmosphere for 30 min to eliminate surface adsorbed substances. Then the background scan of the sample was performed. Methyl benzoate was bubbled into the in-situ cell under an Ar atmosphere for 40 min. Next, the cell was purged with Ar at 35 °C for 30 min, and the adsorption spectra of methyl benzoate were recorded. Finally, the temperature was increased to 300 °C, and the adsorption spectra of methyl benzoate were recorded at increasing temperature. The in-situ reaction was conducted following the same procedure with H2 (10% H2-Ar mixed gas) introduced during the heating process.
Overlayer formation of the catalysts was investigated using hydrogen temperature-programmed reduction (H2-TPR) on VDsorb-92i. 50 mg of catalyst was treated at 300 °C for 30 min under flowing Ar at 30 sccm. After cooling to room temperature, the catalyst was exposed to 10% H2-Ar (30 sccm) and heated to 800 °C at a ramp rate of 10 °C/min to determine the reducibility of the catalysts.
The absorption and desorption of H2 and ethyl p-methylbenzoate on the catalysts were investigated using temperature-programmed desorption (TPD) on VDsorb-92i. 50 mg of catalyst was loaded into a quartz U-reactor and pre-treated at 300 °C for 30 min in a flowing 10% H2-Ar (30 sccm) to simulate the in-situ reduction process. After cooling to room temperature, the catalyst was heated to 300 °C for 30 min under flowing Ar at 30 sccm to eliminate surface adsorbed substances. After the temperature dropped to 30 °C, the H2 or ethyl p-methylbenzoate was bubbled onto the catalysts for 40 min under a flow of Ar. The sample was then purged by Ar for 40 min to remove the physically adsorbed substances, and the temperature was heated to 800 °C at a heating rate of 10 °C·min−1.
Computational methods
The spin-polarized density functional theory (DFT) calculations were performed by VASP (version number: 5.4.4)55, using the Perdew–Burke–Ernzerh (PBE) functional within the generalized gradient approximation (GGA)56. The VDW correction (PBE-D3) was also included. The core-valence electron interaction was described by the projector augmented wave (PAW) method57, and the valence electronic states were expanded in the plane-wave basis sets with a cutoff energy of 450 eV. According to the experimental characterization results, the surface configurations of Cu (111), hcp-Co (101) and fcc-Co (111) were modeled separately as the p (2 × 2), p (2 × 2) and p (2 × 2) periodic slabs, and the vacuum between slabs was 15 Å to exclude the mutual influence. During structural optimization, the bottom two layers were fixed, and all other atoms were fully relaxed. For these surface slabs, a 2 × 2 × 1 k-point mesh was used. The transition states were searched using a constrained optimization scheme58. The force threshold for the optimization and transition state search was 0.05 eV/Å. The adsorption energy of surface species was defined via the following formula (2):
where \({E}_{{surf}}\), \({E}_{X}\), and \({E}_{\frac{X}{{surf}}}\) are the energies of the optimized catalyst surface, X in the gas phase, and X adsorbed on the catalyst surface, respectively. The more negative the \({E}_{{ads}}\left(X\right)\) value is, the more strongly the species X binds to the surface. Note that the energy reference of the H atom is half that of H2.
Data availability
Relevant data supporting the key findings of this study are available within the article and the Supplementary Information file. All raw data generated during the current study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Jie, X. et al. Microwave-initiated catalytic deconstruction of plastic waste into hydrogen and high-value carbons. Nat. Catal. 3, 902–912 (2020).
Duan, J. et al. Coking-resistant polyethylene upcycling modulated by zeolite micropore diffusion. J. Am. Chem. Soc. 144, 14269–14277 (2022).
Borrelle, S. et al. Predicted growth in plastic waste exceeds efforts to mitigate plastic pollution. Science 369, 1515–1518 (2020).
Lau, W. et al. Evaluating scenarios toward zero plastic pollution. Science 369, 1455–1461 (2020).
Han, W. et al. One-pot catalytic conversion of polyethylene wastes to gasoline through a dual-catalyst system. Chem 11, 102340 (2025).
Martín, A., Mondelli, C., Jaydev, S. & Pérez-Ramírez, J. Catalytic processing of plastic waste on the rise. Chem 7, 1487–1533 (2021).
Ran, H., Zhang, S., Ni, W. & Jing, Y. Precise activation of C–C bonds for recycling and upcycling of plastics. Chem. Sci. 15, 795–831 (2024).
Zhang, W. et al. Low-temperature upcycling of polyolefins into liquid alkanes via tandem cracking-alkylation. Science 379, 807–811 (2023).
Du, J. et al. Efficient solvent- and hydrogen-free upcycling of high-density polyethylene into separable cyclic hydrocarbons. Nat Nanotechnol 18, 772–779 (2023).
Ran, H., Sun, X., Zheng, M. & Jing, Y. Synergistic catalysis for recycling and upcycling of plastics. ACS Catal 15, 8551–8585 (2025).
Zhang, F. et al. Polyethylene upcycling to long-chain alkylaromatics by tandem hydrogenolysis/aromatization. Science 370, 437–441 (2020).
Zhao, B., Hu, Z., Sun, Y., Hajiayi, R., Wang, T. & Jiao, N. Selective upcycling of polyolefins into high-value nitrogenated chemicals. J. Am. Chem. Soc. 146, 28605–28611 (2024).
Chu, M., Tu, W., Zhang, Q., Huang, W., Chen, J. & Ma, D. Co-recycling of plastics and other waste materials. Nat. Rev. Clean Technol. 1, 320–332 (2025).
Feng, J. et al. Micelles cascade assembly to tandem porous catalyst for waste plastics upcycling. Angew. Chem. Int. Ed. 63, e202405252 (2024).
Li, X. et al. Efficient metallic Ni as a bifunctional electrocatalyst for integrating continuous PET plastic upcycling with hydrogen production. Appl. Catal. B Environ. 371, 125211 (2025).
Lee, K., Jing, Y., Wang, Y. & Yan, N. A unified view on catalytic conversion of biomass and waste plastics. Nat. Rev. Chem. 6, 635–652 (2022).
Li, M. & Zhang, S. Tandem chemical depolymerization and photoreforming of waste PET plastic to high-value-added chemicals. ACS Catal 14, 2949–2958 (2024).
Chu, M., Liu, Y., Lou, X., Zhang, Q. & Chen, J. Rational design of chemical catalysis for plastic recycling. ACS Catal 12, 4659–4679 (2022).
Zhang, S. et al. Depolymerization of polyesters by a binuclear catalyst for plastic recycling. Nat. Sustain. 6, 965–973 (2023).
Kratish, Y. & Marks, T. Efficient polyester hydrogenolytic deconstruction via tandem catalysis. Angew. Chem. Int. Ed. 134, e202112576 (2022).
Wu, Y. et al. Catalytic degradation of polyethylene terephthalate using a phase-transitional zirconium-based metal-organic framework. Angew. Chem. Int. Ed. 61, e202117528 (2022).
Hu, Y. et al. Highly efficient depolymerization of waste polyesters enabled by transesterification/hydrogenation relay under mild conditions. Angew. Chem. Int. Ed. 62, e202312564 (2023).
Cao, J. et al. Depolymerization mechanisms and closed-loop assessment in polyester waste recycling. Nat. Commun. 15, 6266 (2024).
Gao, Z., Ma, B., Chen, S., Tian, J. & Zhao, C. Converting waste PET plastics into automobile fuels and antifreeze components. Nat. Commun. 13, 3343 (2022).
Cheng, J. et al. Selective upcycling of polyethylene terephthalate towards high-valued oxygenated chemical methyl p-methyl benzoate using a Cu/ZrO2 catalyst. Angew. Chem. Int. Ed. 63, e202319896 (2024).
Sun, Z. et al. Value-added upcycling of PET to 1,4-cyclohexanedimethanol by a hydrogenation/hydrogenolysis relay catalysis. Angew. Chem. Int. Ed. 63, e202408561 (2024).
Zhu, Y., Mao, Z., Wu, W., Han, B. & Mei, Q. Selective asymmetric hydrogenation of waste polyethylene terephthalate via controlled sorption through precisely tuned moderate acid sites. J. Am. Chem. Soc. 147, 10662–10677 (2025).
Guo, Z. et al. Hydrogenating polyethylene terephthalate into degradable polyesters. Angew. Chem. Int. Ed. 64, e202418157 (2025).
Zou, Q., Long, T., Fang, R., Zhao, X., Wang, F. & Li, Y. Atomic Cu-O-Zr sites for highly selective production of p-xylene from tandem upcycling of PET and CO2. Angew. Chem. Int. Ed. 64, e202507309 (2025).
Lu, S., Jing, Y., Feng, B., Guo, Y., Liu, X. & Wang, Y. H2-free plastic conversion: converting PET back to BTX by unlocking hidden hydrogen. ChemSusChem 14, 4242–4250 (2021).
Jing, Y. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a Ru/Nb2O5 catalyst. Angew. Chem. Int. Ed. 60, 5527–5535 (2021).
Murali, V. et al. Selective one-pot chemical recycling of PET waste to xylene monomers: insights into a Ru/TiO2 catalyst design and interfacial dynamics in a biphasic system. Green Chem 27, 2203–2219 (2025).
Ye, M. et al. Ruthenium/TiO2-catalyzed hydrogenolysis of polyethylene terephthalate: reaction pathways dominated by coordination environment. Angew. Chem. Int. Ed. 62, e202301024 (2023).
Guo, Y., Jing, Y., Xia, Q. & Wang, Y. NbOx-based catalysts for the activation of C–O and C–C bonds in the valorization of waste carbon resources. Acc. Chem. Res. 55, 1301–1312 (2022).
Kim, S. et al. Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts. Green Chem. 21, 3715–3743 (2019).
Wei, J. et al. Hydrodeoxygenation of oxygen-containing aromatic plastic wastes to liquid organic hydrogen carriers. Angew. Chem. Int. Ed. 62, e202310505 (2023).
Xiang, S. et al. A unique Co@CoO catalyst for hydrogenolysis of biomass-derived 5-hydroxymethylfurfural to 2,5-dimethylfuran. Nat. Commun. 13, 3657 (2022).
Hongkailers, S., Jing, Y., Wang, Y., Hinchiranan, N. & Yan, N. Recovery of arenes from polyethylene terephthalate (PET) over a Co/TiO2 catalyst. ChemSusChem 14, 4330–4339 (2021).
Shao, Y. W. et al. The quantitative conversion of polyethylene terephthalate (PET) and Coca-Cola bottles to P-Xylene over Co-based catalysts with tailored activities for deoxygenation and hydrogenation. Green Chem 25, 10513–10529 (2023).
Goodman, E., Schwalbe, J. & Cargnello, M. Mechanistic understanding and the rational design of sinter-resistant heterogeneous catalysts. ACS Catal 7, 7156–7173 (2017).
Li, W. et al. Crystallographic dependence of CO2 hydrogenation pathways over HCP-Co and FCC-Co catalysts. Appl. Catal. B Environ. 315, 121529 (2022).
Yan, Y. Promoted deep oxidation of m-xylene and inhibited the generation of carbon-deposited species by Ce modified Co3O4: The key role of modulating internal electron transport pathway. Appl. Catal. B Environ. 365, 124864 (2025).
Zhao, Q. et al. Novel monolithic catalysts derived from in-situ decoration of Co3O4 and hierarchical Co3O4@MnOx on Ni foam for VOC oxidation. Appl. Catal. B Environ. 265, 118552 (2020).
Cui, M. et al. Asymmetric site-enabled O-O coupling in Co3O4 for oxygen evolution reaction. ACS Catal 14, 16353–16362 (2024).
Jing, Y., Shakouri, M., Liu, X., Hu, Y., Guo, Y. & Wang, Y. Breaking C–C bonds and preserving C–O bonds in aromatic plastics and lignin via a reversing bond energy cleavage strategy. ACS Catal 12, 10690–10699 (2022).
Ding, J. et al. Unraveling dynamic structural evolution of single atom catalyst surface-enhanced infrared absorption spectroscopy. J. Am. Chem. Soc. 147, 9601–9609 (2025).
Wei, Y. L. et al. Phase transition induced hydrogen activation for enhanced furfural reductive amination over a cocu bimetallic catalyst. Chem. Sci. 15, 20338–20345 (2024).
Dai, Z. et al. Surface engineering on bulk Cu2O for efficient electrosynthesis of urea. Nat. Commun. 16, 3271 (2025).
Li, Z. et al. Oxidation of reduced ceria by incorporation of hydrogen. Angew. Chem. Int. Ed. 58, 14686–14693 (2019).
Xie, L., Liang, J., Jiang, L. & Huang, W. Effects of oxygen vacancies on hydrogenation efficiency by spillover in catalysts. Chem. Sci. 16, 3408–3429 (2025).
Lin, Z. et al. Unlocking the potential of oxide-based catalysts for CO2 photo-hydrogenation: oxygen vacancies promoted C─O bond cleavage in key intermediates. Adv. Mat. 37, 2408906 (2025).
Li, A. et al. Active Cu0-Cuσ+ sites for the hydrogenation of carbon-oxygen bonds over Cu/CeO2 catalysts. ACS Catal. 12, 1315–1325 (2022).
Shirayama, K., Jin, X. & Nozaki, K. Selective hydrogenation of aldehydes under syngas using CeO2-supported Au nanoparticle catalyst. J. Am. Chem. Soc. 146, 14086–14094 (2024).
Ngu, J., Najmi, S., Selvam, E., Vance, B., Yang, P. & Vlachos, D. G. Catalytic deconstruction of organic additive-containing plastics. Nat. Chem. Eng. 2, 220–228 (2025).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Alavi, A., Hu, P., Deutsch, T., Silvestrelli, P. & Hutter, J. CO oxidation on Pt(111): an ab initio density functional theory study. Phys. Rev. Lett. 80, 3650–3653 (1998).
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (22572083) (Y. J.), the Fundamental Research Funds for the Central Universities (491914380001 (Y. J., J. S., X. C.), 491914380008 (Y. J., X. C.)), Nanjing University International Collaboration Initiative (X. C.), Independent research project of State Key Laboratory of Water Pollution Control and Green Resource Recycling (Y. J.), and start-up funding for high-level talent at Nanjing University (491916002207) (Y. J.). We thank the BL11U beamlines (https://cstr.cn/31131.02.HLS.CSS) of the Catalysis and Surface Science Endstation in the National Synchrotron Radiation Laboratory (NSRL) in Hefei (https://cstr.cn/31131.02.HLS) for providing beam time, technical support and assistance in data collection and analysis to support this work.
Author information
Authors and Affiliations
Contributions
Y.J., X.C., F.M., and S.L. conceived and supervised the project. Y.J., W.N., and H.R. designed the experiments. W.N., H.R., Y.L., P.Z., T.G., and J.S. conducted the experiments and analyzed the data. Z.L. performed the DTF calculations. W.N., H.R., and R.W. performed the LCA and TEA. W.N. wrote the paper. Y.J., X.C., B.L., W.N., and H.R. revised the manuscript. All authors participated in discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ni, W., Ran, H., Wang, R. et al. Stepwise hydrogen spillover–engineered synergistic sites enable near-quantitative conversion of waste PET to p-xylene. Nat Commun 17, 2128 (2026). https://doi.org/10.1038/s41467-026-68990-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68990-4
