
Abstract
Crabgrasses (Digitaria spp.) rank among the most invasive upland weeds globally, exhibiting remarkable ecological plasticity. To uncover the genomic basis underlying their weediness and environmental adaptability, we generate a telomere-to-telomere (T2T) reference genome of Digitaria sanguinalis, along with its diploid progenitor D. radicosa and tetraploid progenitor D. milanjiana. In addition, we re-sequence 579 accessions collected from fields of 24 grain-producing provinces in China. A phenotypic classification system for Digitaria species is developed by integrating genome-scale data with detailed morphological observations. Phylogenomic and comparative genomic analyses resolve the evolutionary history of Digitaria and reveal lineage-specific expansions in herbicide-associated gene families. Extensive sympatric introgression is observed within Digitaria, correlated with environmental selection. Through dose-response assay of 196 accessions, we identify 19 candidate genes associated with herbicide resistance, including DsSOH1. Haplotype analysis indicates that the resistant allele at DsSOH1 locus originated from D. ciliaris, suggesting a possible evolutionary contribution of introgression to the distribution of resistance-associated alleles. Together, our findings provide genomic insights into the evolutionary success of D. sanguinalis and offer resources for studying polyploid evolution and environmental adaptation as well as precision weed management in agroecosystems.
Similar content being viewed by others


Introduction
Weeds are ecologically resilient organisms that thrive in agricultural ecosystems, exhibiting broad adaptability1,2,3. Digitaria sanguinalis (L.) Scop., commonly known as large crabgrass, is one of the world’s worst weeds. It is widely distributed from tropical to temperate regions and thrives in both cultivated and no-tillage farming systems4,5. At high densities, this species can cause yield losses above 90% and is frequently found in soybean, maize, and sorghum fields6,7. The genus Digitaria comprises over 220 species, whose identification is complicated by extensive phenotypic convergence8,9,10. This taxonomic ambiguity has led to inconsistent assessments of species distributions, ultimately hindering the development and implementation of precise weed management strategies.
With widespread and intensifying use of agrochemicals, herbicide resistance has emerged as a global threat to effective weed control, currently affecting 273 weed species worldwide11,12,13. Herbicide resistance typically arises through two primary mechanisms14. The first is target-site resistance (TSR), which results from mutations or amplification of herbicide target genes. The second is non-target-site resistance (NTSR), which is conferred by enhanced metabolic detoxification or sequestration. This process involves major detoxification enzyme families such as cytochrome P450s, Glutathione transferases (GSTs), Aldo-keto reductases (AKRs) and ATP-binding cassette transporters (ABC transporters)15. For D. sanguinalis, resistance has been documented to acetolactate synthase (ALS), photosystem II, Acetyl CoA carboxylase (ACCase), and 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS) inhibitor herbicides16,17,18,19. The ALS-inhibitor herbicide nicosulfuron has been widely used for large crabgrass control since its registration in China in the 1990s18. In recent years, however, widespread resistance to nicosulfuron has emerged across numerous D. sanguinalis populations, involving both TSR and NTSR mechanisms20,21,22. This escalating resistance evolution is a growing concern, posing a significant threat to the effectiveness and sustainability of current chemical weed management strategies. However, the molecular and evolutionary basis of NTSR remains unclear due to the lack of high-quality genomic resources.
Polyploidization is widespread across the plant kingdom and is particularly prevalent among weedy species, where it often contributes to increased resistance to stress23,24,25. The presence of multiple homoeologous genomes within a single nucleus introduces regulatory complexity, including coordinated gene expression and extensive epigenomic remodeling26,27. In grasses, polyploidization has repeatedly facilitated the acquisition of stress resilience28,29. A well-known example is hexaploid wheat (Triticum aestivum), which exhibits increased fitness by combining root sodium retention, mediated by HKT1.5 from diploid Aegilops tauschii, with enhanced seed germination from tetraploid emmer wheat29. Patterns of gene retention and loss following polyploidization are also shaped by selection30. Disease-resistance genes have undergone significant contraction in barnyardgrass (Echinochloa crus-galli), in contrast to their expansion in wheat31, suggesting divergent evolutionary trajectories driven by ecological pressures32. D. sanguinalis is hexaploid (2n = 6× = 54). Therefore, investigating the evolutionary consequences of polyploidization is crucial for understanding the genetic basis of its ecological success and remarkable adaptability across agroecosystems.
Introgression is increasingly recognized as a powerful evolutionary force across diverse taxa, particularly in response to rapid environmental changes, including anthropogenic stressors such as herbicide exposure and pollution33. While natural interspecific hybridization has been documented in Digitaria, the functional implications of such hybridization events remain poorly understood34,35,36. In European aspen (Populus tremula), adaptive introgression has been shown to facilitate adaptation to high latitudes37. Another notable example is the Gulf killifish (Fundulus grandis), which acquired pollution tolerance through recent introgression of aryl hydrocarbon receptor loci from F. heteroclitus38,39. Similarly, adaptive introgression may serve as a key mechanism underpinning the wide geographic distribution and rapid evolutionary response of large crabgrass to increasing herbicide selection pressure.
In this study, to investigate the evolutionary trajectory and adaptive mechanisms of Digitaria, we assemble T2T genomes of hexaploid D. sanguinalis, and its tetraploid progenitor D. milanjiana and diploid progenitor D. radicosa. We also conduct large-scale genomic analyses of 579 Digitaria accessions and perform herbicide dose-response assays on a representative panel of 196 accessions. Our results reveal that introgression from closely related species may facilitate ecological adaptation in D. sanguinalis, including the acquisition of NTSR-associated haplotypes, which may have accelerated the recent evolution of herbicide resistance. These findings advance our understanding of Digitaria adaptive evolution, and will be helpful for development of precision weed management strategies.
Results
Genomic landscape of Digitaria reference genomes
A representative D. sanguinalis accession (#YJ2023) was collected from agricultural fields in Shandong Province, China, and its genome was sequenced, as well as its tetraploid progenitor, D. milanjiana (accession #DZ2), and diploid progenitor, D. radicosa (accession #YZGJ2). Cytological analysis confirmed its hexaploid status (2n = 6× = 54) (Supplementary Fig. 1), and k-mer analysis (k = 21, peak depth = 34) of short-read data estimated a genome size of 1.24 Gb (Supplementary Fig. 2), consistent with flow cytometry results (1.35 pg/1 C) for D. sanguinalis (Supplementary Fig. 3). The heterozygosity was estimated at 0.05%.
For de novo assembly of D. sanguinalis, we employed complementary long-read technologies: 81× coverage PacBio HiFi reads (N50 = 15.9 kb) and 88× coverage Nanopore ultra-long reads (N50 = 100.2 kb). A chromosome-level assembly was generated using Hi-C data (79× coverage) to scaffold the initial contigs. In total, 419 contigs were anchored into 27 scaffolds, yielding a final assembly size of 1.35 Gb. To delineate subgenomes, we identified subgenome-specific k-mers and clustered homeolog-differentiating scaffolds, enabling consistent partitioning into three distinct subgenomes (Supplementary Fig. 4). Three subgenomes were designated as C, D, and E, based on the markedly low mapping rate (19.72%) and genome coverage (17.53%) with D. exilis, supporting its distinct genomic origin40. Following error correction and scaffold ordering, a chromosome-level high-quality assembly was generated with subgenome sizes of 453.06 Mb (CH, subgenome C in the hexaploid), 419.87 Mb (DH), and 474.80 Mb (EH), respectively. The assembly contains only two unresolved gaps (Supplementary Fig. 5). Assessment with BUSCO (v5.6.1, Poales lineage dataset) showed 99.30% completeness of conserved genes41. Genome annotation was performed using a combination of homology-based, transcript-based, and ab initio prediction approaches, and 114,996 gene models were identified after filtering 709.61 Mb (51.80%) of repetitive sequences. Conserved centromeric regions containing tandem repeats (detailed below) were identified on all 27 chromosomes. Telomeric arrays (TTTAGGGn) were resolved at 50 of 54 chromosomes, comprising 23 fully terminal chromosomes (telomeres at both ends) and 4 partially terminal chromosomes (single telomere detected) (Supplementary Data 1). rDNA sequences were identified on seven chromosomes (Supplementary Table 1). Through nation-wide sampling in China, we identified the diploid (2n = 2× = 18) D. radicosa, and tetraploid (2n = 4× = 36) D. milanjiana, as the progenitors of D. sanguinalis (details provided in the next section) (Fig. 1a). In parallel, two high-quality genomes for D. radicosa (489.39 Mb, CC genome) and tetraploid D. milanjiana (908.85 Mb, with subgenome sizes of 421.50 Mb (DT, subgenome D in the tetraploid) and 469.45 Mb (ET)) were also generated in this study (Supplementary Table 2; Supplementary Note 1).

a Morphological features of the three Digitaria species: D. radicosa (#YZGJ2), D. milanjiana (#DZ2), and D. sanguinalis (#YJ2023), shown from left to right. b Genome architecture of the three Digitaria species. Track i represents the chromosomes; tracks ii-iv depict the density of annotated genes (ii), transposable elements (iii), and GC content (iv), respectively; track v illustrates syntenic relationships. C, genome of D. radicosa; DT and ET, subgenomes of D. milanjiana; CH, DH, and EH, subgenomes of D. sanguinalis. c Phylogenetic tree constructed with single-copy orthologs using ASTRAL among Poaceae genomes. Gray nodes denote speciation events out of Digitaria, while red and blue nodes mark polyploidization and speciation events within Digitaria, respectively. Subgenome labels (e.g., A, B, C, D and E) correspond to inferred ancestral contributions in polyploids. S. viridis, Setaria viridis; E. indica, Eleusine indica; O. sativa, Oryza sativa. d Relative abundance of stress-associated gene families across Poaceae genomes. Gene family sizes were standardized as Z-scores and visualized as a gradient heatmap. Gene families associated with abiotic and biotic stress responses are indicated in blue and red, respectively. Gene families previously implicated in non-target-site herbicide resistance are highlighted in green. FAR1, Far-red impaired response 1 DNA-binding domain; Hsp70, Heat shock protein 70 family; P450, Cytochrome P450 family; ABC transporter, ATP-binding cassette transporter; GRAS, GAI-RGA- and -SCR; AP2, Apetala2; NAM C-terminal, No Apical Meristem Protein C-terminal domain; UDP/GT, UDP-glucoronosyl and UDP-glucosyl transferase; D-mannose binding lectin, D-mannose binding lectin domain; NB-ARC, nucleotide-binding adapter shared by APAF-1, R proteins, and CED-4. O. sativa, Oryza sativa; E. haploclada, Echinochloa haploclada; E. crus-galli, Echinochloa crus-galli; E. oryzicola, Echinochloa oryzicola; S. italica, Setaria italica; P. hallii, Panicum hallii; D. exilis, Digitaria exilis; D. radicosa, Digitaria radicosa; D. milanjiana, Digitaria milanjiana; D. sanguinalis, Digitaria sanguinalis. E. crus-galli AH, BH, and CH, subgenomes of E. crus-galli; E. oryzicola AT and BT, subgenomes of E. oryzicola; D. exilis AT and BT, subgenomes of D. exilis; D. milanjiana DT and ET, subgenomes of D. milanjiana; D. sanguinalis CH, DH, and EH, subgenomes of D. sanguinalis. Source data are provided as a Source Data file.
To assess the assembly quality of three Digitaria reference genomes, we first mapped Illumina paired-end reads to their respective assemblies. Sequencing reads from diploid, tetraploid, and hexaploid Digitaria accessions were uniformly mapped to the D. sanguinalis reference genome, with high mapping rates of 99.51%, 99.48%, and 99.58%, respectively (Supplementary Table 3). RNA-seq reads showed normal alignment ratios to their respective genomes (94.12% for D. radicosa, 89.81% for D. milanjiana, and 93.32% for D. sanguinalis) (Supplementary Data 2). The genome assembly index LAI scores were 14.41, 17.30, and 15.62 for the three genomes, respectively, comparable to those of Arabidopsis thaliana (TAIR10) and Vitis vinifera42,43. We also estimated base-level accuracy and completeness of these assemblies and high assembly consensus quality values, 58.70 (99.74%), 54.51 (99.74%), and 50.08 (99.60%), were achieved for the three genomes, respectively (Supplementary Table 3). For continuity, we detected potential assembly gaps with low-confidence read supports using CRAQ, and high scores were also obtained for the three assemblies44 (Supplementary Tables 3 and 4). Taken together, these results suggest that the three assemblies are of high quality in terms of continuity, completeness, and accuracy.
Whole-genome alignments revealed extensive synteny between D. sanguinalis and its diploid/tetraploid progenitors, with conserved macro-collinearity across 95.0% of the genome (Fig. 1b; and Supplementary Fig. 5). The results also provide independent validation of our chromosome-scale assembly quality.
We calculated the synonymous substitutions per synonymous site (Ks) for orthologous gene pairs among the Digitaria species and other grass species to estimate their divergence times (Fig. 1c). The Ks peak for orthologs between D. radicosa and Oryza sativa was 0.57, corresponding to an estimated divergence time of ~45 million years ago (mya) (Fig. 1c), while Digitaria diverged from Eleusine ~35 mya, and subsequently from Setaria around 17 mya. This phylogenetic branching timeline was consistent with previous estimates45,46 (Supplementary Fig. 6). The two lineages with diagnostic spikelet arrangement phenotypes, D. exilis and D. sanguinalis, diverged at 12 mya. A large-scale amplification of Gypsy-type transposable elements was observed in the D. sanguinalis genome compared to D. exilis (Supplementary Fig. 7a). Further analysis of subgenome differentiation in D. sanguinalis resolved three ancestral lineages: CH diverged around 6.9 mya, followed by the bifurcation of DH and EH at ~6.4 mya. Using insertion polymorphisms of long terminal repeat retrotransposons, calibrated against synonymous mutation rates, we estimated that the tetraploidization event leading to D. milanjiana occurred around 0.9 mya, while hexaploidization in D. sanguinalis followed at ~0.4 mya. Interestingly, DH and EH in D. sanguinalis exhibit similar patterns to those in D. milanjiana (Supplementary Fig. 8). In addition, a maximum-likelihood (ML) phylogeny based on a concatenated matrix of 2030 single-copy orthologs among the 14 (sub)genomes was reconstructed, and a coalescent-based phylogenetic analysis was also performed, integrating individual gene trees (Fig. 1c; and Supplementary Fig. 9). These divergence estimates were concordant with our Ks-based molecular dating analyses, with robust statistical support at all key nodes.
Using domain-based gene family quantification across 18 genomes, we observed significant lineage-specific contraction in biotic stress-responsive gene families, highlighting differences between polyploid upland and paddy weeds as well as crops (Fig. 1d). Consistent with evolutionary patterns observed in other weeds31, Digitaria exhibited pronounced contraction of NB-ARC domain-containing genes (median 252 ± 48.7 copies vs. 447 ± 94.0 in crops; two-tailed t-test, p = 0.002) and D-mannose-binding lectin genes (61 ± 9.1 vs. 116 ± 22.8 in crops; two-tailed t-test, p = 0.001) compared to crops. The decay in defense-related genes likely reflected ecological trade-offs that favor growth in ruderal habitats, where biotic stress responses may be reduced in importance31,47. Interestingly, comparative analyses revealed a significant expansion of UDP-glucuronosyl/glucosyl transferase (UDP/GT) genes in Digitaria (162 ± 28.5) relative to Echinochloa species (64 ± 13.1; two-tailed t-test, p = 1.63e-5). Furthermore, gene families such as GAI-RGA-SCR (GRAS, 67.0 ± 5.6 vs. 43.5 ± 7.3; two-tailed t-test, p = 0.008) also exhibited expansion (Supplementary Data 3). These expansions may have provided a genomic foundation for adaptation under modern weed management systems. UDP/GTs were significantly enriched on Chr3 in Digitaria (Fisher’s test, p < 0.01) (Supplementary Fig. 10), which also harbors the highest transposon density across the genome (Supplementary Fig. 7b). Several flood-adapted gene families, such as the anaerobic-response Apetala2, showed marked expansion in Echinochloa (copy number: 182.0 ± 12.3 vs. 159.0 ± 9.9 in D. sanguinalis; two-tailed t-test, p = 0.008), likely reflecting adaptation to prolonged submergence (Supplementary Data 3). Compared with ancestral genomes, D. sanguinalis exhibited a lineage-specific amplification of far-red impaired response 1 DNA-binding genes (avg. 71.3 copies vs. 8.5 copies in D. exilis, 59.5 copies in D. milanjiana, and 54.0 copies in D. radicosa) likely associated with its mat-forming growth habit and the shaded microhabitats it occupies (Fig. 1d). Concurrently, D. exilis showed a distinctive expansion in Heat shock protein 70, consistent with its adaptation to extreme thermal environments in west African provenances (Fig. 1d).
Population structure and demography of Digitaria species
To investigate the genetic diversity and population structure of Digitaria, we re-sequenced the genomes of 579 Digitaria accessions collected over the past decade from a wide range of habitats across China (Fig. 2a; and Supplementary Fig. 11; Supplementary Data 4). Based on read mapping rates and genome coverage to the D. sanguinalis reference genome (Fig. 2b; and Supplementary Data 4), combined with morphological traits and genome size estimates (flow cytometry and k-mer analysis) (Supplementary Fig. 12; and Supplementary Data 4), the 579 accessions were classified into two major clades defined by distinct spikelet arrangements (Supplementary Note 2). The first clade, the ternate-type group, includes D. exilis, D. ischaemum, and D. violascens, species with primarily European distributions. The second clade, the binate-type group, comprises D. ciliaris, D. bicornis, and D. sanguinalis, which collectively represent the most globally invasive agricultural weeds48,49. On average, D. bicornis showed a 76.82% mapping rate with average coverage of 65.44%, 83.89%, and 85.12% across the CH, DH, and EH, respectively (Supplementary Fig. 13). D. ciliaris achieved a 94.63% mapping rate, with CH, DH, and EH coverage of 95.36%, 84.20%, and 85.94%, respectively (Supplementary Fig. 13). D. sanguinalis accessions showed the highest mapping rates, averaging 97.46%, with CH, DH, and EH coverage of 98.11%, 98.04%, and 97.03%. In particular, D. milanjiana and D. radicosa had differential subgenome coverage patterns (Fig. 2b). For example, D. radicosa exhibited a mapping rate of 95.05%, with subgenome compartment coverage of CH: 95.63%, DH: 2.07%, and EH: 1.80%, while D. milanjiana showed a mapping rate of 93.17%, with CH, DH, and EH coverage of 7.96%, 86.57%, and 87.75%, respectively (Supplementary Data 4). This asymmetric subgenome coverage provides genomic evidence supporting their ancestral contributions (i.e., tetraploid and diploid progenitor) to the allohexaploid genome of D. sanguinalis. In contrast, ternate-type Digitaria species consistently exhibited low mapping rates (~18.2%) and subgenome coverage (~13.6%), consistent with Ks-based molecular divergence estimates (Figs. 1c and 2b), indicating a more distant evolutionary relationship with the binate-type clade.

a Geographic distribution of Digitaria accessions used in this study. The primary collection region is shown in detail, with an inset in the upper-left corner displaying the full geographic extent of sampling across China. Different species are represented by distinct symbols, while D. sanguinalis varieties are shown as colored circles. Range boundaries of major taxa are shaded. b Species identification based on mapping rates and coverage of reads aligned to the D. sanguinalis genome (#YJ2023). c Unrooted maximum-likelihood tree of D. sanguinalis, D. ciliaris, and D. bicornis accessions. d A rooted maximum-likelihood tree of D. sanguinalis accessions based on genome-wide SNPs, using D. ciliaris as an outgroup. Varieties and putative admixed populations are color-coded. e Phenotypic variation in seed morphology (aspect ratio and 1000-seed weight) across Digitaria species (n = 9 for D. ischaemum, n = 16 for D. violascens, n = 5 for D. bicornis, n = 51 for D. ciliaris, n = 485 for D. sanguinalis) and D. sanguinalis varieties (n = 111 for var. pubescens, n = 105 for var. sanguinalis, n = 78 for var. parvispicula, n = 35 for var. glabra). Central dots represent medians, and whiskers indicate the interquartile ranges. Statistical significance was assessed by two-tailed t-tests. f Demographic inference of D. sanguinalis varieties. Time points T1-T4 denote estimated divergence events. Nₑ, effective population size; mya, million years ago. Source data are provided as a Source Data file.
Our nationwide sampling effort covered 25 provinces across China, spanning from Hainan (18.3°N) in the tropical south to Heilongjiang (47.3°N) in the subarctic north (Fig. 2a). D. sanguinalis was the dominant species (78.90%) in the sampling population, with notable prevalence in major agricultural regions such as Henan, Shandong, and Shanxi (Supplementary Data 4). In contrast, the majority (68.75%) of D. violascens accessions were collected from northeastern provinces, Heilongjiang and Jilin. Regional differences in diversity were evident. Southern coastal provinces exhibited higher diversity, with Hainan showing the highest Simpson diversity index and Shannon index, 0.60 and 0.99, respectively, followed by Guangxi and Guangdong. In contrast, lower diversity levels were observed in Anhui and Jiangsu, with Simpson index values of 0.07 and 0.11, respectively (Supplementary Table 5).
We called single nucleotide polymorphisms (SNPs) across the CH, DH, and EH, yielding 10.76 million SNPs and 23.76 SNPs/kb for CH, 9.07 million and 21.60 SNPs/kb for DH, and 7.45 million and 15.68 SNPs/kb for EH. These SNPs were used for population structure and phylogenetic analyses of binate-type group (ternate-type accessions were excluded from subsequent analyses due to low mapping rates) (Supplementary Figs. 14 and 15). Within the retained binate-type specimens, each species formed a monophyletic clade in maximum-likelihood phylogenies (Fig. 2c). Meanwhile, we identified phylogenetically intermediate outliers that displayed three convergent signatures of potential hybridization: firstly, they occupied intermediate positions in species phylogeny between species clades (Fig. 2d); secondly, principal component analysis (PCA) positioned them centrally between primary species clusters (e.g., between D. ciliaris and D. sanguinalis) (Supplementary Fig. 15); thirdly, ancestry composition analysis revealed admixed genomic profiles (Supplementary Fig. 14). Together, these findings point to possible historical introgression among Digitaria species, although further analyses are needed to robustly resolve the extent and directionality of these events. Within D. sanguinalis, four distinct varieties and three admixed populations were identified, each showing marked biogeographic specialization across China (Fig. 2b, d; Supplementary Fig. 16). The phylogenetically basal var. glabra was predominantly distributed in northeastern regions and is characterized by the narrowest leaf blades, most compact tiller angles, and the heaviest 1000-seed weight among all varieties (Fig. 2d, e). In contrast, var. parvispicula is dominant in southern provinces and is distinguished by elongated, slender leaves and the lightest grain weight (Fig. 2d, e; and Supplementary Figs. 17 and 18). In the middle-lower Yellow River basin, the remaining two varieties exhibit north-south geographical separation relative to the river (Fig. 2b; and Supplementary Fig. 16). Although morphologically similar, var. pubescens, localized on the northern slopes, is distinguished by heavier seeds and the presence of golden bristles on the lemma surface (Fig. 2b, e; and Supplementary Figs. 17 and 18). Field surveys revealed that admixed populations were found in transitional ecotones between discrete varietal ranges (Fig. 2b; and Supplementary Fig. 16), further supporting their role as dynamic genetic intermediates. This classification into four distinct varieties lays the groundwork for exploring whether historical introgression has contributed to the observed genetic and phenotypic diversity.
By comparing the unfolded joint site frequency spectrum (SFS) of unlinked SNPs, polarized using known ancestral alleles, we inferred that archaic introgression from D. ciliaris into D. sanguinalis started at ~431,653 years before present (yr BP, 95% confidence interval (CI) 350,044−2,420,993 yr BP), following the speciation of D. sanguinalis, which itself was estimated to have occurred ~1.072 mya (95% CI: 712,489-2,921,965 yr BP) (Fig. 2f). Var. glabra diverged from the ancestral D. sanguinalis lineage at ~53,926 yr BP (95% CI: 37,101-1,423,489 yr BP). This was followed by the separation of the common ancestor of var. sanguinalis and var. parvispicula from var. pubescens at ~46,719 yr BP (95% CI: 18,199-1,156,488 yr BP). During this diversification process, introgression was detected between the ancestral var. pubescens and the common ancestor of the other two varieties. The best-fitting demographic model also predicted ongoing, bidirectional introgression between D. ciliaris and multiple D. sanguinalis varieties since the emergence of var. parvispicula around 39,561 yr BP (95% CI: 2,919-438,092 yr BP) (Fig. 2f; and Supplementary Fig. 19; Supplementary Table 6 and 7). Given the weed’s prevalence in upland agricultural systems, these patterns likely reflect human-mediated dispersal. Agricultural practices, especially mechanized harvesting and commercial seed exchange, may have accelerated the nationwide spread of D. sanguinalis and facilitated genetic exchange among historically isolated subspecies. To examine more recent demographic trends over the past 10,000 years, both folded and unfolded SFS were analyzed, and a sharp population bottleneck was detected in var. glabra around 30,000 years ago (Supplementary Fig. 20). For D. ciliaris, a severe and continuous decline in the effective population size began ~1000 years ago, following an earlier bottleneck. In contrast, after recovering from a bottleneck around 5000 years ago, the population sizes of var. sanguinalis and var. parvispicula had remained relatively stable over the past 2000 years.
Overall, nucleotide diversity (π) displayed an asymmetric distribution across the Digitaria genomes, with DH in both D. ciliaris and D. sanguinalis exhibiting the lowest levels of diversity (Supplementary Fig. 21). Except in var. pubescens, EH showed higher diversity than CH, suggesting that var. pubescens underwent distinct genomic alterations leading to elevated diversity. Moreover, the divergence between DH and EH was more pronounced in D. sanguinalis than in other Digitaria species (Supplementary Fig. 9), implying that D. sanguinalis experienced stronger post-origin introgression than other species, with a directional bias favoring CH and EH genomic compartments.
Comparative analyses of population structure across spatially clustered and temporally distinct populations of D. sanguinalis from 2013 to 2023 in the Huang-Huai-Hai agroecosystems revealed clear evidence of population genetic restructuring over time (Supplementary Fig. 22a; and Supplementary Table 8). Accessions collected in 2013 and 2015 were designated as historical groups, whereas geographically proximate accessions collected in 2023 were defined as modern groups. Modern groups exhibited markedly higher admixture diversity compared to historical collections (Supplementary Fig. 22b). While core geographic clusters have retained notable spatial structure, temporal analyses revealed genetic homogenization over time. For example, in GR7, the proportion of the k4 ancestral component declined sharply from 89.47% in 2013 to 31.95% in 2023. Concurrently, the admixture index increased from 0.29 to 0.55, reflecting a substantial rise in genomic intermixing (Supplementary Fig. 23; and Supplementary Table 8).
Sympatric introgression driving local environmental adaptation
Introgression is known to have played a primary role in increasing genetic diversity. We further constructed a ML phylogeny using SNPs from chloroplast genomes to infer the maternal origins of Digitaria topologies (Fig. 3a). Species-level divergence patterns in the tree were congruent with those inferred from the nuclear SNPs, notably with D. violascens and D. ciliaris forming a monophyletic clade (Figs. 2b and 3a). Interestingly, a subset of D. sanguinalis accessions (n = 9) was scattered within the clade containing D. ischaemum and D. violascens, suggesting persistent chloroplast introgression between lineages that diverged more than 10 mya (Fig. 3a, b). Although D. bicornis formed a monophyletic group in the nuclear phylogeny, its chloroplast genomes clustered within Clade 4, which is dominated by D. sanguinalis accessions (95.68%) (Figs. 2d and 3b). This implied a D. sanguinalis origin of the D. bicornis chloroplast genome, likely resulting from historical chloroplast capture through hybridization or horizontal transfer. The chloroplast phylogeny further supports the hypothesis of at least two distinct maternal donors in the origin of D. sanguinalis. One lineage corresponded to the monophyletic Clade 4, while the other branches as a sister group to the D. ciliaris monophyly, collectively forming Clade 3, a topology that was discordant with the nuclear genome (Fig. 3a, b). Both diploid and tetraploid ancestors of D. sanguinalis were positioned within Clade 3, with D. radicosa nested inside the D. ciliaris subclade (Fig. 3a). This placement aligned with the high CH coverage observed in D. ciliaris (95.36%), suggesting shared ancestry of CH between D. ciliaris and D. sanguinalis (Fig. 3a; and Supplementary Fig. 13). Overall, the chloroplast phylogenies indicate extensive interspecific introgression across Digitaria, most likely resulting from historical hybridization events50,51.

a Maximum-likelihood phylogeny of accessions based on chloroplast genome SNPs. D. sanguinalis and its putative progenitors (#YJ2023, #DZ2, and #YZGJ2) are marked with asterisks. b Bar plots summarizing species and varieties composition across major clades in chloroplast phylogeny. c Distribution of introgression intensity between sympatric D. ciliaris and D. sanguinalis population pairs. Red dots indicate mean genome-wide proportions of introgression (PIG, right Y-axis). The violin plots show the distribution of genome-wide fd values across 8517, 9002, 12,958, 14,703, 9673, 9640 and 11,143 sliding windows for populations LF to ZJ, respectively. Box plots show the median (center line), the 25th and 75th percentiles (bounds of the box), and the range of values within 1.5× the interquartile range (whiskers). d Enriched Pfam domains in introgressed regions across sympatric populations. Detoxification-related domains are highlighted in green and heat-shock-related is shown in red. Statistical significance was assessed using Fisher’s exact test with FDR correction. ABC, ATP-binding cassette transporter; Myb-like, Myb-like DNA-binding domain; UDP/GTs, UDP-glucuronosyl and UDP-glucosyl transferase; P450, Cytochrome P450; C2, C2 domain; AP2, Apetala2; Hsp20, Hsp20/alpha crystallin family; D-mannose, D-mannose binding lectin. e Manhattan plot showing SNP associations with minimum temperature of the coldest month (Bio6) based on LFMM analysis. P-values were computed by transforming Z-scores under a standard normal distribution. Dashed horizontal lines denote significance thresholds (blue represents the FDR correction, adjusted p = 0.05; red represents the Bonferroni correction, adjusted p = 0.05). Genes previously validated in Oryza sativa are labeled. f Geographic distribution of DsRZ2 haplotypes in sympatric D. sanguinalis accessions. The background map is shaded according to Bio6 variation. g Frequency of DsRZ2 haplotypes across D. ciliaris and D. sanguinalis varieties. Accessions with missing genotypes at the focal region were excluded. GLA, D. sanguinalis var. glabra; PUB, D. sanguinalis var. pubescens; SAN, D. sanguinalis var. sanguinalis; PAR, D. sanguinalis var. parvispicula. h Proportion of introgression fd distributions on Chr4 in DZ, SJZ and SX. i Stress-responsive genes identified within the 54.2-54.6 Mb region of Chr4. Source data are provided as a Source Data file.
Given sympatric distribution observed for D. ciliaris and D. sanguinalis, we applied the genome-wide D-statistic and fd value to test for introgression between 7 sympatric population pairs of D. ciliaris and D. sanguinalis (Supplementary Fig. 24a, b). All 7 populations exhibited significant signals of introgression (D > 0.11 and Z > 3 across all 7 populations; Supplementary Table 9). The strongest signal was detected in the SX population (D = 0.16), whereas the JN exhibited the weakest evidence of introgression (D = 0.11) (Supplementary Table 9). This pattern was further supported by the same analysis using sliding windows, revealing that SX harbored the greatest proportion of introgression across the genome (PIG) across the genome (Fig. 3c; and Supplementary Table 10). In contrast, eastern populations, including DZ, JN, and LF, displayed fewer introgressed regions, indicating minimal impact of introgression in these lineages (PIG in Fig. 3c).
To assess the potential role of environmental factors in shaping sympatric introgression, we conducted matrix regression analyses (Mantel/partial Mantel tests) on pairwise shared introgressed genomic regions (PSIG), defined as the Jaccard similarity index of introgressed haplotypes between “D. sanguinalis - D. ciliaris” population pairs. Environmental and geographic distance matrices were constructed using data from 7 population pairs (Supplementary Data 5). Both Mantel tests revealed significant negative correlations between PSIG and geographic distance (Pearson’s r = −0.70, p = 0.02), as well as between PSIG and environmental divergence (Pearson’s r = −0.64, p = 0.01) (Supplementary Table 11). However, partial Mantel tests did not detect significant associations when controlling for the alternative variable, likely due to high collinearity between geographic and environmental distances (Pearson’s r = 0.61, p = 1.48e-5) (Supplementary Fig. 24c). To account for the potential confounding effect of population structure on the relationship between environmental distance and PSIG, we performed a partial Mantel test controlling for pairwise genetic divergence (Fst). This analysis revealed a significant negative correlation between environmental distance and PSIG, suggesting that environmental distance explains variation in PSIG beyond what can be attributed to genetic divergence (Supplementary Table 11). Furthermore, correlation analysis between PSIG and specific environmental variables identified a significant association with precipitation (Supplementary Fig. 24d; and Supplementary Table 11).
To investigate the functional relevance of introgressed genomic regions, we performed Pfam domain enrichment analyses across populations (Fig. 3d). The number of introgressed genes varied from 2181 to 3472 among populations, and enrichment profiles were largely population-specific, with 13–25 significantly enriched domains per population and only 14 domains shared across more than three populations (Supplementary Fig. 25). Several functionally relevant domains were recurrently enriched across populations, spanning categories such as detoxification (e.g., ABC transporters, UDP/GTs), signaling (e.g., protein kinases, WD40 repeats), and transcriptional regulation (e.g., Myb-like DNA-binding domains) (Fig. 3d; Supplementary Data 6). The Hsp20 domain, associated with heat stress responses, was uniquely enriched in the SX population, which experiences the highest temperatures among all sampled regions (Fig. 3d; Supplementary Fig. 24a; Supplementary Data 5), suggesting a possible case of environmentally mediated introgression linked to thermal adaptation.
To identify genetic variants associated with environmental adaptation, we conducted genotype-environment association (GEA) analyses using the latent factor mixed model (LFMM), which accounts for background population structure while testing for associations between genotypes and environmental variables52 (Fig. 3e; Supplementary Figs. 26-28). The analysis included 19 environmental variables, comprising 10 temperature-related and 9 precipitation-related factors (Supplementary Data 7). In total, we identified 9437 SNPs significantly associated with one or more environmental variables, corresponding to 2117 genes (Supplementary Figs. 26-28; Supplementary Data 8). These environment-associated variants exhibited a genome-wide distribution, without clustering in specific genomic regions, suggesting that adaptation in Digitaria is governed by polygenic architectures rather than by hard selective sweeps at a handful of loci (Supplementary Figs. 26-28).
Several genes previously implicated in climate adaptation were identified in our analysis as harboring variants significantly associated with environmental variables53 (Supplementary Figs. 26-28; Supplementary Data 8). For example, the gene DsRZ2, which is homologous to OsRZ2, exhibited strong associations with the minimum temperature of the coldest month (Bio6) (Fig. 3e). This gene encodes a protein containing both a zinc knuckle domain (PF00098) and an RNA recognition motif (PF00076), and is known to play a critical role in plant protection against cold and freezing stress54,55. Expression analysis, based on RNA-seq data from root, stem, old leaf, and young leaf tissues, confirmed ubiquitous expression of DsRZ2 with RPKM values ranging from 4.40 to 7.24 (Supplementary Fig. 29a). We identified three major DsRZ2 haplotypes, exhibiting distinct geographic distributions (Supplementary Table 12). Hap1 was fixed (> 90%) in accessions located in the northeast that experience coldest winter temperatures −7.65 °C, whereas Hap3 dominated (87% frequency) in southern populations subjected to −4.77 °C (Fig. 3f; and Supplementary Fig. 29b, c). The observed haplotype frequencies also differed across D. sanguinalis varieties, indicating a possible association with subspecies identity (Fig. 3g). Furthermore, extended haplotype homozygosity (EHH) analysis at the DsRZ2 locus did not exhibit significant differences between haplotypes carrying the T or the C allele, which may result from polygenic additive effects (Supplementary Fig. 30; standardized |iHS| score = 0.72).
Given the recurrent introgression observed in Digitaria, we tested whether local adaptation in D. sanguinalis may have been facilitated by introgression. In ABBA-BABA statistics, significantly elevated fd values were detected at the DsRZ2 locus in sympatric populations (Fig. 3h; and Supplementary Fig. 29d). This indicates that in these D. sanguinalis accessions, genetic variation within an environmentally associated region on Chr4 more closely resembled sympatric D. ciliaris haplotypes than those of allopatric D. sanguinalis populations, supporting a hybrid origin for this ~500 kb genomic segment. This introgressed segment contains a functionally coordinated cluster of six stress-adaptive loci, including RZ2 and four genes previously characterized in rice (OsRALyase, OsBIHD1, OsUBC26, BK-PP2A)56,57,58,59 (Fig. 3h, i). The presence of these genes in an introgressed block suggests that introgression from D. ciliaris acted as a beneficial reservoir of adaptive alleles.
Temporal escalation and geographic spread of ALS-inhibitor resistance
Nicosulfuron has been widely used for weed management since its registration in the 1990s18. To quantify the temporal dynamics of ALS-inhibitor resistance in Digitaria, we performed nicosulfuron single-dose bioassays on accessions collected from the Huang-Huai-Hai agroecosystems in 2013, 2015, and 2023, respectively (Supplementary Table 13). As expected, resistance levels among surveyed populations exhibited a marked increase over a decade. Survival rates (number alive after treatment) following herbicide treatment rose from 36.5% in 2013 to 74.2% in 2023 (Supplementary Table 13), while the mean herbicide control efficacy concurrently declined from 86.0% to 70.7%. To further characterize resistance variation, we conducted dose-response assays on 196 representative accessions, determining GR₅₀ (herbicide dose causing 50% plant growth reduction) and GR₉₀ values (Supplementary Data 9). The GR₅₀ estimates varied by more than 2000-fold, ranging from 0.083 g a.i. ha-1 (observed in accession #15-9 from Anhui) to 168 g a.i. ha-1 (accession #W-21 from Shandong), reflecting substantial inter-population differences in resistance levels.
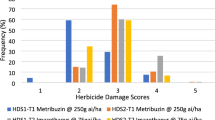
Despite being predominantly collected from ecologically comparable environments, different Digitaria species displayed substantial variation in herbicide resistance (Fig. 4a; Supplementary Data 9). Accessions of D. bicornis, primarily sourced from the Hainan Island, exhibited the lowest GR50 values, indicating high susceptibility to nicosulfuron (Fig. 4a). In contrast, D. ischaemum, D. ciliaris, and D. sanguinalis consistently showed higher GR50 values (Fig. 4a; and Supplementary Data 9). The median GR₉₀ value of the three weeds reached 84 g a.i. ha-1, with 100 g a.i. ha-1, 80 g a.i. ha-1, and 84 g a.i. ha-1 of D. ischaemum, D. ciliaris, and D. sanguinalis accessions, respectively, above the recommended field application dose (60 g a.i. ha-1). These variations in GR50/GR90 values in surveyed accessions have already rendered nicosulfuron weed control ineffective. To assess the temporal dynamics of resistance development, D. sanguinalis accessions collected from the lower-middle Yellow River region (32°−40°N, 108°−120°E) were used in the analysis. Both GR₅₀ (one-tailed t-test, p = 4.26e-2) and GR₉₀ (one-tailed t-test, p = 4.16e-3) values showed positive correlations with collection years, indicating a progressive increase in resistance over time (Fig. 4b). This trend highlights the rapid adaptation of D. sanguinalis populations to escalating herbicide selection pressure within agroecosystems. Accessions exhibiting resistance (GR₉₀ > 60 g a.i. ha-1) were found to be geographically widespread across multiple provinces, highlighting the increased challenges such a distribution poses for effective weed management (Supplementary Fig. 31).

a Nicosulfuron resistance levels across Digitaria species. Circles indicate mean GR₅₀ values (g a.i. ha-1), with error bars representing interquartile ranges. b Trends in nicosulfuron resistance dynamics in D. sanguinalis accessions sampled from 2013 to 2023. Linear regression lines represent the fitted mean response, with shaded bands indicating 95% confidence intervals around the predicted mean. P values from one-tailed t-tests are indicated. c Manhattan plot of SNP associations with nicosulfuron resistance in D. sanguinalis. P values were assessed using a generalized least squares F-test. Dashed horizontal lines denote significance thresholds (yellow represents the FDR correction, adjusted p = 0.05; red represents the Bonferroni correction, adjusted p = 0.05). SNPs near known NTSR-related gene families are highlighted in green. Previously validated genes are highlighted in yellow. d Allele frequency distribution for SNP at Chr20: 34,032,644 across groups with varying resistance levels: high (HR), moderate (MR), low (LR), and susceptible (S). e GR₅₀ values of D. sanguinalis accessions with different genotypes at Chr20: 34,032,644 (CC: n = 126; CA: n = 3; AA: n = 6). Box plots show the median (center line), the 25th and 75th percentiles (bounds of the box), and the range of values within 1.5× the interquartile range (whiskers). Statistical significance was assessed by two-tailed t-tests. f Zoomed-in view of GWAS signals at Chr20: 34.0-34.1 Mb. P-values were assessed using a generalized least squares F-test. Yellow points represent SNPs with adjusted p < 0.05. Bottom panel shows local genomic signatures (left: fd; right: Fst) across LR, MR, and HR groups. S, susceptible groups. Gray rectangles represent 99% bootstrap quantiles, dark green points denote windows with Z-score > 4 and yellow points denote windows with one-tailed p < 0.001 in randomization tests. g Divergence time estimates for resistant group under MSC-I model within a 100-kb window surrounding Chr20: 34,032,644. The orange dotted arrow indicates introgression from D. ciliaris to resistant D. sanguinalis group along with probability. h An unrooted maximum-likelihood tree inferred from SNPs within a 100-kb window flanking Chr20: 34,032,644. Source data are provided as a Source Data file.
To assess potential TSR, we first examined non-synonymous mutations in ALS, the herbicide target gene of nicosulfuron. Whole-genome sequencing of the Digitaria accessions (n = 554) identified 25 non-synonymous mutations in three DsALS gene copies (Supplementary Fig. 32a; and Supplementary Table 14; Supplementary Data 10). The DsALS-C copy, located on the CH, harbored the highest number of mutations (12 on the CH, 7 on the DH, and 6 on the EH). Most mutations (15/25) were present in a heterozygous state across accessions, in contrast to the predominantly homozygous resistance alleles observed in Echinochloa31 (Supplementary Table 14). These alleles also remained at low frequencies (less than 0.16) (Supplementary Table 14; Supplementary Data 10). No shared mutation sites were observed among three DsALS copies, and no accession carried variants in all three copies, either in homozygous or heterozygous state (Supplementary Data 10). In addition, no accessions exhibited abnormal coverage at any of the three DsALS gene copies (avg. coverage: 0.96 for DsALS-C, Shapiro-Wilk test, p > 0.01; 0.95 for DsALS-D, Shapiro-Wilk test, p > 0.01 and 0.92 for DsALS-E, Shapiro-Wilk test, p > 0.01). While no fixed mutations were identified in these copies, the complexity of the hexaploid genome does not preclude the presence of a dominant mutation. Further studies at both the genetic and functional levels will be necessary to establish whether such a mutation underlies TSR in D. sanguinalis.
Gene amplification-mediated dosage effects represent a mechanism for rapid adaptation to intense selection pressures, as illustrated in blackgrass (Alopecurus myosuroides), and bamboo rats60,61. To determine whether NTSR in D. sanguinalis involves gene amplification, we assessed variation in coverage for six key resistance-related gene families (cytochrome P450s, GSTs, UDP/GTs, AKRs, NB-ARC, and ABC transporters). Most accessions showed coverage levels for resistance-related gene families similar to those of single-copy genes, although 6 to 73 accessions exhibited high-coverage genes across different gene families (Supplementary Fig. 32b). However, correlation analysis between resistance levels and NTSR gene coverage revealed no association with resistance (Supplementary Fig. 32b; and Supplementary Data 11). Overall, no evidence was found to support metabolic resistance mediated by gene amplification in D. sanguinalis, though further investigation is needed to fully exclude this possibility.
To identify genes either constitutively expressed or induced by herbicide, we performed time-course RNA sequencing on leaf tissue from a resistant (R; accession #21-17) and a susceptible (S; accession #15-2) D. sanguinalis accession treated with nicosulfuron (Supplementary Data 12). In the R accession, 152 genes exhibited induced upregulation following herbicide treatment, including AKR1, UGT706D1, CYP81A6, and ABCG43, which are involved in reactive oxygen species (ROS) scavenging and stress response processes62,63,64,65. We identified DsSOH1, encoding a short-chain dehydrogenase/reductase (SDR), as a candidate resistance gene. This gene family is widely conserved across plants and implicated in detoxification and abiotic stress responses66,67. It exhibited nicosulfuron-inducible upregulation specifically in the R but not in the S accession (Supplementary Fig. 33). A total of 149 genes exhibited constitutive differential expression, comprising two genes (CYP75B3 and CYP92C21) with characterized homologs in rice involved in biotic stress responses68,69. Additional analyses were described in the Supplementary Note 3 (see Supplementary Figs. 34–36).
Adaptive introgression enables evolutionary escape from herbicide
To identify genomic loci underpinning NTSR, we conducted a genome-wide association study (GWAS) using 141 accessions based on their GR₅₀ values under nicosulfuron treatment70. A total of 40 resistance-associated SNPs (rSNPs) were identified, corresponding to 19 genes, including three homologs (DsCYP81A6, DsAKR4C16 and DsABCC1) functionally validated to confer non-target-site herbicide resistance in other grass species64,71,72 (Fig. 4c). rSNPs were identified 1898 bp upstream of DsSOH1 and 1891 bp downstream of DsCYP81A6, both of which exhibited differential expression in response to nicosulfuron treatment (Fig. 4c; Supplementary Fig. 33).
Herbicide resistance levels were quantitatively assessed using the Resistance Index (RI). Based on RI values, accessions were categorized into four distinct resistance groups: Susceptible (S), Low Resistance (LR), Moderate Resistance (MR), and High Resistance (HR) (Supplementary Data 9). The rSNP located near DsSOH1 was present in 40% of HR accessions but was absent in both LR and S groups (Fig. 4d). Phenotypic assessments further demonstrated that accessions carrying the alternate allele (A) exhibited significantly higher herbicide resistance compared to those with the reference allele (C) (Fig. 4e). To trace the origin of the haplotype at DsSOH1, we conducted ABBA-BABA analysis using the topology (((P1, P2), P3), O), with D. bicornis, susceptible D. sanguinalis, resistant D. sanguinalis, and D. ciliaris were assigned to O, P1, P2, and P3, respectively (Supplementary Fig. 37a). A prominent fd peak was detected specifically at the DsSOH1 locus in the HR group, suggesting introgression from D. ciliaris, whereas the MR and LR groups exhibited progressively weaker signals of admixture (Fig. 4f). The DsSOH1 variant was not fixed in the HR group, with an observed allele frequency of 0.40, supporting a scenario of recent adaptive introgression (Fig. 4d). Concurrently, elevated genome-wide Fst values revealed population subdivision within D. sanguinalis (Fig. 4f). The regions with the highest Fst differentiation between resistant and susceptible populations coincided precisely with the fd peak interval, strongly implicating introgression from D. ciliaris at the DsSOH1 locus (Fig. 4f). Although EHH analysis revealed difference between the A and C alleles, the |iHS| value (1.39, within the 90th percentile of the genome-wide distribution) did not reach the conventional threshold for detecting strong recent positive selection (Supplementary Fig. 37b).
To clarify the ancestry of the introgressed genomic region, we employed Bayesian Phylogenetics and Phasing (BPP v4.0)73 under an explicit introgression model (Fig. 4g). Parameter estimates supported a topology in which the DsSOH1 haplotypes in resistant D. sanguinalis accessions originated from recent introgression with D. ciliaris (φ = 0.30, 2.5% HPD = 0.24, 97.5% HPD = 0.36), while alternative scenarios were statistically rejected (Supplementary Data 13). Moreover, divergence time estimates indicate that speciation between D. sanguinalis and D. ciliaris substantially predated the inferred introgression event (divergence τ = 0.002010 vs. introgression divergence τ = 0.000056), providing further confirmation that resistance alleles originated via admixture. Consistent with the wide geographic distribution of resistant D. sanguinalis accessions, maximum-likelihood phylogenies constructed from introgressed SNPs consistently grouped resistant accessions within clades of sympatric D. ciliaris (Fig. 4h). This recurrent phylogenetic pattern suggests that the NTSR-associated haplotype at DsSOH1 likely evolved through parallel, recent introgression from neighboring D. ciliaris populations.
To investigate the correlation between introgression frequency from D. ciliaris and herbicide resistance levels in D. sanguinalis, we compared the fd values at rSNPs to the genome-wide background. The HR group exhibited significantly higher introgression frequencies at rSNPs compared to the genomic background (Mann-Whitney U test, p = 0.02), a pattern not detected in the MR or LR groups (Supplementary Fig. 37c). In addition, the HR group exhibited the greatest cumulative length of introgressed windows across the genome (HR vs. MR, Mann-Whitney U test, p = 1.60e-2; HR vs. LR, Mann-Whitney U test, p = 3.90e-5) (Supplementary Fig. 37d). These findings suggest that introgression from D. ciliaris may be associated with the emergence and spread of NTSR in D. sanguinalis. Interestingly, we observed a higher frequency of rSNPs in accessions collected in 2023 compared to those collected in 2013 (Supplementary Fig. 37e), which may be correlate with the elevated herbicide resistance observed in recent populations.
Discussion
Polyploidization has played a pervasive role in plant evolution74,75,76. Although chromosome-level assemblies are now available for at least 10 hexaploid species, their diploid and tetraploid progenitors have been resolved at comparable resolution in only a few cases, mostly in wheat and oats77,78. In contrast to the large genomes of wheat (17 Gb) and oats (11 Gb), the compact genomes of D. sanguinalis and its ancestors serve as an effective model system for studying how polyploidization influences genome evolution, particularly the structural dynamics of repetitive elements. Here, we generated near-complete assemblies of D. sanguinalis and its two putative progenitors, providing a valuable resource for exploring genome evolution in the Digitaria complex. These assemblies, especially those of the ancestral materials, could be further improved by integrating ultra-long reads and complementary sequencing approaches.
Accurate taxonomic resolution of Digitaria species forms a foundation for basic research and precision weed management9,79. However, species classification remains challenging due to extensive morphological convergence among over 220 described Digitaria species80,81,82. To address these ambiguities, we employed an integrative approach that combined phenotypic traits, such as genome size, 1000-grain weight, and aspect ratio, with genomic characteristics, including whole-genome mapping rates and coverage depth profiles (Figs. 2b, d, and e). This multidimensional strategy enabled reliable species discrimination and also revealed ecotypic differentiation within D. sanguinalis, where geographically distinct populations exhibited unique genomic and phenotypic signatures. For example, D. sanguinalis var. parvispicula shows significantly expanded leaf area indices, likely enhancing photosynthetic capacity under high-temperature and high-precipitation regimes. Collectively, our results provide a robust taxonomic framework for the genus Digitaria, resolving long-standing classification ambiguities.
Evolution of herbicide resistance in populations of numerous weed species underscores the urgent need to understand the genetic basis of resistance and its underlying evolutionary dynamics, so as to inform effective management strategies12,83. Although TSR is a common mechanism of herbicide resistance13,84, our analysis of 81 D. sanguinalis resistant accessions did not detect any known resistance-conferring mutations or coverage variation at target genes (Supplementary Fig. 32a; and Supplementary Table 14; Supplementary Data 10). This observation is consistent with previous reports in Chinese resistant Digitaria populations16,18,21. This finding presents a paradox, given the long-standing hypothesis that polyploid genomes are expected to promote TSR evolution due to the presence of multiple gene copies85. Nevertheless, in polyploid species, the effect of target site mutations can be masked or diluted by co-existing wild-type alleles. As a result, the effectiveness of TSR may depend on several factors, including the ploidy level, the number of mutated alleles, the relative expression levels of homologs carrying the mutations, and the dominance of the resistance allele86.
NTSR poses a potentially greater threat to agricultural systems, due to its polygenic nature and complex evolutionary trajectories14,15,87. In our study, we identified 40 rSNPs, a pattern consistent with findings in blackgrass, where mesosulfuron-methyl and clodinafop resistance are primarily NTSR-driven88,89. In our study, rSNPs were present at low frequencies (mean allele frequency = 0.063), suggesting substantial spatial heterogeneity in resistance architectures. This likely reflects adaptation to divergent agricultural selection regimes, influenced by regional cropping systems and herbicide application histories. In particular, the shift toward sequential or combination treatments, e.g., pre-emergence soil herbicides such as pyroxasulfone or oxadiazon, followed by post-emergence nicosulfuron or mesotrione, may have reduced the efficacy of individual TSR mutations and intensified selection for polygenic NTSR mechanisms90,91,92. Furthermore, some low-frequency resistance variants may function as compensatory alleles, mitigating potential fitness costs associated with primary resistance mechanisms24. The functional relevance of these variants will be further evaluated in future studies by integrating key phenotypic covariates, such as relative growth rate. In conclusion, our findings highlight a compelling case in which NTSR has become the dominant resistance strategy in D. sanguinalis under complex herbicide selection regimes.
Although introgression is increasingly recognized as a key mechanism of adaptive evolution, well-documented cases of beneficial introgression remain relatively rare33,93. In our study, we detected recurrent post-divergence introgression between D. ciliaris and D. sanguinalis, two species with overlapping distributions across ecologically heterogeneous regions. This frequent gene flow may facilitate environmental adaptation in D. sanguinalis, particularly in relation to cold tolerance (Fig. 3h). Comparable patterns of adaptive introgression have been reported in oaks (Quercus spp.), where introgression between sympatric species contributes to climatic adaptation94,95. Given the broad geographic overlap of many weedy species, such processes may also be widespread in globally distributed genera, such as Echinochloa (including E. crus-galli) and Setaria (including S. italica), where recurrent hybridization and local selection pressures likely drive adaptation in response to heterogeneous environments worldwide.
While introgression events in weedy species have been increasingly reported96,97, our findings suggest that adaptive introgression between closely related weedy species may facilitate the rapid evolution of herbicide resistance. Our analyses identified introgression signals flanking rSNPs, and revealed a potential correlation between the frequency of introgressed haplotypes at rSNPs and the herbicide resistance levels observed in D. sanguinalis accessions (Supplementary Fig. 37). These results support the hypothesis that closely related species can serve as genetic reservoirs for adaptive traits in weeds, consistent with previous observations of crop-to-weed introgression contributing to herbicide resistance98,99,100. In waterhemp (Amaranthus tuberculatus), introgression has been implicated in shaping the landscape-scale distribution of herbicide resistance101,102. By contrast, no comparable introgression signals associated with herbicide resistance have been detected in barnyardgrass31. Instead, population structure analysis reveals distinct subspecies differentiation in barnyardgrass, which likely driven by predominant self-fertilization and limited interspecific genetic exchange. Both crabgrass and waterhemp are sympatric with closely related species, providing opportunities for gene exchange103 (Supplementary Fig. 11). Many other weedy taxa also co-occur with closely related species, such as weedy rice, Amaranthus, and Setaria. Collectively, these cases suggest that introgression likely plays a significant role in facilitating weed adaptation to agricultural environments.
In summary, this study report genome assemblies for diploid, tetraploid, and hexaploid Digitaria species, establishing essential genomic resources for global accession profiling, herbicide resistance research, and the investigation of evolutionary mechanisms underlying weed adaptation. Crucially, we demonstrate that introgression may serve as an important source of adaptive genetic variation, offering insights to support the development of more integrated and sustainable weed management strategies.
Methods
Genome sequencing and assembly
Voucher specimens of the three sequenced species were deposited in the Herbarium of Zhejiang University (HZU), with accession numbers HZU60147516 (D. sanguinalis, #YJ2023), HZU60147511 (D. milanjiana, #DZ2), and HZU60147514 (D. radicosa, #YZGJ2).
For D. sanguinalis, total genomic DNA was extracted from young leaves using the cetyltrimethylammonium bromide (CTAB) method. High-molecular-weight DNA was prepared using a nuclei method104 for Nanopore ultralong sequencing and library preparation, followed by sequencing on a PromethION platform. ~116.98 Gb of ultralong reads (N50 > 100 kb) were de novo assembled with NextDenovo v2.5.2 using default parameters105. In parallel, single-molecule real-time (SMRT) sequencing libraries were prepared following PacBio protocols and sequenced on the Sequel II platform using the circular consensus sequencing approach. HiFi reads and ultralong reads were co-assembled using hifiasmv0.19.8-r603, and the contiguity of initial assembly was improved by merging with the initial NextDenovo assembly using Quickmerge106,107, with validation based on long-read mapping depth. The assembly was further polished by PacBio HiFi and Illumina data via NextPolish2 (v0.2.1) with default parameters108. Hi-C data were processed with YaHS v1.1 to generate a chromosome-scale contact map (Supplementary Fig. 4a), which was manually curated in Juicebox v1.11109,110,111. Subgenome phasing was performed using a k-mer based approach implemented in SubPhaser112.
For D. milanjiana, genomic DNA was extracted from young leaves using the CTAB method. Illumina paired-end libraries were prepared according to the manufacturer’s protocol (Illumina, USA). PacBio long reads were generated, error-corrected, and assembled into contigs using hifiasm. The high-contiguity D. sanguinalis assembly was used as a reference to anchor and order the contigs, and the contigs were further scaffolded into pseudochromosomes corresponding to the DH and EH using Ragtag v2.1.0113.
For D. radicosa (#YZGJ2), DNA extraction, library preparation, and genome assembly followed the same procedures described for D. milanjiana.
Genome annotation
Repeat families were identified de novo and initially classified using RepeatModeler v1.0.10114 followed by genome-wide repeat annotation performed with RepeatMasker v4.0.7114.
Protein-coding genes were predicted using an integrative approach that combined ab initio prediction, homology-based inference, and transcriptome-supported annotation. Ab initio predictions were generated with Fgenesh and AUGUSTUS v3.2.2, whereas homology-based evidence was obtained using GMAP115,116,117. All evidence types were integrated using EVidenceModeler v1.1.1116,118, resulting in a non-redundant consensus gene set. Gene models were retained only if supported by homology to known proteins, transcript alignments, or by at least two independent ab initio predictions. Low-confidence models, defined as those encoding peptides shorter than 50 amino acids or showing significant similarity to repetitive elements in Repbase (E-value < 1e-5, identity > 30%, and coverage > 25%), were filtered out to improve annotation quality.
Functional annotation of predicted protein-coding performed was conducted using InterProScan v5.24-63.0 across Digitaria spp., Echinochloa spp., O. sativa, S. italica and Panicum hallii40,119,120,121,122. Homologous genes previously validated in rice and maize were annotated using BLAST, retaining hits with an E value < 1e-5 and sequence identity > 50%53,123.
Repetitive elements annotation
Centromeric satellite repeats were predicted using the Tandem Repeat Annotation and Structural Hierarchy (TRASH) pipeline124 in two iterative rounds. In the first round, genomic sequences were partitioned into 1-kb windows, and local k-mer frequencies were calculated to detect repeat-enriched regions using default parameters. The most abundant repeat templates were clustered and extracted using CD-HIT125. In the second round, TRASH was executed with the parameters “--simpleplot --frep 10 --N.max.div 5 --par 5 --seqt,” with the extracted templates provided via the “--seqt” option. Windows were scored based on the proportion of repeated k-mers, and regions exceeding the threshold were classified as repeat-rich. Tandem repeats within these windows were further characterized based on the spacing between identical k-mers, enabling the identification of consensus repeat units, including CEN113, CEN159, and CEN178, hereafter collectively referred to as cenSat.
Ribosomal DNA (rDNA) regions were annotated via BLAST searches against maize rDNA references sequences, including 5S rDNA (DQ351339), 5.8S rDNA (AF019817), and the intergenic spacer (AF013103). Telomeric repeats were identified by scanning chromosome termini for high-copy-number tandem arrays of the canonical monomer “TTTAGGG”.
Genome quality assessment
Assembly quality for each genome was evaluated based on completeness, correctness, and continuity. For completeness, NGS short reads and PacBio HiFi long reads were mapped to their respective assemblies using BWA mem v0.7.17-r1188126 and Minimap2 v2.03127 with default parameters, respectively. Mapping statistics were summarized with Sambamba Flagstat v1.0.1128. Additional completeness evaluations were performed using the LTR Assembly Index (LAI)129 and BUSCO v5.5.0 with the poales_odb 10 database41. Short reads were split into 21-mers using Meryl v1.4 with low frequency k-mers removed (meryl gt 1) prior to generating a k-mer reference library. Then, k-mer based completeness was estimated with Merqury v1.3130. For correctness, base-level accuracy was evaluated by estimating quality values with Merqury, based on the same k-mer library. For continuity, potential assembly gaps and structural inconsistencies were identified using CRAQ44. Long and short reads were aligned against assemblies, and AQI scores were calculated by analyzing clipped alignments to identify local and large-scale errors.
Phylogenetic analysis
Homoeologous exchanges (HEs) in polyploids can distort phylogenetic inference; therefore, candidate HE regions in the D. sanguinalis genome were identified and excluded prior to downstream phylogenetic analyses. ~99× short reads coverage from D. radicosa and 50× from D. milanjiana were mapped to the D. sanguinalis reference genome using Bowtie2 with default settings131. Mapping depths were calculated in 100-kb sliding windows across the genome. For the DH and EH, windows with coverage exceeding 20× and showing higher mapping depth for D. radicosa than D. milanjiana were designated as candidate HE regions; the same approach was applied to the CH. Adjacent candidate HE windows were merged, resulting in four distinct HE regions. Genes located within these regions were excluded from subsequent phylogenetic reconstruction.
Phylogenetic relationships among Digitaria and related species were inferred using three approaches. First, genetic divergence was estimated based on synonymous substitution (Ks) values, and pairwise Ks peak distributions were used to infer relative divergence times. Second, a concatenated alignment of 2,030 single-copy orthologs, identified by OrthoFinder132 and excluding HE-associated genes, was used to construct a ML tree. Amino acid sequences were aligned using MAFFT v7.310133 and trimmed with TrimAl under default parameters134. The ML tree was generated with IQ-TREE v1.6.12135 with 1000 bootstrap replicates and the best-fit substitution model (GAMMA + JTT + F4) as determined by ModelFinder. Third, 2,030 single gene trees were inferred individually using RAxML under the best-fitting amino acid substitution models, and a coalescent-based species tree was subsequently estimated with ASTRAL v5.7.8136,137.
Collection and phenotyping of Digitaria accessions
A total of 579 Digitaria accessions were collected from 24 provinces across China from 2013 to 2023 (Supplementary Data 4). Seeds collected prior to 2023 were preserved at the Plant Protection Research Institute, Shandong Academy of Agricultural Sciences. In 2023, 496 accessions were grown under uniform field conditions at the Jiyang Research Station of the Shandong Academy of Agricultural Sciences (Jinan), with five representative individuals planted per accession for phenotypic evaluation. The geographic distribution of collected accessions was visualized using R v4.3.1. Based on detailed collection-site metadata, 272 accessions were further classified into three ecological habitat types: natural habitats (n = 40; riverbanks or wilderness), agricultural habitats (n = 221; active croplands), and disturbed habitats (n = 11; parks or roadsides).
Definition of spatial groups and comparisons
Accessions collected in 2013 or 2015 within a of 100-km radius were clustered into historical groups using the “great_circle” method in the geopy package, with the centroid of each cluster designated as the groups’ location. Historical groups with at least four accessions were retained for temporal comparison. To create modern groups, we selected modern accessions in close proximity (< 100 km) to the historical groups. Finally, 11 groups comprising 167 accessions were used in the analysis (Supplementary Fig. 22). For each spatial group, the normalized Shannon entropy was calculated as a diversity score using R package entropy based on k = 6 from the admixture analysis. Nucleotide diversity and Tajima’s D were estimated with VCFtools v0.1.17138 in non-overlapping 100-kb windows. Heterozygosity was calculated for each accession.
Resequencing and variant calling
Genomic DNA was extracted from fresh leaves using a standard CTAB-based protocol. Paired-end resequencing libraries were prepared and sequenced on the DNBSEQ T7 platform. Raw reads were quality-filtered using fastp v0.24.2139, and high-quality paired-end reads were mapped to D. sanguinalis reference genome (#YJ2023) using Bowtie2 with default parameters131. Whole-genome variant calling and filtering were performed using an integrated pipeline140. To minimize false positives due to high sequence similarity between subgenomes, resequencing reads of YJ2023 were realigned to its reference, and variants detected in this process were excluded from the final dataset. Variants were further filtered by the minor allele frequency (MAF) greater than 0.01 and missing rate less than 30%. Functional annotation of all high-confidence variants was conducted using SnpEff v3.652141.
Population structure and genetic diversity
Species identity of each accession was initially assessed by calculating read mapping rates to the YJ2023 reference genome and estimating genome coverage using sambamba v1.5 and bamdst128,142. Population structure was inferred with FastStructure143 based on genome wide SNPs and synonymous SNPs from CH, DH and EH, with k values ranging from 2 to 9. Phylogenetic relationships were reconstructed with FastTreeMP144 based on SNPs from 499 accessions (excluding D. violascens and D. ischaemum due to low genome coverage), using 51 D. ciliaris accessions as the outgroup; branch support was assessed with 1000 bootstrap replicates. Trees were visualized using iTOL v7 (http://itol.embl.de)145. PCA was conducted in PLINK v1.90b6.20146 using a linkage-disequilibrium pruned SNP set (10 SNPs per 50-kb sliding window, r² <0.5). Nucleotide diversity (π) was calculated with VCFtools v0.1.17138 in non-overlapping 20-kb, 50-kb and 100-kb windows, and genome-wide or subgenome-level diversity was reported as the mean π across all windows within each population.
Demographic history inference based on site frequency spectrum (SFS)
To reconstruct the demographic history of Digitaria and evaluate the role of introgression during species and lineage divergence, we performed composite ML inference based on the site frequency spectrum (SFS). Joint folded two-dimensional SFSs (2D-SFSs) were generated from four-fold synonymous SNPs using easySFS.py (https://github.com/isaacovercast/easySFS). Eight demographic scenarios were designed to test for the occurrence and timing of introgression between lineages and species (Supplementary Fig. 18), with divergence time priors informed by fossil calibrations in TimeTree 5147. Likelihood estimation for each scenario was conducted using fastsimcoal2148, with 100,000 coalescent simulations per likelihood estimation (-n 100,000) and 40 expectation-conditional maximization (ECM) cycles (-L 40). Model selection was performed using the Akaike information criterion (AIC), calculated as AIC = \(2k-2\mathrm{ln}(L)\) (MaxEstLhood), where k represents the number of estimated parameters and MaxEstLhood is the ML function value for the model. To avoid convergence to local optima, each analysis was repeated at least twice, and the best-supported model based on AIC was re-run 100 times to obtain refined parameter estimates. Subsequently, 100 independent DNA polymorphism datasets were simulated as joint SFSs conditional on estimated demographic parameters. ML analysis was then applied to each joint SFS over 40 ECM cycles to obtain confidence intervals (CIs) for final estimates.
Changes in effective population size over time were further inferred using Stairway Plot v2.0149, based on both folded and unfolded SFSs. Population-specific SFSs were constructed and folded from the same SNP dataset used for fastsimcoal2.
Phylogeny of chloroplast genomes
The reference chloroplast genome of YJ2023 was assembled de novo using GetOrganelle v1.7.7.1150. For other accessions, chloroplast-derived reads were extracted from paired-end resequencing data with GetOrganelle and mapped to YJ2023 chloroplast reference genome. Variants were called using the GATK pipeline, resulting in 4,742 high-quality chloroplast SNPs after filtering for MAF 0.05 and missing rate 0.2 across 579 Digitaria accessions. A ML tree was constructed in IQtree with 1500 bootstrap replicates135, and the tree was rooted with S. italica as the outgroup.
Detection of sympatric introgression between D. ciliaris and D. sanguinalis
Sympatric population pairs were defined based on pairwise geographic distances calculated with the “great_circle” method in the geopy package, with a threshold of 150-km. Populations pairs comprising at least two D. ciliaris and eight D. sanguinalis accessions were retained for downstream analyses. Introgression in sympatric population pairs was assessed using D-statistics151,152, and the extent of introgressed genomic regions was quantified using the modified fd statistic153. The phylogenetic topology (((P1, P2), P3), O) was applied, with D. bicornis designated as the outgroup (O), sympatric D. sanguinalis as P2, sympatric D. ciliaris as P3, and allopatric D. sanguinalis accessions as P1. Under the null hypothesis, ABBA and BABA site patterns are expected to occur at equal frequencies due to incomplete lineage sorting, whereas an excess of ABBA sites indicates introgression between P2 and P3. To minimize sample bias, eight individuals were randomly selected for P1 and P2, and three for P3 and the outgroup.
Introgression was evaluated in two steps. First, D-statistics were calculated genome-wide. To account for linkage disequilibrium, significance was assessed using a block jackknife approach151, in which the genome was divided into 1-Mb blocks, and one block was sequentially removed at a time to estimate the mean and variance of D. Second, introgressed genomic regions were identified using sliding windows of 10-, 50-, and 100-kb, with corresponding step sizes of 5-, 25-, and 50-kb, respectively. Windows containing fewer than 200, 100, or 20 SNPs (for 100-, 50-, and 10-kb windows, respectively) were excluded. Both D and fd statistics were computed with “ABBABABAwindows.py” and Fst values for the same windows were calculated with “popgenWindows.py”153. According to the filtering criteria used by Zhou et al154., fd values outside the 0-1 range were set to zero. The overall introgression level was quantified as the Proportion of Introgression across the Genome (PIG) and Pairwise Shared Introgressed Genomic regions (PSIG) was used to quantify shared introgression among sympatric populations. Regions with fd > 0.6 were defined as significantly introgressed, and genes within these regions were subjected to functional enrichment analysis.
Functional enrichment analysis of introgressed genes
Gene families were annotated based on Pfam domains identified in the YJ2023 genome. For enrichment analysis, all Pfam domains in YJ2023 were used as the background set. To test whether the Pfam domains found within introgressed regions represented a non-random sampling, we applied Fisher’s exact test to compare their frequencies against the genome-wide background. Multiple testing correction was performed using the Benjamini-Hochberg and domains with adjusted p-values < 0.05 were considered significantly enriched. Fold enrichment was calculated as the ratio of each Pfam domain’s frequency in introgressed regions to its frequency within the background.
Environmental factors and correlation with introgression
To investigate the relationship between introgression and environmental variation, 91 environmental variables were retrieved from the WorldClim database (http://www.worldclim.org/) (Supplementary Data 5). These variables encompassed four primary categories, temperature, precipitation, wind speed, and solar radiation, as well as 19 additional bioclimatic variables. Pairwise environmental distance matrices for the primary categories were calculated using Euclidean distances with the R package “ecodist”155, and a separate matrix was generated from the 19 bioclimatic variables. Longitude and latitude were also used to generate distance matrices. Geographic distances among populations were computed using the “great_circle” method in the “geopy” package, and statistical significance in downstream analyses was assessed using 10,000 permutations.
Mantel and partial Mantel tests were conducted using “ecodist” to assess correlations between PSIG and pairwise geographic, environmental and Fst distances.
Identification of environment-associated genetic variants
Variants with MAF > 0.01 and missing rate > 0.2 were retained, yielding a dataset of 3,981,523 SNPs for downstream analyses. Associations between allele frequencies and 19 environmental variables were initially assessed using a univariate latent factor mixed model (LFMM) implemented in the R package LEA v3.14.0156. Latent factors were determined based on the ancestry clusters inferred from FastStructure, consistent with results from the demographic history analysis. For each environmental variable, five independent runs were conducted, with 5000 burn-in iterations followed by 10,000 sampling iterations. Median p values from five independent runs were calculated and adjusted for multiple testing using both the false discovery rate (FDR, 5%) and the Bonferroni method.
Single-dose bioassays on Digitaria
A single-dose herbicide bioassay was conducted on Digitaria accessions (n = 53 in 2013, n = 56 in 2015, n = 52 in 2023) at the 2-3 leaf stage. Plants were grown in 9-cm diameter pots filled with sterilized potting soil and maintained under controlled environmental conditions (30/25 °C day/night, 16 h of photoperiod) until herbicide application. A commercial formulation of nicosulfuron (field-recommended dose: 60 g a.i. ha-1) was applied using an enclosed cabinet sprayer calibrated to deliver 450 L ha-1 at a pressure of 0.4 MPa (ASS-5, Information Technology Research Center, Beijing, China). Untreated control (0 g a.i. ha-1) was also included for comparison. Each treatment, including the control, was replicated four times with one pot per replicate.
Survival rate (%) was calculated as:
Ns represents the number of surviving plants, and NT represents the total number of plants per pot. Plants were classified as surviving if they exhibited active regrowth or retained green tissue.
Aboveground biomass from each pot was harvested, oven-dried at 70 °C for 72 h, and weighed. Control efficacy (%) was calculated as the percentage of biomass reduction relative to the untreated control:
Bt is the biomass in the treatment, and Bc is the biomass in the control.
Whole-plant resistance bioassay
Seeds of 196 Digitaria accessions were collected, air-dried, and stored at 4 °C prior to use. Seeds were sown in moist loam soil in 9-cm diameter pots, covered with 1 cm of soil, and grown under greenhouse conditions (30/25 °C day/night, 16 h of photoperiod) with sub-irrigation.
Herbicide resistance to nicosulfuron was evaluated using whole-plant bioassays. At the three-leaf stage, ten seedlings were foliar treated with the commercial formulation of nicosulfuron at seven doses, 0, 30, 60, 120, 240, 480, and 960 g a.i. ha-1 (field-recommended dose: 60 g a.i. ha-1). Applications were performed using an enclosed cabinet sprayer (ASS-5, Information Technology Research Center, Beijing, China) calibrated to deliver 450 L ha-1 at 0.4 MPa. Plant survival and aboveground dry biomass were assessed 21 days after treatment. Each treatment included four biological replicate pots (10 seedlings per pot) and was independently repeated.
Dose-response relationships for biomass reduction (expressed as a percentage of untreated controls) were analyzed by nonlinear regression in SigmaPlot v12.5 (Systat Software Inc.), fitting a four-parameter log-logistic model:
where Y is the response (percentage of control), X is the herbicide dose, C and D are the lower and upper asymptotes, GR50 is the herbicide dose required to plant growth reduction by 50%, and b is the slope of the curve. A mixed-model ANOVA was conducted to assess differences in percent control and biomass reduction across treatments. Resistance indices (RIs) for individual accessions were calculated as the ratio of their GR₅₀ values to the average GR₅₀ of susceptible accessions, which were defined as those controlled at the recommended field dose (GR₉₀ <60 g a.i. ha⁻¹). Resistance levels were categorized into four groups based on RI values: S (susceptible, RI < 2), LR (low resistant, RI > 2 and RI < 4), MR (moderate resistant, RI > 4 and RI < 10), and HR (high resistant, RI > 10).
Herbicide resistance analyses
ALS genes were identified in the D. sanguinalis genome using BLASTP. The predicted ALS protein sequences were aligned with orthologous sequences from O. sativa and A. thaliana using MAFFT133, and known resistance-associated sites were annotated across the three DsALS copies. A comprehensive catalog of causative mutations was then established for all accessions (Supplementary Data 10). Genealogical relationships among haplotypes were reconstructed using HapNetworkView based on variant profiles across the three ALS copies157.
NTSR-related genes were identified based on Pfam domain annotations. Sequencing depth for each annotated gene was calculated, and gene family abundance was estimated as the sum of average depths across all loci, using custom scripts. Coverage was normalized by dividing the average read depth of each gene family by the mean sequencing depth of 2030 single-copy genes. Accessions with sequencing depth < 5 were excluded to minimize bias in coverage estimation.
Plant material and nicosulfuron treatment for RNA-seq
The nicosulfuron-resistant (#21-17, GR50 = 122.53 g a.i. ha-1) and susceptible (#15-2, GR50 = 0.59 g a.i. ha-1) D. sanguinalis accessions were selected for RNA-seq analysis. Plants were grown in 9-cm diameter pots filled with moist loam soil, covered with a 1-cm soil layer, and maintained under greenhouse conditions (30/25 °C day/night, 16 h of photoperiod) with sub-irrigation at the Jiyang Research Station of the Shandong Academy of Agricultural Sciences (Jiyang, Shandong, China).
At the three-leaf stage, seedlings were treated with 2 g a.i. ha-1 nicosulfuron based on the GR50 value distribution of susceptible populations from dose-response assays, to assess herbicide response. Each treatment included three biological replicates, with three individual plants pooled to constitute one replicate. Leaf tissues were collected at three time points: 0 h (untreated control), 6 h (early response), and 24 h (late response) post-treatment. Samples were immediately frozen in liquid nitrogen and stored at −80 °C until RNA extraction and sequencing.
RNA sequencing and transcriptome analysis
Total RNA was extracted from 18 samples, comprising three time points (0 h, 6 h, and 24 h) for each accession following nicosulfuron treatment, with three biological replicates per time point. Messenger RNA was purified using poly-T oligo-attached magnetic beads, and sequencing libraries were constructed according to the DNBSEQ standard protocol. Libraries were pooled based on effective concentration and target data yield. The 5′ ends of the libraries were phosphorylated and circularized, followed by rolling circle amplification to generate DNA nanoballs, which were subsequently loaded onto a flow cell for sequencing on the DNBSEQ-T7 platform.
After trimming adapter sequences and filtering out low-quality reads, clean reads were mapped to the YJ2023 reference genome using Hisat2 v2.1.0158 and gene expression levels were quantified by StringTie v2.2.1159 with default parameters. DEGs were identified with the pyDESeq2 package v0.5.2160, with significance thresholds set at |log2(fold change)| ≥ 1 and FDR-adjusted p-value < 0.05. GO enrichment analysis was performed using the clusterProfiler package161.
GWAS of nicosulfuron resistance
GWAS were conducted on 141 D. sanguinalis accessions using GR50 values using EMMAX70. SNPs were filtered with MAF > 0.05 and missing genotype rate <0.1. A pairwise genetic distance matrix, derived from simple matching coefficients of SNPs, was used to define the variance-covariance structure of random effects in the linear mixed model. Median p-values from five independent runs were calculated and adjusted for multiple testing using both the false discovery rate (FDR, 5%) and the Bonferroni correction. Manhattan plots were generated using the R package “qqman”162 for visualization of association signals.
To examine selection pressures acting on adaptive variants associated with herbicide resistance and climatic adaptation, extended haplotype homozygosity (EHH) was assessed for strongly associated loci, and the integrated haplotype score (iHS) was calculated for common variants using hapbin v1.3.0163.
Introgression analysis at resistance associated loci
To assess potential introgression from D. ciliaris into resistant D. sanguinalis accessions, we applied the ABBA-BABA test under a four-taxon topology of the form ((P1, P2), P3, O), where P1 represented the susceptible D. sanguinalis population, P2 comprised resistant accessions (including HR, MR, and LR), P3 corresponded to D. ciliaris, and D. bicornis served as the outgroup (Supplementary Fig. 37a). We estimated the fd values across the genome with sliding windows of 10-, 50-, and 100-kb and respective step sizes of 5-, 25-, and 50-kb. Windows containing fewer than 200, 100, or 20 SNPs (for 100-, 50-, and 10-kb windows, respectively) were excluded. For each window, we also calculated Fst between susceptible and resistant D. sanguinalis accessions. To identify windows with significantly elevated introgression, we performed two-tailed Z-tests and defined outlier windows as those with |Z-score | > 4. To establish significance thresholds for Fst, we conducted randomization tests by permuting the group labels, generating a null distribution from which one-tailed p-values were derived (e.g., one-tailed p = 0.001 corresponds to the 0.1% or 99.9% quantile range).
To assess the phylogenetic structure and potential introgression events, we applied coalescent-based modeling using BPP v4.6.273. Analyses were based on a 100-kb genomic region flanking the rSNP (Chr20: 34,032,644). First, to test for the presence of introgression under the MSC-I model, we implemented the A00 method, which estimates the posterior probabilities of divergence times and introgression probabilities (Supplementary Data 13). This approach allowed us to quantify the likelihood of introgression between D. ciliaris and resistant D. sanguinalis accessions within this focal genomic region. In parallel, we used the A11 method to evaluate whether the hypothesized species tree topology receives the highest posterior probability. All analyses were conducted using three independent MCMC runs with 10,000 burn-in iterations followed by 100,000 samples drawn every 10 iterations to ensure convergence and robustness.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genome assemblies and annotations for the three Digitaria species generated in this study have been deposited in National Genomics Data Center (NGDC) database under the following accessions: GWHGGEX00000000 (https://ngdc.cncb.ac.cn/gwh/Assembly/98807/show) (D. sanguinalis, #YJ2023), GWHGGEY00000000 (https://ngdc.cncb.ac.cn/gwh/Assembly/98808/show) (D. radicosa, #YZGJ2) and GWHGGEZ00000000 (https://ngdc.cncb.ac.cn/gwh/Assembly/98809/show) (D. milanjiana, DZ2). HiFi sequencing, Hi-C, and RNA-Seq data used for genome assembly and annotation are available under NGDC project accession PRJCA044252. Raw resequencing data for all accessions have been deposited in the National Center for Biotechnology Information (NCBI) under accession PRJNA1296823. The other reference genomes used in this study were retrieved from NGDC or NCBI under accessions PRJCA003883, PRJNA265547 (https://www.ncbi.nlm.nih.gov/bioproject/265547), PRJNA931988, PRJNA953663, PRJEB36539, PRJNA250527, and PRJNA32913. Resequencing data of green foxtail were downloaded from the European Nucleotide Archive (ENA) under accession PRJNA633413. Map base layers were obtained from the official Tianditu platform (https://www.tianditu.gov.cn/). Source data are provided with this paper.
Code availability
All custom scripts used in this study are available at GitHub (https://github.com/Ne0tea/DigitariaPop).
References
Mahaut, L. et al. Weeds: against the rules? Trends Plant Sci. 25, 1107–1116 (2020).
Sharma, G., Barney, J. N., Westwood, J. H. & Haak, D. C. Into the weeds: new insights in plant stress. Trends Plant Sci. 26, 1050–1060 (2021).
Stewart, C. N. Becoming weeds. Nat. Genet. 49, 654–655 (2017).
Burton, G. P. et al. Phylogenetics, evolution and biogeography of four Digitaria food crop lineages across West Africa, India, and Europe. Biorxiv 2025 3, 645812 (2025).
Holm, L. G., Plucknett, D. L., Pancho, J. V. & Herberger, J. P. The World’s Worst Weeds. (University Press, Honolulu, Hawaii, 1977).
Oreja, F. H., Batlla, D. & de la Fuente, E. B. Digitaria sanguinalis seed dormancy release and seedling emergence are affected by crop canopy and stubble. Weed Res. 60, 111–120 (2020).
Oreja, F. H., Bastida, F. & Gonzalez-Andújar, J. L. Simulación de estrategias de control para la toma de decisión de Digitaria sanguinalis en soja resistente a glifosato. Cienc. E Investig. Agrar. 39, 299–308 (2012).
Boonsuk, B., Chantaranothai, P. & Hodkinson, T. R. A taxonomic revision of the genus Digitaria (Panicoideae: Poaceae) in mainland southeast Asia. Phytotaxa 246, 248–282 (2016).
Kok, P. D. F., Robbertse, P. J. & van Wyk, A. E. Systematic study of Digitaria section Digitaria (Poaceae) in southern Africa. South Afr. J. Bot. 55, 141–153 (1989).
Touafchia, S. et al. Evolutionary history, traits, and weediness in Digitaria (Poaceae: Panicoideae). Bot. J. Linn. Soc. 203, 1–19 (2023).
Délye, C. Unravelling the genetic bases of non-target-site-based resistance (NTSR) to herbicides: a major challenge for weed science in the forthcoming decade. Pest Manag. Sci. 69, 176–187 (2013).
Heap, I. The international herbicide-resistant weed database. http://www.weedscience.org/ (2025).
Powles, S. B. & Yu, Q. Evolution in action: plants resistant to herbicides. Annu. Rev. Plant Biol. 61, 317–347 (2010).
Kreiner, J. M., Stinchcombe, J. R. & Wright, S. I. Population genomics of herbicide resistance: adaptation via evolutionary rescue. Annu. Rev. Plant Biol. 69, 611–635 (2018).
Yuan, J. S., Tranel, P. J. & Stewart, C. N. Non-target-site herbicide resistance: a family business. Trends Plant Sci. 12, 6–13 (2007).
Guan, Y., Liu, L., Zou, Y., Yang, C. & Ji, M. Involvement of P450s in the metabolic resistance of Digitaria sanguinalis (L.) scop. to ALS-inhibiting herbicides. Pestic. Biochem. Physiol. 204, 106038 (2024).
Laforest, M. et al. Acetyl-CoA carboxylase overexpression in herbicide-resistant large crabgrass (Digitaria sanguinalis). Pest Manag. Sci. 73, 2227–2235 (2017).
Wang, R. et al. ALS gene overexpression and enhanced metabolism conferring Digitaria sanguinalis resistance to nicosulfuron in China. Front. Plant Sci. 14, 1290600 (2023).
Yanniccari, M. et al. A novel EPSPS pro-106-his mutation confers the first case of glyphosate resistance in Digitaria sanguinalis. Pest Manag. Sci. 78, 3135–3143 (2022).
Li, J., Li, M., Gao, X. & Fang, F. A novel amino acid substitution Trp574Arg in acetolactate synthase (ALS) confers broad resistance to ALS-inhibiting herbicides in crabgrass. Pest Manag. Sci. 73, 2538–2543 (2017).
Mei, Y., Si, C., Liu, M., Qiu, L. & Zheng, M. Investigation of resistance levels and mechanisms to nicosulfuron conferred by non-target-site mechanisms in large crabgrass (Digitaria sanguinalis L.) from China. Pestic. Biochem. Physiol. 141, 84–89 (2017).
Zhao, B. et al. Target-site mutation and enhanced metabolism endow resistance to nicosulfuron in a Digitaria sanguinalis population. Pestic. Biochem. Physiol. 194, 105488 (2023).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Rutland, C. A., Hall, N. D. & McElroy, J. S. The impact of polyploidization on the evolution of weed species: historical understanding and current limitations. Front. Agron. 3, 626454 (2021).
Soltis, P. S., Marchant, D. B., Van De Peer, Y. & Soltis, D. E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 35, 119–125 (2015).
McDaniel, S. F. Local adaptation, recombination, and the fate of neopolyploids. N. Phytol. 244, 32–38 (2024).
Shimizu, K. K. Robustness and the generalist niche of polyploid species: genome shock or gradual evolution? Curr. Opin. Plant Biol. 69, 102292 (2022).
Menardo, F. et al. Hybridization of powdery mildew strains gives rise to pathogens on novel agricultural crop species. Nat. Genet. 48, 201–205 (2016).
Yang, C. et al. Evolution of physiological responses to salt stress in hexaploid wheat. Proc. Natl. Acad. Sci. 111, 11882–11887 (2014).
Cheng, F. et al. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4, 258–268 (2018).
Wu, D. et al. Genomic insights into the evolution of Echinochloa species as weed and orphan crop. Nat. Commun. 13, 689 (2022).
Ye, C.-Y. et al. The genomes of the allohexaploid Echinochloa crus-galli and its progenitors provide insights into polyploidization-driven adaptation. Mol. Plant 13, 1298–1310 (2020).
Taylor, S. A. & Larson, E. L. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3, 170–177 (2019).
Carnahan, H. L. & Hill, H. D. Cytology and genetics of forage grasses. Bot. Rev. 27, 1–162 (1961).
Yasue, T. Studies on hybrids in Digitaria II. The F1 plants, Digitaria ciliaris HERS. x D. ischaemum MUHL. Jpn. J. Breed. 6, 265–271 (1957).
Yasue, T. Studies on hybrids in Digitaria.: I. Technique of hybridization. Jpn. J. Breed. 6, 197–201 (1956).
Rendón-Anaya, M. et al. Adaptive introgression facilitates adaptation to high latitudes in european aspen (Populus tremula L.). Mol. Biol. Evol. 38, 5034–5050 (2021).
Oziolor, E. M. et al. Adaptive introgression enables evolutionary rescue from extreme environmental pollution. Science 364, 455–457 (2019).
Reid, N. M. et al. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science 354, 1305–1308 (2016).
Abrouk, M. et al. Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate. Nat. Commun. 11, 4488 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40, D1202–D1210 (2012).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat. Commun. 14, 6556 (2023).
Huang, W. et al. A well-supported nuclear phylogeny of Poaceae and implications for the evolution of C4 photosynthesis. Mol. Plant 15, 755–777 (2022).
Zhang, L. et al. Phylotranscriptomics resolves the phylogeny of Pooideae and uncovers factors for their adaptive evolution. Mol. Biol. Evol. 39, msac026 (2022).
Brown, J. K. M. & Rant, J. C. Fitness costs and trade-offs of disease resistance and their consequences for breeding arable crops. Plant Pathol. 62, 83–95 (2013).
Ngom, A. et al. Révision du genre Digitaria Haller (Poaceae) au Sénégal : proposition d’une clé de détermination pour une meilleure identification des espèces. Int. J. Biol. Chem. Sci. 10, 58–88 (2016).
Webster, R. D. Taxonomy of Digitaria section Digitaria in North America (poaceae: paniceae). SIDA Contrib. Bot. 12, 209–222 (1987).
Feng, M., Augstein, F., Kareem, A. & Melnyk, C. W. Plant grafting: molecular mechanisms and applications. Mol. Plant 17, 75–91 (2024).
Gaut, B. S., Miller, A. J. & Seymour, D. K. Living with two genomes: grafting and its implications for plant genome-to-genome interactions, phenotypic variation, and evolution. Annu. Rev. Genet. 53, 195–215 (2019).
Frichot, E., Schoville, S. D., Bouchard, G. & François, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 30, 1687–1699 (2013).
Yao, W., Li, G., Yu, Y. & Ouyang, Y. funRiceGenes dataset for comprehensive understanding and application of rice functional genes. GigaScience 7, gix119 (2018).
Kim, J. Y. et al. Zinc finger-containing glycine-rich RNA-binding protein in Oryza sativa has an RNA chaperone activity under cold stress conditions. Plant Cell Environ. 33, 759–768 (2010).
Xu, T., Han, J. H. & Kang, H. Structural features important for the RNA chaperone activity of zinc finger-containing glycine-rich RNA-binding proteins from wheat (Triticum avestivum) and rice (Oryza sativa). Phytochemistry 94, 28–35 (2013).
Liu, X. et al. Rice ubiquitin-conjugating enzyme OsUBC26 is essential for immunity to the blast fungus Magnaporthe oryzae. Mol. Plant Pathol. 22, 1613–1623 (2021).
Zhao, J.-L. et al. Mutual regulation of receptor-like kinase SIT1 and B’κ-PP2A shapes the early response of rice to salt stress. Plant Cell 31, 2131–2151 (2019).
Luo, H., Song, F. & Zheng, Z. Overexpression in transgenic tobacco reveals different roles for the rice homeodomain gene OsBIHD1 in biotic and abiotic stress responses. J. Exp. Bot. 56, 2673–2682 (2005).
Yang, Q. et al. NOA1 functions in a temperature-dependent manner to regulate chlorophyll biosynthesis and Rubisco formation in rice. PLOS ONE 6, e20015 (2011).
Cai, L. et al. The blackgrass genome reveals patterns of non-parallel evolution of polygenic herbicide resistance. N. Phytol. 237, 1891–1907 (2023).
Klure, D. M., Greenhalgh, R., Orr, T. J., Shapiro, M. D. & Dearing, M. D. Parallel gene expansions drive rapid dietary adaptation in herbivorous woodrats. Science 387, 156–162 (2025).
Guo, R. et al. A novel aldo-keto reductase gene, OsAKR1, from rice confers higher tolerance to cadmium stress in rice by an in vivo reactive aldehyde detoxification. J. Hazard. Mater. 470, 134212 (2024).
Oda, K., Otani, M., Uraguchi, S., Akihiro, T. & Fujiwara, T. Rice ABCG43 is Cd inducible and confers Cd tolerance on yeast. Biosci. Biotechnol. Biochem. 75, 1211–1213 (2011).
Pan, L. et al. CYP81A68 confers metabolic resistance to ALS and ACCase-inhibiting herbicides and its epigenetic regulation in Echinochloa crus-galli. J. Hazard. Mater. 428, 128225 (2022).
Peng, M. et al. Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 8, 1975 (2017).
Chatterjee, Y., Tomar, S., Mishra, M., Pareek, A. & Singla-Pareek, S. L. OsLdh7 overexpression in rice confers submergence tolerance by regulating key metabolic pathways: anaerobic glycolysis, ethanolic fermentation and amino acid metabolism. Plant Cell Environ. 48, 2804–2820 (2025).
Nakazono, M. et al. Expression of a gene encoding mitochondrial aldehyde dehydrogenase in rice increases under submerged conditions. Plant Physiol. 124, 587–598 (2000).
Chen, S., Sun, B., Shi, Z., Miao, X. & Li, H. Identification of the rice genes and metabolites involved in dual resistance against brown planthopper and rice blast fungus. Plant Cell Environ. 45, 1914–1929 (2022).
Li, W. et al. Overexpression of the homoterpene synthase gene, OsCYP92C21, increases emissions of volatiles mediating tritrophic interactions in rice. Plant Cell Environ. 44, 948–963 (2021).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Chen, W. et al. PfGSTF2 endows resistance to quizalofop-p-ethyl in Polypogon fugax by GSH conjugation. Plant Biotechnol. J. 23, 216–231 (2025).
Goldberg-Cavalleri, A., Onkokesung, N., Franco-Ortega, S. & Edwards, R. ABC transporters linked to multiple herbicide resistance in blackgrass (Alopecurus myosuroides). Front. Plant Sci. 14, 1082761 (2023).
Flouri, T., Jiao, X., Rannala, B. & Yang, Z. A Bayesian implementation of the multispecies coalescent model with introgression for phylogenomic analysis. Mol. Biol. Evol. 37, 1211–1223 (2020).
Ren, R. et al. Widespread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Mol. Plant 11, 414–428 (2018).
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Wood, T. E. et al. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. 106, 13875–13879 (2009).
Tiwari, V. K., Saripalli, G., Sharma, P. K. & Poland, J. Wheat genomics: genomes, pangenomes, and beyond. Trends Genet 40, 982–992 (2024).
Zhang, H. et al. Super-pangenome analyses across 35 accessions of 23 Avena species highlight their complex evolutionary history and extensive genomic diversity. Nat. Genet. 57, 2276–2288 (2025).
Sharma, M. L. & Sharma, K. Cytological studies in the north indian grasses. Cytologia (Tokyo) 44, 861–872 (1979).
Georgia, A. E. & Georgia, A. E. A. Manual of Weeds, with Descriptions of All the Most Pernicious and Troublesome Plants in the United States and Canada, Their Habits of Growth and Distribution, with Methods of Control. https://doi.org/10.5962/bhl.title.29396. (The Macmillan Company, New York, 1916).
Henrard, J. T. Monograph of the Genus Digitaria. Agron. J. 42, 631–631 (1950).
Vietmeyer, N. et al. Lost Crops of Africa: Volume I: Grains. 2305 https://doi.org/10.17226/2305. (National Academies Press, Washington, D.C., 1996).
Luo, Q. & Liu, Y.-G. Breeding herbicide-resistant rice using CRISPR-cas gene editing and other technologies. Plant Commun. 6, 101172 (2025).
Délye, C., Jasieniuk, M. & Corre, V. L. Deciphering the evolution of herbicide resistance in weeds. Trends Genet 29, 649–658 (2013).
Otto, S. P. & Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 34, 401–437 (2000).
Yu, Q., Ahmad-Hamdani, M. S., Han, H., Christoffers, M. J. & Powles, S. B. Herbicide resistance-endowing ACCase gene mutations in hexaploid wild oat (Avena fatua): insights into resistance evolution in a hexaploid species. Heredity 110, 220–231 (2013).
Baucom, R. S. Evolutionary and ecological insights from herbicide-resistant weeds: what have we learned about plant adaptation, and what is left to uncover? N. Phytol. 223, 68–82 (2019).
Délye, C. et al. Geographical variation in resistance to acetyl-coenzyme a carboxylase-inhibiting herbicides across the range of the arable weed Alopecurus myosuroides (black-grass). N. Phytol. 186, 1005–1017 (2010).
Kersten, S. et al. Standing genetic variation fuels rapid evolution of herbicide resistance in blackgrass. Proc. Natl. Acad. Sci. 120, e2206808120 (2023).
Comont, D. et al. Evolution of generalist resistance to herbicide mixtures reveals a trade-off in resistance management. Nat. Commun. 11, 3086 (2020).
Huang, J., Wang, S. & Xiao, Z. Rising herbicide use and its driving forces in China. Eur. J. Dev. Res. 29, 614–627 (2017).
Liu, X. et al. Managing herbicide resistance in China. Weed Sci. 69, 4–17 (2021).
Edelman, N. B. et al. Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599 (2019).
Fu, R. et al. Genome-wide analyses of introgression between two sympatric Asian oak species. Nat. Ecol. Evol. 6, 924–935 (2022).
Ren, Y.-B., Yu, S.-L., Sun, H. & Ma, X.-G. Genomic insights into introgression between Quercus aquifolioides and its sympatric relatives across elevational gradients. Mol. Ecol. 34, e17747 (2025).
Ribeiro, V. H. V., Gallagher, J., Mallory-Smith, C., Barroso, J. & Brunharo, C. A. C. G. Multiple origins or widespread gene flow in agricultural fields? Regional population genomics of herbicide resistance in Bromus tectorum. Mol. Ecol. 34, e17791 (2025).
Wedger, M. J. et al. Recent crop-to-weed adaptive introgression has reshaped the genomic composition and geographical structure of US weedy rice (Oryza spp.). Mol. Ecol. https://doi.org/10.1111/mec.17604 (2024).
Li, L., Li, Y., Jia, Y., Caicedo, A. L. & Olsen, K. M. Signatures of adaptation in the weedy rice genome. Nat. Genet. 49, 811–814 (2017).
Li, L.-F. et al. Porous borders at the wild-crop interface promote weed adaptation in Southeast Asia. Nat. Commun. 15, 1182 (2024).
Zhu, M. et al. Landrace introgression contributed to the recent feralization of weedy rice in east China. Plant Commun. 5, 101066 (2024).
Kreiner, J. M., Tranel, P. J., Weigel, D., Stinchcombe, J. R. & Wright, S. I. The genetic architecture and population genomic signatures of glyphosate resistance in Amaranthus tuberculatus. Mol. Ecol. 30, 5373–5389 (2021).
Meimberg, H., Rice, K. J., Milan, N. F., Njoku, C. C. & McKay, J. K. Multiple origins promote the ecological amplitude of allopolyploid Aegilops (Poaceae). Am. J. Bot. 96, 1262–1273 (2009).
Kreiner, J. M. et al. Multiple modes of convergent adaptation in the spread of glyphosate-resistant Amaranthus tuberculatus. Proc. Natl. Acad. Sci. USA 116, 21076–21084 (2019).
Zhang, M. et al. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat. Protoc. 7, 467–478 (2012).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107 (2024).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res 44, e147 (2016).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Hu, J. et al. NextPolish2: a repeat-aware polishing tool for genomes assembled using HiFi long reads. Genomics Proteom. Bioinforma. 22, qzad009 (2024).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Jia, K.-H. et al. SubPhaser: a robust allopolyploid subgenome phasing method based on subgenome-specific k-mers. N. Phytol. 235, 801–809 (2022).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23, 258 (2022).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 25, 1–4 (2009).
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res 10, 516–522 (2000).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Goff, S. A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100 (2002).
Lovell, J. T. et al. The genomic landscape of molecular responses to natural drought stress in Panicum hallii. Nat. Commun. 9, 5213 (2018).
Zdobnov, E. M. & Apweiler, R. InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Zhang, G. et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554 (2012).
Woodhouse, M. R. et al. A pan-genomic approach to genome databases using maize as a model system. BMC Plant Biol. 21, 385 (2021).
Wlodzimierz, P. et al. Cycles of satellite and transposon evolution in Arabidopsis centromeres. Nature 618, 557–565 (2023).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR assembly index (LAI). Nucleic Acids Res 46, e126 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M. & Katoh, K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res 47, W5–W10 (2019).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yin, J., Zhang, C. & Mirarab, S. ASTRAL-MP: scaling ASTRAL to very large datasets using randomization and parallelization. Bioinformatics 35, 3961–3969 (2019).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2, e107 (2023).
Ye, C.-Y. et al. Genomic evidence of human selection on Vavilovian mimicry. Nat. Ecol. Evol. 3, 1474–1482 (2019).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6, 80–92 (2012).
He, X. et al. Comprehensive fundamental somatic variant calling and quality management strategies for human cancer genomes. Brief. Bioinform. 22, bbaa083 (2021).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLOS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res 52, W78–W82 (2024).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, s13742 (2015).
Kumar, S. et al. TimeTree 5: an expanded resource for species divergence times. Mol. Biol. Evol. 39, msac174 (2022).
Marchi, N., Kapopoulou, A. & Excoffier, L. Demogenomic inference from spatially and temporally heterogeneous samples. Mol. Ecol. Resour. 24, e13877 (2024).
Liu, X. & Fu, Y.-X. Stairway plot 2: demographic history inference with folded SNP frequency spectra. Genome Biol. 21, 280 (2020).
Jin, J.-J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Durand, E. Y., Patterson, N., Reich, D. & Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Martin, S. H., Davey, J. W. & Jiggins, C. D. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Mol. Biol. Evol. 32, 244–257 (2015).
Zhou, Y. et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 52, 1412–1422 (2020).
Goslee, S. C. & Urban, D. L. The ecodist Package for Dissimilarity-based Analysis of Ecological Data. J. Stat. Softw. 22, 1–19 (2007).
Gain, C. & François, O. LEA 3: factor models in population genetics and ecological genomics with R. Mol. Ecol. Resour. 21, 2738–2748 (2021).
Chi, L. et al. HapNetworkView: a tool for haplotype network exploration and visualization. BMC Genomics 26, 52 (2025).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Muzellec, B., Teleńczuk, M., Cabeli, V. & Andreux, M. PyDESeq2: a Python package for bulk RNA-seq differential expression analysis. Bioinformatics 39, btad547 (2023).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. The Innovation 2, 100141 (2021).
Turner, S. D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 3, 731 (2018).
Maclean, C. A., Chue Hong, N. P. & Prendergast, J. G. D. hapbin: an efficient program for performing haplotype-based scans for positive selection in large genomic datasets. Mol. Biol. Evol. 32, 3027–3029 (2015).
Acknowledgements
This work was supported by National Key Research and Development Program (2023YFD1400502). We would like to express our gratitude to Yu Fang, Fanjing Yang, Yutong Liu, Jiaxin Li and Tao Li for their dedicated efforts in sample collection.
Author information
Authors and Affiliations
Contributions
L.F., L.B. and M.L. conceived the study. Y.H. contributed to genomic analyses, pipeline development, and interpretation of evolutionary patterns. J.L. contributed to D. sanguinalis accessions phenotyping and herbicide resistance bioassay. S.Z., K.Y., Zhefu Li, X.G., and R.Z. collected Digitaria accessions, processed genome annotation and conducted cytological analysis on Digitaria species. S.W. and Zuren Li. performed quality control and initial data filtering. L.X. conducted tandem repeat annotation. Y.F. provided insights on phylogenetic modeling and evolutionary inference. B.K.S. and A.M. assisted in the validation of functional genetic variants. Q.Y., F.L. and L.B. supervised the experimental design and contributed to population-level analysis. L.F., L.B., M.L. and D.W. jointly supervised the study. Y.H. wrote the initial manuscript with input from all co-authors. All authors discussed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dana MacGregor and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Li, J., Li, Z. et al. Digitaria genome analyses indicate introgression may drive local adaptation and herbicide resistance. Nat Commun 17, 2669 (2026). https://doi.org/10.1038/s41467-026-69076-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69076-x
