
Abstract
Developing low-cost, high-performance, and chemically recyclable alternatives to bulk polyolefin commodities is critical. Aromatic polyesters synthesized via ring-opening copolymerization (ROCOP) of phthalic anhydride (PA) and olefin-derived epoxides are promising candidates. However, achieving high molecular weight (MW) and chemical recyclability to monomers remain key challenges, especially for epoxides with high isomerization tendency including styrene oxide, 2-vinyloxirane, and isobutylene oxide. Here, we report an organic acid-base pair-mediated strategy for the ROCOP of PA and these bulk-olefin-derived epoxides to produce high-MW (Mn = 105.1–163.3 kDa) polyesters. This strategy effectively suppressed acidic impurities-triggered, PA-mediated self-catalytic isomerization of these acid-sensitive epoxides. The produced high-MW polyesters showed superior mechanical and processing properties comparable to commodity polystyrene as well as high tensile strength (53.1–58.9 MPa) and Young’s modulus (2.16–3.02 GPa). Furthermore, these materials can be chemically recycled into PA and aldehyde monomers under mild conditions, enabling the end-of-life recycling options for ROCOP-derived polyesters.
Similar content being viewed by others
Introduction
Polyolefins have unparalleled advantages including cost-effective and abundant feedstock availability as well as excellent properties. To this end, they have emerged as the most ubiquitous class of synthetic polymers in global manufacturing1,2. However, their carbon-carbon backbones with inherent chemical stability make them resistant to degradation or chemical recycling3. An attractive strategy to address this dilemma is to develop degradable or chemically recyclable alternatives to bulk polyolefin products4.
As an excellent candidate for degradable or chemically recyclable polymer products, polyesters are a class of polymers that feature ester backbones and display lower monomer–polymer equilibria4,5. The ring-opening copolymerization (ROCOP) of epoxides with cyclic anhydrides provides a promising and versatile platform for the synthesis of polyesters6,7,8. And this synthetic route offers two key advantages: (i) most of the monomers are commercially available and low-cost chemicals already used at massive scale in the polymer industry, in which epoxides are specifically accessible via epoxidation of olefin feedstocks9,10; (ii) the broad monomer scope and mild reaction conditions enable polyesters with tunable properties and versatile functionalities. In particular, the production of high-molecular-weight (MW, Mn ≥ 100 kDa) polyesters via ROCOP of phthalic anhydride (PA) with various epoxides has garnered one of the most extensive attentions. This is not only due to the cost-effective advantage of PA (~1000 $/ton)11, but also because the rigid benzene ring backbones ensure excellent thermal and mechanical properties12,13,14.
However, till now, obtaining high-MW polymers is a major challenge for the development of PA-based polyesters, which locks the intrinsic properties of polyesters and hinders the in-depth comparative analysis with commodity polyolefins. Generally, this challenge has been recognized to be caused by the dilemma between catalyst activity and undesired chain transfer, such as transesterification. In recent years, extensive research on catalysts has enabled excellent activity and selectivity in the ROCOP of PA and some epoxides including propylene oxide (PO), cyclohexane oxide (CHO)12,13,14,15,16,17,18. Besides, continuous studies have since leveraged these catalytic systems to copolymerize PA with other similar epoxides like 1-butene oxide (BO), cyclopentene oxide (CPO), and 4-vinyl-1-cyclohexene 1,2-epoxide (VCHO)12,13,14. Recently, our group has developed a series of hydrogen-bond-functionalized imidazole catalysts for the copolymerization of various epoxides with PA, including PO, CHO, VCHO, and phenyl glycidyl ether (Fig. 1a)14. High-MW polyesters (Mn = 100.5–518.5 kDa) were obtained, which was attributed to the reduction of basicity of alkoxide anions by hydrogen-bond, thereby lowering transesterification. These high-MW polyesters exhibited high tensile strength (47.7–60.4 MPa) and Young’s modulus (~2000 MPa) similar to those of polylactic acid or polystyrene (PS). Moving forward, important advancements of PA-based polyesters will include the development of more cost-effective and abundant olefin feedstocks that would amplify the synthetic advantage of this strategy.

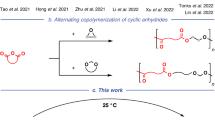
a The high-MW polyesters (Mn = 101.4–384.3 kDa for poly(CHO-alt-PA); Mn = 148.9–518.5 kDa for poly(PO-alt-PA)) have been achieved in the ROCOP of PA and CHO/PO, which was reported in representative previous work12,13,14,17. b Only low-MW polyesters (Mn < 20 kDa for poly(SO-alt-PA) and poly(IBO-alt-PA); the Mn of poly (VIO-alt-PA) is rare) can be produced in the ROCOP of PA and acid-sensitive epoxides (SO, VIO, and IBO), which was reported in previous work14,18,22,23,24,25,26,27,28,29,30,31,32,33,34. c This work developed an acid-base pair-mediated strategy for the ROCOP of PA and acid-sensitive epoxides (SO, VIO, and IBO) to produce high-MW polyesters (Mn = 105.1–163.3 kDa). These high-MW polyesters have high mechanical performance. Further, these materials can be chemically recycled into PA and corresponding anhydride monomers, catalyzed by sulfonic acids/ZnCl2.
Styrene oxide (SO), 2-vinyloxirane (VIO), and isobutylene oxide (IBO) are promising comonomers synthesized via the epoxidation of bulk olefin feedstocks (styrene: ~1100 $/ton19, butadiene: ~1400 $/ton20, and isobutylene: ~1200 $/ton21). However, despite advances in catalyst development including imidazole-based catalysts reported by our group, only low-MW (Mn < 20 kDa) polyesters have been obtained (Fig. 1b)14,18,22,23,24,25,26,27,28,29,30,31,32,33,34. These low-MW polyesters are far from sufficient for further development of practical materials. It is assumed that there might be an unaware cause contributing to the low-MW product limitation. Notably, a well-known reaction, i.e., isomerization of epoxides wherein acid catalysis triggers Meinwald rearrangement forming carbocation intermediates and aldehydes, has often been overlooked in ROCOP studies35,36,37. The aldehyde byproducts may also act as additional initiators or chain transfer agents through tautomerism, leading to low-MW polyesters. It was reported that diacids, either derived from unpurified residues or formed by the reactions of residual diols/water with cyclic anhydrides, are inevitably present in the copolymerization and may serve as the acid catalysts38. Compared to PO (methyl group) or CHO (cyclohexyl ring), SO (phenyl group), VIO (vinyl group), and IBO (gem-dimethyl group) are acid-sensitive and more prone to undergo such rearrangement and form aldehydes under acid catalysis, owing to the greater stability of their corresponding carbocations25,28,34,38. Additionally, the aldehydes may result in the ring opening of cyclic anhydrides39,40, yielding more carboxylic acids during copolymerization to further amplify the isomerization. These potential side reactions make the acid tolerance of epoxides a key limiting factor for MW. Collectively, this potential self-catalytic isomerization process could accelerate the production of aldehydes, resulting in uncontrolled polymerization and low-MW products. It was hypothesized that high-MW polyesters could be accessible by eliminating the content of diacids to concomitantly suppress epoxide isomerization in the ROCOP of PA and acid-sensitive epoxides.
Acid-base pairs, benefiting from an extraordinary tunability of acid and base components, have achieved remarkable successes in the polymerization of polar monomers8,41,42,43. Taking advantage of their modularity and synergistic mechanism, we envision that a bulky base may serve as an efficient proton trap to eliminate free acid and further suppress isomerization as well as prevent undesired nucleophilic initiation processes, whereas a weak acid may act as a mild monomer activator to minimize the tendency of isomerization.
Herein, we develop an organic acid-base pair-mediated strategy for the ROCOP of acid-sensitive epoxides and PA (Fig. 1c). This strategy effectively suppressed isomerization and achieved high MW for aromatic polyesters, including poly(SO-alt-PA), poly(VIO-alt-PA), and poly(IBO-alt-PA). The polymerization mechanism was systematically investigated through control experiments, intermediate characterization, polymer analysis, and kinetic studies. Furthermore, the structure-property relationships for these high-MW polyesters were established, revealing critical correlations between their chemical architectures and material characteristics. Additionally, a mechanism-inspired depolymerization of these polyesters was investigated using sulfonic acids or ZnCl2 as catalysts, achieving degradable, chemically recyclable, and strong thermoplastics with the recycled products PA and aldehyde monomers.
Results
Epoxide/PA ROCOP catalysis
To test our assumption, a series of sterically hindered bases with gradient basicity and mild acids were used on the ROCOP of SO and PA (see Supplementary Section “Synthesis of (thio)urea catalysts” and “NMR spectra of catalysts” pages 7–13 for synthesis and characterization of (thio)urea catalysts). The bases were evaluated with thiourea TU-I at a molar ratio of acid/base/PA/SO = 1:5:500:900 and a temperature of 80 °C (Table 1, entries 1–9). The excess base was used to fully quench acidic species in the system (see Supplementary Figs. 17–20 for detected impurities and their content). The yield of phenylacetaldehyde (Supplementary Fig. 26), an isomerization product of SO, was drastically reduced from 5.4% to 0.1%, and even became undetectable as the basicity of bases pKa(MeCN) increased from ~14.13 to 42.744,45. However, bases with excessively strong basicity (pKa(MeCN) ≥ 33.5) could induce isomerization of SO into acetophenone (Supplementary Fig. 27), and even decrease MW. Compared to the polymerization without the addition of base (Supplementary Table 1, entry 1), the resulting polyesters exhibited higher MW 8.5–26.6 kDa. Among the bases tested, phosphazene bases tBu-P1, tBu-P1’, and tBu-P2 could afford higher-MW polyesters, which might be attributed to their moderate basicity and weaker nucleophilicity. Based on the base screening results, the phosphazene base tBu-P1’ was the best choice for a faster polymerization rate and narrower MW distribution (Ð = 1.13) (Supplementary Fig. 21). Then acids were further screened with the optimal base tBu-P1’ under identical conditions (Table 1, entries 10–12), where triethylborane (TEB) yielded the highest MW of 68.5 kDa (Ð = 1.24) with the fastest polymerization rate. To our surprise, all acids produced polyesters with higher MW and narrower MW distribution than those in polymerization without acid (Supplementary Table 1, entry 2). Interestingly, even when combined with TEB, the base tBu-P1’ still produced the highest-MW polymer (Supplementary Table 1, entries 3–10, see Supplementary Section “Effect of acid-base pairs (TEB/bases) on the copolymerization of SO and PA” page 26 for discussion). These screening results demonstrated that acids and bases play a synergistic role in achieving high-MW polyesters, both of which are indispensable.
With the optimal acid-base pair in hand, the effects of the amount of tBu-P1’ and TEB, as well as the polymerization temperature, were investigated to synthesize higher-MW polyesters. It was observed that as the loading of tBu-P1’ increased from 0.6% to 1%, the MW could be raised gradually from 34.9 to 68.5 kDa, with a rapid narrowing of the MW distribution from 1.78 to 1.24 (Table 1, entry 12, and Supplementary Table 1, entries 11 and 12). These results showed that an excess of tBu-P1’ was required to fully suppress isomerization and achieve high MW. Interestingly, even when the loading of tBu-P1’ was further increased to 1.4%, there was only a slight decline in the MW (Supplementary Table 1, entry 13), indicating enhanced operability of the polymerization process. Remarkably, a polyester with a high MW up to 163.3 kDa (Ð = 1.27) was obtained by lowering the polymerization temperature to 60 °C and reducing the TEB loading to 0.1% (Table 1, entries 13 and 14), representing a rare example of a MW of poly(SO-alt-PA) exceeding 100 kDa (Fig. 2). Compared with previous work for this copolymerization (Fig. 2)14,18,22,23,24,25,26,27,28,29,30,31,32,33, the TEB/tBu-P1’ catalyst in this work exhibited superior MW control, achieving a Mn 8.5 times greater than the Cr(III)/PPNCl catalyst. Moreover, the TU-I/tBu-P1’ catalyst produced polyester with a Mn threefold higher than Cr(III)/PPNCl catalyst (Supplementary Table 1, entry 18).

Based on the success of the ROCOP of SO and PA, this polymerization strategy was further extended to achieve high MW for other acid-sensitive epoxides (VIO and IBO). Under optimized conditions (TEB/tBu-P1’/PA/VIO = 1.5:3.5:1000:1400, 45 °C, 65 h), a poly(VIO-alt-PA) polymer with a MW up to 105.1 kDa (Ð = 1.50) was afforded (Table 1, entry 15; see Supplementary Table 2 for optimization details). No (E)-but-2-enal, an isomerization product of VIO, was detected in the polymerization (Supplementary Fig. 28). It presented a rare example where the MW of poly(VIO-alt-PA) was higher than 50 kDa. However, the TU-I/tBu-P2 catalyst showed superior catalytic performance for the copolymerization of IBO and PA, producing a poly(IBO-alt-PA) with a high MW of 125.2 kDa and a Ð of 1.42 (Table 1, entry 16; see Supplementary Table 3 for optimization details)34. The isomerization of IBO yielding isobutyraldehyde (Supplementary Fig. 29) was also effectively suppressed by this catalyst. This distinct catalytic behavior was likely due to the inability of IBO containing a gem-dimethyl group to isomerize to ketone, thereby enabling the use of the stronger base tBu-P2. Additionally, the more sterically hindered tertiary alkoxide anions may bind more tightly to thiourea TU-I than to TEB with a bulkier triethyl-substituted structure, thus reducing side reactions. These compelling results conclusively established the robust efficacy of the proposed polymerization strategy for producing high-MW polyesters from acid-sensitive epoxides and PA (see Supplementary Fig. 24 for SEC traces of these high-MW polyesters). Some bimodal SEC traces may be due to the coexistence of monofunctional and difunctional initiators in the polymerization system (Supplementary Fig. 25)13,15,46, where the generated phenylacetaldehyde and residual diacids/diols/water act as the monofunctional and difunctional initiators (Supplementary Figs. 38–42), respectively.
Mechanism study
To demonstrate the acidic impurities-triggered, PA-mediated self-catalytic isomerization mechanism of epoxides, control experiments were carried out on the ROCOP of SO and PA. It was observed that SO remained stable under 80 °C for 24 h alone, but underwent isomerization after the addition of PA, forming 4.7% phenylacetaldehyde and low-MW (Mn = 3.5 kDa) copolymer (Table 2, entries 1 and 2). Notably, the phenylacetaldehyde yield increased significantly to 7.1% after the addition of 0.5 mol% phthalic acid (relative to SO), accompanied by PA conversion increasing from 54% to 88% (Table 2, entry 3). However, only 0.2% of phenylacetaldehyde was formed after the addition of phthalic acid alone (Table 2, entry 4), likely due to phthalic acid annihilation via the nucleophilic addition with SO. Similar phenomena could also be observed when monomethyl phthalate was added (Table 2, entry 5). These results indicated that phthalic acid residues or monophthalate could induce SO isomerization to phenylacetaldehyde, with the process being reactivated by PA. It was found that generated phenylacetaldehyde could lead to the ring opening of PA via aldol condensation-derived water, forming phthalic acid (Supplementary Figs. 30–32). This reaction also enabled alternating copolymerization between phenylacetaldehyde and PA, yielding oligomers end-capped with carboxylic acid groups (Supplementary Fig. 33). Moreover, the hydrolysis experiment of the oligomer obtained (Table 2, entry 3) revealed that the produced phenylacetaldehyde was able to initiate the ring opening of SO, then generating carboxylic acid by reaction with PA (Supplementary Figs. 34–36). These findings directly confirmed the self-catalytic nature of SO isomerization mediated by PA. Consequently, these carboxylic acids and phenylacetaldehyde spontaneously initiated the ROCOP of SO and PA to give low-MW polymers even without initiators or catalysts.
Therefore, a substantial excess of the base tBu-P1’ (tBu-P1’: acidic species (phthalic acid + H₂O) ≈ 13:1 molar ratio) was necessary to effectively inhibit the isomerization side reaction, reducing the yield of phenylacetaldehyde to 0.3% and achieving higher MW (Mn = 13.0 kDa) (Table 2, entry 6). These effects could be attributed to the base tBu-P1’ quenching phthalic acid by an acid-base neutralization reaction (Fig. 3a), as supported by control experiments (Table 2, entries 4, 7, and 8). Moreover, the base tBu-P1’ could also reduce the Lewis acidity of TEB, thus decreasing the isomerization of SO (Table 2, entries 9 and 10).

a 1H NMR spectra (500 MHz, CDCl3) of phthalic acid/tBu-P1’ (2:1 molar ratio) and tBu-P1’ (phthalic acid is insoluble in CDCl3). b Polymerization kinetics of PA and SO catalyzed by TEB/tBu-P1’ (TEB/tBu-P1’/PA/SO = 1:5:500:1800, 80 °C, [PA]0 = 1.85 M, [SO]0 = 6.48 M). kp, rate constants of PA. kp’, rate constants of SO. c The graph shows the content of hydrolysis products of poly((S)-SO-alt-PA) ((S)-1-phenylethane-1,2-diol:(R)−1-phenylethane-1,2-diol = 72:28) determined by high performance liquid chromatography analysis with a chiral column. d The plausible isomerization mechanism of SO to phenylacetaldehyde mediated by PA. e The plausible chain propagation process for the ROCOP of SO and PA catalyzed by TEB/tBu-P1’.
TEB, except for its well-studied roles in activating monomers and stabilizing alkoxide anions during copolymerization16,47,48,49, also played an important role in suppressing the formation of phenylacetaldehyde (Table 2, entry 11). This phenomenon may result from the quenching of phthalic acid by TEB with three electronegative ethyl groups (Supplementary Fig. 37)50,51.
To confirm the chain initiation process, a poly(SO-alt-PA) oligomer was synthesized under these conditions (TEB:tBu-P1’:PA:SO:toluene = 1:5:500:900:1800, 80 °C, 11.5 h). The matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was used for the analysis of the end groups. The MALDI-TOF MS spectrum of this oligomer showed that multiple distributions of polymer chains, initiated by residual diacids/diols/water and generated phenylacetaldehyde (see Supplementary Fig. 39 for possible chain ends). No peaks of polymer chains initiated by the bulky-base tBu-P1’ with weak nucleophilicity were observed (Supplementary Figs. 38–42).
To study the chain propagation process, a polymerization kinetic experiment was conducted (Supplementary Section “Kinetics procedure” page 45). The reaction kinetics revealed a zero-order dependence on PA concentration and first-order dependence on SO concentration, with calculated rate constants of 0.329 mol L–1 h–1 (PA) and 0.063 h–1 (SO) (Fig. 3b). The kinetic results indicated that the rate-determining step may involve the nucleophilic attack of carboxylate anions on TEB-activated SO. Furthermore, the regioselectivity of this process was confirmed by the predominant attack on the methylene carbon (CH2), yielding 72% of the configuration-preserved hydrolysis product (S)−1-phenylethane-1,2-diol, compared to 28% configuration-inverted (R)−1-phenylethane-1,2-diol generated from the methine carbon (CH) (Fig. 3c; see Supplementary Section “Evaluation of the regiochemistry of the copolymerization of SO/PA using (S)-SO” page 46 for more details).
Based on the above mechanistic experiments, the isomerization mechanism of SO mediated by PA was proposed (Fig. 3d). The process has two pivotal steps, involving the generation of phenylacetaldehyde and carboxylic acids. SO underwent a carbocation intermediate and subsequent Meinwald rearrangement to yield phenylacetaldehyde, catalyzed by carboxylic acids35,36. This process was first triggered by the phthalic acid derived from both residues and the reactions of residual diols/water with PA. The production of carboxylic acids occurred through multiple routes: (i) nucleophilic addition of PA with the resulting phenylacetaldehyde, (ii) hydrolysis of PA with water sourced from phenylacetaldehyde aldol condensation, (iii) alcoholysis of PA driven by nucleophilic addition of SO with phenylacetaldehyde/carboxylic acids. Critically, phenylacetaldehyde-mediated carboxylic acids further catalyze SO isomerization, thereby accelerating phenylacetaldehyde generation. These processes established an acidic impurities-triggered, PA-mediated self-catalytic isomerization cycle for SO.
Combined with reported literature and our experimental results, the polymerization mechanism was proposed16,52,53. The initiation process was triggered by the residual diacids/diols/water that were deprotonated by the base tBu-P1’. Moreover, the formed phenylacetaldehyde could also initiate the ring opening of SO and PA during polymerization (Fig. 3d and Supplementary Fig. 39). This process could be effectively inhibited by both the acid-base neutralization reaction of tBu-P1’ and the protonolysis reaction of TEB, contributing significantly to the production of high-MW polymers. However, excessive basicity may induce the isomerization of SO to acetophenone (Table 2, entries 12 and 13, see Supplementary Section “Study on the strong base-catalyzed isomerization mechanism from SO to acetophenone” pages 47 and 48 for plausible isomerization mechanism), thereby potentially hindering MW. During the propagation process, as shown in Fig. 3e, the carboxylate anions preferentially attacked the methylene carbon (CH2) in TEB-activated SO due to the less steric hindrance, which became the rate-determining step. The alkoxide anions were bound to TEB, leading to enhanced stability and improved MW.
Polyester’s structure-property relationships
With these high-MW polyesters in hand, their intrinsic properties and in-depth investigations of their structure-property relationships were accessible. To better understand this relationship of poly(IBO-alt-PA) with gem-dimethyl groups, the poly(PO-alt-PA) (Mn = 94.0, Ð = 1.44) with methyl groups was prepared according to the ref. 14.
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were used to evaluate the thermal performance of these high-MW polyester samples (Supplementary Table 4), including P-SO-PA−132, P-VIO-PA-105, P-IBO-PA-125, and the sample Pc-IBO-PA-125 after solution crystallization (polyester sample names: P-epoxide-anhydride-Mn, SEC, “c” means that the polymer sample is crystalline). The DSC curves revealed that P-SO-PA-132, P-VIO-PA-105, and P-IBO-PA-125 were amorphous plastics, exhibiting glass transition temperatures (Tg) ranging from 53 to 94 °C (Fig. 4a). For Pc-IBO-PA-125, a melting peak with a melting point of 125 °C was observed only in the first heating run, indicating its semicrystalline nature and slow crystallization kinetics. This result was corroborated by wide-angle X-ray diffraction with a crystallinity degree (Xc) of 42% (Supplementary Fig. 51). Compared to P-VIO-PA-105 with vinyl side groups (Tg = 53 °C), the P-SO-PA-132 featuring bulky phenyl rings had a higher Tg value of 94 °C. TGA analysis showed that all the polyesters show thermal decomposition temperatures exceeding 250 °C, with P-SO-PA-132 displaying the highest thermal decomposition temperature at 5% weight loss (Td(5%) = 302 °C) (Supplementary Fig. 52). In contrast, the P-IBO-PA-125 showed the lowest Td(5%) with a value significantly lower than that of P-PO-PA-94, which may be attributed to the enhanced β-H thermal elimination of polyesters by an additional methyl group (Supplementary Figs. 53–55)54,55.

a Second heating run DSC curves of high-MW polymers at the rate of 10 °C min−1, first heating run DSC curve of the polyester sample Pc-IBO-PA−125 at the rate of 20 °C min−1. b Rheological characterization of high-MW polyesters. c Representative stress−strain curves of high-MW polymers at the rate of 5 mm min–1. d Ultimate tensile strength and Young’s modulus of high-MW polymers. e A radar chart summarizes the ultimate tensile strength, water contact angle, Young’s modulus, Tg, Td(5%), and viscous flow temperature (Tf) of the polyester samples (P-SO-PA−132, P-VIO-PA−105, and P-IBO-PA−125) and the selected commodity PS resin (PS−110). Error bars represent standard deviation, n = 3–6 independent replicates. f Comparison of the ultimate tensile strength and Tg value of the polyester sample P-SO-PA−132 with reported ROCOP-derived polyesters and commodity polyester materials (see Supplementary Table 6 for more details).
The rheological properties of polymers play a very important role in their processing behavior and end-use performance. To elucidate this relationship, the polyesters’ molecular dynamics and extent of entanglement were further investigated by oscillatory shear rheology. The variable frequency rheology indicated that all the polyesters showed the typical thermoplastic behavior (Fig. 4b), confirming their excellent melt-processability. Notably, these polymers displayed pseudoplastic (shear-thinning) behavior within the tested frequency range, as evidenced by the monotonic decrease in complex viscosity with increasing shear frequency (Supplementary Fig. 60). This behavior suggested that processing performance could be improved through elevated shear rates. These polyesters showed significant distinction in their molecular dynamics (Supplementary Table 5), which could be characterized through two key relaxation time parameters: entanglement relaxation time (τₑ) and terminal relaxation time (τt), related to the segmental motion and polymer chains’ motion, respectively. The P-SO-PA-132 with phenyl ring side groups exhibited the slowest molecular dynamics with a τₑ of 26.7 ± 3.5 s and a τt of 13.0 ± 3.7 h, indicating potential superior anti-stress relaxation ability in use. In contrast, the P-VIO-PA-105 featuring enhanced segmental motion by vinyl side groups showed significantly shorter relaxation time (τₑ = 0.56 ± 0.10 ms, τt = 0.55 ± 0.21 s) than P-IBO-PA-125 with gem-dimethyl groups (τₑ = ~10.26 ms, τt = 35.68 ± 4.73 s), which may demonstrate more efficient internal stress relaxation during processing. P-IBO-PA-125 with gem-dimethyl groups showed slightly longer relaxation time compared to P-PO-PA-94 (Supplementary Table 4, entry 4). These results from molecular dynamics generally showed the opposite trend of decreasing Tg values as the polyester backbones became more flexible. The extent of entanglement was evaluated by entanglement molecular weight (Me) calculated using the tube model equation (Supplementary Section “Measurements” page 6). The P-SO-PA-132 with the slowest molecular dynamics showed the highest Me of 19.4 ± 2.9 kDa. Similarly, the Me (11.6 ± 1.7 kDa) of P-IBO-PA-125 was slightly higher than that of P-PO-PA-94 (8.8 ± 1.3). However, P-VIO-PA-105 displayed a Me of 16.3 ± 2.4 kDa, higher than that of P-IBO-PA-125. These results indicated Me values mainly related to polymer molecular dynamics, where slower dynamics lead to higher Me13,56, except for P-VIO-PA-105.
To study the mechanical performance, dumbbell-shaped specimens were fabricated from hot pressing and underwent tensile testing (Supplementary Section “Measurements” page 5). No obvious changes were observed in P-VIO-PA-105 before and after processing, as confirmed by 1H NMR spectra (Supplementary Fig. 49) and SEC traces (Supplementary Fig. 50). The tensile testing revealed that all the polyester samples are strong and rigid thermoplastics with a high ultimate tensile strength (51.9–58.9 MPa) and Young’s modulus (2.16–3.02 GPa) (Fig. 4d, Supplementary Table 4). Among these, both P-VIO-PA-105 with vinyl groups and P-IBO-PA-125 with gem-dimethyl groups showed comparable ultimate tensile strength ~53 MPa, surpassing that of P-PO-PA-94 (methyl groups). These results may be attributed to enhanced intermolecular interaction resulting from vinyl groups and gem-dimethyl groups. Notably, P-SO-PA-132 featuring bulky phenyl ring groups displayed the strongest ultimate tensile strength 58.9 ± 0.7 MPa, ~1.2-fold greater than that of P-PO-PA-94 (methyl groups). A yield point was observed in P-VIO-PA-105 and P-IBO-PA-125 with an elongation at break of ~3.5%, indicating improved ductility. Additionally, the crystalline Pc-IBO-PA-125 showed the highest Young’s modulus reaching 3.02 ± 0.26 GPa.
The wettability of polyester materials was characterized through water contact angle measurements. The results showed that all the polymers were hydrophilic with water contact angles varying from 74 ± 1° to 80 ± 1° (Supplementary Fig. 65). Intriguingly, the crystalline Pc-IBO-PA-125 exhibited an increased water contact angle from 74 ± 1° to 86 ± 2°, revealing that crystallization may promote the surface exposure of hydrophobic polymer groups. The MW dependence of polymer performance was also studied by poly(SO-alt-PA). Surprisingly, both the thermal and mechanical performance of this polyester exhibited significant enhancement as the MW increased from 63.8 to 132.6 kDa (Fig. 4a, c, and d). Especially, the ultimate tensile strength of P-SO-PA-132 showed a ~150% enhancement. These performance improvements were due to the intricate polymer chain entanglements induced by the elevated MW14,57.
To assess the potential of these polyester materials as alternatives to conventional polyolefins, commodity PS resin (Mn = 110.3 kDa, Ð = 2.08) was purchased, with its corresponding properties characterized under the same testing protocols. As shown in Fig. 4e, the three polyester materials exhibited superior mechanical and processing performance as well as enhanced hydrophilicity compared to PS. Their tensile behavior indicated these materials could serve as suitable substitutes for conventional PS in terms of their tensile properties. Notably, the poly(SO-alt-PA) copolymer not only exhibited excellent mechanical and processing performance, but its thermal performance and hydrophilicity were also comparable to PS, making it an ideal alternative for PS.
Chemical recycling of polyesters
Addressing synthetic challenges for high-MW polyesters should be pursued in parallel with evaluating their chemical recycling—the achievement of this dual objective represents a pivotal step toward advancing the future circular plastics economy58,59. Nevertheless, direct methods for chemical recycling of ROCOP-derived polyesters back to their monomers or other reactive monomers remain scarce, particularly for aromatic polyesters, due to their depolymerization thermodynamic barriers60. Inspired by the isomerization mechanism whereby these epoxides may form carbocation intermediates under acid catalysis, it was hypothesized that these polyesters may exhibit similar reactivity. Under acid catalysis, the phthalate anions would leave, generating carbocation intermediates, thereby facilitating the depolymerization of polyesters into monomers.
The poly(IBO-alt-PA) was used as a model to investigate chemical recycling. Excitingly, this polymer underwent efficient depolymerization with >99% conversion using a typical p-methylbenzenesulfonic acid catalyst (5 wt%) at 150 °C for 4 h (Fig. 5a, and Supplementary Table 7, entry 1). The depolymerization products were identified as PA (94% isolated yield) and isobutyraldehyde (60% isolated yield) by 1H NMR (Supplementary Figs. 67 and 68) and chromatography-mass spectrometry (Supplementary Fig. 69). When ZnCl2 was employed as a Lewis acid catalyst, excellent isolated yields of PA (91%, Fig. 5c) and isobutyraldehyde (86%, Fig. 5d) were achieved (Supplementary Table 7, entry 2). Similarly, the poly(SO-alt-PA) was also successfully depolymerized into PA with high conversion (>99%) and isolated yield (92–94%) using this method (see Supplementary Table 7 for more details). The low yield of phenylacetaldehyde was due to its extreme thermal instability, leading to trimer formation (Supplementary Fig. 72) or polymerization (Supplementary Figs. 74 and 75)61. The depolymerization of poly(VIO-alt-PA) with vinyl groups could also give a good yield (84%) of PA (see Supplementary Table 7 for more details). The low yield of (E)-but-2-enal was due to this polymer was prone to cross-linking under the depolymerization conditions. It should be pointed out that the aldehyde products can not only serve as polymerization monomers40,61, but also undergo copolymerization with anhydrides (Supplementary Fig. 33)39,62. This depolymerization method represents a rare example where ROCOP-derived polyesters with depolymerization thermodynamic barriers could be directly converted into their cyclic anhydrides and other reactive monomers by chemical recycling under mild conditions (120–170 °C)60.

a Photo of the reaction setup. b Physical state of the crude product of PA with 94% isolated yield. c Physical state of the PA after recrystallization with 91% isolated yield. d Physical state of the recovered isobutyraldehyde with 86% isolated yield. e The residual solid after depolymerization with >99% conversion.
Discussion
In summary, we have developed an organic acid-base pair-mediated strategy in the synthesis of high-MW polyesters from the ROCOP of acid-sensitive epoxides and PA. This strategy produced polyesters with high MW (Mn = 105.1–163.3 kDa), including poly(SO-alt-PA), poly(VIO-alt-PA), and poly(IBO-alt-PA) by addressing the longstanding acidic impurities-triggered, PA-mediated self-catalytic epoxides isomerization barrier. The acid-base pair catalysts could effectively catalyze the ROCOP while quenching the acidic species during the polymerization, thereby suppressing epoxide isomerization and achieving high MW. These high-MW polyesters exhibit excellent mechanical performance with high tensile strength (53.1–58.9 MPa) and Young’s modulus (2.16–3.02 GPa), due to polar polymer backbones and low Me (11.6–19.4 kDa). These polyesters also show hydrophilicity, tunable Tg (53–94 °C), and low processing viscosity. Moreover, these polyesters can be efficiently chemically recycled into PA and aldehyde monomers under mild acid-catalyzed conditions.
Methods
General copolymerization procedure
Polymerizations were performed under an argon atmosphere. To a dried Schlenk tube equipped with a magnetic stirrer were added PA (1.48 g, 10 mmol), tBu-P1’ (31.2 mg, 0.1 mmol), TEB (1 M in THF, 20 μL, 0.02 mmol), SO (2.16 g, 18 mmol), and then heated to 80 °C for 5.5 h. The reaction mixture was quenched by cooling to room temperature and diluted with 0.5 mL DCM. A sample (>0.1 mL) was taken from the solution for the analysis of the conversion of PA, and the yields of isomerization products by 1H NMR (500 MHz, CDCl3). And the resulting polymer was precipitated from ethanol only once and dried in vacuo at 50 °C for 24 h.
General depolymerization procedure
Representative procedure for the reactive distillation depolymerization of poly(IBO-alt-PA). Under argon atmosphere, to a 50 mL round bottom flask containing a stir bar was added poly(IBO-alt-PA) (1.00 g), followed by acid catalyst (50.0 mg). The flask was fitted short-path distillation head, and a tared 50 mL round bottom flask was used to collect the distillation products. The apparatus was maintained at ambient pressure and heated to 150 °C. The receiving flask was placed in an ice-water bath to minimize isobutyraldehyde evaporation and prevent polymerization during the extended reaction times. The reaction apparatus was brought to room temperature, the sublimated PA adhering to the flask wall was scraped off, and was recrystallized with ethyl acetate. The received products were weighed to calculate isolated yields. The residual solid was dissolved in DCM, followed by the addition of 1,3,5-trimethylbenzene as an internal standard to quantify polyester conversion.
Data availability
The polymerization kinetics data, high performance liquid chromatography data, nuclear magnetic resonance data, and properties test data that support the findings of this study are available in Figshare under accession code of https://doi.org/10.6084/m9.figshare.30740780. The remaining data necessary to support the conclusions of this paper are provided in the main text and the Supplementary Information. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Hustad, P. D. Frontiers in olefin polymerization: reinventing the world’s most common synthetic polymers. Science 325, 704–707 (2009).
Stürzel, M., Mihan, S. & Mülhaupt, R. From multisite polymerization catalysis to sustainable materials and all-polyolefin composites. Chem. Rev. 116, 1398–1433 (2016).
Tardy, A., Nicolas, J., Gigmes, D., Lefay, C. & Guillaneuf, Y. Radical ring-opening polymerization: Scope, limitations, and application to (bio)degradable materials. Chem. Rev. 117, 1319–1406 (2017).
Coates, G. W. & Getzler, Y. D. Y. L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 5, 501–516 (2020).
Shi, C., Quinn, E. C., Diment, W. T. & Chen, E. Y. Recyclable and (bio)degradable polyesters in a circular plastics economy. Chem. Rev. 124, 4393–4478 (2024).
Longo, J. M., Sanford, M. J. & Coates, G. W. Ring-opening copolymerization of epoxides and cyclic anhydrides with discrete metal complexes: structure-property relationships. Chem. Rev. 116, 15167–15197 (2016).
Plajer, A. J. & Williams, C. K. Heterocycle/heteroallene ring-opening copolymerization: Selective catalysis delivering alternating copolymers. Angew. Chem. Int. Ed. 61, e202104495 (2022).
Ryzhakov, D. et al. Organo-catalyzed/initiated ring opening co-polymerization of cyclic anhydrides and epoxides: an emerging story. Polym. Chem. 12, 2932–2946 (2021).
Kamata, K. et al. Efficient epoxidation of olefins with ≥99% selectivity and use of hydrogen peroxide. Science 300, 964–966 (2003).
Jalil, A. et al. Nickel promotes selective ethylene epoxidation on silver. Science 387, 869–873 (2025).
Phthalic Anhydride Price Index, can be Found Under https://m.Chemicalbook.Com/priceindex_cb7414905.Htm (2025) (accessed 4 June 2025).
Yang, L. et al. Highly active aggregate catalysts for the synthesis of high-molecular-weight polyesters via copolymerization of epoxides and cyclic anhydrides. Polym. Chem. 15, 2482–2491 (2024).
Jannsen, N., Poon, K. C., Craze, A., Gao, C. & Williams, C. K. Controlled catalysis delivering high molecular weight polyesters as recyclable alternatives to polystyrenes. Angew. Chem. Int. Ed. 64, e202505070 (2025).
Xie, Z. et al. Record-high-molecular-weight polyesters from ring-opening copolymerization of epoxides and cyclic anhydrides catalyzed by hydrogen-bond-functionalized imidazoles. J. Am. Chem. Soc. 147, 12115–12126 (2025).
Xie, R. et al. Record productivity and unprecedented molecular weight for ring-opening copolymerization of epoxides and cyclic anhydrides enabled by organoboron catalysts. Angew. Chem. Int. Ed. 60, 19253–19261 (2021).
Zhang, J., Wang, L., Liu, S. & Li, Z. Phosphazene/Lewis acids as highly efficient cooperative catalyst for synthesis of high-molecular-weight polyesters by ring-opening alternating copolymerization of epoxide and anhydride. J. Polym. Sci. 58, 803–810 (2020).
Li, B., Wang, K., Li, Z. & Li, Z. Controlled ring-opening copolymerization of epoxides and cyclic anhydrides through bifunctional thiourea-quaternary ammonium salt catalysts. Polym. Chem. 15, 3893–3900 (2024).
Chen, C.-M. et al. Alkali metal carboxylates: Simple and versatile initiators for ring-opening alternating copolymerization of cyclic anhydrides/epoxides. Macromolecules 54, 713–724 (2021).
Styrene Price Index, can be Found Under https://www.Chemicalbook.Com/priceindex_cb3415111.Htm (2025) (accessed 4 June 2025).
Butadiene Price Index, can be Found Under https://www.Chemicalbook.Com/priceindex_cb2733269.Htm (2025) (accessed 4 June 2025).
Isobutylene Price Index, can be Found Under https://www.Chemicalbook.Com/priceindex_cb4763080.Htm (2025) (accessed 4 June 2025).
Ji, H.-Y., Chen, X.-L., Wang, B., Pan, L. & Li, Y.-S. Metal-free, regioselective and stereoregular alternating copolymerization of monosubstituted epoxides and tricyclic anhydrides. Green Chem. 20, 3963–3973 (2018).
Ji, H.-Y., Wang, B., Pan, L. & Li, Y.-S. Lewis pairs for ring-opening alternating copolymerization of cyclic anhydrides and epoxides. Green Chem. 20, 641–648 (2018).
Li, H., Luo, H., Zhao, J. & Zhang, G. Well-defined and structurally diverse aromatic alternating polyesters synthesized by simple phosphazene catalysis. Macromolecules 51, 2247–2257 (2018).
Hosseini Nejad, E., Paoniasari, A., Koning, C. E. & Duchateau, R. Semi-aromatic polyesters by alternating ring-opening copolymerisation of styrene oxide and anhydrides. Polym. Chem. 3, 1308–1313 (2012).
Darensbourg, D. J., Poland, R. R. & Escobedo, C. Kinetic studies of the alternating copolymerization of cyclic acid anhydrides and epoxides, and the terpolymerization of cyclic acid anhydrides, epoxides, and CO2 catalyzed by (salen)CrIIICl. Macromolecules 45, 2242–2248 (2012).
Liu, Y., Guo, J.-Z., Lu, H.-W., Wang, H.-B. & Lu, X.-B. Making various degradable polymers from epoxides using a versatile dinuclear chromium catalyst. Macromolecules 51, 771–778 (2018).
Abbina, S., Chidara, V. K. & Du, G. Ring-opening copolymerization of styrene oxide and cyclic anhydrides by using highly effective zinc amido–oxazolinate catalysts. ChemCatChem 9, 1343–1348 (2017).
Bukowski, W., Bukowska, A., Sobota, A., Pytel, M. & Bester, K. Copolymerization of phthalic anhydride with epoxides catalyzed by amine-bis(phenolate) chromium(III) complexes. Polymers 13, 1785–1802 (2021).
Diment, W. T. et al. Catalytic synergy using Al(III) and group 1 metals to accelerate epoxide and anhydride ring-opening copolymerizations. ACS Catal. 11, 12532–12542 (2021).
Li, J. et al. Enantioselective resolution copolymerization of racemic epoxides and anhydrides: efficient approach for stereoregular polyesters and chiral epoxides. J. Am. Chem. Soc. 141, 8937–8942 (2019).
Tsai, C.-Y., Huang, M.-C., Lin, M.-L., Su, Y.-C. & Lin, C.-C. Well-defined and highly effective nickel catalysts coordinated on tridentate sno schiff-base derivatives for alternating copolymerization of epoxides and anhydrides. Inorg. Chem. 61, 19870–19881 (2022).
Li, Y. et al. Synthesis of alternating polyesters using a dual catalytic system of alkali metal alkoxides and crown ether. Eur. Polym. J. 220, 113497 (2024).
Hu, L. et al. Alternating copolymerization of isobutylene oxide and cyclic anhydrides: A new route to semicrystalline polyesters. Macromolecules 54, 6182–6190 (2021).
Khanppnavar, B. et al. Structural basis of the Meinwald rearrangement catalysed by styrene oxide isomerase. Nat. Chem. 16, 1496–1504 (2024).
Pocker, Y., Ronald, B. P. & Anderson, K. W. A mechanistic characterization of the spontaneous ring opening process of epoxides in aqueous solution: Kinetic and product studies. J. Am. Chem. Soc. 110, 6492–6497 (1988).
Xu, J. et al. Asymmetric catalytic vinylogous addition reactions initiated by Meinwald rearrangement of vinyl epoxides. Angew. Chem. Int. Ed. 60, 14521–14527 (2021).
Harrold, N. D., Li, Y. & Chisholm, M. H. Studies of ring-opening reactions of styrene oxide by chromium tetraphenylporphyrin initiators. Mechanistic and stereochemical considerations. Macromolecules 46, 692–698 (2013).
Zhang, X., Guo, W., Zhang, C. & Zhang, X. A recyclable polyester library from reversible alternating copolymerization of aldehyde and cyclic anhydride. Nat. Commun. 14, 5423–5430 (2023).
Martin, B. Y., Schutz, L. & Claverie, J. P. Mechanistic insights on the anionic polymerization of aliphatic aldehydes. Macromolecules 54, 9165–9173 (2021).
McGraw, M. L. & Chen, E. Y. X. Lewis pair polymerization: perspective on a ten-year journey. Macromolecules 53, 6102–6122 (2020).
Hong, M., Chen, J. & Chen, E. Y. Polymerization of polar monomers mediated by main-group Lewis acid-base pairs. Chem. Rev. 118, 10551–10616 (2018).
Xu, J. et al. Ionic h-bonding organocatalysts for the ring-opening polymerization of cyclic esters and cyclic carbonates. Prog. Polym. Sci. 125, 101484–101508 (2022).
Kaljurand, I. et al. Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pka units: Unification of different basicity scales. J. Org. Chem. 70, 1019–1028 (2005).
Baumgartner, L. M., Dennis, J. M., White, N. A., Buchwald, S. L. & Jensen, K. F. Use of a droplet platform to optimize Pd-catalyzed C–N coupling reactions promoted by organic bases. Org. Process Res. Dev. 23, 1594–1601 (2019).
Abel, B. A., Lidston, C. A. L. & Coates, G. W. Mechanism-inspired design of bifunctional catalysts for the alternating ring-opening copolymerization of epoxides and cyclic anhydrides. J. Am. Chem. Soc. 141, 12760–12769 (2019).
Zhang, Y.-Y. et al. Organoboron-mediated polymerizations. Chem. Soc. Rev. 53, 3384–3456 (2024).
Zhang, J., Wang, L., Liu, S. & Li, Z. Synthesis of diverse polycarbonates by organocatalytic copolymerization of CO2 and epoxides: from high pressure and temperature to ambient conditions. Angew. Chem. Int. Ed. 61, e202111197 (2022).
Wang, J. et al. Tug-of-war between two distinct catalytic sites enables fast and selective ring-opening copolymerizations. Angew. Chem. Int. Ed. 61, e202208525 (2022).
Brown, H. C. & Hébert, N. C. Protonolysis of triethylborane with carboxylic acids. J. Organomet. Chem. 255, 135–142 (1983).
Liang, H. & Morken, J. P. Stereospecific transformations of alkylboronic esters enabled by direct boron-to-zinc transmetalation. J. Am. Chem. Soc. 145, 9976–9981 (2023).
Zhao, M. et al. One-step synthesis of linear and hyperbranched CO2-based block copolymers via organocatalytic switchable polymerization. Macromolecules 56, 2379–2387 (2023).
Gao, T. et al. Toward fully controllable monomers sequence: binary organocatalyzed polymerization from epoxide/aziridine/cyclic anhydride monomer mixture. J. Am. Chem. Soc. 146, 25067–25077 (2024).
Pohl, H. A. The thermal degradation of polyesters. J. Am. Chem. Soc. 73, 5660–5661 (1951).
Buxbaum, L. H. The degradation of poly(ethylene terephthalate). Angew. Chem. Int. Ed. 7, 182–190 (1968).
Rosetto, G., Vidal, F., McGuire, T. M., Kerr, R. W. F. & Williams, C. K. High molar mass polycarbonates as closed-loop recyclable thermoplastics. J. Am. Chem. Soc. 146, 8381–8393 (2024).
Hester, H. G., Abel, B. A. & Coates, G. W. Ultra-high-molecular-weight poly(dioxolane): enhancing the mechanical performance of a chemically recyclable polymer. J. Am. Chem. Soc. 145, 8800–8804 (2023).
Korley, L. T. J., Epps, T. H., Helms, B. A. & Ryan, A. J. Toward polymer upcycling—adding value and tackling circularity. Science 373, 66–69 (2021).
Vidal, F. et al. Designing a circular carbon and plastics economy for a sustainable future. Nature 626, 45–57 (2024).
Smith, M. L., McGuire, T. M., Kerr, R. W. F. & Williams, C. K. From polymers to rings and back again: chemical recycling of polyesters to macrolactones. Angew. Chem. Int. Ed. 64, e202423478 (2025).
Kubisa, P., Neeld, K., Starr, J. & Vogl, O. Polymerization of higher aldehydes. Polymer 21, 1433–1447 (1980).
Zhang, X., Feng, X., Guo, W., Zhang, C. & Zhang, X. Chemically recyclable polyvinyl chloride-like plastics. Nat. Commun. 15, 8536–8545 (2024).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China, Fund for Distinguished Young Scholars (No. 52325301, received by X.P.), CAS Project for Young Scientists in Basic Research (No. YSBR-094, received by X.P.), the National Natural Science Foundation of China (No. 52203017, received by C.H.), and the International Partnership Program of Chinese Academy of Sciences (029GJHZ2023017MI, received by X.P.). We thank Yihan Zhou and Hao Wang (Changchun Institute of Applied Chemistry, Chinese Academy of Sciences) for MALDI-TOF MS analysis of polymer samples.
Author information
Authors and Affiliations
Contributions
X.P. and X.C. conceived the project. C.H., X.P., and X.C. directed the research. Z.X. designed and performed the polymerization experiments related to polymer synthesis, polymer characterizations, catalyst synthesis, and characterizations. Z.X. and Z.Y. designed and conducted the mechanism experiments. Z.X. and M.N. conducted the physical property measurements. Z.X., Y.Z., and T.Y. performed the chemical recycling experiments. Z.X., Z.Y., C.Y., and X.P. wrote the initial paper. All authors contributed to the data analysis and discussions and the revised paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mina Mazzeo, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, Z., Yang, Z., Hu, C. et al. Acid-base pair-mediated copolymerization of acid-sensitive epoxides and cyclic anhydride for synthesizing recyclable thermoplastics. Nat Commun 17, 2668 (2026). https://doi.org/10.1038/s41467-026-69201-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69201-w
