
Abstract
GLT25D1 O-galactosylates hydroxylysine residues in collagen and is essential for collagen maturation and function. Dysfunctions of GLT25D1 cause various tissue disorders. Despite its biological significance, the action mechanism of GLT25D1 remains enigmatic. Here we report the cryo-EM structures of human GLT25D1 and its ternary complex with UDP and hydroxylated acceptor substrates, revealing a bi-lobe architecture for the GLT25D1 monomer that organizes into dimeric and hexameric oligomers. The N-lobe of GLT25D1 contains a high-affinity UDP-galactose binding site, and the C-lobe is the catalytic domain of the enzyme. The structures together with biochemical analyses unravel the key recognition of the consensus “Hyl-Gly” motif from collagen acceptor substrates and associated catalytic mechanism. We further demonstrate that GLT25D1 mutations linked to cerebral small vessel disease and musculoskeletal defects adversely affect its function via distinct mechanisms. Our findings elucidate the molecular mechanism underlying collagen glycosylation and provide a molecular framework for understanding GLT25D1-related diseases.
Similar content being viewed by others


Introduction
Collagen, as the most abundant protein in all animal tissues and the primary constituent of the extracellular matrix (ECM), plays a crucial role in maintaining tissue structure, mechanical integrity, and cellular functions1,2,3. Its dysregulation is linked to numerous human diseases, including cancer, arthritis and more than 40 hereditary disorders. One defining feature of collagen is its O-linked glycosylation, a modification that critically regulates collagen properties and functions. During collagen biosynthesis, specific hydroxylysine (Hyl) residues are galactosylated by the enzyme GLT25D1 and GLT25D2 (also known as colgalt1 and colgalt2), and some of these modifications can be further glucosylated by lysyl hydroxylase 3 (LH3) (Fig. 1A)4,5,6,7,8. The resulting Gal(β1-O) monosaccharide and less abundant Glc(α1-2)Gal(β1-O) disaccharide are essential for proper collagen maturation and ECM-cell interactions9,10. The significance of collagen glycosylation is further underscored by its ubiquitous presence in all animal collagens and its high conservation across species, from sponges to humans11.

A Schematic representation of the collagen modification pathway. Hydroxylysine residues formed by collagen lysyl hydroxylases (LH/PLODs) at specific sites of nascent procollagen are galactosylated by GLT25D1/2. Some of these modifications undergo further glucosylation by lysyl hydroxylases. B Size-exclusion chromatography analysis of purified human GLT25D1 reveals two distinct non-aggregate elution peaks (10.3 ml and 11.8 ml, marked by dashed lines), indicative of multiple oligomeric states of GLT25D1. Inset: SDS–PAGE analysis of the eluted fractions. C In vitro galactosyltransferase activity of GLT25D1 with various acceptor substrates: gelatin, a peptide containing hydroxylysine Hyl (sequence: SGA-Hyl-GEKGS), a peptide containing lysine K instead of hydroxylysine (SGA-K-GEKGS) and an uncoupled control lacking any acceptor substrate. Activity was assessed by monitoring the production of UDP. Data are represented as mean ± s.d. for three independent replicates, evaluated using an unpaired, two-tailed Student’s t test at a 95% confidence interval (CI), and differences are depicted as ***p < 0.001; ****p < 0.0001. The exact p-values are as follows. For the peptide with hydroxylysine substrate: UDP-Gal vs UDP-Glc (p = 0.0002). For the gelatin substrate: UDP-Gal vs UDP-Glc (p < 0.0001). Mass spectrometric analysis of reaction products generated by GLT25D1 in the presence of a peptide acceptor substrate (SGA-Hyl-GEKGS; Hyl, hydroxylysine) and different donor substrates: D UDP-galactose, E UDP-glucose, F a mixture of UDP-galactose and UDP-glucose. Top row: spectra of no-enzyme controls, showing peaks (z = 3, m/z = 279.4; z = 2, m/z = 418.7; marked with blue asterisks *) corresponding to the unmodified peptide (MW of 835 Da). Bottom row: spectra of GLT25D1 reactions, showing additional peaks (z = 3, m/z = 333.4; z = 2, m/z = 499.7; marked with pink asterisks *) only in (D) and (F) that correspond to the galactosylated peptide (MW of 997 Da). Source data are provided as a Source Data file.
GLT25D1 catalyzes the initial step of collagen glycosylation and is broadly expressed across human tissues, while its homologous isoform, GLT25D2 (55% sequence identity), is restricted only to a few cell types4. GLT25D1 is indispensable for embryonic development and cell growth, typically in a collagen-dependent manner. Its inactivation in C. elegans causes growth defects and morphologic abnormalities12,13; simultaneous inactivation of both GLT25D1 and GLT25D2 impairs osteosarcoma cell viability, accompanied by intracellular accumulation of type I collagen14,15; GLT25D1 disruption in mice and zebrafish causes embryonic lethality, defects in vascular network formation or disorganized muscle fibers16,17. Beyond collagen, several additional substrates have recently been identified, including hepassocin and hepatocyte-derived fibrinogen-related protein 118, adiponectin19,20 and mannose-binding lectin21. GLT25D1 deficiencies have been linked to a spectrum of human diseases, including cerebral small vessel diseases (SVDs), vascular cognitive impairment (VCI), and connective tissue disorder17,22,23,24, highlighting its physiological importance.
GLT25D1, localized in the endoplasmic reticulum (ER), is responsible for transferring galactose from UDP-galactose to hydroxylysine (Hyl) at specific Yaa-positions within the characteristic Gly-Xaa-Yaa motifs of the collagen helical domain, where Yaa is often occupied by Hyl or Hyp (hydroxyproline)5,6,7. It contains two glycosyltransferase domains: a N-terminal CAZy GT2 domain and a C-terminal GT25 domain21. Mutagenesis analysis identified two DxD motifs (residues 166-168; residues 461-463) essential for GLT25D1 activities, located in the N-terminal and C-terminal domains, respectively25. Despite these findings, key mechanistic aspects of GLT25D1–including its substrate recognition, catalytic mechanism, and even the precise location of its active site–remain elusive.
Here, we report the structures of apo-human GLT25D1 as well as its ternary complex with donor and acceptor substrates. Through structure-guided functional analyses, we elucidate the molecular basis of collagen galactosylation by GLT25D1 and provide mechanistic insights into the deleterious mutations of the enzyme that cause various collagen-related human diseases.
Results
Functional characterization of GLT25D1
Human GLT25D1 (30-619 Aa) was successfully expressed using the Pichia pastoris expression system, and it was further purified through immobilized metal affinity chromatography, followed by protease treatment to cleave the purification tag. Subsequent size-exclusion chromatography revealed two main elution peaks, indicating the presence of at least two oligomeric states of GLT25D1 in solution (Fig. 1B). To evaluate whether the recombinant protein was functional, we measured its collagen galactosyltransferase (ColGalT) activity using gelatin as the acceptor substrate. The activity was readily detected in the presence of donor substrate UDP-galactose and Mn2+, whereas no activity was observed with UDP-glucose and Mn2+, demonstrating donor specificity (Fig. 1C).
To further validate and quantitate the specific enzymatic activity of GLT25D1, we selected a native peptide “GAKGEKG” in collagen alpha-1(VII) chain (Uniprot ID: Q02388), which contains two Gly-Xaa-Lys tripeptides (“GAK” and “GEK”) populated in different collagen chains11. We synthesized this designed acceptor peptide SGA-Hyl-GEKGS (referred to as “hydroxylated acceptor peptide” in this study), where hydroxylysine (Hyl, underlined) was positioned next to an alanine residue. Activity assay using this hydroxylated acceptor peptide confirmed the donor preference for UDP-galactose, consistent with the results obtained with the gelatin acceptor (Fig. 1C). In contrast, GLT25D1 exhibited no apparent activity with a control peptide lacking hydroxylysine (Fig. 1C). Utilizing the designed peptide, the reaction product by GLT25D1 was analyzed via UPLC-MS analysis. When GLT25D1 was incubated with UDP-glucose or UDP-galactose, only UDP-galactose can introduce an expected molecular weight increase of 162 Da to the peptide, confirming galactose modification (Fig. 1D, E). Simultaneous incubation with both UDP-galactose and UDP-glucose resulted only in monosaccharide modification (Fig. 1F). These findings are consistent with a previous study4, unequivocally demonstrating that GLT25D1 exclusively possesses galactosyltransferase activity when modifying hydroxylysine in collagen.
Oligomeric GLT25D1 and its overall structure
To gain deeper insights into the molecular mechanism of GLT25D1, we conducted a cryo-electron microscopy (cryo-EM) study (Supplementary Fig. 1 and Supplementary Table 1). The protein sample analyzed was prepared by pooling fractions corresponding to the two major oligomeric peaks isolated by size-exclusion chromatography (Fig. 1B). 2D classification analysis revealed that GLT25D1 exists in two oligomeric states as dimer and hexamer (Fig. 2A), consistent with size-exclusion chromatography analysis (Fig. 1B). 3D reconstruction of the hexameric state allowed us to derive a 3.1-Å resolution map of six GLT25D1 protomers arranged as a trimer of three dimers (designated as P1-P1’; P2-P2’; P3-P3’) (Fig. 2B). This hexamer measures approximately 110 Å in height and 120-150 Å in diameter and has a C3 symmetry axis coinciding with a central axial channel approximately 19 Å wide (Fig. 2B). Further symmetry expansion and local refinement with dimer mask yielded a 3.3-Å resolution map, showing improved density quality of one composite dimer (Supplementary Fig. 1C, E, F). This enabled the de novo building of the atomic model of GLT25D1 homo-hexamer, with most residues resolved except for one unstructured region (residues 561-619).

A Representative Cryo-EM 2D class averages of GLT25D1, showing its dimeric (top) and hexameric (bottom) forms. B Cryo-EM map of hexameric GLT25D1 (contour level 0.012). Left, side view. Right, top view. The six composing protomers (P1-P3; P1’-P3’) are depicted in different colors and labeled accordingly. The C3 symmetry axis is indicated by an arrow. C Domain organization of GLT25D1. The middle linker loop (M-loop, residues 328-340) connects the N-terminal GT2 domain (NGT) and C-terminal GT25 domain (CGT). The NGT domain is preceded by a long N-terminal loop (N-loop, residues 38-51). D Structural architecture of a single GLT25D1 protomer depicted in cartoon representation, with structural elements labeled as in (C). The α-helices flanking the central β-sheets in the NGT and CGT domains are labeled according to the topology diagram (Supplementary Fig. 2). Inset, close-up view of the interactions between the NGT and CGT domains (dashed box). E Atomic model of hexameric GLT25D1 in surface representation (side view). The hexamer is organized as a trimer of dimers with C3 symmetry. The six composing promoters are colored and labeled as in (B). F Cartoon representation of a C2-symmetrical, head-to-head dimeric unit of GLT25D1, highlighted by the box in (E). G Close-up view of the dimer interface of GLT25D1, as indicated by the dashed box in (F). Key residues mediating dimerization are shown as sticks, with polar interactions represented by dashed lines. H Size-exclusion chromatography analysis of wild-type GLT25D1 (blue) and the dimer-disrupting double mutant H113 A/W158A (gold). The mutant shows a shift in oligomer equilibrium towards monomers, indicating impaired dimerization. I Enzymatic activity of the dimer-disrupting mutant H113A/W158A, using peptide with hydroxylysine or gelatin as acceptor substrates. Activities are normalized to the amount of protein. Data are presented as mean ± s.d. for three independent replicates. Statistical significance was determined using ordinary one-way ANOVA, multiple comparison test (Dunnett’s test at 95% CI), and differences are depicted as ***p < 0.001; ****p < 0.0001; ns not significant. The exact p-values are as follows. For the peptide with hydroxylysine substrate: WT vs H113A–W158A (p = 0.0005) and Control (p < 0.0001). For the gelatin substrate: WT vs H113A–W158A (p = 0.6212) and Control (p < 0.0001). Source data are provided as a Source Data file.
Each composite monomer of the GLT25D1 hexamer exhibits a bi-lobe architecture, with both domains adopting the canonical GT-A fold characteristic of glycosyltransferase (Fig. 2C, D)26. The N-terminal glycosyltransferase domain (NGT; residues 51-328) features a central β-sheet (β1-7) flanked by multiple α-helices, forming a Rossmann-like fold topology (Supplementary Fig. 2). Likewise, the C-terminal glycosyltransferase domain (CGT; residues 332-561) is organized around a central six-stranded β-sheet (β8-13) encompassed by several α-helices (Supplementary Fig. 2). The interface between two glycosyltransferase domains (NGT and CGT domains) is mediated by an extended linker (M-loop, residues 328-340). This is further strengthened by multiple interactions among α7, α8, α11, and the loop preceding α11, including polar contacts (E314 with R439; E306 with R446) and hydrophobic packing among M309, H310, L313, M316, A364, F440 and I442 (Fig. 2D).
Molecular basis of GLT25D1 oligomeric assembly
Our results showed that GLT25D1 predominantly exists as dimers and hexamers (Fig. 2A), with the hexamer arranged as a trimer of three dimers (Fig. 2E and Supplementary Fig. 3A–C), suggesting that the dimer is the basic unit for GLT25D1 oligomerization. This dimer is formed by interactions at the NGT domains (“head”) of both protomers, adopting a butterfly-like shape with two-fold symmetry (Fig. 2F). Each “wing” of the head-to-head dimer extends outward from the central interface, with the two outer C-terminal CGT domains spatially separated by approximately 165 Å. The central interface is extensive with a buried surface area of 2942 Å2, involving multiple loops of the NGT domain organized into three layers (Fig. 2G). In the top layer, the two protomers exchange their extended N-terminal loops (N-loop, residues 38-51), where residues S47 and P48 of one protomer forms hydrogen bonds with consecutive alanine residues (A245-A246) of the neighboring protomer. In the middle layer, the close stacking of arginine paring (R53-R53) is further reinforced by a charged interaction between R53 and D160. In the bottom layer, W158 strategically inserts itself into the space between R53 and H113, establishing robust cation-pi and pi-pi interactions that are likely critical for dimerization. Indeed, this is strongly corroborated by the observation that H113A/W158A double mutations resulted in a pronounced shift in the oligomer equilibrium, predominantly favoring monomer formation in solution (Fig. 2H). Interestingly, the galactosyltransferase activity of the H113A-W158A mutant against gelatin was not dramatically affected relative to that of wild-type GLT25D1 (Fig. 2I), suggesting that the oligomerization is not required for at least in vitro catalytic activity.
In the GLT25D1 hexamer, three dimer units are arranged around the three-fold axis (Fig. 2E). This configuration symmetrically assembles C3-related protomers (P1, P2 and P3; P1’, P2’ and P3’) into two stacked and non-equivalent trimer rings, with the bottom ring (P1’, P2’ and P3’) extending further away from the C3 axis. The hexamer is stabilized by crossover contacts of two neighboring dimer units (e.g., P1-P1’ and P3-P3’) and this forms two inter-dimer interacting interfaces (I and II) located exclusively within their NGT domains near the intersection, burying a total surface area of 1924 Å2 (Supplementary Fig. 3D). Specifically, at ‘Interface I’ between the interacting dimers (e.g., P1-P1’ and P3-P3’), W43 on N-terminal loop of protomer P3’ stacks with R223 in the NGT domain of the neighboring protomer P1, which was further strengthened by an interprotomer salt-bridge interaction between E286 and R326 (Supplementary Fig. 3E). At ‘Interface II’, W43 on N-terminal loop of protomer P3 stacks with A245, A246 and R248 in NGT of the adjacent protomer, reinforced by a polar interaction between R42 and N249 (Supplementary Fig. 3F).
Identification of UDP-galactose endogenously bound to NGT domain
Both NGT and CGT have been implicated in catalyzing collagen galactosylation25. Notably, despite no donor ligand being added during sample preparation, densities consistent with a UDP-hexose ligand were observed in an open pocket of the NGT domain (Fig. 3A, B). To identify the co-purified endogenous ligand, we employed LC-ESI-MS/MS analysis. The mass data from the extracts of purified GLT25D1 matched either UDP-galactose or its isomer, UDP-glucose (Supplementary Table 2). Furthermore, the elution profile aligned with UDP-galactose standard, rather than UDP-glucose (Fig. 3C). Consequently, we modeled UDP-galactose, fitting well into the ligand cryo-EM densities (Fig. 3B). These findings strongly suggest the tight and specific binding of UDP-galactose to the NGT domain of GLT25D1. Indeed, in our model, the UDP-galactose is held in place by a rich network of interactions, forming a highly conserved cavity suited for its accommodation (Fig. 3B and Supplementary Fig. 4). Specifically, the uridine moiety is sandwiched between R61 and V143 and further stabilized by hydrogen bonds with Y126 side chain and L59 main chain. The diphosphate moiety is stabilized by K133 and an Mn2+ ion, where the ion was coordinated by the DXD motif (D166 and D168). Additionally, the galactose moiety is packed with W135 and engaged with a series of polar contacts with surrounding R147, D265 and H235.

A Structure of GLT25D1 with an endogenous UDP-galactose ligand (orange sphere) bound to the NGT domain. B Close-up view of the UDP-galactose binding pocket in the NGT domain, showing residues involved in ligand binding (cyan sticks). Potential hydrogen bonds are indicated by dashed lines. Inset, modeled UDP-galactose (orange stick) superimposed with its corresponding EM density (gray mesh; contour level 0.022). C Liquid chromatography-tandem mass spectrometry chromatogram of GLT25D1 protein extracts (black; bottom), compared with standards of UDP-galactose (gold; top) and UDP-glucose (blue; middle). D Relative enzymatic activity of wild-type (WT) GLT25D1 and mutants targeting the UDP-galactose binding site, as described in (B). Activities were normalized to the amount of protein. Data are presented as mean ± s.d. for three independent replicates. Statistical significance was determined using ordinary one-way ANOVA, multiple comparison test (Dunnett’s test at 95% CI) and differences are depicted as **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant. The exact p-values are as follows. For the peptide with hydroxylysine substrate: WT vs Control (p < 0.0001), W135A (p < 0.0001), R147A (p = 0.9998), D166A (p < 0.0001) and D265A (p < 0.0001). For the gelatin substrate: WT vs Control (p < 0.0001), W135A (p = 0.0749), R147A (p = 0.0010), D166A (p < 0.0001) and D265A (p = 0.0030). Source data are provided as a Source Data file.
Surprisingly, mutational analysis did not support this UDP-galactose binding site in the NGT domain as the ColGalT catalytic site. We generated GLT25D1 mutants with alanine substitutions in the NGT domain, targeting residues involved in uridine stabilization (R61, V143), ion coordination (D166, D168), and galactose interaction (W135, R147, H235, D265). Among the subset of mutants that could be successfully purified (Supplementary Fig. 5; see discussion for explanations), their residual activities varied considerably, ranging from wild-type levels (R147A) to minor reductions (D265A, W135A), with only D166A exhibiting the most substantial decrease in catalytic activity (Fig. 3D). Collectively, these findings strongly suggest that the ColGalT catalytic site resides outside the NGT domain.
Ternary complex structure of GLT25D1 with bound substrates
To localize the ColGalT catalytic site, we expressed and purified the isolated NGT domain (residues 30-333) and CGT domain (residues 332-619) of GLT25D1 and assessed their individual catalytic activities through two methods. First, in assays monitoring UDP generation, the NGT domain exhibited no enzymatic activity, whereas the CGT domain readily showed activity with both gelatin and hydroxylated peptide substrates (Fig. 4A). Second, mass spectrometric reaction product analysis in the presence of hydroxylated peptide substrate confirmed no modification by the NGT domain but significant modification by the CGT domain (Supplementary Fig. 5H). These results indicate that the ColGalT catalytic site resides within the CGT domain, consistent with our mutational analysis of the NGT domain (Fig. 3D).

A Enzymatic activity of the isolated NGT and CGT domains of GLT25D1, assessed using acceptor peptide (containing the hydroxylysine) or gelatin as acceptor substrates. Activities were normalized to the amount of protein. Data are represented as mean ± s.d. from three independent replicates. Statistical significance was determined using ordinary one-way ANOVA, multiple comparison test (Dunnett’s test at 95% CI), and differences are depicted as ***p < 0.001; ****p < 0.0001; ns, not significant. The exact p-values are as follows. For the peptide with hydroxylysine substrate: WT vs Control (p < 0.0001), CGT (p = 0.9702) and NGT (p < 0.0001). For the gelatin substrate: WT vs Control (p < 0.0001), CGT (p = 0.0009) and NGT (p < 0.0001). B Ternary complex structure of GLT25D1 (blue) with acceptor peptide (yellow) and UDP (cyan sphere), bound at the CGT domain. The NGT domain contains UDP-galactose (orange sphere), similar to that in Fig. 3A. Inset, density map (gray mesh; contour level 0.022) for modeled UDP (cyan stick) and Mn2+ (purple sphere). C Ligand-induced conformational change in GLT25D1. Superposition of the GLT25D1 apo-state (light orange; shown in cartoon with transparent surface) and the ternary complex (blue cartoon) structures reveals ordering of a 20-residue loop (Y561-E580) adjacent to the CGT active site, which is disordered in the apo structure. D Close-up view of the UDP binding pocket in the CGT domain. Key residues lining the UDP binding site are shown as sticks. D522, the catalytic base identified from later analysis (Fig. 5), is also shown. H-bonds, salt bridges and metal coordination are indicated as dashed lines. E Relative activity of wild-type (WT) GLT25D1 and UDP-binding site mutants, as described in (D). Activities were normalized to the amount of protein. Data are presented as mean ± s.d. for three independent replicates. Statistical significance was determined using ordinary one-way ANOVA, multiple comparison test (Dunnett’s test at 95% CI), and differences are depicted as ****p < 0.0001. The exact p-values are as follows. For the peptide with hydroxylysine substrate: WT vs Control (p < 0.0001), R354A (p < 0.0001), D436A (p < 0.0001), D437A (p < 0.0001), S569A (p < 0.0001) and T571A (p < 0.0001). For the gelatin substrate: WT vs Control (p < 0.0001), R354A (p < 0.0001), D436A (p < 0.0001), D437A (p < 0.0001), S569A (p < 0.0001) and T571A (p < 0.0001). Source data are provided as a Source Data file.
To investigate the molecular basis of substrate recognition and catalysis, we incubated GLT25D1 (pooled from the 11.8 mL SEC peak in Fig. 1B) with UDP-galactose, hydroxylated peptide acceptor (as that tested in Fig. 1D) as well as Mn2+, and performed cryoEM analysis (Supplementary Fig. 6). The resulting complex structure retained the UDP-galactose:Mn2+ in the NGT domain as observed in the apo enzyme, but revealed additional densities for UDP:Mn2+ and acceptor peptide in the CGT domain, clearly confirming the CGT domain as the ColGalT catalytic domain (Fig. 4B and Supplementary Figs. 7 and 8). Notably, comparing the substrate-bound and apo states of the CGT domain revealed the ordering of a 20-residue loop (Y561-E580; termed the ‘lid loop’) near the CGT active site, which is disordered in the apo structure (Fig. 4C, Supplementary Fig. 7A, B). Two highly conserved residues, H560 and R351, function to stabilize this ordered loop through backbone stacking at its beginning and a salt bridge with D570 at its middle, respectively (Supplementary Fig. 7C). Alanine substitutions of either residue compromised enzymatic activity, underscoring the functional importance of this loop (Supplementary Fig. 7C, D). Indeed, enclosed by this rearranged lid loop, UDP is bound within an embedded pocket of the CGT domain, lined with a series of conserved residues (Fig. 4D and Supplementary Fig. 9). Specifically, the uracil moiety of UDP is held in position by a hydrogen bond to G377 main chain and by stacking between C412 and L348 while the ribose moiety forms hydrogen bonds with D436. The pyrophosphate moiety is stabilized by a Mn2+ metal ion, coordinated by D437. Additional stabilization of pyrophosphate moiety comes from contacts with R354 and residues S569 and T571 from the lid loop. Substituting these ligand-binding site residues individually to alanine markedly reduced enzyme activity (Fig. 4E). In particular, D436A, D437A, S569A and T571A nearly abolished activity (Fig. 4E).
Acceptor peptide binding mode and catalytic mechanism
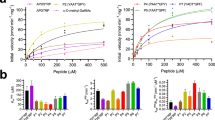
Collagens feature a (Gly-Xaa-Yaa)n repeating pattern and thereby GLT25D1 specifically galacosylates hydroxylysine (Hyl) residues within the “Xaa-Hyl-Gly” consensus sequence6. This is evident from the sequence analysis of confirmed galactosylation sites in human collagen type II (Fig. 5A)27. Structural analysis of the ternary complex (GLT25D1/acceptor peptide/UDP) uncovered the molecular basis for the recognition of this consensus sequence. In the CGT active site, EM densities corresponding to a strand of four residues were identified, enabling the modeling of an acceptor peptide “Ala-Hyl-Gyl-Glu” (Fig. 5B, C). The acceptor peptide interacts with GLT25D1 primarily via its amide backbone, mediated by conserved GLT25D1 residues on both sides of the peptide. Specifically, K470, Y494, and W495 form one side of the binding interface, while Y561 and T562 define the opposite side (Fig. 5B). These residues form a cleft connecting the binding sites of acceptor and donor substrates. Notably, the narrowest constriction of the cleft, measuring 6.8 Å in width, is defined by K470 and T562, with the β-carbon of T562 positioned merely 4.1 Å away from the α-carbon of glycine (at subsite +1) of the acceptor peptide (Fig. 5D). This geometry specifically accommodates the invariant glycine residue following Hyl, while excluding bulkier residues. Furthermore, the invariant hydroxylysine (Hyl) is stabilized within this cleft by the “YW” motif (Y494 and W495), with its ε-amino group potentially anchored by E523 (Fig. 5B). This configuration positions the 5-hydroxyl group of hydroxylysine in close proximity to the β-phosphate of UDP in CGT active site, appropriate for galactose transfer. Indeed, mutagenesis experiments support these structural insights: the W495A mutation abolished enzymatic activity; K470A, Y494A, Y561A and E523A mutations markedly reduced GLT25D1 activity (Fig. 5E). Interestingly, alanine substitution of T562 preserved activity (Fig. 5E), suggesting that its interaction with acceptor peptide may rely more on the β-carbon of this residue (Fig. 5D).

A Sequence conservation of the 14 confirmed galactosylation sites in human collagen type II (generated by WebLogo). The logo size is proportional to the level of conservation. The hydroxylysine (Hyl) and its following glycine residues are absolutely conserved. B Close-up view of the CGT active site in the ternary complex structure of GLT25D1 (in gray surface rendering) with collagen acceptor peptide (yellow stick) and UDP (cyan stick). The acceptor peptide is positioned within a binding cleft, adjacent to UDP. GLT25D1 residues lining the acceptor-binding interface are shown as blue sticks. Yellow dashed lines indicate potential hydrogen bond or salt bridge. C The acceptor peptide (yellow) and its surrounding GLT25D1 residues (blue) are superimposed with their corresponding densities (gray mesh; contour level 0.022). D Side view of the acceptor peptide binding cleft, rotated by 45 degrees relative to that in (B). Pink dashed lines indicate distances between groups as labeled. E Relative activity of wild-type (WT) GLT25D1 and mutants targeting residues in the acceptor binding site as well as catalytic residue D522 as identified in (C). Activities were normalized to the amount of protein. Data are presented as mean ± s.d. for three independent replicates. Statistical significance was determined using ordinary one-way ANOVA, multiple comparison test (Dunnett’s test at 95% CI) and differences are depicted as **p < 0.01; ****p < 0.0001; ns not significant. The exact p-values are as follows. For the peptide with hydroxylysine substrate: WT vs Control (p < 0.0001), K470A (p < 0.0001), Y494A (p < 0.0001), W495A (p < 0.0001), Y561A (p < 0.0001), T562A (p < 0.0001), D522A (p < 0.0001) and E523A (p < 0.0001). For the Gelatin substrate: WT vs Control (p < 0.0001), K470A (p < 0.0001), Y494A (p < 0.0001), W495A (p < 0.0001), Y561A (p = 0.9997), T562A (p < 0.0001), D522A (p < 0.0001) and E523A (p = 0.0037). Source data are provided as a Source Data file. F Proposed SN2 single-displacement reaction mechanism of GLT25D1. Specifically, D522 deprotonates the 5-hydroxyl group of hydroxylysine, activating it as a nucleophile to attack the anomeric carbon (C1) of the galactose moiety.
The structure of the ternary complex also provides valuable insights into the catalytic mechanism of GLT25D1. Inspection of the active site within the CGT domain revealed that D522, an invariant residue conserved from humans to sponges, is located within 4 Å of the acceptor hydroxyl group (Fig. 5B and Supplementary Fig. 4). This proximity ideally positions D522 as the catalytic base, primed to activate the nucleophile, consistent with an SN2-type inverting catalytic mechanism (Fig. 5F)26. Similar mechanisms have been observed in other glycosyltransferases, such as POMGnT1, POGLUT1 and POFUT2, where aspartate or glutamate serves as a general base28,29,30. Indeed, site-directed mutagenesis of D522 completely abrogated enzymatic activity, underscoring its essential role in catalysis, as demonstrated by both UDP generation assays (Fig. 5E) and mass spectrometric analysis of reaction products (Supplementary Fig. 5I). Furthermore, we observed that the ColGalT activity requires Mn2+ for both the full-length GLT25D1 and its truncated CGT domain (Supplementary Fig. 5J, K), consistent with previous findings31,32,33. This highlights the critical role of Mn2+ in catalysis, likely by facilitating the departure of the diphosphate leaving group, as proposed for other glycosyltransferases26. Collectively, these structural and functional analyses conclusively define the active site of GLT25D1 and identify its key residues involved in collagen galactosylation (Figs. 4 and 5).
Molecular insights into pathogenic mutations of GLT25D1
Deficiencies in GLT25D1 have been linked to a spectrum of human diseases, with several potential pathogenic mutations identified in cerebral small vessel disease (SVD; L151R, A154P, E366Rfs, G377R, G209fs, and G209_K212del), vascular cognitive impairment (VCI; R471W) and musculoskeletal defects (W135R)17,22,23,24. To elucidate the mechanistic impacts of these mutations, we mapped them onto the GLT25D1 ternary complex structure (Fig. 6A). This analysis revealed that the SVD-related frameshift mutations (E366Rfs and G209fs) result in truncated GLT25D1 proteins lacking the catalytic CGT domains, likely rendering the enzyme inactive (Fig. 2C). Additionally, the SVD-associated point mutations L151R and A154P, located in helix α3, are expected to disrupt the domain’s core fold stability (Fig. 6A). Specifically, the L151R mutation introduces a large, charged residue into a hydrophobic core, while A154P is likely to impair helical structure and packing. These destabilizing effects are consistent with the dramatically reduced expression levels observed for both mutants (Fig. 6B), in agreement with a previous study22. Further analysis revealed that SVD mutant G209_K212del and musculoskeletal-defect-related mutant W135R are situated near the bound UDP-galactose in the NGT domain (Fig. 6A). Functional assays demonstrated that G209_K212del preserves half the normal activity, whereas W135R almost abolishes the enzymatic function (Fig. 6C). Lastly, the SVD-associated mutation G377R and VCI-related mutation R471W are located within the active site of CGT domain (Fig. 6A). Residue G377 is directly involved in donor ligand binding (Fig. 4D), while R471 sits next to the critical acceptor-binding residue K470 (Fig. 5B). Indeed, neither G377R nor R471W appeared to markedly affect protein expression levels (Fig. 6B), but G377R almost eliminates GLT25D1 activity and R471W leads to a substantial reduction in enzymatic function (Fig. 6C).

A Pathogenic mutations in GLT25D1 associated with SVD (L151R, A154P, G209_K212del and G377R), VCI (R471W), and musculoskeletal-defect-related (W135R) are mapped onto the ternary structure of GLT25D1 in complex with acceptor peptide and UDP. Mutated residues are displayed as color-coded sticks and highlighted with transparent surfaces. CGT active site contains the acceptor peptide (yellow cartoon) and UDP (cyan sticks), while the NGT domain contains the endogenous UDP-galactose (magenta sticks). B Relative expression levels of wild-type (WT) GLT25D1 and pathogenic mutants analyzed by western blotting with an anti-GLT25D1 antibody. All experiments were independently performed in triplicate with similar results. C Relative enzymatic activity of wild-type (WT) GLT25D1 and pathogenic mutants. Activities were normalized to the amount of protein. Data are presented as mean ± s.d. for three independent replicates. Statistical significance was determined using ordinary one-way ANOVA, multiple comparison test (Dunnett’s test at 95% CI), and differences are depicted as ****p < 0.0001. The exact p-values are listed as follows. For the peptide with hydroxylysine substrate: WT vs Control (p < 0.0001), W135R (p < 0.0001), G209_K212del (p < 0.0001), G377R (p < 0.0001) and R471W (p < 0.0001). For the gelatin substrate: WT vs Control (p < 0.0001), W135R (p < 0.0001), G209_K212del (p < 0.0001), G377R (p < 0.0001) and R471W (p < 0.0001). Source data are provided as a Source Data file.
Dysfunctions of collagen and its glycosylation have increasingly been recognized as critical factors in cancer progression and metastasis10,34,35. Motivated by this association, we identified cancer-associated GLT25D1 mutations and analyzed them by mapping onto the ternary complex structure. These mutations are distributed across both the NGT and CGT domains (Supplementary Fig. 10), consistent with the functional importance of each domain. Notably, four cancer-associated mutations (R147H, R147C, D168H, and G377D) are situated at key ligand-binding interfaces and are likely to impair GLT25D1 function: R147H, R147C, and D168H in the NGT domain (Fig. 3B), and G377D in the CGT domain (Fig. 4D). Moreover, GLT25D1 alterations have been linked to tumorigenesis in several cancer types, including malignant mammary tumors and kidney renal clear cell carcinoma36,37. These findings highlight the broader implications of GLT25D1 dysfunction, extending its relevance to cancer biology.
Discussion
GLT25D1 initiates collagen glycosylation by catalyzing hydroxylysine galactosylation, a process critical for collagen maturation and functional integrity. In this study, we report the cryo-EM structures of human GLT25D1, and its ternary complex with donor and hydroxylated acceptor substrates. This reveals a distinctive bi-domain architecture of GLT25D1, composed of two GT-A (NGT and CGT) domains. Functional and structural analysis allowed us to pinpoint the ColGalT catalytic site within the CGT domain and to elucidate the molecular determinants for collagen galactosylation.
Regarding acceptor substrate recognition, we demonstrated that GLT25D1 accommodates acceptor peptide via a narrow binding cleft, which excludes secondary structural elements other than loops (Fig. 5B–D). This is consistent with the enrichment of glycine and proline residues in collagen chain, which imparts flexibility5. Moreover, we discovered that the hydroxylysine and the adjacent invariant glycine residues, forming a “Xaa-Hyl-Gly” motif, serve as key recognition features (Fig. 5B–D). This likely underpins a universal mechanism for GLT25D1-mediated collagen recognition and defines its sequence specificity.
As for catalytic mechanism, we identified residue D522 of the “DE” motif (Asp522 and Glu523) as a base catalyst, facilitating the activation of 5-hydroxyl group of hydroxylysine for nucleophilic attack (Fig. 5F). Concurrently, a manganese (II) ion, coordinated by residue D437 of “EDD” motif (Glu435, Asp436 and Asp437), stabilize the donor β-phosphate and primes it for catalysis (Fig. 5F). These findings support an inverting SN2 catalytic mechanism for collagen galactosylation26. Notably, the binding of UDP to the active site triggers conformational ordering of a lid-like loop (lid loop; Y561-E580) (Fig. 4C). Within this loop, residues S569 and T571 of the “STDE” motif (Aa 569-572) interact with the donor β-phosphate (Fig. 4D). This restructured lid loop also contributes to the binding of the acceptor peptide through residues Y561 and T562 (Fig. 5B and Supplementary Fig. 7B). This suggest a concerted binding mode for the donor and acceptor substrates; thereby the dynamic transition between the open and closed conformations of the lid loop could facilitate efficient substrate binding and product release38. Importantly, the CGT active site and its associated motifs are highly conserved across metazoan species, from sponges to humans (Supplementary Figs. 4 and 9), highlighting a unified mechanism for substrate recognition and catalysis and underscoring the evolutionary importance of collagen glycosylation.
Interestingly, in the NGT domain, we identified an endogenous UDP-galactose ligand, whose presence was confirmed by both cryo-EM density and LC-MS/MS analysis (Fig. 3B, C). However, multiple lines of evidence indicate that the NGT domains lacks catalytic activity (Figs. 3D and 4A; Supplementary Fig. 5H). Consistently, our cryo-EM analysis of the ternary complex further reveals that the acceptor peptide substrate binds specifically to the CGT domain rather than the NGT domain (Fig. 4B). Additionally, although a negatively charged residue (D265) is located near the anomeric carbon (Fig. 3B)—a position often associated with catalytic base function in glycosyltransferases26—alanine substitution at this site only mildly impaired enzymatic activity (Fig. 3D), ruling out its role in catalysis. Despite being catalytically inactive, the NGT domain contains a deeply embedded ligand-binding pocket that mediates extensive interactions with UDP-galactose (Fig. 3B and Supplementary Fig. 8A–C), suggesting that the ligand binding may contribute to enzyme structural stability. Supporting this, half of our designed mutants targeting this binding pocket (R61A, V143A, D168A and H235A) could not be purified, a result that may be influenced by the stabilizing effect of UDP-galactose, though other factors such as protein misfolding could also contribute. Among the purified mutants, variable effects on catalytic activity were observed (Fig. 3D), implying potential long-range allosteric effects. To investigate this further, we performed enzyme kinetics assays (Supplementary Fig. 11A). The kinetic parameters obtained for the wild-type (WT) GLT25D1 (Supplementary Fig. 11B) were consistent with other studies25,39. Notably, the NGT mutant D166A revealed a ~2.3-fold increase in Km, a ~1.6-fold decrease in kcat, and a ~3.8-fold reduction in catalytic efficiency (kcat/Km) compared to the wild-type enzyme (Supplementary Fig. 11B), supporting a potential allosteric regulatory mechanism. To gain deeper mechanistic insights, we performed molecular dynamics (MD) simulations of GLT25D1 in the apo state and in complex with UDP-galactose bound to the NGT domain (Supplementary Fig. 11C–E). The apo form showed elevated flexibility across the protein, as reflected by broadly increased root-mean-square fluctuation (RMSF) values (Supplementary Fig. 11C), indicating structural destabilization in the absence of ligand. Consistently, thermal stability analysis demonstrated that ligand-binding mutations in the NGT domain exhibited a more pronounced destabilizing effect compared to those in the CGT domain (Supplementary Fig. 12). In contrast, the MD analysis showed that UDP-galactose binding at the NGT domain restricted global flexibility while selectively increasing RMSF in three localized regions (ΔRMSF < 0; Supplementary Fig. 11D): region 1 (residues 330–342), which connects the NGT and CGT domains; and regions 2 and 3 (residues 375–385 and 387–405, respectively), which line the substrate-binding site in CGT domain (Supplementary Fig. 11E). These regions are spatially connected: β8 in region 1 runs parallel to β9 immediately preceding region 2, and regions 2 and 3 are directly linked. These observations suggest the presence of allosteric communication between the ligand-binding sites in the NGT and CGT domains. Consistent with this notion, kinetic analysis also indicates that the NGT domain influences the CGT domain, as evidenced by a lower Km of the CGT domain toward gelatin compared to that of the wild-type enzyme (Supplementary Fig. 11A). Notably, the high conservation of the ligand binding pocket of NGT domain, second only to the CGT active site (Supplementary Fig. 9), further underscores its functional importance, warranting more in-depth investigation into its non-catalytic roles.
Our findings revealed that GLT25D1 forms an elongated dimer through interactions between the NGT domains, positioning two CGT catalytic sites approximately 130 Å apart (Fig. 2F). This spatial organization may facilitate simultaneous galactosylation of multiple hydroxylysine residues along the extended collagen chain, enhancing catalytic efficiency. Consistently, LH3, another enzyme involved in collagen glycosylation, has also been reported to form an elongated dimeric structure40, in contrast with the parallel dimerization mode for several bi-domain glycosyltransferases involved in the synthesis of polysaccharides with alternating sugar units (such as EXTL3, LARGE1, and EXT1-EXT3)41,42,43. Additionally, we also uncovered a distinctive hexameric assembly of GLT25D1, comprising a trimer of head-to-head dimers (Fig. 2E). Given the widely known triplex helix feature of collagen, it is conceivable that this hexameric organization could enable multiple catalytic centers to simultaneously act on three collagen chains, thereby ensuring the high efficiency required for proper collagen glycosylation and assembly.
The molecular characterization of disease-linked GLT25D1 mutations improves our understanding of the pathogenic mechanism (Fig. 6 and Supplementary Fig. 10) and provides a foundation for developing precise biochemical diagnosis to probe GLT25D1 dysregulation in collagen-related diseases. The distinct structural and functional deficiencies caused by specific variants may correlate with varying clinical severities and subtypes, awaiting further investigation. From a therapeutic perspective, these mechanistic insights suggest that restoring enzymatic activity—either through stabilization of misfolded proteins, or gene-based approaches—could represent viable treatment strategies. Moreover, the association of GLT25D1 dysfunction with both inherited vascular/musculoskeletal disorders and tumors highlights the potential of targeting GLT25D1 in both rare diseases and oncology.
During the preparation or review of this manuscript, two related studies on GLT25D1 were published: Peng et al. described cryo-EM structures of the GLT25D1–LH3 complex bound to UDP or UDP-galactose, while De Marco et al. used the crystallography method to determine the structures of GLT25D1 bound to UDP or UDP-galactose39,44. These independent works reflect the rapid progress in the field, complementing and enhancing one another. In particular, all three works consistently confirmed the dimeric architecture of GLT25D1 and revealed the unexpected binding of UDP-galactose in the NGT domain. Beyond these consensus findings, our study provides several distinct contributions. Most importantly, we distinctly present GLT25D1 in a ternary complex with UDP/UDP-galactose and a designed hydroxylysine-containing acceptor substrate, supported by detailed structure-based functional characterization. Such a ternary complex is essential for a deeper understanding of a protein-targeting glycosyltransferase like GLT25D1, yet was not available in the other two studies. This approach has enabled us to distinctly define the precise catalytic mechanism and elucidate the molecular determinants governing collagen recognition. Furthermore, in addition to the dimeric form consistently reported across all three studies, we have identified a hexameric assembly of GLT25D1, an architectural feature not previously described in this enzyme family. We also performed a detailed functional analysis of disease-related mutations, clarifying their mechanistic impact on GLT25D1 dysfunction—an aspect not explored in the other studies. Comparative analysis also highlights several discrepancies in metal ion identity and substrate modeling among these independently determined structures (Supplementary Fig. 13). Regarding the metal ion, our cryo-EM sample for the ternary complex was prepared in the presence of 1 mM MnCl2, and accordingly, we modeled a Mn²⁺ ion in the ligand binding sites of both the NGT and CGT domains. De Marco et al. reported a Ca²⁺ ion at the equivalent site of the NGT domain and a Mn2+ ion in the CGT domain. It is possible that metal ion occupancy may be influenced by cellular or experimental conditions, and further investigation will be needed to determine the physiologically relevant cofactor. Regarding ligand modeling, there are also variations. In the NGT domain, the galactose moiety of UDP-galactose adopts a favorable conformation in our structure, consistent with that reported by Peng et al., but distinct from the conformation reported by De Marco et al. (Supplementary Fig. 13A–C). The UDP moiety, however, is consistent between our model and that of De Marco et al., whereas Peng et al. modeled the uracil group in a flipped orientation (Supplementary Fig. 13A–C). In the CGT domain, the UDP orientation is consistent between our study and De Marco et al., but again differs from the flipped uracil conformation reported by Peng et al. (Supplementary Fig. 13A–C).
Collectively, our study provides a molecular foundation to understand the molecular basis of collagen galactosylation, detailing GLT25D1’s substrate specificity, catalytic mechanism, and structural assembly. This not only enhances our understanding of collagen glycosylation but also sheds light on how pathogenic GLT25D1 mutations contribute to diseases associated with defective collagen modification.
Methods
Expression and purification of human GLT25D1
The gene encoding human GLT25D1 (GenBank ID: NM_024656.4) was amplified from the cDNA of 293 T cells. The secreted form of GLT25D1, encompassing residues 30-619, was cloned into the pPICZ C vector (Thermo Fisher Scientific) with a C-terminal HRV3C protease recognition site followed by a 10×His tag. Site-directed mutagenesis was performed using an inverse PCR strategy45, with the plasmid harboring the wild-type GLT25D1 gene as the template and corresponding mutation-specific primers. The resulting PCR products were treated with DpnI (NEB; 37 °C, 1 h) to digest the methylated template, phosphorylated with T4 polynucleotide kinase (NEB; 37 °C, 30 min), and ligated using T4 DNA ligase (NEB; 16 °C, 16 h). The circularized DNA was then transformed into E. coli DH5α competent cells (Sangon Biotech). Clonies with related mutations were verified by PCR and confirmed by DNA sequencing.
For expression, the recombinant plasmids were linearized by restriction enzyme PmeI and subsequently transformed into Pichia pastoris X33 (Thermo Fisher Scientific, C18000) competent cells via electroporation. Transformants were sequentially inoculated in YPD and BMGY media at 28 °C. When OD₆₀₀ reached 2-6, cells were harvested by centrifugation at 300 × g for 5 min. The BMGY medium was then removed, and the cell pellet was resuspended in BMMY medium. Protein expression was induced at 24 °C for 72 h, with 0.5% (v/v) methanol added every 24 h, following Pichia Expression Kit USER GUIDE (Invitrogen, Publication Number MAN0000012).
For protein purification, the collected cells were lysed in buffer A (25 mM HEPES/NaOH, 500 mM NaCl, pH 8.0). The supernatant was collected by centrifugation at 15,000 × g for 30 min. Protein purification was performed using an ÄKTA pure chromatography system equipped with a 5 mL column of nickel nitrilotriacetic acid (Ni-NTA) resin (GE Healthcare). The column was sequentially washed with 10 column volumes each of buffer A containing 20 mM, 50 mM, and 100 mM imidazole, respectively. The target proteins were eluted with buffer A supplemented with 500 mM imidazole. To remove the C-terminal 10×His tag, the eluted protein was digested with HRV 3 C protease (Genscript) at a 1:20 (w/w) protease-to-protein ratio, followed by overnight incubation at 4 °C. The digested protein was desalted with a HiPrep™ 26/10 column (GE Healthcare), concentrated and further purified by size-exclusion chromatography on a Superdex 200 Increase 10/300 column (GE Healthcare) equilibrated in buffer A. The collected fractions were concentrated to 1–2 mg/mL for cryo-EM grid preparation. Protein concentration was quantified by A280 using NanoDrop Spectrophotometers, with an extinction coefficient of 107635 M–1 cm–1 and a molecular weight of 68.3 kDa, respectively.
For activity analysis, wild-type and mutant proteins were purified by affinity chromatography as described above. The purified proteins were then desalted and exchanged into buffer A (25 mM HEPES/NaOH, 500 mM NaCl, pH 8.0) using a HiPrep™ 26/10 column (GE Healthcare) and concentrated to 0.5 mg mL⁻¹ for subsequent assays.
Glycosyltransferase activity assays
Glycosyltransferase activity was assayed using the UDP-GloTM Glycosyltransferase Assay (Promega). The hydroxylated peptide SGA-Hyl-GEKGS (where Hyl denotes hydroxylysine) was sourced from Shenzhen PepBiotic. Its synthesis involved the prior preparation of the hydroxylysine building block, which was subsequently incorporated into the peptide sequence via solid-phase peptide synthesis (SPPS)46. The corresponding non-hydroxylated peptide SGA-K-GEKGS was obtained from GenScript Biotech. The standard reaction mixture (25 µL) contained 0.625 mM peptide, 0.7 µM (0.05 mg/ml) of wild-type or mutated GLT25D1 and a reaction buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1 mM MnCl2. The reaction was initiated by adding UDP-galactose or UDP-glucose (Promega) to a final concentration of 0.5 mM. The reaction was carried out at 37 °C for 1 h. For assays utilizing gelatin as the substrate, 1.5 mg/mL gelatin (solubilized through heating denaturation at 95 °C for 10 min) was added to the reaction mixture, according to a previous study47. After the reaction, the enzymatic reaction mixture was mixed with an equal volume of UDP-GloTM Detection Reagent (15 μL) in white polystyrene, 384-well assay plates (Perkin-Elmer). The plates were incubated at 25 °C for 1 h, and luminescence was recorded using a Pherastar FS system (BMG Labtech). To ensure accurate activity comparisons across variants, SDS-PAGE band intensity quantification was used to normalize the enzyme amounts for WT GLT25D1 and its mutants. All experiments were performed in triplicate. Uncoupled control reactions were under identical conditions but in the absence of the acceptor substrate. The wild-type (WT) activity against gelatin is normalized to WT activity against the hydroxylysine peptide. For kinetic analysis, the reaction contained 0.7 μM of GLT25D1 or its variants, 500 μM of the UDP-Gal, and gelatin at concentrations ranging from 0.5 mg/mL to 30 mg/mL. After incubation at 37 °C for 5 min, the reactions were analyzed using the UDP-Glo luminescence assay (Promega) as described above. Kinetic parameters (Km, kcat, and kcat/Km) and their standard errors were derived by fitting the data to the Michaelis–Menten equation via nonlinear regression in GraphPad Prism 8. All experiments were performed in triplicate.
LC-MS analysis of enzymatic products
For LC-MS analysis of enzymatic products, enzymatic reactions were prepared by combining 1.7 μM GLT25D1 or its mutant variants with 1.25 mM hydroxylated peptide substrate, and 0.5 mM either UDP-Galactose or UDP-Glucose in a reaction buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1 mM MnCl2. The reactions were carried out at 37 °C for 3 h. Following the enzymatic reaction, each sample was concentrated by nitrogen blow-down evaporation and subsequently reconstituted in a methanol/water (80:20, v/v) solution. The samples were then centrifuged at 15,000 × g for 20 min at 4 °C, and the resulting supernatants were analyzed using a UHPLC-MS system following a previous study48,49. The analysis was performed using a Vanquish UHPLC system (Thermo Scientific) coupled to Q-ExactiveTM Plus mass spectrometer (Thermo Scientific). A Hypersil Gold Vanquish column (Thermo Scientific, 2.1 × 100 mm, 1.9 μm) was used for chromatographic separation, with a gradient mobile phase of solvent A (0.1% formic acid in water) and solvent B (acetonitrile) at a flow rate of 0.3 mL/min. The gradient elution conditions were as follows: 10% B (0−2 min), linear increase to 50% B (2–10 min), 80% B (10–13 min), 95% B (13–14 min), and return to 10% B (14–18 min). Mass spectrometric detection was conducted in positive ion mode with a spray voltage of 3.5 kV and a heated capillary temperature of 320 °C. MS scans were performed over ranges of m/z 70-1500, with a resolution of 70,000, an automatic gain control (AGC) target of 1 × 106 and a maximum injection time of 100 ms. The MS experiments were run once per sample.
Cryo-EM data acquisition
For single-particle cryo-EM analysis, the grids (Quantifoil R1.2/1.3 Au, 300 mesh) were glow-discharged using a PELCO EasiGlow unit at (15 mA for 60 s). Then a 3 µL aliquot of purified GLT25D1 (pooled from the 10.3-ml and 11.8-mL SEC peak in Fig. 1B) at a concentration of 1.2 mg/mL was applied to glow-discharged grids. The grids were blotted for 3–4 s at 100% humidity and flash-frozen in liquid ethane using a FEI Vitrobot Mark IV System (Thermo Fisher Scientific). For the ternary complex sample, GLT25D1 (1.2 mg/mL; pooled from the 11.8-ml SEC peak in Fig. 1B) was mixed with 0.8 mM UDP-galactose and 1 mM hydroxylated acceptor peptide, supplemented with 1 mM MnCl2. The mixture was incubated at 4 °C for 30 min before freezing the cryo-EM grids as described above.
Cryo-EM data were collected on a FEI Titan Krios electron microscopy operated at 300 kV equipped with a Gatan K3 Summit camera and a GIF Quantum energy filter (slit width 20 eV). Automated data acquisition was performed with FEI EPU. Micrographs were recorded under super-resolution counting mode in a physical pixel size of 1.076 Å for GLT25D1 sample and 0.855 Å for ternary complex sample. Defocus values varied from −1.2 μm to −3 μm. A total exposure of 1.3 s was dose-fractionated into 32 frames, yielding a total accumulated fluence of 50 electrons per square angstrom.
Image processing and 3D reconstruction
The dose-fractionated movies collected were motion-corrected and dose-weighted with MotionCor250. The contrast transfer function (CTF) parameters for individual micrographs were determined using CTFFIND451. Micrographs displaying poor quality, as assessed by manual inspection, were excluded from further analysis.
For the GLT25D1 dataset, the image-processing steps were performed using RELION-352, and processing workflow is presented in Supplementary Fig. 1. A set of 1500 particles was manually selected to generate 2D class templates for reference-based automatic particle picking. The automatic picking yielded in 773,881 particles from 1192 micrographs. Two rounds of reference-free 2D classification were performed to remove contaminants and low-quality particles, resulting in a cleaned set of 720,616 particles. 3D classification was performed using an ab initio map generated by RELION-3 as the initial reference model. The 3D class displaying clear hexamer features (125,162 particles) was selected. Subsequent refinement with C3 symmetry, combined with CTF refinement and polishing, resulted in a map with an overall resolution of 3.1 Å. This reconstruction revealed weak density within the outer C-terminal domain (CTD) regions, suggesting resolution loss due to symmetry averaging. To address this, we expanded the C3-symmetry-imposed dataset (125,162 particles) with the symmetry expansion tool in RELION, increasing particle count to 375,486. Subsequently, focused refinement was performed on this expanded dataset, using a focused mask encompassing a single GLT25D1 dimer. This strategy generated a map with significantly improved density in both NTD and CTD domains, achieving an overall resolution of 3.3 Å.
For the ternary complex dataset, image processing was performed using a combination of cryoSPARC53 and RELION-352. The corresponding workflow is provided in Supplementary Fig. 6. Initially, a total of 13,403,413 particles were automatically picked from 14,079 micrographs, using a blob-based particle picking algorithm in cryoSPARC. Multiple rounds of 2D classification were performed, and class averages displaying recognizable secondary structures were selected. The resulting subset (3,033,718 particles) was subject to multiclass ab initio reconstruction followed by heterogeneous refinement. The dominant class (1,896,674 particles) with high-resolution features was selected and imported into RELION-3 for further processing. Subsequent refinement with C2 symmetry yielded a map with an overall resolution of 2.9 Å. We noted the outer C-terminal domain (CTD) exhibiting relatively weak density due to an elongated dimer shape, which hindered the precise identification of bound substrates. To address this, we expanded the C2-symmetry-imposed dataset using the symmetry expansion tool in RELION. The expanded set (3,793,348 particles) underwent one round of 3D classification without alignment, using a focused mask around a single GLT25D1 subunit. A 3D class displaying the best density features in the CTD region was selected for 3D refinement with a focused monomer mask, resulting in a map of the ternary complex at an overall resolution of 3.2 Å.
For both datasets, the overall resolutions were estimated based on the gold-standard Fourier shell correlation 0.143 criterion54. Local resolution distribution was estimated using ResMap55.
Model building and refinement
The GLT25D1 dimer was initially built from the map refined with a focused dimer mask (GLT25D1 dataset; Supplementary Fig. 1). The initial monomeric model was generated using AlphaFold 356 and two copies of this model were fitted into the EM densities as rigid bodies in Chimera to generate a preliminary dimer model57. It was subsequently improved by interactive rebuilding in Coot58 and iterative refinement in PHENIX with secondary structure and geometry restraints applied59. For the hexameric GLT25D1 structure, three copies of the GLT25D1 dimer were docked into the consensus map refined with C3 symmetry (GLT25D1 dataset; Supplementary Fig. 1). Rigid-body adjustments of each monomer were performed in Chimera to slightly optimize the fit. The ternary complex structure was built based on the map refined with a focused monomer mask (ternary complex dataset; Supplementary Fig. 6).
The final models were validated using MOLPROBITY60. The illustrated figures were prepared using PyMOL (Schrödinger, LLC)61, Chimera and ChimeraX57,62. Detailed statistics for 3D reconstructions and model refinements are provided in Supplementary Table 1.
Identification of the endogeneously bound ligand in GLT25D1
Purified GLT25D1 protein was digested overnight with trypsin at 30 °C. For ligand extraction, four volumes of ice-cold HPLC-grade methanol (Thermo Fisher Scientific) were added to the digested sample, followed by thorough mixing and incubation on crushed ice for 20 min. The crude extracts were then centrifuged at 13,000 × g for 15 min at 4 °C. The supernatant was collected, evaporated under vacuum and resuspended in 200 µL of Milli-Q water. Detection and identification of the analytes were conducted with UPLC-MS/MS method63,64. Chromatographic separation was performed on an ACQUITY UPLC H-Class plus system (Waters, Wilmslow, UK) equipped with a BEH amide column (2.1 × 100 mm, 1.7μm; Waters, Wilmslow, UK). An injection volume of 5 µL was used, and the mobile phase consisted of solvent A (50 mM ammonium formate solution, pH = 3.6) and solvent B (acetonitrile) at a flow rate of 0.4 mL/min. The elution was maintained at (79% solvent A):(21% solvent B). Ultra-pure UDP-sugar standards (Promega) were used for calibration. Mass spectrometric analysis was conducted in negative ion mode on a TQ-XS system (Waters, Wilmslow, UK) equipped with an electron spray ionization (ESI) source. Optimal multiple reaction monitoring (MRM) conditions were established for analyte detection. Two individual transitions were monitored for each analyte under the following parameters: ion spray voltage, 3.5 kV; auxiliary gas pressure, 5 arbitrary units; ion transfer tube temperature, 350 °C; ion source temperature, 150 °C. Detailed values of collision energy, cone voltage, and transitions for the MRM mode are provided in Supplementary Table 2.
Differential scanning fluorimetry
The thermostability of wild-type and GLT25D1 mutants was evaluated using differential scanning fluorimetry65. For each assay (20 µL in total volume) prepared in a 96-well PCR plate, 2 µg of purified protein samples were mixed with 5×SYPRO Orange (Sigma Aldrich; 1:1000 dilution of the stock) in buffer (25 mM HEPES, pH 8.0, 500 mM NaCl). Thermal denaturation profiles were determined by monitoring fluorescence over a temperature gradient on a QuantStudioTM 3 Real-Time PCR Instrument (Applied Biosystems). The incubation was started at 25 °C for 2 min, followed by a linear temperature ramp from 25 °C to 90 °C at a rate of 1 °C per min. Melting curve analysis was performed using Protein Thermal Shift Software v1.4 (Applied Biosystems). The melting temperature (Tm) was calculated from the inflection point of the melting curve (derivative melting point). All experiments were conducted in quadruplicate, and the mean values with the standard deviation of the respective Tm values were presented.
Western blots
Wild-type and mutated GLT25D1 constructs were generated and expressed in Pichia cells as described above. Briefly, transformed yeast cells were induced in BMMY medium containing 0.5% (v/v) methanol for 24 h. Cells were then harvested, lysed, and equal amounts of protein were separated on 12% SDS-PAGE gels and electroblotted onto a nitrocellulose membrane (MiliporeSigma). For immunodetection, the membrane was probed with a mouse anti-GLT25D1 primary antibody (1:1000 dilution; cat no. 16768-1-AP, Proteintech) followed by incubation with a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG secondary antibody (1:5000 dilution; cat no. SA00001-1, Proteintech). GAPDH level was used as a loading control for all samples66, and was detected with a rabbit anti-GAPDH primary antibody (1:5000 dilution; cat no. 10494-1-AP, Proteintech) and an HRP-conjugated goat anti-rabbit IgG secondary antibody (1:5000 dilution; cat no. SA00001-2, Proteintech). Protein bands were visualized using the Odyssey infrared imaging system (LI-COR Biosciences), and densitometric analysis was performed to quantify protein expression levels. The uncropped and unprocessed scans are provided in the Source Data file.
Molecular dynamics simulations
Molecular dynamics (MD) simulations were carried out using GLT25D1 structure, both with and without UDP-Galactose molecule bound to the NGT domain. Force field parameters for GDU were generated using Antechamber. The simulations were performed with the Amber20 software package, employing the ff19SB force field for the protein and the TIP3P water model. System preparation was conducted using the LEaP module, where the protein–ligand complex was solvated in a truncated octahedral water box with a minimum buffer distance of 12 Å from the solute to the box edge. Sodium and chloride ions were added to neutralize the system and adjust the ionic strength to ~150 mM.
Energy minimization was performed in two stages: first, with positional restraints on the solute using the conjugate gradient algorithm for 2000 steps, followed by an unrestrained minimization of the entire system for 2500 steps (1000 steps of steepest descent and 1500 steps of conjugate gradient). The minimized system was then gradually heated from 0 to 300 K over 100 ps under constant-volume (NVT) conditions, with weak restraints on the solute. Finally, 100 ns production MD simulations were conducted under constant-pressure (NPT) conditions using the pmemd.CUDA engine in Amber20. All simulations were performed with four independent replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-63937 (Dimer of GLT25D1), EMD-63938 (Hexamer of GLT25D1), and EMD-63936 (Ternary complex of GLT25D1). The atomic coordinates have been deposited in the Protein Data Bank (PDB) under accession codes 9U7I (Dimer of GLT25D1), 9U7J (Hexamer of GLT25D1), and 9U7H (Ternary complex of GLT25D1). This study analysed several protein structures publicly available from the PDB under the accession codes 8ZGE and 9EVJ. The initial coordinate files and final output coordinate files of the MD simulation have been provided in a public repository, Zenodo, under [https://doi.org/10.5281/zenodo.18220626]67. Source data are provided with this paper.
References
Shoulders, M. D. & Raines, R. T. Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 (2009).
Bielajew, B. J., Hu, J. C. & Athanasiou, K. A. Collagen: quantification, biomechanics, and role of minor subtypes in cartilage. Nat. Rev. Mater. 5, 730–747 (2020).
Myllyharju, J. & Kivirikko, K. I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 (2004).
Schegg, B., Hülsmeier, A. J., Rutschmann, C., Maag, C. & Hennet, T. Core glycosylation of collagen is initiated by two beta(1-O)galactosyltransferases. Mol. Cell Biol. 29, 943–952 (2009).
Hennet, T. Collagen glycosylation. Curr. Opin. Struct. Biol. 56, 131–138 (2019).
Yamauchi, M. & Sricholpech, M. Lysine post-translational modifications of collagen. Essays Biochem. 52, 113–133 (2012).
Ito, S. & Nagata, K. Quality control of procollagen in cells. Annu. Rev. Biochem. 90, 631–658 (2021).
Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 21, 729–749 (2020).
Stawikowski, M. J., Aukszi, B., Stawikowska, R., Cudic, M. & Fields, G. B. Glycosylation modulates melanoma cell α2β1 and α3β1 integrin interactions with type IV collagen. J. Biol. Chem. 289, 21591–21604 (2014).
Lauer-Fields, J. L., Malkar, N. B., Richet, G., Drauz, K. & Fields, G. B. Melanoma cell CD44 interaction with the alpha 1(IV)1263-1277 region from basement membrane collagen is modulated by ligand glycosylation. J. Biol. Chem. 278, 14321–14330 (2003).
Ramshaw, J. A. M., Shah, N. K. & Brodsky, B. Gly-X-Y tripeptide frequencies in collagen: a context for host-guest triple-helical peptides. J. Struct. Biol. 122, 86–91 (1998).
Kamath, R. S. et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237 (2003).
Simmer, F. et al. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 1, E12 (2003).
Baumann, S. & Hennet, T. Collagen accumulation in osteosarcoma cells lacking GLT25D1 collagen galactosyltransferase. J. Biol. Chem. 291, 18514–18524 (2016).
Terajima, M. et al. Role of glycosyltransferase 25 domain 1 in type I collagen glycosylation and molecular phenotypes. Biochemistry 58, 5040–5051 (2019).
He, L. et al. Down-regulation of GLT25D1 inhibited collagen secretion and involved in liver fibrogenesis. Gene 729, 144233 (2020).
Geister, K. A. et al. Loss of function of Colgalt1 disrupts collagen post-translational modification and causes musculoskeletal defects. Dis. Model. Mech. 12, dmm037176 (2019).
Mori, K., Suzuki, T., Miura, K., Dohmae, N. & Simizu, S. Involvement of LH3 and GLT25D1 for glucosyl-galactosyl-hydroxylation on non-collagen-like domain of FGL1. Biochem. Biophys. Res. Commun. 560, 93–98 (2021).
Richards, A. A. et al. Adiponectin multimerization is dependent on conserved lysines in the collagenous domain: evidence for regulation of multimerization by alterations in posttranslational modifications. Mol. Endocrinol. 20, 1673–1687 (2006).
Webster, J. A. et al. Collagen beta (1-O) galactosyltransferase 1 (GLT25D1) is required for the secretion of high molecular weight adiponectin and affects lipid accumulation. Biosci. Rep. 37, BSR20170105 (2017).
Liefhebber, J. M., Punt, S., Spaan, W. J. & van Leeuwen, H. C. The human collagen beta (1-O)galactosyltransferase, GLT25D1, is a soluble endoplasmic reticulum-localized protein. BMC Cell Biol. 11, 33 (2010).
Miyatake, S. et al. Biallelic COLGALT1 variants are associated with cerebral small vessel disease. Ann. Neurol. 84, 843–853 (2018).
Teunissen, M. W. A. et al. Biallelic variants in the COLGALT1 gene cause severe congenital porencephaly: a case report. Neurol. Genet 7, e564 (2021).
Mönkäre, S. et al. Genetic analysis reveals novel variants for vascular cognitive impairment. Acta Neurol. Scand. 146, 42–50 (2022).
Perrin-Tricaud, C., Rutschmann, C. & Hennet, T. Identification of domains and amino acids essential to the collagen galactosyltransferase activity of GLT25D1. PLoS ONE 6, e29390 (2011).
Lairson, L. L., Henrissat, B., Davies, G. J. & Withers, S. G. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev. Biochem 77, 521–555 (2008).
Van den Steen, P. E. et al. Generation of glycosylated remnant epitopes from human collagen type II by gelatinase B. Biochemistry 43, 10809–10816 (2004).
Kuwabara, N. et al. Carbohydrate-binding domain of the POMGnT1 stem region modulates O-mannosylation sites of alpha-dystroglycan. Proc. Natl. Acad. Sci. USA 113, 9280–9285 (2016).
Yu, H. et al. Structural analysis of Notch-regulating Rumi reveals basis for pathogenic mutations. Nat. Chem. Biol. 12, 735–740 (2016).
Chen, C. I. et al. Structure of human POFUT2: insights into thrombospondin type 1 repeat fold and O-fucosylation. EMBO J. 31, 3183–3197 (2012).
Spiro, M. J. & Spiro, R. G. Studies on the biosynthesis of the hydroxylsine-linked disaccharide unit of basement membranes and collagens. II. Kidney galactosyltransferase. J. Biol. Chem. 246, 4910–4918 (1971).
Myllylä, R., Risteli, L. & Kivirikko, K. I. Assay of collagen-galactosyltransferase and collagen-glucosyltransferase activities and preliminary characterization of enzymic reactions with transferases from chick-embryo cartilage. Eur. J. Biochem. 52, 401–410 (1975).
Kivirikko, K. I. & Myllylä, R. Collagen glycosyltransferases. Int. Rev. Connect. tissue Res. 8, 23–72 (1979).
Guo, H. F. et al. A collagen glucosyltransferase drives lung adenocarcinoma progression in mice. Commun. Biol. 4, 482 (2021).
Leitinger, B. Pulling the strings of tumor collagen. Nat. Cancer 3, 9–10 (2022).
de Oliveira, J. T. et al. Coordinated expression of galectin-3 and galectin-3-binding sites in malignant mammary tumors: implications for tumor metastasis. Glycobiology 20, 1341–1352 (2010).
Liu, S. W., Yu, Y., Wang, Y., Zhu, B. Y. & Han, B. M. COLGALT1 is a potential biomarker for predicting prognosis and immune responses for kidney renal clear cell carcinoma and its mechanisms of ceRNA networks. Eur. J. Med. Res. 27, 122 (2022).
Qasba, P. K., Ramakrishnan, B. & Boeggeman, E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem Sci. 30, 53–62 (2005).
De Marco, M. et al. Molecular structure and enzymatic mechanism of the human collagen hydroxylysine galactosyltransferase GLT25D1/COLGALT1. Nat. Commun. 16, 3624 (2025).
Scietti, L. et al. Molecular architecture of the multifunctional collagen lysyl hydroxylase and glycosyltransferase LH3. Nat. Commun. 9, 3163 (2018).
Wilson, L. F. L. et al. The structure of EXTL3 helps to explain the different roles of bi-domain exostosins in heparan sulfate synthesis. Nat. Commun. 13, 3314 (2022).
Leisico, F. et al. Structure of the human heparan sulfate polymerase complex EXT1-EXT2. Nat. Commun. 13, 7110 (2022).
Katz, M. & Diskin, R. Structural basis for matriglycan synthesis by the LARGE1 dual glycosyltransferase. PLoS ONE 17, e0278713 (2022).
Peng, J. et al. The structural basis for the human procollagen lysine hydroxylation and dual-glycosylation. Nat. Commun. 16, 2436 (2025).
Silva, D., Santos, G., Barroca, M., Costa, D. & Collins, T. Inverse PCR for site-directed mutagenesis. Methods Mol. Biol. 2967, 223–238 (2023).
Wu, H. et al. Chemical synthesis and biological evaluations of adiponectin collagenous domain glycoforms. J. Am. Chem. Soc. 143, 7808–7818 (2021).
Myllylä, R., Risteli, L. & Kivirikko, K. I. Collagen glucosyltransferase. Partial purification and characterization of the enzyme from whole chick embryos and chick-embryo cartilage. Eur. J. Biochem. 61, 59–67 (1976).
Maliepaard, J. C. L., Damen, J. M. A., Boons, G. & Reiding, K. R. Glycoproteomics-compatible MS/MS-based quantification of glycopeptide isomers. Anal. Chem. 95, 9605–9614 (2023).
Falck, D. & Wuhrer, M. GlYcoLISA: antigen-specific and subclass-specific IgG Fc glycosylation analysis based on an immunosorbent assay with an LC-MS readout. Nat. Protoc. 19, 1887–1909 (2024).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, 22 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63 (2014).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Pettersen, E. F. et al. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. Struct. Biol. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. Struct. Biol. 66, 12–21 (2010).
Yuan, S., Chan, H. C. S., & Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 7, e1298 (2017).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Yu, H. W. et al. Greater chemical signaling in root exudates enhances soil mutualistic associations in invasive plants compared to natives. New Phytol. 236, 1140–1153 (2022).
Hou, H. et al. Sensitive and high-throughput isomer-specific analysis of human milk oligosaccharides using UPLC-MS/MS and its application to secretor status assignment. Carbohydr. Polym. 352, 123154 (2025).
Wu, T. et al. Protein-adaptive differential scanning fluorimetry using conformationally responsive dyes. Nat. Biotechnol. 43, 106–113 (2025).
Wu, Y. H. et al. Glyceraldehyde-3-phosphate dehydrogenase: a universal internal control for Western blots in prokaryotic and eukaryotic cells. Anal. Biochem. 423, 15–22 (2012).
Sun, H. et al. Molecular basis of collagen galactosylation by GLT25D1. Zenodo https://doi.org/10.5281/zenodo.18220626 (2026).
Acknowledgements
We thank all staff members of the Cryo-EM Center, Southern University of Science and Technology, for their assistance in data collection. This work was mainly supported by the National Natural Science Foundation of China (92478121 to H.Y.), the Shenzhen Medical Research Fund (A2303054 to X.L.) and the Natural Science Fund for Distinguished Young Scholars of Hubei Province (2024AFA044 to M.Z.). Other funds include National Natural Science Foundation of China (92053112, 31971148 to H.Y., 32100575 to M.Z., 0204K0010 to X.H.), Shenzhen Medical Research Fund (B2302039 to M.J.Z.), the Fundamental Research Funds for the Central Universities (5003510141, 5003510056 to H.Y. and 5003510112 to M.Z.), Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (GZB20240245 to X.H.) and Science, Technology and Innovation Committee of Shenzhen General Program (JCYJ202308070093505010 to X.L.). This work was also supported by the advanced computing resources provided by the Supercomputing Center of the USTC.
Author information
Authors and Affiliations
Contributions
M.Z., M.J.Z., X.T.L. and H.Y. conceived and designed experiments. H.S., M.Z. and X.T.L. performed most of the experiments. Y.S., D.Z., Z.H., P.Y., Y.Y., J.L., P.L., X.H. and Y.K. helped with the sample preparation and functional experiments. J.C. and C.W. performed the molecular dynamics simulations. H.S., M.Z., M.J.Z., X.T.L. and H.Y. analyzed the data. M.Z., M.J.Z. and H.Y. wrote the manuscript with the help of all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks James Blaza, Ramon Hurtado-Guerrero, David Kwan, Guang-Yu Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, H., Zhang, M., Shi, Y. et al. Molecular basis of collagen galactosylation by GLT25D1. Nat Commun 17, 2426 (2026). https://doi.org/10.1038/s41467-026-69234-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69234-1
