
Abstract
Influenza A viruses (FLUAV) utilize sialic acid to enter host cells via the envelope’s hemagglutinin (HA), and its affinity for host-specific sialic acid linkage configuration is a major host range determinant. However, some FLUAV subtypes (H17, H18, H19) use the major histocompatibility complex class II (MHCII) as an entry receptor instead of sialic acid (SA), challenging our knowledge about FLUAV tropism and interspecies transmission potential. Here, we show that H3N2 viruses can use MHCII as an alternative entry receptor in a host-specific manner, and adaptation of human viruses to pigs increases affinity for the MHCII swine leukocyte antigen (SLA). By using two prototypic human-seasonal (hVIC/11) and swine-adapted (sOH/04) H3N2 viruses we found that expression of the human (HLA) but not the swine MHCII conferred replication of hVIC/11 in desialylated, non-susceptible cells. Further, expression of SLA in non-susceptible cells conferred susceptibility to infection by sOH/04. Introduction of point mutations near the hVIC/11 HA receptor-binding site (RBS) allowed the use of both human and swine MHCII. Our findings revealed that MHCII can serve as a sialic acid-independent entry receptor to H3N2 FLUAV in a host-specific manner, with potential implications for the viral pathogenesis and adaptation to a new species.
Similar content being viewed by others

Introduction
Sialic acid (SA) is the canonical receptor used by influenza A viruses (FLUAV). These SAs on the cell surface are recognized by the viral hemagglutinin (HA) protein to promote entry and begin the replication cycle1,2. The SA structure is linked to FLUAV host range since avian-origin FLUAVs mainly recognize SA linked to galactose by an α2,3 linkage (α2,3-SA) while mammalian-origin FLUAVs recognize α2,6-linked SA (α2,6-SA)3. Therefore, mutations in the HA protein in or near the receptor-binding site (RBS) can modify the binding preference of the virus4,5,6 and may lead to a switch in the host specificity3. By contrast, recently discovered bat-origin H17 and H18 HA subtypes, as well as duck-origin H19, were shown to exclusively use the Major Histocompatibility Complex class II (MHCII) as an entry receptor7,8,9. This interaction is completely independent of SA as multiple glycan array studies have shown H17-H19 fail to bind to all glycans tested9,10. Interestingly, H17 viruses displayed a species-specific affinity for MHCII in which the human leukocyte antigen isotype DR (HLA-DR) allowed efficient infection of non-permissive cells compared to cells expressing the swine homologue (SLA-DR)8. Conserved residues in the α1, α2, and β1 domains of the HLA-DR molecule were shown to be critical for H18N11 infection of human cells11. More recently, human H2N2 and related avian H2N2 FLUAV were shown to possess dual receptor specificity for SA and MHC II, with entry via MHC class II being independent of sialic acid12. This suggests that MHCII could play a role in the host range of FLUAV, although the ability of other SA-binding HA subtypes to use MHCII as an entry receptor remains unknown.
Pigs are a natural host of FLUAV and have historically been pointed out as important intermediary hosts for the generation of viruses with pandemic potential13. Frequent spillover of FLUAV between humans and swine, followed by subsequent evolution of a few of these strains in the new host, has impacted the epidemiology of circulating strains in both species14,15. We previously showed that adaptation of a human-origin H3N2 virus to pigs selected for a single HA mutation (A138S, H3 numbering) that increased affinity for porcine alveolar macrophages (PAMs) in vivo compared to the original HA16. This ultimately led to apoptosis and depletion of this cell population in the lungs of infected pigs. Additionally, we have observed that a swine (sOH/04) but not a human-adapted (hVIC/11) H3N2 virus can efficiently infect alveolar macrophages in vivo17,18. Considering that PAMs express high quantities of MHCII on the cell surface, we investigated the ability of hVIC/11 and sOH/04 to use HLA-DR or SLA-DR as receptors to initiate infection. We found that both viruses can use MHCII as an entry receptor in addition to SA in a host-specific manner. Moreover, the introduction of point mutations in the hVIC/11 HA RBS changed the virus host range and allowed utilization of the SLA-DR. These mutations also affected SA binding, suggesting that the MHCII-HA interaction is mediated by the RBS or its surroundings and that mutations in or near this pocket have a dual impact on the specificity of the virus for SA and MHCII.
Results
MHCII is needed for the infection of porcine alveolar macrophages (PAMs) by swine-adapted H3N2 FLUAV in the absence of SAs
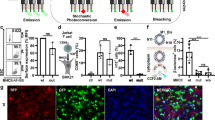
To investigate whether SLA-DR-mediated entry plays a role in FLUAV infection in vivo, we leveraged samples from a previous study16 that showed significant tropism and replication differences between human-origin (hVIC/11) and swine-adapted (sOH/04) FLUAVs in the lungs of pigs. Analysis of lung samples from sOH/04-infected pigs revealed a strong colocalization between SLA-DR and FLUAV HA antigens, suggesting that SLA-DR+ cells might be susceptible to infection with the swine-adapted FLUAV in vivo (Fig. 1a). hVIC/11 was not included in the analysis as it fails to infect swine lungs16. However, it cannot be ruled out that the co-localization observed was due to PAMs’ intrinsic phagocytic activity. To further investigate the role of MHCII during FLUAV infection, we performed infections using alveolar macrophages collected and stored from naïve pigs in a previous study16. Since SA is the primary receptor used by H3N2 FLUAV, depletion of these receptors from PAMs was needed to evaluate the role of MHCII during FLUAV entry. Two methods were evaluated to remove SA on the cell surface: chemical oxidation and enzymatic SA removal with the NanI neuraminidase from C. perfringens, known to cleave both 2,3- and 2,6-Neu5Ac19 and Neu5Gc20. SA oxidation with sodium periodate (NaIO4) was highly cytotoxic (Supplementary Fig. 1a) while SA removal using neuraminidase treatment showed little cytotoxicity (Supplementary Fig. 1b). After a 24-h incubation with 100 mU/mL of neuraminidase (100% cell viability), most of α2,3 and α2,6 SA was successfully removed from PAMs (Fig. 1b and Supplementary Fig. 1c). Further, the neuraminidase treatment did not affect SLA-DR expression in these cells (Fig. 1c). Inoculation of untreated PAMs with sOH/04 and hVIC/11 resulted in replication of both viruses, although hVIC/11 displayed lower titers overall compared to sOH/04 (Fig. 1d, e), particularly at 48 h post infection (hpi). Neuraminidase treatment partially reduced replication of sOH/04 and hVIC/11. Infection of PAMs was significantly reduced only when both SA and SLA-DR were made unavailable by a neuraminidase treatment coupled with an anti-SLA-DR antibody incubation, in a dose-dependent manner (Fig. 1f). Incubation with a control antibody (anti-GFP) did not affect replication in desialylated PAMs (Fig. 1g). These results suggest that FLUAV can infect PAMs in a SA-independent manner, and these infections might be dependent on MHCII availability.

a Representative (n = 3 different animals) right cranial lung samples from mock and sOH/04-infected pigs were deparaffinized, rehydrated and stained for SLA-DR (green) and HA (red) detection. Nuclei were stained using DAPI (blue). Scale bar represents 30 μm. b Neuraminidase treatment efficiently removed sialic acid from the surface of PAMs. Macrophages were incubated with 100 mU/mL of neuraminidase, and sialic acid was stained at 24 h post-treatment. Scale bar represents 30 μm. Imaging was performed three independent times. c SLA-DR expression in desialylated and non-desialylated PAMs was evaluated at 24 h post-neuraminidase treatment by immunofluorescence using an αSLA-DR specific antibody. Scale bar represents 30 μm. Representative images were selected after three independent experiments. PAMs were left untreated or desialylated and further infected ex vivo for 1 h at an MOI of 0.01 of either sOH/04 (blue, d) or hVIC/11 (orange, e) and viral titers in the supernatant were determined at 0, 12, 24, 48, and 72 hpi. Data are presented as the mean ± SEM of three independent experiments. f MHCII availability is critical for sOH/04 infection of desialylated PAMs. 24 h after deacetylation, PAMs were incubated with concentrations ranging 0–10 μg/mL of an αSLA-DR specific antibody or with 10 μg/mL of an anti-GFP control antibody (g) for 1 h before infection. PAMs were infected at an MOI of 0.1 for 1 h, and then cells were supplemented with fresh media containing the desired antibody concentration, and viral titers were measured at 48 hpi. Data are presented as the mean ± SEM of three independent experiments. L.D. limit of detection.
Transient expression of MHCII in desialylated cells enables FLUAV infection and replication
To confirm that FLUAV uses MHCII as an entry receptor, we transfected HEK-293T cells with plasmids encoding the alpha and beta chains of either HLA-DR or SLA-DR. HEK-293T cells were selected due to their high transfection efficiency21. Transient expression of MHCII was demonstrated by immunofluorescence analysis (Supplementary Fig. 2a). Subsequently, transfected cells were treated with exogenous neuraminidase (Supplementary Fig. 2b), and after a 24-h neuraminidase treatment, both α2,6-SA and α2,3-SA were detected on the cell surface (Supplementary Fig. 3). MHCII+ HEK-293T cells bearing or lacking SAs were inoculated with sOH/04 and hVIC/11. Under SA-depleted conditions, FLUAV only infected MHCII-expressing cells (Fig. 2a). This was further confirmed by flow cytometry (Supplementary Fig. 4a). Moreover, neuraminidase treatment completely prevented FLUAV infection when cells were transfected with an empty plasmid (Supplementary Fig. 4a, b). Levels of FLUAV-positive cells among those bearing SA were similar regardless of MHCII expression (Supplementary Fig. 4c). When only MHCII-expressing cells were considered (Fig. 2b, c), we found a clear trend in which hVIC/11 preferably infected HLA-DR-expressing cells. Similarly, transient expression of SLA-DR in desialylated cells significantly increased the affinity of sOH/04 to higher levels than when HLA-DR was present. Quantitatively, hVIC/11 infected 17.9% of HLA-DR+ cells vs 5.1%of SLA-DR-expressing cells (Fig. 2d). On this line, sOH/04 preferentially infected SLA-DR+ cells in which 22.7% of cells were infected, contrasting with the 6.7% of infected cells when HLA-DR was present (Fig. 2d). Control infections performed in untreated cells or desialylated cells transfected with an empty plasmid supported that the transfection process alone did not allow FLUAV infection of non-sialilated cells (Fig. 2e). Notably, sOH/04 infected a higher percentage of cells when SLA-DR was the only available receptor compared to conditions where only SA was present (Fig. 2e, p = 0.02). Alignment of the HLA-DR and SLA-DR alpha and beta chains showed multiple amino acid differences between the two homologue genes that could account for this effect (Supplementary Fig. 5). As a negative control, we used the 2009 pandemic H1N1 A/California/04/2009 virus (pH1N1), a subtype previously shown not to use MHCII as an entry receptor12. pH1N1 successfully infected untreated HEK-293T cells but failed to infect desialylated HEK-293T (Supplementary Fig. 6a), even when HLA-DR or SLA-DR were expressed (Supplementary Fig. 6b, c). After recognition of SA, FLUAV is internalized in endosomes that are further acidified, allowing the virus particle disassembly and genome release into the cytoplasm22. Therefore, we evaluated if FLUAV follows a similar route after interacting with MHCII. Prevention of endosomal acidification by ammonium chloride (NH4Cl)23 (Fig. 2f) and bafilomycin A124 (Fig. 2g) strongly reduced FLUAV infection of desialylated MHCII+ HEK-293T cells, suggesting that upon interaction with MHCII, FLUAV follows a similar endocytic pathway as when SA is used as a receptor.

a HEK-293T cells were transfected with either HLA-DRA + HLA-DRB1 or SLA-DRA + SLA-DRB1. MHCII-expressing cells and cells transfected with an empty plasmid were desialylated at 24 h post-transfection for 24 h. Cells were infected with either sOH/04 or hVIC/11 and infections were incubated for 24 h before processing. Subsequently, cells were stained for MHCII (pink), sialic acid (red), and FLUAV (green). Co-localization between MHCII and FLUAV can be seen as white. Scale bar= 30 μm. Images are representative of three independent experiments. Transient expression of MHCII allows FLUAV entry into desialylated HEK-293T cells. Cells were transfected with either HLA-DR (b) or SLA-DR (c) and further desialylated prior to infection with hVIC/11 or sOH/04. At 24 hpi, cells were fixed and stained for MHCII and FLUAV. The percentage of FLUAV-positive cells was determined by flow cytometry (n = 3 independent experiments). d Percentage of FLUAV-positive cells among desialylated MHCII-expressing cells determined by flow cytometry represented as the mean ± SEM of three independent experiments. sOH/04 is shown in blue while hVIC/11 can be seen in orange. e Percentage of infected HEK-293T cells (regardless of MHCII expression) determined by flow cytometry. Data are presented as the mean ± SEM of three independent experiments. f MHCII-expressing cells were incubated with 20 mM NH4Cl for 1 h prior to infection with either sOH/04 (blue) or hVIC/11 (orange) at an MOI of 1. Infections were maintained at 37 °C for 24 h in presence of NH4Cl and intracellular vRNA was determined at 24 hpi by RT-qPCR. Data are presented as the mean ± SEM of n = 3 independent experiments. g Transfected HEK-293T cells were incubated with 10 nM Bafilomycin A1 before infection and were subsequently infected at an MOI of 1 with sOH/04 (blue) or hVIC/11 (orange). Intracellular FLUAV vRNA was quantified at 24 hpi by RT-qPCR. Data are presented as the mean ± SEM of three independent experiments. L.D. limit of detection. Untreated: non-transfected, non-desialylated cells. Empty vector: desialylated cells transfected with an empty pCAGGS plasmid. HLA-DR: desialylated cells expressing HLA-DR. SLA-DR: desialylated cells expressing SLA-DR.
HEK-293T cells allow entry of FLUAV but they do not support efficient FLUAV replication. Therefore, to assess whether MHCII allows replication of FLUAV in desialylated airway epithelial cells, we used A549 cells due to their high transfection efficiency, high permissibility to FLUAV replication, and as a representative of the primary target cells of FLUAV in mammalian hosts. To fully assess the SA independence of MHCII-mediated entry, we suppressed cytidine monophosphate N-acetylneuraminic acid synthetase (CMAS) expression by shRNA silencing. CMAS is an enzyme that catalyzes the activation of Neu5Ac to CMP-Neu5Ac, and its suppression has been shown to abolish FLUAV infection in A549 cells25. Lentivirus-mediated shRNA-CMAS transduction efficiently prevented SA incorporation on the cell surface (Fig. 3a and Supplementary Fig. 7a, b). These shRNA-CMAS cells were non-susceptible to either hVIC/11 or sOH/04 infection, and transfection with a shRNA-resistant CMAS cDNA construct (shRNA-CMAS + CMAS) restored the susceptibility of the cells to FLUAV infection alongside SA presentation on the surface (Supplementary Fig. 7c, d). Cells transduced with a scrambled shRNA control (shRNA-scrambled) showed no differences in SA content and FLUAV replication compared to the parental A549 cells. SLA-DR and HLA-DR expression was consistently observed on 70% of the cells on average (Fig. 3b, c). CMAS repression prevented H3N2 FLUAV replication. However, MHCII expression granted replication in a species-specific manner: HLA-DR expression allowed replication of hVIC/11 in shRNA-CMAS cells (Fig. 3d) while SLA-DR expression enabled replication of sOH/04 to similar levels as the shRNA-CMAS + CMAS control group (Fig. 3e). Interestingly, sOH/04 also replicated in HLA-DR+ cells, although titers at all timepoints were lower compared to infection in SLA-DR+ cells. We then tested whether anti-MHCII antibodies inhibit FLUAV replication in desialylated MHCII+ A549 cells. The anti-HLA-DR antibody strongly reduced hVIC/11 titers in shRNA-CMAS HLA-DR+ cells (Fig. 3f), while the anti-SLA-DR antibody showed a similar effect in sOH/04 titers in shRNA-CMAS SLA-DR+ cells (Fig. 3g). Finally, we evaluated if infection of MHCII+ cells can induce cell death. hVIC/11 replication led to a significant increase in shRNA-CMAS HLA-DR+ cells mortality at 48 hpi (Fig. 3h), similar to the shRNA-scrambled and shRNA-CMAS + CMAS control groups. Conversely, sOH/04 replication resulted in a significant increase in cell death in SLA-DR+ cells, although it also affected HLA-DR+ cells’ viability (Fig. 3i). Altogether, these results show that MHCII can be used as an entry receptor, and MHCII-mediated entry of FLUAV in cells lacking SA leads to productive infection and cell death.

a A549 cells were transduced with a lentivirus encoding a shRNA targeting CMAS (shRNA-CMAS) or a shRNA-scrambled negative control. α2,6-SA and α2,6-SA levels on the cell surface were evaluated by lectin staining and flow cytometry. b MHCII expression in A549 cells was evaluated by flow cytometry. Cells were transfected with equimolar quantities of either HLA-DRA + HLA-DRB1 or SLA-DRA + SLA-DRB1 and MHCII expression was assessed at 48 h post-transfection. c MHCII expression levels on shRNA-CMAS A549 cells were quantified at 48 h post transfection by flow cytometry. shRNA-CMAS cells transfected with an empty plasmid (blue), HLA-DR-expressing plasmids (black), SLA-DR-expressing plasmids (yellow), or reconstituted with a shRNA-resistant CMAS cDNA (white) were infected with either hVIC/11 (d) or sOH/04 (e). Viral titers were measured in the supernatant at 0, 12, 24, 48, and 72 hpi by RT-qPCR. Infection of shRNA-scrambled A549 cells was included as both shRNA and transduction controls (red). L.D= limit of detection. Data are presented as the mean ± SEM of three independent experiments. Similarly, shRNA-scrambled or shRNA-CMAS cells were transfected with either HLA-DR or SLA-DR, were incubated with 10 μg/mL of an anti-HLA-DR, anti-SLA-DR, or a control antibody (anti-GFP) for 1 h prior to infection with hVIC/11 (f) or sOH/04 (g). At 48 hpi viral titers in the supernatant were quantified by RT-qPCR. Data are presented as the mean ± SEM of three independent experiments. The effect of FLUAV replication in either shRNA-scrambled or shRNA-CMAS MHCII-expressing cells on the cell viability was evaluated for hVIC/11 (h) and sOH/04 (i). Cells were transfected and infected for further determination of cell viability at 48 hpi. For figures panels h and i, data is presented as the mean ± SEM of n = 3 independent experiments. Mock infection: non-infected cells. Empty vector: Cells transfected with an empty pCAGGS plasmid. HLA-DR: Cells expressing HLA-DR. SLA-DR: Cells expressing SLA-DR. CMAS: Cells transfected with a shRNA-resistant CMAS cDNA.
Mutations near the sialic acid receptor binding site of H3N2 FLUAV HA alter species-specificity of MHCII-mediated entry
H2N2 FLUAV has been shown to interact with MHCII via the HA1 domain12. Comparison of hVIC/11 and sOH/04 HA revealed several amino acid differences in the HA1 domain (Supplementary Fig. 8), and no obvious pattern could be detected that could account for differences in their MHCII binding preference. Nonetheless, we have previously shown that adaptation of hVIC/11 to pigs selected for three mutations within the HA RBS: A138S, V186G, and F193Y (H3 numbering), which resulted in increased replication in SA-bearing swine cells18. These mutations spontaneously appeared individually in different mutant viruses after infection of pigs with hVIC/1118. All these residues are located in the surroundings of the SA-binding pocket, although they do not directly interact with SA (Fig. 4a). Furthermore, the A138S amino acid change significantly increases affinity for PAMs in vivo16. Therefore, we evaluated the ability of mutants hVIC/11-A138S, hVIC/11-V186G, and hVIC/11-F193Y to utilize either HLA-DR or SLA-DR as a receptor in vitro. Infection of desialylated MHCII+ HEK-293T cells revealed a preference for SLA-DR over HLA-DR (Fig. 4b, c). When only MHCII+ cells were considered, a similar trend was observed, with all three mutant viruses using both HLA-DR and SLA-DR as entry receptors, but with a statistically significant higher preference for the swine homologue (Fig. 4d). Quantification of FLUAV-positive cells among total cells (regardless of MHCII expression) supported that infection of desialylated cells is completely abolished even when cells are transfected with an empty plasmid (Fig. 4e). Evaluation of the replication capacity of the viruses in MHCII-transfected, shRNA-CMAS A549 cells, revealed an increase of the viral titers overtime in both HLA-DR- and SLA-DR-expressing cells, with no differences observed between the MHCII from both species (Fig. 4f–h). This contrasts with hVIC/11 (Fig. 3d), which failed to efficiently replicate in SLA-DR-expressing cells without SA. Overall, these data demonstrate that the introduction of swine-adapting point mutations near the receptor binding site of hVIC/11 increases the affinity of the virus for SLA-DR. This is consistent with previous reports suggesting that these mutations represent an advantage for the virus in the swine host18. Additionally, we tested whether these mutations could also affect the SA-binding preference of the virus. By performing a solid-phase SA receptor-binding assay, we observed that hVIC/11-A138S and hVIC/11-F193Y exhibited higher affinity for α2,6 SA compared to hVIC/11 (Fig. 5a, b, d), although no changes were seen for α2,3 binding. Contrary, hVIC/11-V186G (Fig. 5c) had a reduced binding to both α2,3 and α2,6 compared to all the mutants, including the parental hVIC/11. Taken together, these results demonstrate that mutations in the proximity of the SA RBS can have a dual effect and impact MHCII recognition and SA binding at the same time.

a hVIC/11 HA trimer model made using PyMOL based on previously published crystal structure (PDB: 4WE856). Sialic acid RBS is highlighted in pink while residues 138, 186, and 193 (H3 numbering) are shown in red. HEK-293T cells transfected with HLA-DR- (b) or SLA-DR-expressing plasmids (c) and then desialylated with 100 mU/mL of neuraminidase. Subsequently, cells were infected at an MOI of 0.01 with hVIC/11-A138S, hVIC/11-V186G, or hVIC/11-F193Y. At 24 hpi cells were stained for MHCII (APC) and FLUAV (Alexa488). d FLUAV-positive cells among MHCII-expressing HEK-293T cells was determined by gating live, MHCII+ cells only. Percentages were obtained by flow cytometry. Data is presented as the mean ± SEM of n = 3 independent experiments. e Total amount of FLUAV-infected HEK-293T cells was determined by multi-color flow cytometry. Data are presented as the mean ± SEM of three independent experiments. To evaluate virus replication in A549 cells, shRNA-scrambled (red), shRNA-CMAS cells transfected with an empty plasmid (blue), shRNA-CMAS cells transfected with HLA-DR-encoding plasmids (black), shRNA-CMAS cells transfected with SLA-DR-expressing plasmids (yellow), and shRNA-CMAS cells transfected with a shRNA-resistant CMAS cDNA (white) were infected at an MOI of 0.01 with either hVIC/11-A138S (f), hVIC/11-V186G (g), or hVIC/11-F193Y (h). Viral titers in the supernatant were quantified at 0, 12, 24, 48, and 72 hpi by RT-qPCR. Data are presented as the mean ± SEM of three independent experiments. L.D. limit of detection.

Purified hVIC/11 (a), hVIC/11-A138S (b), hVIC/11-V186G (c), and hVIC/11-F193Y (d) were normalized to 100 HAU and adsorbed in fetuin-coated plates. Adsorbed viruses where then incubated with different concentrations Neu5Acα2-3Galβ1-4GlcNAcβ-PAA-biotin (α2,3 SA) or Neu5Acα2-6Galβ1-4GlcNAcβ-PAA-biotin (α2,6 SA). Data are presented as the mean ± SEM of three independent experiments.
Discussion
Recent methodological advances have increased our understanding of the tropism and receptor-specificity of FLUAV beyond the SA linkage to galactose, including the description of numerous non-canonical entry receptors26. However, these newly identified molecules often rely on SA to act as a functional receptor27,28, with the exception of a subset of phosphoglycans29. Bat-derived H17 and H18 and duck-derived H19 HAs were recently shown to be unable to bind SA and instead use MHCII as a SA-free entry mechanism9,10,30. Interestingly, MHCII was suggested to promote entry of H2N2 FLUAV into dendritic cells31, which was further recently demonstrated12. In this report, we found that MHCII acts as a functional SA-independent entry receptor for two prototypic H3N2 viruses from human- and swine-origin, and that binding is likely mediated by the vicinity of the HA RBS. To our knowledge, this is the first report of MHCII-mediated infection of group 2 HAs. Moreover, affinity for different MHCII homologs seems to be in line with the host from which the virus originated, implying a role of the MHCII complex in the adaptation process of FLUAV to a new host.
Infection of neuraminidase-treated PAMs revealed that the swine-adapted sOH/04 was still able to replicate, but replication was inhibited by blocking SLA-DR. Findings were similar for the human-seasonal hVIC/11, although replication was lower. Previous literature has shown that human and swine alveolar macrophages exhibit similar levels of α2,3- and α2,6-SA32,33. Hence, it is likely that the observed differences in affinity for these swine cells correlate with the ability of the viruses to recognize the SLA-DR more than the affinity for host-specific SA conformations. Moreover, our findings suggest that binding to MHCII in addition to SAs increases the affinity of FLUAV to PAMs but not in an additive effect. Although we were unable to completely remove SA from PAMs, the neuraminidase treatment, coupled with an anti-SLA-DR ab incubation, completely blocked sOH/04 infection. This suggests that the remaining SA was not enough to initiate an infection. A recent report showed that replication of a seasonal H3N2 was not boosted by HLA-DR expression in the presence of SA12. This is in line with our results, in which neither HLA-DR nor SLA-DR enhanced viral replication when SA was present. However, because only a limited number of viruses were tested, it remains unclear whether other H3N2 viruses can utilize MHCII, especially since MHCII binding can vary within a subtype (e.g., H2N2 viruses12). Our data points to SA remaining the primary receptor for these viruses. Since a complete absence of sialylated glycans is unlikely in the swine respiratory tract, MHCII could favor infectivity when glycan-mediated binding is suboptimal, such as during adaptation of human-origin viruses to swine hosts. Glycan array experiments with hVIC/11 HA and its A138S, V186G, F193Y variants show preferential binding to extended human-like tri-LacNAc α2,6 structures34 which are uncommon in swine35, making HA-glycan interactions inefficient. In this context, engagement of MHCII provides a selective alternative entry route, rescuing infectivity and supporting early rounds of replication. This is particularly relevant in macrophages, which express robust levels of MHCII and have a glycan landscape with fewer of both elongated and α2,6-sialylated glycans to support HA binding36. This is supported by enhanced infection of sOH/04 in human-origin cells expressing SLA-DR compared to the same cells containing SA only (Fig. 2e), although the virus did not reach higher titers by using SLA-DR compared to SA (Fig. 3e). This mechanism is consistent with replication of hVIC/11 A138S and sOH/04 in swine alveolar macrophages16, suggesting that MHCII serves as an additional entry factor when glycan-HA compatibility is limited, enabling selective infection of MHCII-positive cells. Further, since our previous findings revealed that MHCII-mediated hVIC/11-A138S infection of macrophages can induce cell death16, this could be a mechanism used by FLUAV to deplete AMs in the lungs16,37 and reduce their antiviral activity38,39,40,41; hence, facilitating virus spread in the respiratory tract. Recent studies have shown that human ciliated, secretory, and goblet epithelial cells can support MHCII-mediated infection12. Identifying analogous MHCII–expressing epithelial cells in the swine respiratory tract would further support this model. Future studies beyond the scope of this research will be needed to confirm this hypothesis. Further, the mechanism by which MHCII-mediated entry occurs and how it differs from the canonical SA-triggered entry remains unknown. However, our data showed that MHCII-mediated entry and infection were disrupted by NH4Cl and bafilomycin A1. These compounds prevent endosomal acidification42 and acid lysosome formation43, respectively, and inhibit FLUAV infection by preventing fusion with the endosome membrane and viral particle disassembly44. Hence, our data suggest that upon MHCII-mediated internalization, FLUAV follows a similar trafficking pathway as occurs with SA-triggered entry.
Similar to H17, H18, and H19 viruses that can use a wide range of MHCII homologs from different bat species, humans, swine, and chickens8, the H3N2 strains used here were shown to utilize both HLA-DR and SLA-DR as entry receptors in HEK-293T cells. As observed in PAMs, this promiscuity may have implications for the amplified tropism of H3N2 viruses that would facilitate host jumps. However, the human-adapted hVIC/11 virus exhibited a preference for HLA-DR, and similarly, the swine-adapted sOH/04 preferentially used SLA-DR. The same pattern was observed in shRNA-CMAS A549 cells. Such host-specificity could have implications on the host range of influenza viruses, but the ability to still bind to MHCII from other species, even if less efficiently, could play a role in facilitating the adaptation to a new host.
In a previous report, we found that the A138S mutation in a hVIC/11 HA increased affinity for PAMs in vivo but did not reach the same level of affinity as the swine-adapted sOH/0416. This effect is consistent with the dual usage of SLA-DR by hVIC/11-A138S and the inability of hVIC/11 to efficiently replicate in SLA-DR-positive cells. Other mutations near the RBS (V186G and F193Y) were also found after transmission in pigs and resulted in a fitness advantage for the viruses in swine respiratory cells18. Here, introduction of these single point mutations in the HA, near the RBS of hVIC/11, modified the MHCII recognition pattern of the virus and allowed utilization of both HLA-DR and SLA-DR. Although in silico and in vitro analyses suggest that the HA binds to MHCII via the head domain11,12, the specific site on the HA protein responsible for MHCII recognition by H17-H19 remains unknown. Our data are in agreement with these observations and suggest that the interaction between MHCII and H3 is mediated by the HA RBS surroundings, as the residues evaluated here do not directly interact with SA but may affect conformation of the pocket45. Interestingly, a recent report found that antibodies targeting the H2 RBS can also inhibit MHCII binding12, which supports that residues near the HA RBS play a role in MHCII binding. Furthermore, we found that all three mutations also affected SA binding, demonstrating that mutations in the HA RBS can have a dual impact on MHCII and SA recognition. However, it is possible that residues 138, 186, and 193 (H3 numbering) do not directly interact with MHCII but simply induce conformational changes in the RBS that could indirectly favor or prevent recognition of MHCII. Crystallization of the HA-MHCII complex in future studies will be critical to understand the molecular basis of this interaction.
Overall, our results demonstrate that prototypic H3N2 viruses can use the MHCII complex as an alternative entry receptor, resulting in a dual receptor preference (SA and MHCII), and binding to MHCII is likely mediated by the surroundings of the HA RBS. Similar to SA, the affinity of the viruses for MHCII seems to be host-specific and adaptation of human viruses to pigs selects for mutations in the HA that enhance affinity for SLA-DR. Interestingly, during the early stages of adaptation, the virus is still able to utilize both HLA-DR and SLA-DR, enabling the virus to expand its host range. Further, mutations occurring in the vicinity of the HA RBS can affect the affinity of the HA for both SAs and MHCII receptors simultaneously. This is particularly important as MHCII might act as a tropism determinant that selectively promotes infection of antigen-presenting cells and ultimately favors virus replication in the lungs.
Methods
Ethics statement
Animal studies described in this report were approved by the Institutional Animal Care and Use Committee at the University of Georgia (protocol A2019 03-031-Y3-A9). 3-week-old healthy cross-bred pigs were obtained from Midwest Research Swine Inc. (Gibbon, MN) and housed in animal biosafety level 2 conditions according to the Guide for the Care and Use of Agricultural Animals in Research and Teaching. At the end of the study, animals were euthanized following the American Veterinary Medical Association (AVMA) guidelines. Sex was not a variable considered in the study, but both sexes were included.
Cells and viruses
Madin-Darby canine kidney (MDCK) were a kind gift from Dr. Robert Webster. Human lung carcinoma (A549) and human embryonic 293 T (HEK-293T) cells were purchased from the American Type Culture Collection (ATCC). Cells were maintained in Dulbecco’s Modified Eagle's Medium (DMEM, Sigma-Aldrich, St Louis, MO) supplemented with 2mM L-glutamine (Sigma-Aldrich, St Louis, MO), 10% fetal bovine serum (FBS, Sigma-Aldrich, St Louis, MO), and 1% antibiotic/antimycotic (Sigma-Aldrich, St Louis, MO). Cells were incubated at 37 °C under 5% CO2.
FLUAV viruses in this study were generated using an 8-plasmid reverse genetic system as previously described46,47 using an isogenic backbone. HA sequences were confirmed by Sanger sequencing, and viral stocks were made in MDCK cells in opti-MEM containing 1 μg/ml of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Worthington Biochemicals, Lakewood, NJ) at 37 °C. Viral titers were determined by TCID50 using the Reed and Muench method48.
Generation of A549 cytidine monophosphate N-acetylneuraminic acid synthetase (CMAS) deficient cells
CMAS expression was silenced by using a shRNA lentiviral vector. Inverted, self-complementary hairpin DNA oligos targeting the CMAS mRNA (shRNA-CMAS: 5’-AAGAGTGCTTGAAGAGAGTGG-3’) were cloned into a pLKO.1 plasmid (kindly donated by David Root, Addgene plasmid #10878) under the control of the human RNA polymerase III U6 promoter49. To generate lentiviruses encoding CMAS shRNA, or the scrambled shRNA negative control (kindly donated by David Sabatini50, addgene plasmid # 1864), HEK-293T cells were co-transfected with pCMVdr8.2 dvpr, pCMV-VSV-G, and the corresponding pLKO.1 shRNA plasmid. Supernatants were collected at 24, 48, and 72 h post-transfection. Supernatants were pooled, and cell debris was discarded by centrifugation at 500 x g for 5 min and further filtration through a 0.45 μm filter. Lentiviruses were concentrated using the Lenti-X concentrator (Takara Bio, Mountain View, CA) for 6 h and resuspended in 1/100th of the original volume according to the manufacturer’s instructions. A549 cells were transduced with pLKO.1-shRNA-CMAS-puro- or pLKO.1-scrambled-shRNA-puro-encoding lentiviruses with 8 μg/mL polybrene (Sigma-Aldrich, St Louis, MO). At 48 h post-transduction, cells were incubated with media containing 1 μg/mL puromycin (Thermo Fisher Scientific, Waltham, MA). Successful CMAS silencing was confirmed by measuring SA levels on the cell surface using flow cytometry and immunofluorescence as described below. Flow cytometry data was analyzed with FlowJo v10.10.0 and confocal images with ImageJ v1.54 p and Fiji v2.16.0.
Porcine alveolar macrophage isolation
Swine alveolar macrophages were collected and isolated as previously described51,52 with minor modifications. Three-week-old crossbred pigs were obtained from Midwest Research Swine Inc. (Glencoe, MN). Animals were housed under biosafety level 2 conditions at the University of Georgia. After a 7-days acclimatation period, animals were confirmed negative for anti-FLUAV antibodies by competitive ELISA. For euthanasia, pigs were anesthetized with a cocktail of ketamine (6 mg/kg), xylazine (3 mg/kg), and telazol (6 mg/kg), and an intravenous pentobarbital overdose (Euthasol, 87 mg/kg). Lungs were aseptically collected, and alveolar macrophages were harvested by rinsing the lungs twice with calcium-free phosphate-buffered saline (PBS). Samples were pelleted at 500 x g for 10 min at 4 °C and washed twice with calcium-free PBS. Subsequently, the pellet was resuspended in RPMI-1640 media (Sigma-Aldrich, St Louis, MO) supplemented with 10% FBS, 1% antibiotics/antimycotics, 2 mM L-glutamine, and 1% non-essential amino acids. Cells were passed through a 40 μm cell strainer and then incubated for 2 h at 37 °C in 10-cm petri dishes. After incubation, non-adherent cells were discarded, and PAMs were transferred to a new petri dish and incubated at 37 °C under 5% CO2 using the media described above. Further characterization by flow cytometry was performed as previously described16,53. These cells exhibited a CD172aposCD163highMHCIIpos phenotype.
Tissue immunofluorescence
Right cranial lobe samples were collected from infected (sOH/04) and non-infected pigs in neutral-buffered formalin and paraffin embedded in a previous study16. For SLA-DR and HA detection, samples were deparaffinized and stained as previously described54 with minor modifications. Briefly, lung sections were deparaffinized and rehydrated, followed by an antigen retrieval step in citrate buffer (10 mM sodium citrate, pH 6.0) for 40 min. Samples were permeabilized with 0.3% Triton-X100 (Sigma-Aldrich, St Louis, MO) and blocked with 5% Bovine Serum Albumin (BSA, Sigma-Aldrich, St Louis, MO) for 1 h. FLUAV was detected using an anti-Multi-Hemagglutinin (H3N2) polyclonal antibody (eEnzyme, IA-PAN4-0100, Gaithersburg, MD), while an anti-pig SLA class II DR clone 2E9/13 (Bio-Rad, MCA2314GA, Hercules, CA) for MHCII detection was used. Samples were then incubated with a secondary 594-conjugated anti-rabbit antibody in a 1:1000 dilution for HA detection and an Alexa 488-conjugated anti-mice antibody in a 1:1000 dilution for SLA-DR. After a 1-h incubation, tissue samples were stained with 0.5 μg/mL 4’,6-diamine-2-phenylindole (DAPI, Sigma-Aldrich, St Louis, MO) for 15 min. Finally, samples were mounted on glass slides with mounting media (Vector Laboratories, Newark, CA) and imaged using a ZEISS LSM 900 confocal microscope. Images were analyzed with ImageJ v1.54 p and Fiji v2.16.0.
Sialic acid removal of PAMs and HEK-293T cells
α2,3 and α2,6-SA were removed from the cell surface by incubating cells with 100 mU/mL of neuraminidase from Clostridium perfringens (Sigma-Aldrich, St Louis, MO) for 24 h prior to infection. After infection, fresh neuraminidase was added, and new neuraminidase was added every 24 h post-infection. Sialic acid hydrolysis was confirmed by immunofluorescence and flow cytometry using fluorescein-conjugated SNA (Vector Laboratories, Newark, CA) and biotinylated MALII (Vector Laboratories, Newark, CA). Flow cytometry data were analyzed with FlowJo v10.10.0.
Viral growth kinetics
Isolated PAMs were seeded in a 12-well plate at a density of 1.5 × 105 cells/well. Similarly, A549 cells were seeded in a 6-well plate at a density of 5 × 105 cells/well. All cells were incubated at 37 °C overnight and then infected at an MOI of 0.01 for 1 h at 37 °C. After infection, plates were washed three times with PBS and cells were supplemented with Opti-MEM (Thermo Fisher Scientific, Waltham, MA) containing 0.1 μg/mL of TPCK-treated trypsin. Supernatant aliquots were collected at 0, 12, 24, 48, and 72 h post-infection and kept at –80 °C until processing. Viral RNA was extracted using the MagMax-96 AI/ND viral RNA (Thermo Fisher Scientific, Waltham, MA) isolation kit according to manufacturer’s instructions. Purified RNA was used as template for RT-qPCR using the Quantabio qScript XLT One-Step RT-qPCR ToughMix kit (Quantabio, Beverly, MA) and specific primers targeting the M segment of the virus (Supplementary Table 1). FLUAV titers were determined by comparison to a TCID50eq standard curve of an exact virus match of known titer previously determined using the Reed and Muench method.
HLA-DR and SLA-DR antibody blocking
PAMs were seeded in 12-well plates at a density of 1.5 × 105 cells/well while A549 cells were plated in a 6-well plate at a density of 5 × 105 cells/well. Plates were incubated overnight at 37 °C. The next day, cells were incubated for 1 h at 4 °C with an anti-SLA-DR (Bio-Rad, Hercules, CA) or an anti-HLA-DR (Thermo Fisher Scientific, Waltham, MA) antibody at concentrations ranging from 1 to 10 μg/mL or an anti-GFP antibody as a control (Sigma-Aldrich, St Louis, MO). After incubation, cells were infected at an MOI of 0.1 for 1 h at 37 °C in the presence of the indicated antibody concentrations. Finally, cells were washed three times with PBS and supplemented with Opti-MEM containing 0.1 μg/mL of TPCK-treated trypsin and the desired antibody concentrations. Samples were collected at 48 hpi for further virus titration.
Plasmids
HLA-DRA (GenBank accession number: NM_019111.5), HLA-DRB1 (GenBank accession number: NM_002124.4), SLA-DRA (GenBank accession number: MF498819.1), and SLA-DRB1 (GenBank accession number: MF498811.1) gene fragments were synthetized (Twist Bioscience, San Francisco, CA) and amplified by PCR to introduce EcoRI and BglII restriction sites using the primers listed in Supplementary Table 1. pCAGGS plasmid and MHCII fragments were digested using EcoRI and BglII restriction enzymes (New England Biolabs, Ipswich, MA) according to the manufacturer’s instructions for 1 h at 37 °C for further ligation and transformation. Plasmid sequences were confirmed by whole plasmid sequencing.
MHCII expression in A549 and HEK-293T cells
Cells were seeded at a density of 8 × 105 cells/well in 6-well plates and were incubated at 37 °C until a 70% confluence was reached. MHCII subunits were transfected using equimolar quantities of either HLA-DRA and HLA-DRB1 or SLA-DRA and SLA-DRB1 and TransIT LT1 (Mirus Bio, Madison, WI) in a 1:2 ratio DNA: TransIT. HLA-DR and SLA-DR complexes expression was assessed by flow cytometry and immunofluorescence as described below. At 24 h post-transfection, cells were desialylated as described above, and non-sialylated cells were infected at 48 h post-transfection. Flow cytometry data were analyzed with FlowJo v10.10.0 and confocal images with ImageJ v1.54 p and Fiji v2.16.0.
MHCII-mediated entry assessment
HEK-293T cells were seeded and transfected as described above. At 24 h post-transfection, HEK-293T cells were desialylated with 100 mU/mL of neuraminidase from Clostridium perfringens for 24 h at 37 °C. At 48 h post-transfection, cells were infected at an MOI of 0.1 for 1 h at 37 °C. After incubation, the virus-containing supernatant was discarded, and fresh media supplemented with 0.1 μg/mL TPCK-treated trypsin and 100 mU/mL of neuraminidase was added. Neuraminidase was not included in the non-desialylated control groups. Finally, infections were incubated for 24 h at 37 °C for further processing.
Flow cytometry analysis
Twenty-four hours after infection, supernatants from HEK-293T-infected cells were collected, and cells were trypsinized and centrifuged at 500 x g for 5 min. Cells were resuspended in PBS supplemented with 5% FBS and stained with the LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Cells were then fixed with 4% paraformaldehyde for 15 min at room temperature, followed by a 15-min permeabilization with 0.3% Triton-X (Sigma-Aldrich, St Louis, MO). Afterwards, cells were washed three times and incubated for 1 h with the following primary antibodies: Alexa 647-conjugated anti-SLA-DR clone 2E9/13 (Bio-Rad, Hercules, CA), anti-HLA-DR clone LN3 (Thermo Fisher Scientific, Waltham, MA) at a 1:200 dilution in PBS, and an anti-Multi-Hemagglutinin (H3N2) polyclonal antibody (eEnzyme, IA-PAN4-0100, Gaithersburg, MD) at a 1:500 dilution. Following incubation, cells were washed three times with PBS. For simultaneous detection of FLUAV and MHCII, samples were incubated for 1 h with an Alexa 488-conjugated anti-human secondary antibody and an APC-conjugated anti-mice secondary antibody, respectively (Thermo Fisher Scientific, Waltham, MA), both in a 1:1000 dilution. Finally, cells were washed three times with PBS and analyzed using a NovoCyte Quanteon flow cytometer system (Agilent, Santa Clara, CA) using the gating strategy exemplified in Supplementary Fig. 9. Data were analyzed using FlowJo version 10.10.0 (FlowJo, Ashland, OR).
Immunofluorescence staining
HEK-293T and A549 cells were plated in poly-D-Lysine-coated glass slides and incubated at 37 °C until an 80% confluence was reached. Cells were transfected and desialylated as described above. Cells were infected at an MOI of 0.01 and incubated for 24 h at 37 °C as described above. After infection, cells were fixed with 4% formaldehyde for 30 min at room temperature, followed by a 10-min permeabilization with 0.3% Triton-X100 and a 1-h blocking step using 3% bovine serum albumin (BSA, Thermo Fisher Scientific, Waltham, MA) and 0.1% Triton-X100 in PBS. HLA-DR, SLA-DR, and FLUAV were stained using the specific antibodies described above in a 1:200 dilution for 1 h at room temperature in 3% BSA and 0.1% Triton-X100 in PBS. After incubation, samples were washed three times with PBS and incubated with the secondary antibodies described above in a 1:1000 dilution with 0.5 μg/mL 4’,6-diamine-2-phenylindole (DAPI, Sigma-Aldrich, St Louis, MO), 3% BSA, and 0.1% Triton-X100 in PBS for 1 h. Finally, cells were washed 5 times with PBS and mounted on glass slides using mounting media. For SA staining, samples were incubated with either biotin-conjugated or fluorescein-labeled SNA and biotin-conjugated MAL II lectins in a 1:250 dilution in PBS for 30 min prior to permeabilization. After incubation with lectins, cells were incubated for 30 min with a secondary Alexa 594-conjugated streptavidin in a 1:1000 dilution. Samples were imaged using a ZEISS LSM 900 confocal microscope. Images were analyzed with ImageJ v1.54 p and Fiji v2.16.0.
FLUAV entry blockage and cell viability assay
HEK-293T cells were plated and transfected as described above. 1 h before infection, cells were incubated with 20 mM of NH4Cl (Sigma-Aldrich, St Louis, MO) or 10 nM Bafilomycin A1 (Cell Signaling Technologies, Danvers, MA). After incubation, cells were infected at an MOI of 0.1 as described above and kept at 37 °C for 24 h in the presence of the desired inhibitor. At 24 hpi, cells were washed three times with PBS, and total cellular RNA was extracted using the MagMax-96 AI/ND RNA (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. RNA samples were normalized to 1 μg, and FLUAV M segment copy number was determined by RT-qPCR using the Quantabio qScript XLT One-Step RT-qPCR ToughMix kit (Quantabio, Beverly, MA) and a standard curve of an M-cloned segment plasmid of known concentration.
Cellular viability was assessed using the CellTiter Glo Cell Viability kit (Promega, Madison, WI) according to the manufacturer’s instructions.
MHCII-mediated replication in A549 cells
Parental, shRNA-scrambled, and shRNA-CMAS A549 cells were seeded and transfected as described above. At 48 h post-transfection, cells were infected at an MOI of 0.01 for 1 h at 37 °C. After incubation, the supernatant was discarded and replaced with media containing 0.1 μg/mL TPCK-treated trypsin. Time points were collected at 0, 12, 24, 48, and 72 h post-infection, and viral titers were determined by RT-qPCR using a standard curve generated from 10-fold serial dilutions of a virus stock with a known TCID50 titer.
Solid-phase binding assay
Virus affinity for α2,3 and α2,6 SA was assessed as previously described55. Briefly, viruses were purified by ultracentrifugation at 140,000 x g for 3 h using a 20% sucrose cushion. Pelleted viruses were resuspended in TNE buffer (0.01 M Tris, 0.001 M EDTA, 0.1 M NaCl, pH 7.2). Samples were normalized to 128 HAU and adsorbed in fetuin-coated plates at 4 °C overnight. Unbound virus was washed using 0.02% Tween-80 in PBS (washing buffer), and plates were blocked for 2 h with desialylated BSA (BSA-NA). After blocking, plates were washed three times with washing buffer and incubated with different concentrations of Neu5Acα2-3Galβ1-4GlcNAcβ-PAA-biotin (α2,3 SA, Glycotech, Gaithersburg, MD) or Neu5Acα2-6Galβ1-4GlcNAcβ-PAA-biotin (α2,6 SA, Glycotech, Gaithersburg, MD) in 0.02% Tween-80, 0.1% BSA-NA, and 2 μM oseltamivir in PBS (reaction buffer). After a 1-h incubation, plates were washed five times with washing buffer and incubated with HRP-conjugated streptavidin (Thermo Fisher Scientific, Waltham, MA) in a 1:1000 dilution for 1 h at 4 °C. Finally, plates were washed five times with washing buffer and incubated with TMB (Thermo Fisher Scientific, Waltham, MA) for 10 min. Reaction was stopped using a stop solution (Thermo Fisher Scientific, Waltham, MA), and absorbance was measured at 450 nm using a Synergy HTX Multi-Mode Microplate Reader Agilent BioTek, Santa Clara, CA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 10 software. Data is presented as the mean ± standard error of the mean of at least three independent experiments unless stated otherwise in the figure legend. Each data point represents the average of three technical replicates for a total of three biological replicates per experiment. P values were obtained by ordinary two-way ANOVA with Tukey’s multiple comparison test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data relevant to this study are available within the manuscript and have been provided as a source data file and supplementary information. Additionally, the following databases were used in this study: Protein Data Bank (PDB, https://www.rcsb.org/) and National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/) Source data are provided with this paper.
References
Zhao, C. & Pu, J. Influence of host sialic acid receptors structure on the host specificity of Influenza viruses. Viruses 14 https://doi.org/10.3390/v14102141 (2022).
Samji, T. Influenza A: understanding the viral life cycle. Yale J. Biol. Med. 82, 153–159 (2009).
Suzuki, Y. et al. Sialic acid species as a determinant of the host range of influenza A viruses. J. Virol. 74, 11825–11831 (2000).
Glaser, L. et al. A single amino acid substitution in 1918 Influenza virus hemagglutinin changes receptor binding specificity. J. Virol. 79, 11533–11536 (2005).
Nobusawa, E., Ishihara, H., Morishita, T., Sato, K. & Nakajima, K. Change in receptor-binding specificity of recent human influenza A viruses (H3N2): a single amino acid change in hemagglutinin altered its recognition of sialyloligosaccharides. Virology 278, 587–596 (2000).
Obadan, A. O. et al. Flexibility in vitro of amino acid 226 in the receptor-binding site of an H9 subtype influenza A virus and its effect in vivo on virus replication, tropism, and transmission. J. Virol. 93 https://doi.org/10.1128/jvi.02011-18 (2019).
Giotis, E. S. et al. Entry of the bat influenza H17N10 virus into mammalian cells is enabled by the MHC class II HLA-DR receptor. Nat. Microbiol. 4, 2035–2038 (2019).
Karakus, U. et al. MHC class II proteins mediate cross-species entry of bat influenza viruses. Nature 567, 109–112 (2019).
Karakus, U. et al. H19 influenza A virus exhibits species-specific MHC class II receptor usage. Cell Host Microbe https://doi.org/10.1016/j.chom.2024.05.018 (2024).
Sun, X. et al. Bat-derived influenza hemagglutinin H17 does not bind canonical avian or human receptors and most likely uses a unique entry mechanism. Cell Rep. 3, 769–778 (2013).
Olajide, O. M. et al. Evolutionarily conserved amino acids in MHC-II mediate bat influenza A virus entry into human cells. PLoS Biol. 21, e3002182 (2023).
Karakus, U. et al. MHC class II proteins mediate sialic acid independent entry of human and avian H2N2 influenza A viruses. Nat. Microbiol. https://doi.org/10.1038/s41564-024-01771-1 (2024).
Ito, T. et al. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J. Virol. 72, 7367–7373 (1998).
Rajao, D. S. et al. Novel reassortant human-like H3N2 and H3N1 influenza A viruses detected in pigs are virulent and antigenically distinct from swine viruses endemic to the United States. J. Virol. 89, 11213–11222 (2015).
Nelson, M. I. et al. Introductions and evolution of human-origin seasonal influenza A viruses in multinational Swine populations. J. Virol. 88, 10110–10119 (2014).
Cárdenas, M. et al. Amino acid 138 in the HA of an H3N2 subtype influenza A virus increases affinity for the lower respiratory tract and alveolar macrophages in pigs. PLoS Pathog. 20, e1012026 (2024).
Kawasaki, T., Ikegawa, M. & Kawai, T. Antigen presentation in the lung. Front. Immunol. 13, 860915 (2022).
Mo, J. S. et al. Transmission of human influenza A virus in pigs selects for adaptive mutations on the HA gene. J. Virol. 96, e0148022 (2022).
Li, J. H. & McClane, B. A. The sialidases of type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl. Environ. Micro 80, 1701–1709 (2014).
Medley, B. J. et al. A “terminal” case of glycan catabolism: structural and enzymatic characterization of the sialidases of Clostridium perfringens. J. Biol. Chem. 300, 107750 (2024).
Dekevic, G., Tasto, L., Czermak, P. & Salzig, D. Statistical experimental designs to optimize the transient transfection of HEK 293T cells and determine a transfer criterion from adherent cells to larger-scale cell suspension cultures. J. Biotechnol. 346, 23–34 (2022).
Edinger, T. O., Pohl, M. O. & Stertz, S. Entry of influenza A virus: host factors and antiviral targets. J. Gen. Virol. 95, 263–277 (2014).
Yoshimura, A. & Ohnishi, S. Uncoating of influenza virus in endosomes. J. Virol. 51, 497–504 (1984).
Aganovic, A. pH-dependent endocytosis mechanisms for influenza A and SARS-coronavirus. Front. Microbiol. 14, 1190463 (2023).
Kyawe, P. P. et al. CRISPR editing of candidate host factors that impact influenza A virus infection. Microbiol. Spectr. 13, e0262724 (2025).
Karakus, U., Pohl, M. O. & Stertz, S. Breaking the convention: sialoglycan variants, coreceptors, and alternative receptors for influenza A virus entry. J. Virol. 94 https://doi.org/10.1128/JVI.01357-19 (2020).
Fujioka, Y. et al. A Ca(2+)-dependent signalling circuit regulates influenza A virus internalization and infection. Nat. Commun. 4, 2763 (2013).
Eierhoff, T., Hrincius, E. R., Rescher, U., Ludwig, S. & Ehrhardt, C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 6, e1001099 (2010).
Byrd-Leotis, L. et al. Influenza binds phosphorylated glycans from human lung. Sci. Adv. 5, eaav2554 (2019).
Zhu, X. et al. Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc. Natl. Acad. Sci. USA 110, 1458–1463 (2013).
Hargadon, K. M. et al. Major histocompatibility complex class II expression and hemagglutinin subtype influence the infectivity of type A influenza virus for respiratory dendritic cells. J. Virol. 85, 11955–11963 (2011).
Nelli, R. K. et al. Comparative distribution of human and avian type sialic acid influenza receptors in the pig. BMC Vet. Res. 6, 4 (2010).
Yu, W. C. et al. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J. Virol. 85, 6844–6855 (2011).
Cárdenas, M. et al. Vaccine-induced antigenic drift of a human-origin H3N2 Influenza A virus in swine alters glycan binding and sialic acid avidity. Preprint at https://doi.org/10.64898/2025.12.10.693614 (2025).
Byrd-Leotis, L. et al. Shotgun glycomics of pig lung identifies natural endogenous receptors for influenza viruses. Proc. Natl. Acad. Sci. USA 111, E2241–E2250 (2014).
Hinneburg, H. et al. High-resolution longitudinal N- and O-glycoprofiling of human monocyte-to-macrophage transition. Glycobiology 30, 679–694 (2020).
David, C. et al. Influenza induces depletion of alveolar macrophages in mice but not in humans. Rev. Mal. Respir. 41, 202 (2024).
Wein, A. N. et al. IL-36 gamma protects against severe influenza infection by promoting lung alveolar macrophage survival and limiting viral replication. J. Immunol. 201, 573–582 (2018).
Tate, M. D., Pickett, D. L., van Rooijen, N., Brooks, A. G. & Reading, P. C. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J. Virol. 84, 7569–7580 (2010).
Kim, H. M. et al. Alveolar macrophages are indispensable for controlling influenza viruses in the lungs of pigs. J. Virol. 82, 4265–4274 (2008).
Tumpey, T. M. et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79, 14933–14944 (2005).
Rossman, J. S., Leser, G. P. & Lamb, R. A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 86, 10950–10960 (2012).
Yeganeh, B. et al. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L270–L286 (2015).
Batishchev, O. V. et al. pH-dependent formation and disintegration of the influenza A virus protein scaffold to provide tension for membrane fusion. J. Virol. 90, 575–585 (2016).
Lee, P. S. et al. Receptor mimicry by antibody F045-092 facilitates universal binding to the H3 subtype of influenza virus. Nat. Commun. 5, 3614 (2014).
Hoffmann, E., Krauss, S., Perez, D., Webby, R. & Webster, R. G. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20, 3165–3170 (2002).
Neumann, G. et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 96, 9345–9350 (1999).
REED, L. J. & MUENCH, H. A simple method of estimating fifty percent endpoints12. Am. J. Epidemiol. 27, 493–497 (1938).
Moffat, J. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 (2006).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
Mayer, P. & Lam, C. Porcine alveolar macrophages, isolation, morphological and functional characteristics. Zentralbl Vet. A 31, 59–71 (1984).
Goatley, L. C., Nash, R. & Netherton, C. L. Primary macrophage culture from porcine blood and lungs. Methods Mol. Biol. 2503, 63–72 (2022).
Maisonnasse, P. et al. The respiratory DC/macrophage network at steady-state and upon influenza infection in the swine biomedical model. Mucosal Immunol. 9, 835–849 (2016).
Zaqout, S., Becker, L. L. & Kaindl, A. M. Immunofluorescence staining of paraffin sections step by step. Front Neuroanat. 14, 582218 (2020).
Matrosovich, M. N. & Gambaryan, A. S. Solid-phase assays of receptor-binding specificity. Methods Mol. Biol. 865, 71–94 (2012).
Yang, H. et al. Structure and receptor binding preferences of recombinant human A(H3N2) virus hemagglutinins. Virology 477, 18–31 (2015).
Acknowledgements
We thank the University Research Animal Resources personnel at the University of Georgia for assistance with animal care. This research was supported by an Agriculture and Food Research Initiative grant no. 2020-67015-31563/project accession no. 1022827 from the USDA National Institute of Food and Agriculture to D.S.R. Funding was also provided, in part, by the National Pork Board to D.S.R. under Project #21-085 to D.R.P. and by CRIPT (Center for Research on Influenza Pathogenesis and Transmission), a National Institute of Allergy and Infectious Diseases (NIAID) funded Center for Influenza Research and Response (CEIRR, contract 75N93021C00014 to D.R.P. and A.G-S. D.S.R. is also funded by an Agriculture and Food Research Initiative grant no. 2022-67015-37205/project accession no. 1028058 from the USDA National Institute of Food and Agriculture. D.R.P. is also funded by GRANT12901999, project accession no. 1022658 from the National Institute of Food and Agriculture (NIFA), U.S. Department of Agriculture. D.R.P. receives additional support from the Georgia Research Alliance and the Caswell S. Eidson endowment funds from The University of Georgia. This study was partly supported by resources and technical expertise from the Georgia Advanced Computing Resource Center, a partnership between the University of Georgia’s Office of the Vice President for Research and the Office of the Vice President for Information Technology. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
M.C., D.S.R., and C.J.C. designed the experiments. M.C. performed the cloning, growth kinetics, immunofluorescence, and MHCII expression experiments. M.C. performed the cell viability assays. M.C. and S.C. performed the flow cytometry experiments. M.C. analyzed the data. M.C. and D.S.R. wrote the manuscript. C.J.C., A.G.S., and D.R.P. edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The A.G.-S. laboratory has received research support from Avimex, Dynavax, Pharmamar, 7Hills Pharma, ImmunityBio and Accurius, outside of the reported work. A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Castlevax, Amovir, Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Pagoda, Accurius, Esperovax, Applied Biological Laboratories, Pharmamar, CureLab Oncology, CureLab Veterinary, Synairgen, Paratus, Pfizer, Virofend and Prosetta, outside of the reported work. A.G.-S. has been an invited speaker in meeting events organized by Seqirus, Janssen, Abbott, Astrazeneca and Novavax. A.G.-S. is an inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work. The other authors declare no conflict of interest.
Peer review
Peer review information
Nature Communications thanks James Paulson, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cardenas, M., Compton, S., Caceres, C.J. et al. MHC class II functions as a host-specific entry receptor for representative human and swine H3N2 influenza A viruses. Nat Commun 17, 2560 (2026). https://doi.org/10.1038/s41467-026-69267-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69267-6
