
Abstract
Organic electrosynthesis is a versatile and evergreen tool for constructing chemical compounds. However, the study of highly active electrodes has not received enough attention, which limits the further development of organic electrosynthesis. This work introduces a bottom-up route to prepare chitin-derived composite carbon aerogel electrodes (CCAEs), which can be directly used as electrodes in organic electrosynthesis systems. Various metal nanoparticles, such as Pt, Pd, RuO2, Cu and Ni, are well confined in these free-standing and porous CCAEs (M-CCAEs). The linear sweep voltammetry and in-situ Raman tests under electrochemical conditions show that RuO2-CCAEs possess good electrochemical oxidation ability for chlorine anions and good stabilizing effect on the generated chlorine radicals, which can serve as a mediator for the electrochemical C(sp3)-H activation. The combination of M-CCAEs with mediators achieves a series of electrochemical oxidative C(sp3)-H chlorination, bromination, nitration and etherification. Moreover, M-CCAEs promote the electrochemical hydrogen isotope exchange reaction of some important drug molecule structures, such as Ibuprofen, Diclofenac and Zolpidem.
Similar content being viewed by others


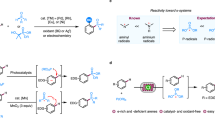
Introduction
Synthetic electrochemistry was selected as one of the 2023 top ten emerging technologies in chemistry by the International Union of Pure and Applied Chemistry, which aimed to showcase the potential of the chemical sciences to foster the well-being of society and the sustainability of our planet1. Organic electrosynthesis has developed phenomenally for more than 170 years since Kolbe Electrolytic Synthesis was reported2. Recently, organic electrosynthesis has ushered in a new wave of development due to its unique reactivity and intrinsic advantages in safety, atom economy, and energy efficiency3,4,5,6,7,8,9,10,11,12,13. Consequently, electrosynthesis has been increasingly adopted in chemical manufacturing to enhance process efficiency while lowering costs and greenhouse gas emissions, in line with requirements for carbon peaking and carbon neutrality14. However, some grand challenges still need to be overcome, including the transformation of substrates with high redox potential and the synthesis of important chemicals with high value15,16,17,18. Alkyl is the ubiquitous structural motif in organic chemistry, and most of the top-selling commercial drugs contain an alkyl moiety19. The C(sp3)–H bonds usually have high bond-dissociation energy and oxidation potential, which makes their direct electrochemical activation challenging. Indirect electrolysis using hydrogen-atom transfer (HAT) mediator to activate C(sp3)–H bonds has got some breakthroughs in recent years (Fig. 1a)20. For example, in 2016, Baran and coworkers reported allylic C–H oxidation using tetrachloro-N-hydroxyphthalimide (Cl4NHPI) as a mediator and tBuOOH as the oxygen source21. Subsequently, they used quinuclidine as a HAT mediator in the electrochemical oxidation of C(sp3)–H bonds to ketones or alcohols22. In 2018, Stahl and coworkers showed I2 was an effective radical trap for electrochemical NHPI-mediated oxidation of methylarene C–H bonds to benzyl iodides23. In 2020, Lambert group and Xu group reported photoelectrochemical C(sp3)–H functionalization of ethers and alkanes using trisaminocyclopropenium (TAC) ion and chlorine anion as the mediator, respectively24,25. Then, the Ackermann group, our group, and Ye group achieved the electrochemical azidation, arylation, and amination of C(sp3)–H bonds by employing Mn complex catalyst, 4-chloro-N-isopropylbenzenesulfonamide, and sulfate anion as the mediator, respectively26,27,28. These reported works focus on searching for new catalytic systems and mediators for realizing target reactions, and nearly all reactions use commercially available metal plates and carbon electrodes as electrodes.

a Indirect electrolysis for C(sp3)–H bond functionalization. b Modified electrodes derive from traditional electrodes. c This work: prepare composite carbon aerogel electrodes decorated with metal nanoparticles (M-CCAEs) from chitin and apply them to electrochemical C(sp3)–H bond functionalization.
Traditional electrodes usually have good stability but have lower activity and worse selectivity in some reactions due to their size effect and inherent properties. With an increasing global energy demand, electrocatalytic reactions, such as hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), have attracted extensive attention, and various advanced electrocatalysts have been reported to lower the reaction overpotentials29. Organic electrosynthesis needs advanced electrodes as well to realize further development30,31. So, we are interested in preparing highly active electrodes to promote the effect of the mediator, such as lowering its overpotential, to improve the Faradaic efficiency, yield, and selectivity of target reactions. Recently, modified electrodes using traditional electrodes as supports have shown good activity in organic electrosynthesis (Fig. 1b). For example, in 2018, Zhang and coworkers prepared NiSe nanorod arrays as an anode through the direct selenization of commercially available nickel foam, and used this NiSe anode for the electrochemical oxidation of primary amines into the corresponding nitriles32. Subsequently, they prepared a series of modified electrodes, such as Ni2P nanosheet arrays anode, copper nanowire arrays cathode, and Pd-P alloy nanoparticle networks cathode for various electrochemical reactions, such as semi-dehydrogenation of tetrahydroisoquinolines, deuteration of halides, and semi-hydrogenation (deuteration) of alkynes33,34,35. In 2020, Berlinguette and coworkers prepared a Pd membrane cathode for reductive deuteration of alkynes, aldehydes, and imines36. Very recently, our group reported a nitrogen-doped Ru carbon felt electrode for electrocatalytic reductive deuteration and deuterodefluorination of (hetero)arenes to give perdeuterated and saturated deuterocarbon products37. Although modified electrodes have shown impressive results, there are rare reports about using modified electrodes to promote the effect of mediators for target reactions, and modified electrodes still can’t cast off the shackles of support materials, which may lead to poor uniformity and stability. Facing the fast improvement of the electrosynthesis level, the novel and matched electrodes are extremely urgent. The rational design and preparation of electrodes through the bottom-up method, rather than modifying existing electrodes, is a promising strategy38. Nevertheless, conductivity, intensity, modifiability, uniformity, universality, activity, and stability are all daunting tasks for preparing electrodes.
Porous carbon materials are suitable supports for nanoelectrodes because they can enhance mass transfer and metal-support interaction39,40,41. Biomass is a cheap and readily available source for porous carbon materials42,43. Cellulose and chitin are the two most abundant biopolymers on Earth. However, they are usually regarded as waste at the end of their lifecycle and directly contribute to CO2 emissions44. Especially for chitin, approximately 6–8 million tons of crab, shrimp, and lobster shells are generated annually by the global seafood processing industries45. These shell wastes are typically disposed of in landfills or discharged into the ocean, leading to a substantial loss of potentially valuable resources. Utilizing abundant and renewable biomass resources to synthesize new materials is environmentally friendly and economically beneficial for a sustainable future and circular economy46,47,48,49. Chitin possesses naturally fixed oxygen and nitrogen in the form of hydroxyl, ether, and amide groups, providing abundant coordination sites for anchoring active metal species50. This property can improve the stability of heterogeneous catalysts in organic solvent systems51, which is essential for electrodes in organic electrosynthesis.
In this work, we demonstrate a facile synthetic route to fabricate composite carbon aerogel electrodes (CCAEs) derived from chitin. Commercially available chitin is dissolved in aqueous alkali and mixed with pre-prepared chitin microspheres decorated with metal ions (M-CMs). These uniform composite chitin solutions with metal ions (M-CCSs) undergo gelation to afford composite chitin hydrogels decorated with metal ions (M-CCHs), which are converted to composite chitin aerogels decorated with metal ions (M-CCAs) through freeze-drying. Finally, composite carbon aerogel electrodes decorated with metal nanoparticles (M-CCAEs) are obtained by calcinating M-CCAs (Fig. 1c). These free-standing nanoelectrodes promote the effect of mediators to exhibit good catalytic performance in electrochemical oxidative C(sp3)–H chlorination, bromination, nitration, etherification, and H/D exchange reactions of some important drug molecules. Several representative electrochemical redox reactions also show the broad application of M-CCAEs.
Results
The preparation of nanoelectrodes
We optimized the preparation methods following the key factors, i.e., conductivity, intensity, modifiability, uniformity, universality, activity, and stability, of free-standing nanoelectrodes (see “Materials preparation” in “Methods”).
Cellulose and chitin aerogels were obtained by screening the dissolution, gelation, and freeze-drying methods, respectively (Fig. S1). These free-standing aerogels were carbonized to conductive carbon aerogels at different calcination temperatures (referred to as CAs-natural polymer-T). The conductivity improved with increasing temperature (Fig. S2). Raman spectroscopy also proved the same trend (Fig. S3). The intensity ratios of the D band to the G band (ID/IG) decreased, which indicated that the corresponding graphitization degree and conductivity grew. The natural polymer-aerogels lost about 80% of weight, which were revealed by thermogravimetric analysis (Fig. S4). Moreover, X-ray photoelectron spectroscopy (XPS) showed that both the ratios of N/C and O/C decreased with increasing temperature (Fig. S5). The content of the C element gradually increased to enhance intrinsic conductivity. Meanwhile, the N and O species were the potential coordination and defect active sites (Figs. S6 and S7). CAs-natural polymer-800/900 were selected to test the mechanical strength due to their better conductivity. The CAs-chitin-800 showed a relatively high tensile strength of 40 MPa in dry conditions (Fig. S8). By comprehensive consideration, CAs-chitin-800 stands out due to the advantages of conductivity, mechanical strength, weight loss, and heteroatomic content, which are essential for electrode materials (Figs. 2a, S9 and S10).

a A radar plot comparing CAs-natural polymer-T. b SEM image of composite carbon aerogels-chitin electrodes (CCAEs). c Nitrogen adsorption and desorption isotherms of CCAEs and Pt-CCAEs. d Nitrogen adsorption DFT pore-size distributions of CCAEs and Pt-CCAEs.
Modifying CAs-natural polymer-800 with metal nanoparticles (NPs) is a pragmatic approach to improving their electrocatalytic activity. Based on the route above, H2PtCl6 was initially added to natural polymer solutions to obtain Pt-CAs-natural polymer-800. Atomic absorption spectrometer results showed the Pt content of Pt-CAs-cellulose-800 was much less than that of Pt-CAS-chitin-800, which indicated the coordination ability of cellulose was weaker than that of chitin (Table S1; Fig. S11). Chitin possessed N and O, while cellulose only contained the O. Then, the transmission electron microscopy (TEM) picture showed that the distribution and size of Pt NPs were not uniform (Figs. S12 and S13), which was not conducive to the repeatability of this preparation method. The reason might be that the metal salts were not mixed well with highly viscous chitin solutions.
To overcome the problem above, we loaded metal ions on chitin microspheres (CMs) in advance and added Pt-CMs into a pure chitin solution to get corresponding Pt-composite carbon aerogels [referred to as Pt-CCAEs (Pt-composite carbon aerogel electrodes); Fig. 1c]. In TEM and SEM pictures, the distribution and size of Pt NPs were well controlled (Figs. S14 and S15). Pt-CCAEs had a higher Pt loading of 1.2 wt% (Table S2; Fig. S11). CMs provided good three-dimensional support to prevent the composite chitin aerogels from collapsing during freeze-drying (Figs. S16 and S17) and gave CCAEs with plentiful porous structures after calcination (Fig. 2b). In addition, the Brunauer-Emmett-Teller (BET) specific surface area of Pt-CCAEs was slightly lower than that of CCAEs because Pt nanoparticles could tightly anchor in micropores without obvious influence on their basic structure (Fig. 2c, d). The micropores could effectively confine Pt NPs in specific channels to prevent aggregation and maintain their high catalytic activity. Remarkably, the large surface area and various abundant pores of the CCAEs were favorable for the permeation and transportation of the reaction species during the electrolytic process (Fig. 2b).
The atomic structure of Pt-CCAEs showed that individual metal NPs were evenly dispersed throughout the carbon nanofibers, which were imaged by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Figs. 3a and S18). Notably, the Pt NPs were surrounded by single-atom Pt. The energy dispersive X-ray spectroscopy (EDS)-mapping further verified the homogeneous distribution of Pt NPs, single-atom Pt, and relative elements C/O/N in the framework (Fig. 3b). Here, the oxidation state of Pt species in Pt-CCAEs was evaluated by the X-ray absorption spectroscopy (XAS) (Figs. 3c and S19; Table S3). The X-ray absorption near-edge structure spectrum of Pt-CCAEs showed the edge energy at 11565 eV, very near that of Pt foil (11564 eV). The white line intensity is lower than PtO2 and close to Pt foil, which accords with the XPS result (Fig. S20). These results proved that the well-dispersed ultra-fine Pt NPs and single Pt atoms anchored on the carbon nanofiber were constructed successfully.

a HAADF-STEM images of Pt-CCAEs. b Energy dispersive X-ray spectroscopy (EDS) elemental mapping results of Pt-CCAEs. c Pt L3-edge EXAFS experimental data for Pt-CCAEs. d HAADF-STEM images of Pd-CCAEs. e EDS elemental mapping results of Pd-CCAEs. f Pd K-edge EXAFS experimental data for Pd-CCAEs. g HAADF-STEM images of RuO2-CCAEs. h EDS elemental mapping results of RuO2-CCAEs. i Ru K-edge EXAFS experimental data for RuO2-CCAEs.
Subsequently, to exhibit the compatibility of this preparation method, we changed the metal precursors to make a series of electrodes, such as Pd-CCAEs, Ni-CCAEs, and Cu-CCAEs (Figs. S21 and S22; Table S2). Remarkably, we used RuO2 NPs anchored on TiO2 instead of metal salts anchored on chitin microspheres to make RuO2-CCAEs (RuO2/TiO2-composite carbon aerogel electrodes). The morphology and structure of TiO2 nanoparticles attached to the hybrid nanofiber were seen by SEM (Figs. S23 and S24; Table S2). The FT-IR and XRD patterns also showed that metal nanoparticles were uniformly dispersed on the carbon layer without structural changes (Figs. S25 and S26). The high-resolution HAADF-STEM images showed that the crystalline Pd NPs and RuO2 NPs were successfully dispersed in the carbon layer after pyrolysis (Figs. 3d, g, S27 and S28). There was an apparent nanocrystalline surface between TiO2 and RuO2 nanoparticles. Meanwhile, the heterointerface existed between amorphous carbon and TiO2. The EDS mapping revealed the homogeneous distribution of Pd NPs and related elements C/O/N in the framework (Fig. 3e). The RuO2 NPs were surrounded by not only C/O/N but also Ti (Figs. 3h and S29). The extended X-ray absorption fine structure (EXAFS) spectra indicated the existence of Pd-Pd bonds of Pd NPs and the Ru-Ru/Ru-O bonds of RuO2 NPs by fitting and comparing them with standard substances (Figs. 3f, i, S30 and S31; Table S3). The XPS characterization also supported the results (Figs. S32 and S33). The nanoparticles of Ni-CCAEs and Cu-CCAEs were evenly scattered on the porous carbon surface in the TEM images (Fig. S34). The XPS patterns showed Ni and Cu species mainly focused on metallic Ni(0) and Cu(0) characteristics (Figs. S35 and S36).
The electrocatalytic activity of nanoelectrodes
HER and OER were conducted to test the redox catalytic performance of M-CCAEs. A typical three-electrode system was employed, and free-standing M-CCAEs were directly used for tests without being ground or adhered to any support (see “Electrocatalytic test” in “Methods”). Figure S37a shows linear sweep voltammetry (LSV) curves of M-CCAEs and Pt/C. Pd-CCAEs gave an overpotential of 43 mV at a current density of 10 mA cm−2 in 0.5 M H2SO4, which was close to Pt/C-CP (Pt/C supported on carbon paper) with an overpotential of 25 mV (Fig. S37b). Pd-CCAEs had the smallest value of 58 mV dec−1 in all M-CCAEs, which indicated that Pd-CCAEs possessed a good capacity for hydrogen evolution as the cathode (Fig. S39a). Chronoamperometry tests examined the electrochemical stability of Pd-CCAEs at overpotentials, ηj = 10 = 43 mV. As shown in Fig. S37c, Pd-CCAEs exhibited good stability with negligible decay after 10 h.
The OER performances were also evaluated in a 0.5 M H2SO4 solution. Figure S38a shows LSV curves of M-CCAEs and benchmark RuO2-CP at scanning rates of 5 mVs−1. RuO2-CCAEs possessed an ultralow overpotential (ηj = 10) value of 288 mV and had a low activation energy to overcome the slow reaction kinetics in acidic environments (Fig. S38b). As expected, the Tafel slope of RuO2-CCAEs was calculated as 83.0 mV dec−1 (Fig. S39b), very close to the benchmark sample RuO2-CP. The catalytic activity at ηj = 10 of 288 mV showed good stability after 10 h of chronoamperometry measurements (Fig. S38c). The remarkable OER activity of RuO2-CCAEs was possibly related to the synergistic effect of multifunctional defects, including N-doping, oxygen vacancy, and RuO2 nanoparticles.
Because M-CCAEs show good activities in HER and OER. We speculate these nanoelectrodes may also perform well in activating some electrochemical mediators. Chlorine anion is a good mediator for alkane C(sp3)–H activation, i.e., Cl anion is oxidized into Cl radical, which can abstract a hydrogen atom from the C(sp3)–H bond to achieve activation52. This strategy has been applied in both photochemical and electrochemical synthesis25,53,54. Fig. 4a shows LSV curves of all M-CCAEs in the Cl anion oxidation reaction (COR), and RuO2-CCAEs gave a relatively low overpotential of 188 mV (Fig. 4b). Moreover, RuO2-CCAEs could be stably utilized for 10 h without noticeable activity loss (Fig. S40). Therefore, the combination of RuO2-CCAEs and Cl anion may be a good electrocatalytic system for C(sp3)–H activation.

a Polarization curves of M-CCAEs and control electrodes for chlorine anion oxidation reaction. b The overpotentials to reach 10 mA cm−2 of M-CCAEs and control electrodes for chlorine anion oxidation reaction. c The Faradaic efficiencies of electrochemical oxidative cyclohexane chlorination on various electrodes in 0.3 M HCl + Cyclohexane + CH3CN. d Cyclic voltammetry experiments of HCl and cyclohexane using a glassy carbon electrode as the working electrode at scan rates of 100 mV s−1. e In situ Raman spectroscopy of RuO2-CCAEs in 0.3 M HCl + CH3CN with various potentials.
The organic electrosynthesis application of nanoelectrodes
Then, we tested the performance of these nanoelectrodes in organic electrosynthetic reactions (see “Electrosynthetic reactions” in “Methods”). Alkyl halides are important coupling partners in organic synthesis, providing highly flexible avenues for diversification55. Classical halogenation of alkanes needs harsh conditions, such as using excessive corrosive Cl2 and Br2 as reagents. Recently, the chlorination of alkanes has undergone fast development, which involves new catalysts and mild catalytic systems56,57,58,59. However, electrochemical chlorination of alkanes has not attracted enough attention. So, we proposed using Cl anion as both substrate and mediator to achieve the electrochemical chlorination of alkanes under mild and simple electrosynthesis conditions. Cyclohexane and concentrated hydrochloric acid (HCl (12.0 mol/L)) were chosen as model substrates to study the chlorination of alkanes (Figs. 4c and S41). After investigations about electrolytes, currents, solvents, and temperatures, the desired product, cyclohexyl chloride 1, was obtained with 55% Faradaic efficiency using platinum plates as both anode and cathode. Then, we improved the reaction efficiency by changing the electrodes. Graphite rods were relatively better than platinum plates. Initial nanoelectrodes CAEs had the same Faradaic efficiency as graphite rods. It was encouraging that 76% Faradaic efficiency was obtained by using CCAEs as electrodes. However, there was no apparent difference between Pt-CCAEs and CCAEs. We speculated that Pt-CCAEs were not suitable as anode and cathode simultaneously. Indeed, using Pt-CCAEs as anodes and CCAEs as cathodes was a better combination. Inspired by these results, we searched for more electrode combinations. Pt-CCAEs as anode and Pd-CCAEs as cathode afforded 90% Faradaic efficiency. Notably, RuO2-CCAEs with good Cl anion oxidation activity as anode and Pd-CCAEs with good HER activity as cathode exhibited the best performance (91% Faradaic efficiency) in all combinations. In addition, other conventional carbon electrodes, such as carbon paper and carbon felt, as well as several prepared carbon felt-supported metal nanoparticle electrodes (Ru-CF, Pt-CF, and Pd-CF), all demonstrated inferior performance compared to RuO2-CCAEs and Pd-CCAEs (see Activity Comparison in the Supplementary Information; Table S4; Fig. S42). Therefore, these free-standing M-CCAEs exhibit excellent activity compared to commonly used electrodes.
In order to test the recyclability of these free-standing nanoelectrodes, the same electrodes of RuO2-CCAEs and Pd-CCAEs were reused for a total of six consecutive experimental cycles of cyclohexane chlorination (see Recyclability Test in Supplementary Information; Table S5; Fig. S43). During those cycles, the electrodes were found to maintain a relatively constant level of activity, with the Faradaic efficiency values varying by no more than 9%. Moreover, ICP-OES analysis of the reaction solution after completion revealed no detectable Ru (Table S6). The used RuO2-CCAEs retained a Ru content of 1.0032 wt%, consistent with that of the freshly prepared electrodes. These results confirm that no metal leaching occurred from the RuO2-CCAEs during the reaction, in agreement with the recycling experiment findings. In addition, anodic chronopotentiometry tests were performed to assess the stability performance of RuO2-CCAEs anode (see Recyclability Test in Supplementary Information; Table S7; Fig. S44), and the anodic potential was maintained at approximately 1.5 V for 50 h, with the Faradaic efficiency decreasing by only 4% after 50 h of electrolysis. The observed results showed the high activity, recyclability, and robustness of M-CCAEs as electrodes for the electrochemical oxidative chlorination of cyclohexane.
To gain more insights into the mechanism of this cyclohexane chlorination promoted by M-CCAEs, we conducted several relevant experiments under the standard reaction conditions (see Mechanism Study in Supplementary Information). Kinetic isotopic effect (KIE) experiments using cyclohexane and cyclohexane-d12 were performed for intermolecular competition and parallel reactions. The KIEs of 1.4 and 1.1 were determined. The results suggested that C–H bond activation might not be involved in the rate-determining step. Moreover, radical-trapping experiments were conducted. When 1,1-diphenylethene was used as the radical-trapping reagent, only 2% yield of the desired product 1 was detected, while the coupling products with Cl radical were obtained. The above results indicated that Cl radicals are probably involved in this transformation. Cyclic voltammetry was used to detect the oxidation potential of substrates (Fig. 4d), suggesting that HCl could be easily oxidized while cyclohexane could not. Two oxidation peaks for HCl are observed, likely corresponding to the formation of Cl radicals and Cl+ species.
All used anodes could oxidize Cl anion to Cl radical under electrochemical conditions, but RuO2-CCAEs as the anode gave the highest Faradaic efficiency. So, we further studied the interaction between Cl radical and RuO2-CCAEs. The in situ Raman spectroscopy of RuO2-CCAEs in HCl solution showed that there was a peak in 505 cm−1 appearing after the potential increased to 1.2 V (Fig. 4e; see In Situ Raman Measurements in Supplementary Information), which meets the result of the CV experiment (Figs. S45 and S46)60,61. This peak is attributed to the Ru-Cl* species, which might be formed by RuO2-CCAEs capturing the Cl radical. Hence, RuO2-CCAEs not only possess the lowest overpotential for the Cl anion oxidation but also stabilize the Cl radical, affording the highest Faradaic efficiency of the cyclohexane chlorination.
The previous reports using Cl anions as the mediator usually produce Cl2 as the intermediate, and light irradiation is required to regenerate Cl radical25,54. We also observed the presence of chlorine gas after the electrochemical oxidative chlorination of cyclohexane was completed (see Mechanism Study in Supplementary Information; Fig. S47). The generation of a small amount of Cl2 may explain why the Faradaic efficiency is approximately 90% rather than 100%. The cyclohexane chlorination remains the dominant reaction pathway, possibly because: (1) In the electrochemical oxidative chlorination of cyclohexane, the high concentration of cyclohexane increases the likelihood that Cl radicals will rapidly react with the cyclohexane. In addition, within this organic solvent system, the electron-transfer rate is slower, and there is no interference from protic solvents. Consequently, Cl radicals exhibit an extended lifetime. This is supported by the square-wave voltammetry (SWV) results: a broader potential window from onset to peak indicates enhanced radical stability, which facilitates more effective H atom abstraction from cyclohexane and subsequent coupling with cyclohexyl radicals, thereby contributing to the higher Faradaic efficiency (Fig. S48). Furthermore, SWV measurements performed on tetraethylammonium chloride (TEACl) also showed two distinct oxidation peaks, providing direct evidence for the presence of Cl radicals. These results are consistent with the CV scan (Fig. 4d), in which two oxidation peaks for HCl are observed, likely corresponding to the formation of Cl radicals and Cl+ species. (2) The reaction operates at a relatively low current density, preventing the potential from rapidly reaching the Cl2 evolution threshold in the organic phase. Anodic chronopotentiometry shows that the anodic potential remains approximately 1.5 V throughout 50 h of electrolysis (see Recyclability Test in Supplementary Information). This stable potential window indicates high selectivity for the desired cyclohexane chlorination and avoids substantial Cl2 generation. (3) In situ Raman spectroscopy provides insights into the adsorption behavior at the electrode surface and interface (see In Situ Raman Measurements in Supplementary Information). The detection of Ru-Cl* active intermediate species suggests that these intermediates play a key role in capturing and stabilizing Cl radical species, thereby suppressing excessive Cl2 evolution. Therefore, we proposed that RuO2-CCAEs effectively stabilize Cl radicals and thus avoid excessive Cl2 production.
Based on the above mechanistic experiments, a plausible mechanism of organic parts was proposed (Fig. S49). Firstly, the Cl anion was oxidized at RuO2-CCAEs to produce the Cl radical, which could be stabilized by RuO2-CCAEs. Then, the Cl radical abstracted an H atom from cyclohexane to form the cyclohexyl radical and Cl anion. Finally, the desired product, cyclohexyl chloride, was obtained by coupling cyclohexyl and Cl radicals. At the same time, concomitant cathodic reduction of the proton at Pd-CCAEs leads to hydrogen evolution. Pd-CCAEs also played an important role in contributing to the higher Faradaic efficiency. The lower overpotential at the cathode can reduce the overall cell voltage, thereby suppressing undesired side reactions such as Cl2 evolution. The above anodic chronopotentiometry tests using Pd-CCAEs as the counter electrode showed that the anodic potential of RuO2-CCAEs remained stable at approximately 1.5 V over 50 h of continuous operation. Moreover, using HCl as both substrate and electrolyte ensures a high concentration of Cl anions, and the efficient H2 evolution at the cathode enhances electrolyte desolvation, which in turn accelerates Cl anion migration toward the anode for activation.
Encouraged by the above results, we next explored the application scopes of electrochemical oxidative C(sp3)–H functionalization (Fig. 5). Firstly, the chlorination of alkanes was studied. The cyclic alkanes were all transformed smoothly into the desired products (1–5). Toluene and its analogs could also be tolerated (6 and 7). Then, we turned our attention to the bromination of alkanes. Hydrobromic acid could not act as a halogen source in this system. We used LiBr as a Br source and calculated the yields based on LiBr. From 5- to 12-membered rings, 84% to 99% yields were obtained (8–12). Toluene also gave the desired product (13). HNO3 was previously reported to activate alkanes under electrolysis conditions62,63. So, we directly used nitric acid as a substrate and mediator to access the nitration of alkanes. Unsatisfactorily, the Faradic efficiency of this transformation was relatively low (14 and 15), which can be attributed to the oxidation potential of HNO3 being higher than that of cyclohexane (see Mechanism Study in Supplementary Information; Figs. S50–S52).

Chlorination or nitration reaction conditions: alkane (2.0 mL), hydrochloric or nitric acid (3.0 mmol), RuO2-CCAEs anode, Pd-CCAEs cathode, constant current = 20 mA, MeCN (8.0 mL), undivided cell, N2, 3 h, room temperature, Faradaic efficiency (FE). Bromination reaction conditions: alkane (2.0 mL), LiBr (0.5 mmol), RuO2-CCAEs anode, Ni plate cathode, constant current = 20 mA, nBu4NBF4 (0.5 mmol), MeCN (8.0 mL), H2O (0.2 mL), undivided cell, N2, 3 h, room temperature, GC yield or 1H NMR yield. Etherification reaction conditions: ether (2.0 mL), alcohol (1.0 mmol), hydrochloric acid (0.2 mmol), RuO2-CCAEs anode, Pd-CCAEs cathode, constant current = 20 mA, nBu4NBF4 (0.5 mmol), MeCN (8.0 mL), undivided cell, N2, 6 h, 0 °C, isolated yield.
After realizing alkane functionalization, we focused on other C(sp3)–H bonds. Tetrahydrofuran (THF) is a commonly used solvent in electrosynthesis with a relatively high oxidation potential. We want to achieve the etherification of THF using alcohol as a coupling partner since it is a good nucleophile. However, alcohols may have lower oxidation potential than THF. We chose THF and benzyl alcohol as model substrates to test their oxidation potential. The oxidation potential of benzyl alcohol is indeed lower than THF (Fig. S53). Nevertheless, the oxidation potential of HCl is lower than that of benzyl alcohol, which can change the oxidation order in the reaction system. So, we used HCl as the mediator, RuO2-CCAEs as the anode, and Pd-CCAEs as the cathode to study the etherification of THF. THF reacted with benzyl alcohol to afford the desired product in 52% yield (16). The control experiments indicated that the yield of etherification product 16 had an apparent decrease without the help of mediator HCl (see Mechanism Study in Supplementary Information). So, HCl played an important role in activating C(sp3)–H bonds. Patureau and coworkers used direct electrolysis of THF to couple with alcohol, so they focused on using 2-phenylethanol with a higher oxidation potential as a substrate, which was less compatible compared with our mediated process64. The electron-donating -OMe group and electron-withdrawing -F group were compatible under the standard conditions (17 and 18). 2-Phenylethanol gave the desired ether product in 80% yields (19). The aliphatic alcohols, such as 1-hexanol, afforded 85% yield (20). In addition, a variety of functional groups, such as chloro-, alkoxy-, alkenyl-, and alkynyl- were all tolerated in this transformation (21–25). Diols such as 1,6-hexanediol showed better reaction efficiency (26). The secondary and tertiary alcohols were also investigated. Cyclohexanol, (R)−1-phenylethanol, and 1-adamantanol proceeded to afford the desired ethers (27–29). 2-Methyltetrahydrofuran, tetrahydropyran, and isochroman gave relatively lower yields (30–32). Moreover, we explored the compatibility of bioactive molecules. L-menthol, dehydroepiandrosterone, and stigmasterol were tolerated in this transformation (33–35). To our delight, carbohydrates such as diacetone-fructose, diacetone-D-glucose, and 2,3,4,6-tetra-O-phenyl-glucopyranose gave the corresponding products in 71, 46, and 50% yields, respectively (36–38).
The deuteration technique has become a hot topic with the launch of a series of deuterated drugs65,66,67. Compared to other deuteration methods, such as reductive deuteration and dehalogenative deuteration, the hydrogen isotope exchange (HIE) reaction is an atomic economy strategy without pre-functionalization68. It is of great significance to realize the HIE reaction of C(sp3)–H bonds due to their wide distribution in best-selling commercial drugs. However, electrochemical synthesis was relatively rarely utilized in HIE reactions of C(sp3)–H bonds69,70,71. We tried to apply the catalytic system of nanoelectrodes with the chloride mediator in the deuterium labeling of drug molecules with D2O or CD3OD as deuterium sources (Fig. 6a). Firstly, the narcotic drug Propanidid, hypoglycemic drug Repaglinide intermediate, and phytohormone indole-3-acetic acid derivative gave 87–90%D deuterium contents (39–41). Common anti-inflammatory and analgesic drugs with a wide range of applications, such as Tolmetin methyl ester, Ibuprofen methyl ester, Loxoprofen methyl ester, and Naproxen methyl ester, also gave the corresponding HIE products (42–45). Drug derivatives with other types of C(sp3)–H bonds, such as Zolimidine (sulfone), Diclofenac (aryl acetamide), Zolpidem (aryl acetamide), Nabumetone (ketone), and Loxoprofen (ketone), were all tolerated in our electrocatalytic system (46–50). It was worth mentioning that we could control the reaction site in α-C–H of ketone and α-C–H of aryl acetate by choosing D2O or CD3OD for Loxoprofen derivatives, respectively (44, 50). It should be noted that this HIE reaction does not proceed in the absence of an applied current (see Control Experiments in Supplementary Information).

a The substrate scope of electrocatalytic HIE reactions of C(sp3)–H bond using M-CCAEs. Reaction conditions: drug (0.3 mmol), D2O (10.0 mmol), Et4NCl (0.25 mmol), RuO2-CCAEs anode, Pd-CCAEs cathode, constant current = 3 mA, DMF (3.0 mL) and THF (3.0 mL), undivided cell, N2, 8 h, room temperature, isolated yield, deuterium incorporation is presented in the brackets, which is determined by proton nuclear magnetic resonance (1H NMR). *CD3OD instead of D2O. b Electrochemical oxidation reactions using M-CCAEs. c Electrochemical reduction reactions using M-CCAEs.
In order to further exhibit the applicability of these nanoelectrodes in organic electrosynthesis, we performed several representative electrochemical redox reactions using our nanoelectrodes (Fig. 6b, c). Firstly, the classic Kolbe reaction proceeded smoothly using RuO2-CCAEs as the anode (51). 4-tert-Butylbenzaldehyde is an important chemical intermediate, and the electrochemical oxidation of 4-tert-butyl toluene to 4-tert-butylbenzaldehyde is a large-scale industrial process in BASF SE. We successfully used CCAEs to obtain 4-tert-butylbenzaldehyde from 4-tert-butyl toluene (52). Subsequently, with Pd-CCAEs as cathode, 4-tert-butyl styrene was reduced to 1-(tert-butyl)−4-ethylbenzene (53). At last, the electrochemical reductive dehalogenation of 4-bromobenzonitrile was achieved by Ni-CCAEs cathode (54).
Discussion
In conclusion, we developed a general route to fabricate free-standing nanoelectrodes from chitin. Our chitin-derived composite carbon aerogel-supported metal nanoparticle electrodes offer several advantages over commonly used electrodes: (1) Low cost and sustainable preparation. Biomass chitin is employed as the carbon support, providing a low-cost and sustainable route that also enables waste valorization. The metal loading is approximately 1 wt%, significantly reducing the cost of preparing these nanoelectrodes. Moreover, CCAEs containing different metal nanoparticles can be readily obtained by simply varying the metal precursors. (2) Favorable structural properties and tunable metal nanoparticles. The large surface area and abundant hierarchical pores of the CCAEs facilitate efficient permeation and transport of reaction species during electrolysis. The rich heteroatomic coordination sites, together with the microporous framework, confine the metal nanoparticles within specific channels, thereby inhibiting aggregation and maintaining excellent catalytic activity. The size of the metal nanoparticles can also be finely controlled. As a result, RuO2-CCAEs exhibit excellent activity in both OER and COR, while Pd-CCAEs show strong activity in HER. (3) Unique ability to stabilize chlorine radicals. RuO2-CCAEs can stabilize Cl radicals under electrochemical conditions, which is crucial for utilizing Cl anion as a mediator in organic electrosynthesis. This property helps suppress excessive Cl₂ formation and ensures high selectivity toward cyclohexane chlorination rather than undesired Cl₂ evolution. Combining these nanoelectrodes with mediators afforded good results in a series of precise electrochemical syntheses in organic solvent systems. Electrochemical oxidative alkane chlorination, bromination, and nitration were achieved using different mediators. The etherification and HIE reaction of C(sp3)–H bonds were realized using RuO2-CCAEs and Cl anion mediator. Several representative electrochemical redox reactions also showed the great application potential of M-CCAEs. (4) High stability and reusability. Both RuO2-CCAEs and Pd-CCAEs can be reused for a total of six consecutive experimental cycles of cyclohexane chlorination with only a slight decrease in Faradaic efficiency. They also sustain continuous electrolysis for 50 h while maintaining a stable anodic potential, demonstrating the excellent durability of the M-CCAEs.
This achievement could be attributed to the multiple structures of our synthesized material, which exhibited the designability and controllability of nanoelectrodes. We believe that our work will stimulate the scientific community to search for new waste-derived materials for a sustainable future and circular economy.
Methods
Materials preparation
A. The synthesis procedure of carbon aerogels-cellulose-temperature (CAs-cellulose-T)
Cellulose solution (5.0 wt%) was prepared by dissolving 3.0 g cotton linter pulp in a 97.0 g mixed solvent under a precooled −10 °C solution (7.0 wt% NaOH/12.0 wt% urea/81.0 wt% water). Low molecular-weight chemical cross-linker epichlorohydrin (ECH, 3.0 mL) was added dropwise to the above bubbles-removed cellulose solution (60.0 g) at −5 °C and homogeneous mixing for 2 h. After cross-linking, the homogeneous solution was obtained by centrifugation and injected into the mold. Then, the mold was put in a 60 °C oven for 30 min to get the cellulose hydrogels, which were washed with deionized water to neutral. The cellulose aerogels were obtained from the corresponding hydrogel by freeze-drying. The cellulose aerogels were placed in the center of a quartz tube furnace and annealed at 600, 700, 800, or 900 °C (2 °C/min) for 2 h with Ar flow to obtain the corresponding carbon aerogels-cellulose-temperature (CAs-cellulose-T) (Fig. S1a).
B. The synthesis procedure of carbon aerogels-chitin-temperature (CAs-chitin-T)
Chitin solution (5.0 wt%) was prepared by dissolving 6.0 g chitin powder in a 94.0 g mixed solvent of 11.0 wt% NaOH/4.0 wt% urea/85.0 wt% water under −35 °C in a cold trap for 4 h and then thawed at room temperature. Low molecular-weight chemical cross-linker epichlorohydrin (ECH, 3.0 mL) was added dropwise to the above bubbles-removed chitin solution (60.0 g) at −5 °C and homogeneous mixing for 2 h. After cross-linking, the homogeneous solution was obtained by centrifugation and injected into the mold. Then, the mold was put in a 60 °C oven for 30 min to get the chitin hydrogels, which were washed with deionized water to neutral. The chitin aerogels were obtained from the corresponding hydrogel by freeze-drying. The chitin aerogels were placed in the center of a quartz tube furnace and annealed at 600, 700, 800, or 900 °C (2 °C/min) for 2 h with Ar flow to obtain the corresponding carbon aerogels-chitin-temperature (CAs-chitin-T) (Fig. S1b).
C. The synthesis procedure of carbon aerogels-cellulose decorated with Pt NPs (Pt-CAs-cellulose-800)
Cellulose solution (5.0 wt%) was prepared by dissolving 3.0 g cotton linter pulp in a 97.0 g mixed solvent under a precooled −10 °C solution (7.0 wt% NaOH/12.0 wt% urea/81.0 wt% water). 8.0 mL of 20 mg mL−1 H2PtCl6 aqueous solution was added into bubbles-removed cellulose solution (60.0 g) dropwise at −5 °C for homogeneous mixing for 2 h. Subsequently, low molecular-weight chemical cross-linker epichlorohydrin (ECH, 3.0 mL) was added dropwise to the above solution at −5 °C and homogeneous mixing for another 2 h. After cross-linking, the homogeneous solution was obtained by centrifugation and injected into the mold. Then, the mold was put in a 60 °C oven for 30 min to get hydrogels, which were washed with deionized water to neutral. The aerogels were obtained from the corresponding hydrogels by freeze-drying. The aerogels were placed in the center of a quartz tube furnace and annealed at 800 °C (2 °C/min) for 2 h with Ar flow to obtain the corresponding carbon aerogels supported Pt nanoparticles (Pt-CAs-cellulose-800).
D. The synthesis procedure of carbon aerogels-chitin decorated with Pt NPs (Pt-CAs-chitin-800)
Chitin solution (5.0 wt%) was prepared by dissolving 6.0 g chitin powder in a 94.0 g mixed solvent of 11.0 wt% NaOH/4.0 wt% urea/85.0 wt% water under −35 °C in a cold trap for 4 h and then thawed at room temperature. 8.0 mL of 20 mg mL−1 H2PtCl6 aqueous solution was added into bubbles-removed chitin solution (60.0 g) dropwise at −5 °C for homogeneous mixing for 2 h. Subsequently, low molecular-weight chemical cross-linker epichlorohydrin (ECH, 3.0 mL) was added dropwise to the above solution at −5 °C and homogeneous mixing for another 2 h. After cross-linking, the homogeneous solution was obtained by centrifugation and injected into the mold. Then, the mold was put in a 60 °C oven for 30 min to get hydrogels, which were washed with deionized water to neutral. The aerogels were obtained from the corresponding hydrogels by freeze-drying. The aerogels were placed in the center of a quartz tube furnace and annealed at 800 °C (2 °C/min) for 2 h with Ar flow to obtain the corresponding carbon aerogel-supported Pt nanoparticles (Pt-CAs-chitin-800).
E. The synthesis procedure of chitin microspheres (CMs)
Chitin microspheres (CMs) with an average diameter of 30 ~ 50 μm were fabricated according to the method in our laboratory. Typically, 6.0 g chitin powder was dissolved in a 100.0 g mixture of 11 wt% NaOH/4 wt% urea/85 wt% water under −35 °C in a cold trap for 4 h, and then thawed at room temperature. Repeating the process of freezing-thawing 2 ~ 3 times generated a transparent chitin solution, which was centrifuged at 0 °C for 10 min to remove the bubbles. A mixture of 250.0 g isooctane and 16.5 g Span 85 was placed in a 1.0 L three-necked flask equipped with a mechanical stirrer, which was stirred at 0 °C for 0.5 h, and then the chitin solution was added quickly. The suspension was kept stirring for 1 h at 0 °C, followed by the addition of 9.0 g Towen 85 and 5.0 g isooctane, which was further stirred for another 1 h at 0 °C. Subsequently, the suspension of emulsion droplets was heated up to 90 °C for 10 min and neutralized with dilute hydrochloric acid (10%) after cooling. The obtained chitin microspheres suspension was washed with ethanol and water several times. Finally, the chitin microspheres were subjected to solvent exchange with tert-butyl alcohol (tBuOH) and lyophilized to afford the white powder.
F. The synthesis procedure of chitin microspheres with metal ions (M-CMs)
0.6 g of nanofibrous chitin microspheres was immersed in 100.0 mL deionized water. 8.0 mL of 25 mg mL−1 H2PtCl6·7H2O aqueous solution was dropwise added into 100.0 mL chitin microspheres suspension and magnetic stirring for 3 h. The mixed suspension was separated by filtration, and the obtained Pt-CM was dried through freeze-drying overnight. Pd-CMs, Ni-CMs, and Cu-CMs were prepared by the same procedure but changing the metal precursor, such as Pd(OAc)2, Ni(acac)2, and Cu(OAc)2.
G. The synthesis procedure of RuO2/TiO2 (60 nm)
8.0 mL of 13 mg mL−1 RuCl3·xH2O was dropwise added into 100.0 mL of 20% H2O2 under ice bath and stirred for 1 h. Then 1.0 g TiO2 (60 nm) was immersed and stirred for 3 h. The obtained materials were separated by centrifugation and dried under vacuum at room temperature overnight. After drying, the as-synthesized precursors were placed in a muffle furnace and annealed at 450 °C (5 °C/min) for 4 h with air flow to obtain RuO2/TiO2.
H. The synthesis procedure of composite carbon aerogel electrodes decorated with metal nanoparticles (M-CCAEs)
0.6 g Pt-CMs was dispersed in 10.0 mL of deionized water and added into bubbles-removed chitin solution (60.0 g) at −5 °C for homogeneous mixing for 2 h. Then, cross-linker ECH (3 mL) was dropwise added into the mixed solution and stirred for 2 h. After cross-linking, the homogeneous solution was obtained by centrifugation and injected into the mold. Then, the mold was put in a 60 °C oven for 30 min to get the composite chitin hydrogels, which were washed with deionized water to neutral. The composite chitin aerogels were obtained from the corresponding hydrogels by freeze-drying. The composite chitin aerogels were placed in the center of a quartz tube furnace and annealed at 800 °C (2 °C/min) for 2 h with Ar flow to obtain the corresponding composite carbon aerogel electrodes supported Pt nanoparticles (Pt-CCAEs). CCAEs, Pd-CCAEs, Ni-CCAEs, Cu-CCAEs, and RuO2-CCAEs were prepared by the same procedure, but changing the M-CMs or RuO2/TiO2 in chitin solution, respectively.
Electrocatalytic test
The electrocatalytic HER, OER, and chlorine anion oxidation reaction measurements were tested on a CHI 760E working station with a standard three-electrode setup. The Ag/AgCl and graphite rod were employed as the reference and counter electrode, respectively. M-CCAEs and other control electrodes (catalysts adhered to carbon paper (CP)) were used as the working electrodes. Control electrodes were prepared as follows: First, all CP substrates were cleaned by sonication treatment in CH3CH2OH, 2 M HCl, and deionized water for 15 min in sequence and finally dried at 80 °C for 12 h. Then, 120 µL of various catalyst inks (5 mg catalyst dispersed in 1 mL of isopropyl alcohol and 160 µL of N-methyl-2-pyrrolidone containing 2.59% PVDF) were pipetted onto the above CP (with a size of 1 cm × 1 cm). After drying, the control electrodes were fabricated. The M-CCAEs are directly used for HER, OER, and chlorine anion oxidation reaction tests with the same size as modified CP electrodes.
Electrosynthesis reactions
A. General procedure for electrochemical oxidative chlorination of cyclohexane
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with RuO2-CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and Pd-CCAEs (2.0 × 1.0 × 0.2 cm3) as the cathode (Fig. S41). Then the cell was charged with nitrogen. Concentrated hydrochloric acid (HCl (12.0 mol/L)) (0.25 mL, 3.0 mmol), cyclohexane (2.0 mL), and MeCN (8.0 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 20 mA under 25 °C for 3 h. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by GC with biphenyl as an internal standard or 1H NMR with CH2Br2 as an internal standard.
B. General procedure for electrochemical oxidative bromination of cyclohexane
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with RuO2-CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and a Ni plate as the cathode (1.5 × 1.5 × 0.03 cm3) as the cathode. Then the cell was charged with argon gas in the glove box. LiBr (43.5 mg, 0.5 mmol) and nBu4NBF4 (164.5 mg, 0.5 mmol) were added in the glove box. After leaving the glove box, the cell was charged with nitrogen. After that, cyclohexane (2.0 mL), MeCN (8.0 mL), and H2O (0.2 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 20 mA under 25 °C for 3 h. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by GC with biphenyl as an internal standard or 1H NMR with CH2Br2 as an internal standard.
C. General procedure for electrochemical oxidative nitration of cyclohexane
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with RuO2-CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and Pd-CCAEs as the cathode (2.0 × 1.0 × 0.2 cm3) as the cathode. Then the cell was charged with nitrogen. Concentrated nitric acid (HNO3 (68 wt%)) (0.2 mL, 3.0 mmol), cyclohexane (2.0 mL), and MeCN (8.0 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 20 mA under 25 °C for 3 h. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by GC with biphenyl as an internal standard.
D. General procedure for electrochemical oxidative etherification of tetrahydrofuran
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with RuO2-CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and Pd-CCAEs (2.0 × 1.0 × 0.2 cm3) as the cathode. Then the cell was charged with argon gas in the glove box. nBu4NBF4 (164.5 mg, 0.5 mmol) was added in the glove box. After leaving the glove box, the cell was charged with nitrogen. Then, benzyl alcohol (108.0 mg, 1.0 mmol), concentrated hydrochloric acid (HCl (12.0 mol/L)) (17 μL, 0.2 mmol), THF (2.0 mL), and MeCN (8.0 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 20 mA under 0 °C for 6 h. When the reaction was finished, the solution was extracted with ethyl acetate. The combined organic layer was dried with Na2SO4, and the solvent was removed with a rotary evaporator. The pure product was obtained by flash chromatography on silica gel using petroleum ether and ethyl acetate as the eluent.
E. General Procedure for electrochemical HIE reaction of C(sp3)–H bonds
An oven-dried, undivided three-necked flask (15 mL) with a stir bar was equipped with RuO2-CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and Pd-CCAEs (2.0 × 1.0 × 0.2 cm3) as the cathode. Then the cell was charged with argon gas in the glove box. Et4NCl (82.8 mg, 0.5 mmol) and Ibuprofen methyl ester (66.0 mg, 0.3 mmol) were added in the glove box. After leaving the glove box, the cell was charged with nitrogen. Then, CD3OD (450 μL, 10.0 mmol), DMF (4.0 mL), and tetrahydrofuran (THF, 2.0 mL) were added. The reaction mixture was stirred and electrolyzed at a constant current of 10 mA under 25 °C for 5.0 h. At the end of the reaction, the reaction was quenched by brine and extracted with EtOAc three times. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum. The desired products were obtained in the corresponding yields after purification by flash chromatography on 200–300 mesh silica gel (PE: EtOAc = 50:1).
F. The procedure for electrochemical oxidation of monomethyl adipate
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with RuO2-CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and a Pt plate (1.5 × 1.5 × 0.03 cm3) as the cathode. Then the cell was charged with argon gas in the glove box. KOH (22.4 mg, 0.4 mmol) was added in the glove box. After leaving the glove box, the cell was charged with nitrogen. Monomethyl adipate (640.0 mg, 4.0 mmol) and MeOH (10.0 mL) were added. The reaction mixture was stirred and electrolyzed at a constant current of 60 mA at 65 °C for 40 min. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by 1H NMR with dibromomethane as an internal standard.
G. The procedure for electrochemical oxidation of 4-tert-butyl toluene
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and a Pt plate (1.5 × 1.5 × 0.03 cm3) as the cathode. Then the cell was charged with nitrogen. After that, 4-tert-butyl toluene (75.0 mg, 0.5 mmol), H2SO4 (98.3 wt%) (55 μL, 1.0 mmol), and ethanol (EtOH) (10.0 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 20 mA at 75 °C for 5 h. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by 1H NMR with dibromomethane as an internal standard.
H. The procedure for the electrochemical reduction of 4-tert-butyl styrene
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and Pd-CCAEs (2.0 × 1.0 × 0.2 cm3) as the cathode. Then the cell was charged with argon gas in the glove box. nBu4NBF4 (164.5 mg, 0.5 mmol) was added in the glove box. After leaving the glove box, the cell was charged with nitrogen. Then, 4-tert-butyl styrene (80.0 mg, 0.5 mmol), H2O (1.0 mL), and MeCN (9.0 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 20 mA under room temperature for 3 h. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by 1H NMR with dibromomethane as an internal standard.
I. The procedure for electrochemical reduction of 1-bromonaphthalene
An oven-dried, undivided three-necked flask (15.0 mL) with a stir bar was equipped with CCAEs (2.0 × 1.0 × 0.2 cm3) as the anode and Ni-CCAEs (2.0 × 1.0 × 0.2 cm3) as the cathode. Then the cell was charged with argon gas in the glove box. 4-Bromobenzonitrile (54.6 mg, 0.3 mmol) and nBu4NBF4 (98.7 mg, 0.3 mmol) were added in the glove box. After leaving the glove box, the cell was charged with nitrogen. Then, triethylamine (60.6 mg, 0.6 mmol) and MeCN (10.0 mL) were successively added. The reaction mixture was stirred and electrolyzed at a constant current of 15 mA under room temperature for 2 h. When the reaction was finished, the solution was extracted with ethyl acetate. The yield was detected by 1H NMR with dibromomethane as an internal standard.
Data availability
All data needed to support the findings of this study are included in the main text or in the Supplementary Information. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Gomollón-Bel, F. IUPAC’s 2023 top ten emerging technologies in chemistry. Chem. Int. 45, 14–22 (2023).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Fu, N., Sauer, G. S., Saha, A., Loo, A. & Lin, S. Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579 (2017).
Huang, X., Zhang, Q., Lin, J., Harms, K. & Meggers, E. Electricity-driven asymmetric Lewis acid catalysis. Nat. Catal. 2, 34–40 (2018).
Zhang, L. et al. Photoelectrocatalytic arene C-H amination. Nat. Catal. 2, 266–373 (2019).
Mo, Y. et al. Microfluidic electrochemistry for single-electron transfer redox-neutral reactions. Science 368, 1352–1357 (2020).
Dong, X., Roeckl, J. L., Waldvogel, S. R. & Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 371, 507–514 (2021).
Chen, H. et al. One-pot bioelectrocatalytic conversion of chemically inert hydrocarbons to imines. J. Am. Chem. Soc. 144, 4047–4056 (2022).
Pulignani, C. et al. Rational design of carbon nitride photoelectrodes with high activity toward organic oxidations. Angew. Chem. Int. Ed. 61, e202211587 (2022).
Han, C. et al. Electrocatalytic hydrogenation of alkenes with Pd/carbon nanotubes at an oil–water interface. Nat. Catal. 5, 1110–1119 (2022).
Kim, J., Jang, J., Hilberath, T., Hollmann, F. & Park, C. B. Photoelectrocatalytic biosynthesis fuelled by microplastics. Nat. Syn. 1, 776–786 (2022).
Luo, L. et al. Selective photoelectrocatalytic glycerol oxidation to dihydroxyacetone via enhanced middle hydroxyl adsorption over a Bi2O3-incorporated catalyst. J. Am. Chem. Soc. 144, 7720–7730 (2022).
Sun, G. Q. et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 615, 67–72 (2023).
Leech, M. C., Garcia, A. D., Petti, A., Dobbs, A. P. & Lam, K. Organic electrosynthesis: from academia to industry. React. Chem. Eng. 5, 977–990 (2020).
Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science 368, 1228–1233 (2020).
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Wang, Q. et al. Electrocatalytic methane oxidation greatly promoted by chlorine intermediates. Angew. Chem. Int. Ed. 60, 17398–17403 (2021).
Fagnani, D. E., Kim, D., Camarero, S. I., Alfaro, J. F. & McNeil, A. J. Using waste poly(vinyl chloride) to synthesize chloroarenes by plasticizer-mediated electro(de)chlorination. Nat. Chem. 15, 222–229 (2023).
The Njarðarson Group. “Top 200 small molecule drugs by sales in 2023”. https://bpb-us-e2.wpmucdn.com/sites.arizona.edu/dist/9/130/files/2024/07/2023Top200SmallMoleculePosterV6.pdf (2023).
Yang, Z., Shi, W., Alhumade, H., Yi, H. & Lei, A. Electrochemical oxidative C(sp3)–H cross-coupling with hydrogen evolution. Nat. Syn. 2, 217–230 (2023).
Horn, E. J. et al. Scalable and sustainable electrochemical allylic C-H oxidation. Nature 533, 77–81 (2016).
Kawamata, Y. et al. Scalable, electrochemical oxidation of unactivated C-H bonds. J. Am. Chem. Soc. 139, 7448–7451 (2017).
Rafiee, M., Wang, F., Hruszkewycz, D. P. & Stahl, S. S. N-hydroxyphthalimide-mediated electrochemical iodination of methylarenes and comparison to electron-transfer-initiated C-H functionalization. J. Am. Chem. Soc. 140, 22–25 (2018).
Huang, H., Strater, Z. M. & Lambert, T. H. Electrophotocatalytic C-H functionalization of ethers with high regioselectivity. J. Am. Chem. Soc. 142, 1698–1703 (2020).
Xu, P., Chen, P. Y. & Xu, H. C. Scalable photoelectrochemical dehydrogenative cross-coupling of heteroarenes with aliphatic C-H bonds. Angew. Chem. Int. Ed. 59, 14275–14280 (2020).
Meyer, T. H., Samanta, R. C., Del Vecchio, A. & Ackermann, L. Mangana(III/IV)electro-catalyzed C(sp3)-H azidation. Chem. Sci. 12, 2890–2897 (2020).
Liu, Y. et al. Time-resolved epr revealed the formation, structure, and reactivity of N-centered radicals in an electrochemical C(sp3)-H arylation reaction. J. Am. Chem. Soc. 143, 20863–20872 (2021).
Zhang, L. et al. Ritter-type amination of C(sp3)-H bonds enabled by electrochemistry with SO42-. Nat. Commun. 13, 4138 (2022).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, 4998 (2017).
Heard, D. M. & Lennox, A. J. J. Electrode materials in modern organic electrochemistry. Angew. Chem. Int. Ed. 59, 18866–18884 (2020).
Wu, X., Wang, Y. & Wu, Z.-S. Design principle of electrocatalysts for the electrooxidation of organics. Chem 8, 2594–2629 (2022).
Huang, Y., Chong, X., Liu, C., Liang, Y. & Zhang, B. Boosting hydrogen production by anodic oxidation of primary amines over a nise nanorod electrode. Angew. Chem. Int. Ed. 57, 13163–13166 (2018).
Huang, C., Huang, Y., Liu, C., Yu, Y. & Zhang, B. Integrating hydrogen production with aqueous selective semi-dehydrogenation of tetrahydroisoquinolines over a Ni2P bifunctional electrode. Angew. Chem. Int. Ed. 58, 12014–12017 (2019).
Liu, C., Han, S., Li, M., Chong, X. & Zhang, B. Electrocatalytic deuteration of halides with D2O as the deuterium source over a copper nanowire arrays cathode. Angew. Chem. Int. Ed. 59, 18527–18531 (2020).
Wu, Y., Liu, C., Wang, C., Lu, S. & Zhang, B. Selective transfer semihydrogenation of alkynes with H2O (D2O) as the H (D) source over a Pd-P cathode. Angew. Chem. Int. Ed. 59, 21170–21175 (2020).
Kurimoto, A., Sherbo, R. S., Cao, Y., Loo, N. W. X. & Berlinguette, C. P. Electrolytic deuteration of unsaturated bonds without using D2. Nat. Catal. 3, 719–726 (2020).
Bu, F. et al. Electrocatalytic reductive deuteration of arenes and heteroarenes. Nature 634, 592–599 (2024).
Fang, Z., Li, P. & Yu, G. Gel electrocatalysts: an emerging material platform for electrochemical energy conversion. Adv. Mater. 32, 2003191 (2020).
Fu, G. et al. Boosting bifunctional oxygen electrocatalysis with 3D graphene aerogel-supported Ni/MnO particles. Adv. Mater. 30, 1704609 (2018).
Miao, Z. et al. Atomically dispersed Fe-Nx/C electrocatalyst boosts oxygen catalysis via a new metal-organic polymer supramolecule strategy. Adv. Energy Mater. 8, 1801226 (2018).
Meng, J., Cheng, C., Wang, Y., Yu, Y. & Zhang, B. Carbon support enhanced mass transfer and metal-support interaction promoted activation for low-concentrated nitric oxide electroreduction to ammonia. J. Am. Chem. Soc. 146, 10044–10051 (2024).
Wu, K. et al. An iron-decorated carbon aerogel for rechargeable flow and flexible Zn-air batteries. Adv. Mater. 32, 2002292 (2020).
Chen, Z. et al. Iron single atom catalyzed quinoline synthesis. Adv. Mater. 33, 2101382 (2021).
Xu, C., Nasrollahzadeh, M., Selva, M., Issaabadi, Z. & Luque, R. Waste-to-wealth: Biowaste valorization into valuable bio(nano)materials. Chem. Soc. Rev. 48, 4791–4822 (2019).
Yan, N. & Chen, X. Sustainability: don’t waste seafood waste. Nature 524, 155–157 (2015).
Liu, T. et al. Chitin-induced dimerization activates a plant immune receptor. Science 336, 1160–1164 (2012).
Suginta, W., Khunkaewla, P. & Schulte, A. Electrochemical biosensor applications of polysaccharides chitin and chitosan. Chem. Rev. 113, 5458–5479 (2013).
Lu, L. et al. Carbon nanofibrous microspheres promote the oxidative double carbonylation of alkanes with CO. Chem 4, 2861–2871 (2018).
Manker, L. P. et al. Sustainable polyesters via direct functionalization of lignocellulosic sugars. Nat. Chem. 14, 976–984 (2022).
Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 31, 603–632 (2006).
Pei, X. et al. Size-controllable ultrafine palladium nanoparticles immobilized on calcined chitin microspheres as efficient and recyclable catalysts for hydrogenation. Nanoscale 10, 14719–14725 (2018).
Sadeghi, M. C. sp3)−H functionalization using chlorine radicals. Adv. Synth. Catal. 366, 2898–2918 (2024).
Shields, B. J. & Doyle, A. G. Direct C(sp3)-H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc. 138, 12719–12722 (2016).
Shi, A., Xie, P., Wang, Y. & Qiu, Y. Photoelectrocatalytic Cl-mediated C(sp3)-H aminomethylation of hydrocarbons by BiVO4 photoanodes. Nat. Commun. 16, 2322 (2025).
Terao, J. & Kambe, N. Cross-coupling reaction of alkyl halides with Grignard reagents catalyzed by Ni, Pd, or Cu complexes with π-carbon ligand(s). Acc. Chem. Res. 41, 1545–1554 (2008).
Liu, W. & Groves, J. T. Manganese porphyrins catalyze selective C−H bond halogenations. J. Am. Chem. Soc. 132, 12847–12849 (2010).
Quinn, R. K. et al. Site-selective aliphatic C-H chlorination using N-chloroamides enables a synthesis of chlorolissoclimide. J. Am. Chem. Soc. 138, 696–702 (2016).
Lv, X. L. et al. A base-resistant metalloporphyrin metal-organic framework for C-H bond halogenation. J. Am. Chem. Soc. 139, 211–217 (2017).
Zhao, M. & Lu, W. Visible light-induced oxidative chlorination of alkyl sp3 C-H bonds with NaCl/Oxone at room temperature. Org. Lett. 19, 4560–4563 (2017).
Lan, J. et al. Efficient electrosynthesis of formamide from carbon monoxide and nitrite on a Ru-dispersed Cu nanocluster catalyst. Nat. Commun. 14, 2870 (2023).
Gao, X. et al. Membrane-free water electrolysis for hydrogen generation with low cost. Angew. Chem. Int. Ed. 64, e202417987 (2025).
Tomat, R. & Rigo, A. Electrochemical oxidation of aliphatic hydrocarbons promoted by inorganic radicals. II. NO3 radicals. J. Appl. Electrochem. 16, 8–14 (1986).
Patra, S., Mosiagin, I., Giri, R., Nauser, T. & Katayev, D. Electron-driven nitration of unsaturated hydrocarbons. Angew. Chem. Int. Ed. 62, e202300533 (2023).
Huang, R., Yu, C. & Patureau, F. W. Electrochemical dehydrogenative acetalization protection of alcohols with tetrahydrofuran. ChemElectroChem 8, 3943–3946 (2021).
Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493–494 (2017).
Keam, S. J. & Duggan, S. Donafenib: first approval. Drugs 81, 1915–1920 (2021).
Mullard, A. First de novo deuterated drug poised for approval. Nat. Rev. Drug Discov. 21, 623–625 (2022).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Li, R. et al. One-pot H/D exchange and low-coordinated iron electrocatalyzed deuteration of nitriles in D2O to α,β-deuterio aryl ethylamines. Nat. Commun. 13, 5951 (2022).
Behera, N. et al. Electrochemical hydrogen isotope exchange of amines controlled by alternating current frequency. Faraday Discuss. 247, 45–58 (2023).
Gao, Q. et al. Electroselective C(sp3)–H deuteration of isoindolinones. Org. Chem. Front. 10, 6212–6218 (2023).
Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2021YFA1500100, A.L.), the National Natural Science Foundation of China (grant no. 22031008, A.L.), the Science Foundation of Wuhan (grant no. 2020010601012192, A.L.), and the Cultivation Project of Nanchang University (F.L.). The authors would like to acknowledge the Center for Electron Microscopy at Wuhan University for their substantial supports to TEM work. The authors would like to acknowledge Prof. Abhishek Dutta Chowdhury from Wuhan University for advice on the manuscript. The authors would like to acknowledge Prof. Lina Zhang from Wuhan University for advice on this work. Mrs. Lina Zhang sadly passed away on October 17th, 2020, as a result of illness. She proposed an alkali urea system and made a great contribution to the dissolving biomass renewable energy.
Author information
Authors and Affiliations
Contributions
L.L., Y.L., and A.L. conceived and designed the project. L.L. and H.L. performed initial discovery and optimization of reaction conditions. Y.L., X.J., and X.P. prepared the catalysts. L.L. and H.L. performed the study of substrate scope. L.L., Y.L., and H.L. performed mechanistic studies. D.Y., Y.-C.C., and J.-L.C. performed the XAS test and analyzed the data. L.L., Y.L., F.L., and A.L. wrote the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hyotaik Kang, Youai Qiu, and Chao Yan for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, L., Li, Y., Li, H. et al. Electrocatalytic C(sp3)-H bond functionalization using biomass-derived electrodes. Nat Commun 17, 2919 (2026). https://doi.org/10.1038/s41467-026-69274-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69274-7
