
Abstract
Recent advancements in artificial intelligence (AI) have revolutionized the field of 3D molecule generation. However, the lack of effective evaluation methods for 3D conformations limits further improvements. Current techniques, in order to achieve the necessary speed for evaluating large number of AI-generated molecules, often rely on empirical geometric metrics that do not adequately capture various conformational anomalies, or on molecular mechanics energy metrics that exhibit low accuracy and lack atomic or torsional details. To address this gap, we propose a two-stage approach that achieves both high speed and quantum mechanical level accuracy. The first stage, termed the validity test, employs an AI-derived force field to identify atoms with elevated energy resulting from implausible neighboring environments. The second stage, known as the rationality test, utilizes a deep learning network trained on data with density functional theory accuracy to detect rotatable bonds with high torsional energies. To demonstrate the functionality of our evaluation system, we applied our approach to five recently reported 3D molecule generation AI models across 102 targets in Directory of Useful Decoys-Enhanced dataset. To facilitate accessibility for the academic community, our method is available as an open-source package.
Similar content being viewed by others


Introduction
Recent advances in deep learning generative models have begun to demonstrate the ability to generate small molecules with 3D conformations based on a given protein pocket1. Both auto-regressive models and diffusion models have shown this capability2,3,4,5,6,7. However, these AI models continue to face challenges in generating physically implausible 3D conformations. A number of studies have reported the production of abnormal conformations, characterized by steric clashes, twisted structures, and the misplacement of hydrogen atoms3,8,9.
To quantify the issues of abnormal conformation and thereby guide AI model training, two types of evaluation methods are frequently adopted: (1) geometry-based and (2) energy-based conformation assessment. Geometric approaches typically examine bond lengths, bond angles, the overlap of Van der Waals radii of two atoms10,11, or the redocking root-mean-squared deviation (RMSD)8. However, these geometric metrics are intrinsically limited by the lack of an energy criterion, which can lead to misleading results. For instance, two conformations with a small RMSD may have substantially different energies. Additionally, geometric approaches may use Kullback-Leibler or Jensen-Shannon divergence to evaluate AI-generated conformations against reference molecules by analyzing geometric features like bond lengths and angles3,6. The reliability of such measurement heavily relies on the representativeness of the reference set. A reference set with limited chemical space coverage can introduce bias, causing misleading results when evaluating molecules that are far from the chemical space covered by the reference set. Energy-based metrics often strive to achieve a balance between accuracy and time efficiency. High-precision calculations, such as density functional theory (DFT)12 techniques, can be quite time-consuming. On the contrary, time-efficient alternatives such as semi-empirical13 and molecular mechanics (MM) methods14,15,16,17 often have relatively low accuracy18,19. More importantly, most current energy-based methods provide only global energies at the molecular level, lacking detailed atom-level or torsion-level energy assessments. This limitation presents a significant challenge for AI algorithm designers, who require detailed insights into the positioning of individual atoms.
In addition to the challenges related to evaluation tactics, the debate over evaluation strategies for AI-generated conformations further complicates the situation. Some studies advocate for pre-refinement evaluation, which involves the direct assessment of AI-generated conformations without MM refinement2,4, while others propose post-refinement evaluation, which involves a preliminary MM optimization with the protein pocket fixed prior to assessment3,7. Both of the pre-refinement and post-refinement evaluations have their pros and cons. Pre-refinement evaluations can reveal model deficiencies that may be concealed by force field optimization; however, they often lack the sensitivity needed for detailed analysis, especially when quantum mechanics (QM) computations are involved. For example, when applying DFT to assess torsion energies in molecular conformations that exhibit abnormal bond lengths, the results are primarily influenced by these irregular bond lengths instead of torsion angles. This situation underscores the necessity of correcting significant anomalies through MM refinement prior to conducting comprehensive assessments. Additionally, a model’s performance in pre-refinement evaluations doesn’t necessarily reflect its overall utility, as medicinal chemists might prefer a model that, despite initial shortcomings, provides more rational conformations after cost-effective force field-based refinement. This emphasizes the complexity of model evaluation in practical applications.
Given the pros and cons of these evaluation strategies and the limitations of existing tactics, there is an urgent need to develop a systematic framework for evaluating AI-generated conformations.
To overcome the limitations in current evaluation methodologies, we propose a two-stage procedure consisting of validity and rationality tests for AI-generated conformations before and after force-field refinement, respectively. For pre-refinement conformations, we determine whether they are valid by detecting abnormal conformations at the atomic level with energy-based metrics. The tool developed for this purpose, named HEAD (high-energy atom detector), utilizes the network of machine learning force fields (MLFFs) to compute the atomic energies corresponding to each atom’s local environment. HEAD shows higher efficiency in detecting abnormal conformations compared to the widely used benchmark method PoseBusters8, which mainly rely on geometry-based metrics. However, a force-field valid conformation may still be irrational because of high torsion energy. To this end, we propose a rationality test designed to quantify the disparity between post-refinement conformations and low-torsional energy conformations. The tool developed for this purpose, named TED (torsional energy descriptor), is mainly composed by a deep learning-based torsion energy prediction model (referred to as the TED-Model henceforth). To train the TED-Model, we developed two data sets, including a pretraining set containing semi-empirical torsion energy data for six million torsion fragments and a training set containing double-hybrid DFT data for 100,000 torsion fragments. TED demonstrates superior accuracy compared to GFN2-xTB when evaluated on a data set of 5000 torsional fragments without information leakage. To illustrate the application of our evaluation system, we evaluated five recently reported 3D molecule generative models including Lingo3DMolv23, Pocket2Mol5, PocketFlow2, TargetDiff6, and PMDM4. Each model generated around 1000 molecules per target across 102 targets from Directory of Useful Decoys-Enhanced (DUD-E) dataset20 and underwent both validity and rationality tests. To facilitate the use of our evaluation system, we have made the HEAD and TED models accessible via link https://github.com/stonewiseAIDrugDesign/HEAD_TED. (The code is also included in Code Ocean capsule21.)
Results
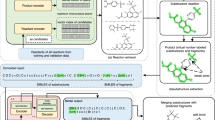
In this section, we first describe the construction of our HEAD and TED modules (Fig. 1a, b), outlining the fundamental logic behind their design and presenting testing results that demonstrate their reliability. We then report an evaluation test in which HEAD and TED were applied to five recently reported 3D molecule generation models: Lingo3DMolv2, TargetDiff, Pocket2Mol, PocketFlow, and PMDM.

a Overview of the HEAD module. It assesses conformation validity by calculating atomic energies (step 1), establishing thresholds for anomaly detection (step 2), and classifying conformations based on energy comparisons against the threshold (step 3). The threshold is denoted as Ec. b Overview of the TED module. It evaluates conformation rationality by decomposing the structure into torsion fragments (step 1), predicting torsion energies for the target rotatable bond within each fragment (step 2), and assigning a binary label based on an energy threshold of 2 kcal/mol (step 3). Details of the torsion energy prediction neural network are provided in Methods.
Development of HEAD
The HEAD module is designed to assess the quality of AI-generated 3D molecular conformations without MM optimization. It directly takes an AI-generated conformation as input and quantitatively identifies atoms responsible for the anomalies of the conformation. This module is developed through three steps, as illustrated in Fig. 1a. The first step involves computing atomic energies using MLFFs. The MLFFs used in this study is ANI-2x22. The second step focuses on establishing thresholds to classify energy values that are significantly higher than normal. This is accomplished by applying the MLFF method to molecules from classical databases to obtain energy distributions for each elemental type. Based on these distributions, thresholds for each element are established. The third step involves comparing the atomic energies in a given molecule with the thresholds of the corresponding elements, resulting in a binary label indicating whether the molecular conformation is abnormal. Detailed information regarding the three steps is provided in the Methods section (HEAD Development).
To evaluate the performance of HEAD, we use PoseBusters8 as a benchmark. PoseBusters is a recently released test suite enabling implausible conformation identification. It utilizes the RDKit23 toolset to evaluate input molecular conformations by analyzing geometry metrics such as bond lengths, bond angles, aromatic ring planarity, double bond planarity, and internal steric clashes. It also employs an energy metric that determines whether the input molecule’s MM-level energy exceeds 100 times the average energy of 50 force field-optimized conformations.
We assessed the performance of HEAD and PoseBusters from three perspectives: (1) recall rate of valid conformations, (2) discriminative ability to distinguish valid from invalid conformations, and (3) speed.
To test the recall rate of valid conformations, we utilized two distinct databases. First, LigBoundConf dataset24, was used as a benchmark for valid conformations of ligand binding with a protein. This dataset includes 8,145 drug-like molecules that are sourced from the Protein Data Bank (PDB)25 and optimized with the OPLS3e force field26 in the presence of binding proteins. Second, the Cambridge Structural Dataset (CSD)27, was used as a benchmark for valid conformations of ligand without protein binding (i.e., apo state). This dataset is comprised of high-quality experimentally determined small molecule crystal structures. Due to the presence of elements not supported by ANI-2x in some molecules from these two datasets, we applied a screening criterion that limits inclusion to molecules composed of commonly occurring elements: hydrogen (H), carbon (C), nitrogen (N), oxygen (O), fluorine (F), sulfur (S), and chlorine (Cl). After applying this criterion, we retained 214,188 molecules from the CSD and 6209 from LigBoundConf.
We considered all conformations in both the CSD and LigBoundConf datasets as valid (positive samples), and assessed the positive recall rate of the testing methods. A low positive recall rate indicates that the method incorrectly classifies valid conformations as invalid. Given the inherent energy differences between bound and apo conformations, we also examined whether the testing methods exhibit different recall rates for bound (LigBoundConf) versus apo (CSD) states. As shown in Fig. 2a, both HEAD and PoseBusters achieve valid ratios exceeding 95% on both datasets, with no notable differences between the LigBoundConf and CSD datasets. This suggests that the criteria applied in both methods for identifying valid conformations are not overly stringent, allowing for effective recognition of experimentally determined conformations in both apo and bound states as valid. In addition, it is noteworthy that, although HEAD and PoseBusters show similar performance, HEAD is around 30 times faster than PoseBusters. This makes the HEAD approach more suitable for high-throughput screening tasks. A detailed speed comparison can be found in Supplementary Table 1.

a Valid conformation recall rates of HEAD and PoseBusters tested using Cambridge Structural Database (CSD) and LigBoundConf. Molecular conformations in CSD and LigBoundConf are all considered as valid, representing valid conformations without and with protein binding, respectively. All bar chart values are obtained from a single deterministic processing of the dataset; repeated runs yield identical results. b Histogram distribution of molecules in GM-5K dataset across ΔE with the bin size of 20 kcal/mol, where \(\Delta {{{\rm{E}}}}={{{{\rm{E}}}}}_{{{{\rm{ori}}}}}^{{{{\rm{DFT}}}}}-{{{{\rm{E}}}}}_{{{{\rm{opt}}}}}^{{{{\rm{DFT}}}}}.\) The red dashed line indicates the threshold value used to classify molecular conformations as valid or invalid. Molecules with ΔE values below the threshold are labeled as valid, while those above the threshold are labeled as invalid. The dynamic adjustment of the threshold value leads to corresponding changes in valid and invalid labels. c Weighted F1 scores of HEAD and PoseBusters across varying labeling thresholds based on ΔE in the GM-5K dataset. The threshold was systematically varied from 0 to 1000 kcal/mol in increments of 1 kcal/mol, resulting in a series of weighted F1 scores. The weighted F1 score is computed by first evaluating the F1 scores for both valid and invalid classes. Subsequently, these F1 scores are averaged considering the number of samples in each class as weights. Panels (d–g) showcase four representative abnormal geometries with their atomic energies, including steric clash (d), twisted ring structure (e), misplacement of a hydrogen atom (f), and valence violation (g). In panel (d), the steric clash between the chlorine atom and a methylene group is circled in red. In each panel, the molecular 3D conformation is shown as sticks, accompanied by a bar chart displaying atomic energies. Atom names are shown on the horizontal axis and energy values on the vertical axis. Atoms exhibiting abnormally high atomic energies are indicated by red arrows in the stick representation and highlighted by red bars in the corresponding bar charts.
Next, we evaluated the discriminative ability of HEAD to distinguish valid from invalid conformations. To do this, we introduced a database, GM-5K, consisting of randomly selected AI-generated molecules from Lingo3DMolv2, TargetDiff, Pocket2Mol, PocketFlow, and PMDM. Because GM-5K dataset contains conformations of varying quality, we employed some calculations to label the conformation quality. Specifically, we employed MMFF94 force field15 for ligand geometry optimization with protein pocket fixed and conducted QM-level (revDSD-PBEP86-D3(BJ)/def2-TZVPP)28,29,30 single point energy computations for the conformations before and after optimization. The energy difference between original and optimized conformations, i.e., \(\Delta {{{\rm{E}}}}={{{{\rm{E}}}}}_{{{{\rm{ori}}}}}^{{{{\rm{DFT}}}}}-{{{{\rm{E}}}}}_{{{{\rm{opt}}}}}^{{{{\rm{DFT}}}}}\), was used as the quality indicator of the original conformation before optimization. GM-5K dataset supported the test of HEAD’s ability to distinguish abnormal conformations from valid ones.
We checked the weighted F1 scores of the HEAD and PoseBusters models on the GM-5K dataset to evaluate their ability to discriminate between valid and invalid 3D molecular conformations. GM-5K contains molecules with ΔE varying from 0 kcal/mol to 1000 kcal/mol, shown as the histogram distribution displayed in Fig. 2b. To compare the discrimination powers of HEAD and PoseBusters, we need to assign a valid-invalid binary label to every molecule in GM-5K based on its ΔE value. Specifically, molecules with ΔE values over a specified threshold should be classified as invalid, while those with values below the threshold should be classified as valid. Since there is no universal threshold for classifying conformation validity, we systematically varied the threshold from 0 to 1000 in increments of 1 kcal/mol. As we adjusted the labeling threshold, as illustrated in Fig. 2b, the corresponding valid and invalid labels were updated accordingly. This process resulted in a set of F1 scores that correspond to the variations of labeling thresholds. The results, plotted in Fig. 2c, shows that HEAD notably outperforms PoseBusters in identifying abnormal conformations with ΔE values between 200 kcal/mol and 600 kcal/mol. For the conformations with ΔE values outside this region, the two methods exhibited comparable performance. Some example cases that can be detected by HEAD but not PoseBusters are shown in Fig. 2d–g. The examples include steric clashes, twisted rings, misplacement of hydrogens, and valence violation. The reason HEAD outperforms PoseBusters in detecting anomalous conformations may be related to the missing definitions of geometric anomalies in PoseBusters. Specifically, anomaly detection is more effective in energy space than in real space. It is highly difficult to enumerate all geometric anomalies in real space. In contrast, all abnormal situations correspond to high-energy responses.
It is also notable that for conformations with relatively small anomalies, indicated by low ΔE values, both PoseBusters and HEAD experience a sharp drop in weighted F1-score (Fig. 2c). This trend indicates the limitations of these methods for detecting anomalies within this range. The potential causes of these limitations will be discussed in the Discussion section.
Subsequently, we sought to assess the capability of the HEAD framework in identifying the quality of ligand-protein pocket interactions. The underlying mechanism relies on using HEAD to calculate atomic-level energy changes for both the ligand and the pocket upon binding. A significant increase in energy during binding indicates the presence of unfavorable interactions between the ligand and the protein pocket. Further methodological details are provided in Methods section (Evaluation of Ligand-Protein Interaction Validity). To quantify HEAD’s performance in this context, we need a ground truth label to indicate the quality of ligand-protein interactions. In this study, we used the binding free energy value for each molecule in the GM-5K dataset to reflect the quality of its interaction with the corresponding binding pocket. The binding free energy values were computed using the molecular mechanics-generalized Born surface area (MM/GBSA) method31,32, as described in the Methods section (GM-5K).
In terms of benchmark technologies, we used two types of methods. First, PoseBusters was used as the representative of the geometric criteria-based method. It assesses ligand-pocket interactions by analyzing atomic-level geometric distances and van der Waals radius overlaps. Second, we noticed that there are several recent studies33,34 using docking software to describe the interactions between ligand and pocket and thereby support the evaluation of AI molecular generation models. We used the work of Ciepliński et al. (2023)33 as a representative of this type of method. It offers a weighted scoring system (Vinardo score) to evaluate ligand-protein interactions based on SMINA35.
To benchmark against PoseBusters, which provides binary labels for clashes between the ligand and the pocket, we consider a positive MM/GBSA binding free energy for the ligand in the pocket as an indicator of unfavorable interactions. In other words, MM/GBSA binding free energy serves as a quantitative measure of the severity of steric incompatibilities between the ligand and the pocket. As shown in the histogram presented in Fig. 3a, 83% of the AI-generated molecules within GM-5K exhibited positive binding free energies, suggesting a prevalence of unfavorable interactions with their respective binding pockets. To evaluate HEAD’s ligand-protein clash detection capabilities in GM-5K, we compared its performance to that of PoseBusters by calculating weighted F1 scores across various binding free energy thresholds for valid (clashes) and invalid (non-clashes) labeling. The results, illustrated in Fig. 3b, indicate that HEAD outperforms PoseBusters in identifying severe clashes (e.g., binding free energy > 100 kcal/mol), while both methods demonstrate comparable performance in detecting mild clashes. To further elucidate the mechanistic basis underlying HEAD’s superiority to PoseBusters, we conducted case analyses of clashes identified by HEAD that were not detected by PoseBusters (Fig. 3c, d). Representative examples in Fig. 3c highlight hydrogen-hydrogen clashes, a category that PoseBusters overlooks due to its reliance on geometric heuristics that consider only heavy-atom distances. In contrast, HEAD’s energy-based framework accounts for both steric and electrostatic contributions from heavy and hydrogen atoms. For example, Fig. 3d illustrates a case in which HEAD detected a lone pair–mediated clash between the ligand O4 atom and the backbone carbonyl oxygen of pocket E377, which was missed by PoseBusters, highlighting HEAD’s enhanced sensitivity to subtle electronic interactions.

Panels (a–d) focus on the comparison between HEAD and PoseBusters using F1 scores, while Panels (e) and (f) compare HEAD with Ciepliński et al. (2023) using Pearson correlations. a Histogram showing the distribution of Molecular Mechanics-Generalized Born Surface Area (MM/GBSA31,32) binding free energies for AI-generated molecules in the GM-5K dataset. The red dashed line indicates the threshold value used to label ligand-protein complexes as either “valid” or “invalid”. Molecules with MM/GBSA binding free energy values below this threshold are labeled as “valid,” while those above the threshold are classified as “invalid”. The dynamic adjustment of the threshold value results in corresponding changes to the valid and invalid labels. b Weighted F1 scores of HEAD and PoseBusters across varying labeling thresholds based on MM/GBSA binding free energy of molecules in the GM-5K dataset. The threshold was systematically varied from 0 to 2000 kcal/mol in increments of 1 kcal/mol, yielding a series of weighted F1 scores. Panels (c) and (d) showcase two representative cases which were detected by HEAD but missed by PoseBusters, including a representative example of ligand-protein clashes involving hydrogens (c), and a case involving lone pair electron clash between two carbonyl groups (d), specifically the ligand O4 atom and the backbone carbonyl oxygen of pocket E377. In each panel, the ligand–protein complex is shown as sticks, accompanied by a bar chart displaying atomic energy difference for ligand upon binding with proteins (\(\Delta {{{\rm{E}}}}={{{{\rm{E}}}}}_{{{{\rm{ligand}}}}}^{{{{\rm{bound}}}}}-{{{{\rm{E}}}}}_{{{{\rm{ligand}}}}}^{{{{\rm{isolated}}}}}\)). Atom names are shown on the horizontal axis and atomic energy differences on the vertical axis. Atoms exhibiting high energies upon binding are highlighted by red bars in the bar charts and indicated by red arrows in the structures, signifying ligand–protein clashes (circled in red). The details of identifying these invalid atoms can be found in Methods section (Evaluation of Ligand-Protein Interaction Validity). e Scatter plot of MM/GBSA binding free energies (log scale) versus Ciepliński’s Vinardo docking scores33 on GM-5K dataset.f Scatter plot of MM/GBSA binding free energies (log scale) versus HEAD’s Ebind values on GM-5K dataset. \({{{{\rm{E}}}}}_{{{{\rm{bind}}}}}={{{{{\rm{E}}}}}_{{{{\rm{complex}}}}}^{{{{\rm{bound}}}}}}-({{{\rm{E}}}}_{{{{\rm{ligand}}}}}^{{{{\rm{isolated}}}}}+{{{{\rm{E}}}}}_{{{{\rm{pocket}}}}}^{{{{\rm{isolated}}}}}).\) PCC stands for Pearson Correlation Coefficient.
To benchmark our method against the work of Ciepliński et al. (2023)33, we first note that their approach employs the Vinardo score to assess the quality of ligand-pocket interactions. The Vinardo score, as implemented in the SMINA software package, represents a weighted sum of ligand-protein steric interactions, hydrophobic contacts, and non-directional hydrogen bonds. Unlike the binary classification label provided by PoseBusters, the Vinardo score is continuous, making the use of F1 scores for performance evaluation inapplicable. Therefore, we employed correlation analysis on the GM-5K dataset by comparing the Pearson correlation between Ciepliński’s Vinardo scores and MM/GBSA binding free energies with that between the binding energy estimates from our HEAD approach (\({{{{\rm{E}}}}}_{{{{\rm{bind}}}}}\) and MM/GBSA reference values. Here, \({{{{\rm{E}}}}}_{{{{\rm{bind}}}}}={{{{{\rm{E}}}}}_{{{{\rm{complex}}}}}^{{{{\rm{bound}}}}}}-({{{\rm{E}}}}_{{{{\rm{ligand}}}}}^{{{{\rm{isolated}}}}}+{{{{\rm{E}}}}}_{{{{\rm{pocket}}}}}^{{{{\rm{isolated}}}}})\). As shown in Fig. 3e, f, the results demonstrate a notable advantage for HEAD, with Ebind exhibiting a Pearson correlation of 0.55 with the MM/GBSA reference values, compared to only 0.14 for Ciepliński’s Vinardo score. This difference indicates that HEAD provides a more reliable and accurate evaluation of ligand-pocket interactions, capturing the nuances of binding energetics more effectively than the Vinardo score.
Next, we conducted a more granular analysis, dividing the GM-5K dataset into drug-like and non-drug-like subsets. Drug-like molecules were defined as those exhibiting a Quantitative Estimate of Drug-likeness (QED) score36 ≥ 0.3 and a Synthetic Accessibility Score (SAS)37 ≤ 5, accounting for 63% of GM-5K molecules. We then compared HEAD and benchmark technologies on both subsets, focusing on ligand conformation and ligand-protein interaction evaluations. The results (Supplementary Information Sec. 2.1) demonstrate that HEAD consistently outperforms benchmarks in both categories for both drug-like and non-drug-like molecules.
Development of TED
The TED module is developed to evaluate the quality of AI-generated 3D molecular conformations after MM-based optimization. It accepts a MM-refined conformation as input, quantitatively predicts the torsion energies for each rotatable bond that excludes hydrogens, and subsequently outputs a binary label indicating whether the input 3D conformation is abnormal. This module works in three steps, as illustrated in Fig. 1b. The first step involves decomposing a given conformation into a series of torsion fragments, each containing a target rotatable bond and its necessary local chemical environment. The second step predicts the torsion energy curve for the target rotatable bond in the torsion fragment using a deep learning model (i.e., TED-Model) that employs an attention mechanism. TED-Model was trained using 6 million torsion fragments with semi-empirical level energy data and fine-tuned using 100,000 torsion fragments with DFT level energy data. The third step assigns a rational-irrational binary label to the conformation by checking whether any of its rotatable bonds have a torsion energy exceeding 2 kcal/mol, a threshold employed in a previous study38. Comprehensive details regarding the development of the TED module, including TED-Model training and dataset construction, are provided in the Methods section (TED Development).
To evaluate the performance of our TED-Model, we compared its predictions with those obtained using the semiempirical GFN2-xTB method13. This assessment used the DFT-5K dataset, which contains 5000 unique torsion fragments not included in the training set of our model to mitigate information leakage. Each torsion fragment in DFT-5K has 24 conformers generated by torsion angle enumeration and labeled with DFT-level energies using approaches described in the Methods section (Torsion Energy Label Preparation).
To assess the alignment of our TED-Model’s predictions with DFT values relative to GFN2-xTB, we computed Pearson correlations on a per-torsion-fragment basis using the DFT-5K dataset. For each torsion fragment, 24 conformers were generated by torsion angle enumeration. We analyzed the correlations between the energies predicted by our model across these 24 conformers and the corresponding DFT energies, as well as the correlations between GFN2-xTB predictions and DFT values. The distribution of these Pearson correlations is presented in Fig. 4a. Our model exhibits a stronger agreement with DFT values, with a per-torsion-fragment average Pearson correlation of 0.84, compared to 0.63 for GFN2-xTB. This superior performance can be attributed to our model’s finetuning using DFT data, which enables it to effectively address scenarios where GFN2-xTB encounters challenges. Specifically, GFN2-xTB exhibits limitations in accurately characterizing anisotropy in electrostatic potential and polarization effects. For instance, the torsion fragment shown in Fig. 4b demonstrates that GFN2-xTB overestimates the torsion energy at ±180°, primarily due to its inadequate treatment of sigma-hole interactions between sulfur and the oxygen of the carbonyl group. Additionally, Fig. 4c demonstrates GFN2-xTB’s tendency to overestimate lone-pair repulsion, leading to an increase in torsion energy at ±180°.

a Histogram distribution of torsion fragments in the DFT-5K dataset across Pearson correlation coefficients between density functional theory (DFT) level energy values and predictions from our TED-Model and GFN2-xTB13. For each torsion fragment in the DFT-5K dataset, torsion energies were computed for 24 conformations derived by enumerating torsion angles with increments of 15°. Pearson correlation coefficients between both methods and DFT were calculated per torsion fragment. b Case illustration of our model’s superior performance to GFN2-xTB’s in the scenario of sigma-hole interactions. There is a sigma-hole associated interaction between the sulfur of the thiazole group and the oxygen of the carbonyl group when the dihedral angle of S-C-C-N is ±180°. c Case illustration of our model’s superior performance to GFN2-xTB’s in the scenario of lone-pair repulsion. There is a lone-pair repulsion between the nitrogen of the thienopyridine group and the oxygen of the carboxy group when the dihedral angle of C-O-C-C is ±180°. For panels (b) and (c), the atoms defining the dihedral angle are highlighted in red.
Evaluation of molecule generative models
In this section, we demonstrated the application of the HEAD&TED system in evaluating AI-generated molecules. Five recently reported AI generative models were included in this assessment. Specifically, Lingo3DMolv2, Pocket2Mol, and PocketFlow were selected to represent autoregressive models, while PMDM and TargetDiff were chosen as representatives of diffusion models. The evaluation was conducted using all 102 protein targets (PDB IDs) from the DUD-E dataset. For each target, models were configured to generate 1000 unique molecules. The binding pocket for each of these 102 PDB IDs was defined by utilizing the co-crystallized ligand and selecting protein residues within a model-specific radius around this ligand. No additional information regarding active compounds or decoys documented in DUD-E was input in the models under evaluation. Some models were unable to generate 1000 unique molecules for some of the 102 targets within reasonable resource consumption; further details can be found in the Supplementary Information Sections 2.2 and 2.3. All the AI-generated molecules were submitted to HEAD for pocket-ligand interaction and conformation validity test. They were then refined using force field OPLS3e26 with protein pockets fixed and then submitted to TED for conformation rationality test.
Prior to evaluating the conformational quality of the generated molecules, we need to emphasize the necessity of eliminating those exhibiting low level of drug-likeness or poor synthetic accessibility. This approach is supported by below observations. Even though the molecules displayed in Supplementary Fig. 1 and Supplementary Table 2 successfully met the criteria set by PoseBusters, HEAD, and TED tests, they were not considered drug candidates due to their low level of drug-likeness. This was reflected in their low Quantitative Estimate of Drug-likeness (QED) scores36 and poor synthetic accessibility, as indicated by high Synthetic Accessibility Scores (SAS)37. Furthermore, we examined the distribution of HEAD and TED passing rates against QED and SAS for molecules produced by the AI models under test (Supplementary Fig. 2). To establish the drug-like region, we defined criteria of a QED score of 0.3 or higher and a SAS score of 5 or lower, which collectively account for over 75% of the molecules listed in DrugBank39. As shown in Supplementary Fig. 2, all the AI models under test have generated molecules which are not drug-like but show high passing rate of HEAD or TED. This observation further emphasizes the necessity of eliminating molecules outside drug-like region before conformation quality evaluation. Otherwise, the evaluation results would be contaminated.
Building on the elimination of molecules with QED lower than 0.3 and SAS higher than 5, we conducted conformation quality evaluation using HEAD&TED. The conformation quality was analyzed on a per-target basis and defined as the percentage of molecules passing the specific test (Fig. 5). Figure 5a displays histograms of target counts versus HEAD ligand-protein interaction evaluation passing rates for each AI model. TargetDiff, Lingo3DMolv2 and PocketFlow demonstrated superior performance, as evidenced by distributions heavily skewed toward high passing rates, with mean values of 86%, 75%, and 77%, respectively. In contrast, other models, including Pocket2Mol and PMDM, exhibited mean passing rates of 40% and 21%, respectively. Regarding the HEAD ligand conformation validity test (Fig. 5b), Lingo3DMolv2 and Pocket2Mol outperformed other models. The TED test indicated that PocketFlow excelled in rationality test compared to other models (Fig. 5c). To further elucidate these findings, we examined the property distribution of molecules generated by different models (Supplementary Fig. 3). The favorable performance of PocketFlow in the TED rationality test may be attributed to its generation of molecules having fewer rotatable bonds, lower molecular weights, and fewer chiral centers compared to other models.

Molecule generative models including Lingo3DMolv23, Pocket2Mol5, PocketFlow2, TargetDiff6, and PMDM4 are involved in this evaluation. a–c Distributions of target counts based on the passing rates of high-energy atom detector (HEAD) for ligand-protein interaction evaluation, HEAD for ligand conformation evaluation, and torsional energy descriptor (TED) for torsion energy evaluation, with histograms presented in panels a–c, respectively. Mean values are indicated with red dashed lines. d Scatter plot of HEAD and TED passing rates on a per-target basis, with points colored according to the AI models. Passing HEAD indicates that both the pocket-ligand interaction and conformation validity tests were successfully passed. e Bubble chart providing a comprehensive evaluation of AI models, where the position of each bubble centroid corresponds to the per-target average of HEAD and TED passing rates. Bubble size encodes the fraction of generated molecules satisfying the drug-likeness thresholds (QED ≥ 0.3; SAS ≤ 5), with the corresponding percentage shown in parentheses. QED stands for Quantitative Estimate of Drug-likeness, and SAS stands for Synthetic Accessibility Score.
The HEAD powered ligand-protein interaction and ligand conformation test reveals the limitations of the AI model, which causes the gap between AI-generated conformations and those refined by force field. However, due to the low cost and high speed of force field-based optimization, this gap can be mitigated by performing such optimization for all AI-generated molecules with pocket fixed. Given the fact that drug designers often aim to design molecules with low torsion strain40, the critical question then becomes whether the force field-refined conformations truly align with the low-torsion energy criteria that drug designers consider acceptable, which is reflected in the TED-powered rationality test. To compile a comprehensive view of these AI molecule generation models, we plotted their HEAD passing rates (valid pocket-ligand interaction and ligand conformation) and TED passing rates on a per-target basis (Fig. 5d). We then considered points from the same AI model as a cluster and used the centroid of each cluster to represent it. These centroids were then transformed into bubbles, with their sizes reflecting the percentage of drug-like molecules, specifically those with a QED no less than 0.3 and an SAS no greater than 5. A larger bubble positioned in the upper right corner of Fig. 5e indicates better overall performance of the model. As observed, no single model outperformed all others comprehensively. However, PocketFlow and Lingo3DMolv2 have the potential to achieve superior performance by improving their validity and rationality, respectively. To further evaluate model performance across different target types, we stratified the results by DUD-E target classes (Supplementary Information Sec. 2.4). PocketFlow, Lingo3DMolv2, and TargetDiff exhibited relatively consistent performance across target classes in both HEAD and TED assessments, whereas PMDM and Pocket2Mol showed variable results.
In addition to its role as an evaluation tool, HEAD-TED can also be employed to establish a unified protocol for screening deep-learning-generated 3D molecules. To this end, we have proposed a multi-stage screening pipeline for AI-generated molecules. This pipeline evaluates molecular candidates based on drug-likeness using QED and SAS metrics, pocket-ligand interaction and ligand conformation validity using HEAD, and conformational rationality using TED, as illustrated in Fig. 6a. We applied this pipeline to the aforementioned molecules generated by five AI molecule models on 102 DUD-E targets. As shown in Fig. 6b, approximately 20% of molecules generated by PocketFlow and Lingo3DMolv2 successfully passed all filters, whereas other models exhibited substantially lower final passing rates.

HEAD strands for a high-energy atom detector. TED stands for torsional energy descriptor. Molecule generative models including Lingo3DMolv23, Pocket2Mol5, PocketFlow2, TargetDiff6, and PMDM4 are involved in this evaluation. a Schematic overview of the sequential screening pipeline for evaluating AI-generated molecules. The pipeline integrates four filters: drug-likeness assessment with passing criteria defined as QED ≥ 0.3 and SAS ≤ 5, pocket-ligand interaction and ligand conformation validity test using HEAD, and conformational rationality test using TED. QED stands for Quantitative Estimate of Drug-likeness. SAS stands for Synthetic Accessibility Score. b Performance comparison of five AI-driven molecular generative models. The average passing rates for molecules generated on DUD-E targets are shown for each model at every screening step. DUD-E stands for the Directory of Useful Decoys-Enhanced dataset20. c Comparison of the final passing rates of different AI models across various target types. Bars represent the average final passing rate of each model for each target class. DUD-E dataset includes the following target types (with the number of targets in parentheses): GPCR (5), Kinase (26), Protease (15), Other Enzymes (36), Nuclear Receptor (11), Cytochrome P450 (2), Miscellaneous (5), and Ion Channel (2). All values are obtained from a single deterministic processing of the dataset; repeated runs yield identical results. Source data including target name, target type, passing rate at each stage of the screening pipeline can be found in supplementary materials.
Further stratification by DUD-E target classes (Fig. 6c) revealed distinct performance trends. PocketFlow demonstrated the most consistent success rates across target classes, whereas PMDM showed considerable variability. The AI models’ performance was most divergent on GPCR targets and most stable on Cytochrome P450 targets.
To evaluate the computational efficiency of HEAD and TED, we performed speed tests on a workstation equipped with a 48-core Intel(R) Xeon(R) Gold 6248 R processor and an NVIDIA V100S GPU. Due to the computational cost of these tests, particularly with repeated runs, we performed the test using the GM-5K dataset. Considering QED/SAS, HEAD, and TED as modules composing a screening pipeline, we performed two types of speed tests: independent module tests and integrated pipeline tests. In independent module tests, each module (QED/SAS, HEAD, and TED) independently processed all the molecules in the GM-5K dataset. Integrated pipeline tests, on the other hand, simulate a realistic screening scenario by running the GM-5K dataset through the complete pipeline, with each module processing only the molecules that pass the preceding filters. Our results demonstrate that HEAD processes molecules at a rate of 6 and 28 molecules per second for ligand-protein interaction and ligand conformation evaluation, respectively (Supplementary Table 3); TED achieves a rate of 8 molecules per second. Furthermore, in the integrated screening pipeline, HEAD and TED accounts for 77% and 22% of the total processing time, respectively (Supplementary Table 4). The integrated pipeline takes 720 seconds to finished screening 5000 molecules.
Discussion
In this work, we divided the evaluation of AI-generated 3D molecular conformations into two stages: validity and rationality tests. The validity test measures how closely AI-generated conformations align with force-field-refined conformations, while the rationality test assesses how closely a force-field-refined AI-generated conformation approaches a low-torsional-energy conformation. To support these evaluations, we introduced two tools: HEAD, powered by an AI-driven force field, and TED, which utilizes an AI torsion energy prediction network. Unlike traditional methods that primarily rely on geometric measures or molecular-level energy metrics, HEAD provides enhanced granularity through atomic-level energy metrics, and TED improves interpretability by identifying rotatable bonds with high torsion energy. Given the critical role that evaluation frameworks play in guiding the iterative development of AI models, we hope that our methodology can facilitate the continuous improvement of AI-based 3D molecular generation models, steering them toward the generation of more valid and rational conformations.
Regarding the limitations of our HEAD & TED approach, we have several points to discuss. First, the HEAD approach demonstrates the ability to distinguish anomalies that deviate significantly from force-field-refined conformations. However, as the degree of anomaly decreases, the discriminative power drops. This reduction may stem from the energy partitioning process during the training of ANI-2x. Specifically, ANI-2x predicts the energy of each atom in a molecular conformation and combines these predictions to obtain the molecule's total energy via simple algebraic summation. All parameters are trained to minimize the difference between the predicted total energy and the ground truth total energy. However, this process does not adequately train the energy distribution process, which poses a potential risk for our HEAD approach. Specifically, if an atom actually has high energy but that energy is distributed among its neighboring atoms, it may go undetected by the HEAD system. The risk of undetected high-energy atoms arising from this issue is relatively high when the molecule's total energy is low. This is consistent with the observation in Fig. 2c that the discriminative power of HEAD decreases along with the decrease of ΔE.
To address this limitation, we introduced the concept of information entropy as a metric for assessing the level of energy distribution among an atom and its spatial neighbors (details in Methods section: Information Entropy). By identifying conformations with high entropy and considering these conformations as invalid, we can detect previously missed abnormal conformations, as demonstrated in Fig. 7a–d. From the perspective of weighted F1 scores (Fig. 7e), we observed an improvement in HEAD’s discriminative power for conformations with small differences from force field refined ones (i.e., low ΔE). However, this improvement is achieved at the cost of reduced weighted F1 scores for high ΔE conformations, as shown in Fig. 7e. The trade-off associated with this entropy-based method highlights the importance of developing solutions from a more fundamental level, such as integrating learnable parameters into the atomic energy summation component of the ANI-2x architecture. This represents a promising direction for future improvements of HEAD approach.

a–d Examples of conformations identified as having high information entropy. In each panel, the molecular 3D conformation is shown as sticks, with the information entropy denoted by H, and accompanied by a bar chart of atomic energies (atom names on the horizontal axis and energy values on the vertical axis). Atoms with relatively high energies that contribute to elevated information entropy are highlighted by red bars and indicated by red arrows. Details of the identification procedure are described in the Methods section (Information Entropy). e Weighted F1 scores demonstrating the performance trade-off of introducing entropy-based metric. While the discriminative power of HEAD improves for conformations with low ΔE, there is a corresponding decrease in weighted F1 scores for conformations with high ΔE. The performance of PoseBusters8 is provided for reference.
Another limitation of our current implementation of HEAD is its reliance on ANI-2x, restricting its applicability to molecules containing only H, C, N, O, F, S, and Cl. To quantify this restriction, we performed a statistical analysis of the GOSTARTM database, a comprehensive resource documenting small molecules found in drug development-related patents since 1960. This analysis revealed that the seven atom types supported by ANI-2x collectively account for approximately 93.6% of the molecules in GOSTARTM database. This suggests that our current implementation of HEAD already covers a substantial portion of the relevant chemical space in medicinal chemistry, ensuring broad applicability in drug discovery. However, we acknowledge that expanding the range of supported atom types is crucial to the method's long-term utility. To that end, we emphasize that HEAD is designed as a flexible framework applicable to any MLFF that employs atomic energy partitioning scheme, not just the ANI-2x model used in this study. For instance, HEAD can be adapted to other MLFFs, such as the MACE-OFF model41. MACE-OFF is a deep-learning-based organic force field designed for fast and accurate interatomic potential predictions, utilizing an equivariant message passing architecture. It was trained on the SPICE dataset42 and able to support 10 atom types, including H, C, N, O, F, P, S, Cl, Br, and I. By using MACE-OFF, our HEAD implementation can cover 98.9% of the molecules in the GOSTARTM database. More importantly, we demonstrated that the transition from ANI-2x to MACE-OFF did not notably compromise performance in terms of ligand-protein interaction evaluation and ligand conformation evaluation (Supplementary Fig. 4a, b). This underscores HEAD’s modular framework and its compatibility with evolving force-field technologies.
Regarding the limitations of TED, it is designed for torsion energies prediction but not for overall strain energy estimation. Specifically, it is important to note that the overall strain energy of a conformation differs from torsion energy, because some relatively long-range intramolecular interactions can offset the energy penalties associated with unfavorable torsion angles and thereby reduce the overall strain energy of the conformation. For instance, Supplementary Fig. 5a presents such an example from the LigBoundConf dataset24. Although this molecule adopts a conformation with high torsion energy, the corresponding torsion angles facilitate the formation of an intramolecular hydrogen bond, which stabilizes the conformation. To test TED’s limitation in distinguishing irrational conformations arising from ligand conformational strain energy issues, we employed the LigBoundConf dataset. This dataset contains bound conformations and corresponding strain energy values for 8145 small molecules extracted from the PDB. The bound ligand conformations were optimized within ligand–protein complexes using Schrödinger’s PrepWizard43 with the OPLS3e force field26. Ligand conformations with heavy-atom RMSD greater than 0.5 Å after minimization were excluded to reduce the underestimation of ligand strain energy caused by the refinement process. Strain energies were calculated at DFT level as the energy difference between these bound conformations and their respective global minimum energy conformations without protein binding. We measured TED's weighted F1 scores on the LigBoundConf dataset to assess its effectiveness at identifying high-strain-energy conformations. The weighted F1 scores were calculated using varying thresholds for rational and irrational labeling (Supplementary Fig. 5b), consistent with our methodology for testing HEAD and PoseBusters on GM-5K, as described in the Results section (Development of HEAD). The results are illustrated in Supplementary Fig. 5c. TED demonstrates a relatively stable but mild discriminative power, having a weighted F1 scores around 0.6. Such limited discriminative power aligns with TED’s focus on assessing torsion quality without accounting for intramolecular interactions. These considerations serve as a reminder to users of TED: when a molecular conformation is deemed irrational by TED, it indicates only that the conformation has high torsion energy. Users should further examine intramolecular interactions for a comprehensive evaluation of strain energy.
Because TED is designed for local torsion energy estimation, not overall strain energy, it does not sample the entire molecule for its global minimum energy conformation. This further contributes to TED’s limitations in classifying high-strain energy conformations. Therefore, we sought alternative tools designed for overall strain energy estimation to complement TED. To this end, we investigated StrainRelief44, a method that quantifies strain energy as the energetic difference between a given conformation and the approximation of its global minimum obtained by force field-based sampling. This method classifies conformations with an estimated strain energy exceeding 16.1 kcal/mol as high-strain. However, our evaluation revealed that the default implementation of StrainRelief lacked the necessary sensitivity to effectively classify high-strain energy conformations in the LigBoundConf dataset. As depicted in Supplementary Fig. 5c, StrainRelief demonstrated a high weighted F1 score (approaching 1) at elevated strain energy thresholds, which then sharply declined towards zero as the threshold decreased. This trend suggests limited discriminative power. Further analysis demonstrated that over 94% of the conformations in the LigBoundConf dataset were classified as low-strain by StrainRelief under its default parameters. To improve its performance, we optimized StrainRelief by systematically varying its high-strain classification threshold. Optimal performance was achieved at a threshold of 5.0 kcal/mol. Nevertheless, even with this optimized configuration, StrainRelief’s performance remained comparable to that of TED (Supplementary Fig. 5c), a method not explicitly designed for comprehensive strain energy estimation. This outcome underscores the critical need for future research focused on developing accurate and efficient methods for calculating overall strain energy.
Another potential direction for improving TED is the data augmentation of the torsion energy prediction model (i.e., TED-Model). Specifically, the input conformers used during training and inference are prepared using the procedure indicated by the red arrows in Supplementary Fig. 6a. This procedure involves an initial sampling to obtain no more than 20 initial conformations driven by ConfGenX45, followed by a systematic enumeration of torsion angles through the rotation of the target rotatable bond for each initial conformation. During model training, this enumeration is then followed by a minimum pooling operation that reduced a set of 20 × 24 conformations to a series of 24 conformations. This pooling process ensures a one-to-one correspondence between the 24 conformations and the corresponding series of 24 torsion energy values, as outlined by the green arrows in Supplementary Fig. 6a. The same sampling and minimum pooling procedures are applied to a specific torsion fragment during model inference. Notably, the minimum pooling operation can be omitted during the preparation of the training data. This omission would result in multiple series of 24 conformations being linked to a single series of 24 torsion energy values. This process is, in fact, a data augmentation that has the potential to accelerate the inference process by eliminating the need for multiple initial conformations of the input torsion fragment. We tested this approach and observed that the data-augmented version of the torsion prediction model is five times faster than the model without data augmentation (Supplementary Information Sec. 2.5). It also achieved a Pearson correlation of 0.82 on DFT-5K, which is only slightly lower than the 0.84 correlation of the non-augmented version.
It’s also important to note that torsion energies can be influenced by the electrostatic potential or dielectric constant of the environment46. For instance, high torsion energy resulting from lone pair electron repulsion may be mitigated by a nearby positive charge47. However, no mature technology currently exists to precisely quantify this influence, posing a challenge for the accurate estimation of ligand torsion energy in the binding pocket. Since the electrostatic potential on the surface of a pocket can be calculated48 or observed through experimental electron density49, an ideal ligand torsion energy prediction model would incorporate such information. Our TED Model, trained with implicit water as a solvent, complements Rai’s vacuum-based torsion energy predictions38 in terms of the environmental dielectric constant, and together they provide a foundation for the future development of torsion energy prediction models sensitive to the dielectric constants or electrostatic potentials of binding pockets.
Regarding the evaluation of AI molecule generative models, our HEAD-TED method specifically quantifies the physical plausibility of ligand conformations within protein binding pockets, providing a critical metric for conformational integrity. While this approach addresses one essential dimension of evaluation, complementary methodologies enhance the comprehensiveness of such analyses. For instance, DrugPose34 evaluates binding mode similarity between generated molecules and experimentally determined co-crystallized reference ligands. Although binding mode similarity exhibits limited correlation with physical plausibility metrics (Supplementary Fig. 7a–f), this parameter aligns with the holistic evaluation strategy proposed in our prior work3,7. Specifically, we advocate that robust molecular generative models should achieve dual objectives: (1) recapitulating known bioactive compounds in both structural topology and binding mode, and (2) producing novel molecular entities with high conformational plausibility. Building upon the insights from HEAD-TED and DrugPose, future research should prioritize developing integrated evaluation platforms that combine diverse metrics for a more comprehensive and predictive assessment of AI-generated molecules.
Methods
Our conformation evaluation system is comprised of two key components: (1) atomic energy-based validity assessment by HEAD and (2) a torsion energy-based rationality assessment supported by TED. In this section, HEAD is described by introducing MLFF architecture and element-wise energy thresholds statistically established from a variety of reference datasets. TED is described from the perspectives of training data preparation and torsion energy prediction model development.
HEAD Development
Preliminary: machine learning force fields
The overview of HEAD is shown in Fig. 1a. Before diving into HEAD method, we briefly introduce MLFFs as the preliminaries. More comprehensive introductions can be found in these review papers50,51,52. MLFFs aim to predict the total energy \({{{{\rm{E}}}}}^{{{{\rm{pred}}}}}\) for a given molecule conformation \(\{\left({{{{\rm{R}}}}}_{{{{\rm{i}}}}},{{{{\rm{S}}}}}_{{{{\rm{i}}}}}\right)\}_{{{{\rm{i}}}}=1,2,..,{{{\rm{N}}}}}\) with \({{{\rm{N}}}}\) atoms, where \({{{{\rm{R}}}}}_{{{{\rm{i}}}}}={\left({{{\rm{x}}}},{{{\rm{y}}}},{{{\rm{z}}}}\right)}_{{{{\rm{i}}}}}^{{{{\rm{T}}}}}\) represents ith atomic positions, and \({{{{\rm{S}}}}}_{{{{\rm{i}}}}}\) represents the scalar feature of ith atom, e.g., atomic number. The models are trained on a large scale of conformations to fit model outputs with total energies obtained from DFT calculations in a supervised manner by minimizing a loss function that typically measures the distance between the predicted energies and DFT energies also with other quantities, such as atomic forces, incorporated22,53,54,55,56. The nearsightedness principle is often assumed for MLFFs, where for ith atom, a receptive field of its neighboring atoms within a cutoff radius is only considered to construct ith atomic environment feature either by pre-designed symmetry-invariant functions57,58,59 or symmetry-equivariant message-passing neural networks41,60 with learnable features. Later, MLFFs output a set of atomic energies (also called site energies in some literature61) corresponding to each atomic environment feature and the total energy can be obtained by a summation over all atomic energies, as shown in Eq. (1).
Such locality property establishes a mapping of each atomic environment feature to its corresponding atomic energy, which enables a liner scaling in system size of different molecules.
Atomic energy extraction
HEAD is built upon MLFFs with one key difference: instead of extracting the total predicted energy Epred of a given molecular conformation, it outputs the local atomic energies {Eipred}i=1,2,…,N for all atoms.
It is important to note that when projecting real space into energy space, all types of unrealistic geometries (e.g., steric clashes, uncommon molecular fragments, twisted structures, misplacement of hydrogen atoms, and excited states) can be reflected as high-energy signals in energy space compared to their realistic counterparts. This offers a comprehensive and rapid assessment, regardless of the unrealistic scenarios present in real space.
In this work, we utilize the ANI-2x model22, which is a variation of the prominent Behler-Parrinello neural network potentials, with modified symmetry functions. ANI-2x is designed for accurate and fast prediction of molecules composed of C, H, O, N, S, F, and Cl, covering a large set of small molecule drugs. Notably, the HEAD approach can, in principle, be applied to other MLFFs.
Statistic element-wise energy thresholds
To distinguish high atomic energies resulting from unrealistic conformations from those of realistic conformations, we establish element-wise energy thresholds. These thresholds are derived from element-wise distributions in energy space, generated by applying MLFFs and extracting atomic energies of molecules in various molecular datasets.
Specifically, we first applied the ANI-2x model to three high-quality datasets: QMugs, OrbNet, and QM962,63,64. The QMugs dataset contains approximately 2 million optimized molecular conformations that are biologically and pharmacologically relevant, generated using the semi-empirical GFN2-xTB method. We used 1.1 million molecules composed of C, H, O, N, S, F, and Cl from OrbNet dataset. The QM9 dataset is a widely used benchmark for MLFFs and includes 134,000 stable organic molecules made up of C, H, O, N, and F.
We then grouped the atomic energies by element type, and the element-wise distributions are presented in Supplementary Information Sec. 2.6. The energy thresholds are initialized based on the computed elbow points derived from the element-wise atomic energies. These thresholds are subsequently refined using a grid search to maximize the average weighted F1 score on the GM-1K dataset. The details of GM-1K dataset are described in Methods section (GM-1K).
HES: high-energy score
A high-energy score (HES) is given to quantitatively measure the level of invalidity. For a given conformation, its HES is defined using Eq. (2),
where \({{{{\rm{Z}}}}}_{{{{\rm{i}}}}}\) is atomic number for ith atom, \({{{{\rm{E}}}}}_{{{{{\rm{Z}}}}}_{{{{\rm{i}}}}}}\) is the atomic energy threshold for that element, \({{{\rm{N}}}}\) is the number of atoms in the conformation. Consequently, \({{{\rm{HES}}}}\) is zero for valid conformation and is larger if the given conformation is more deviated from its valid counterpart. Moreover, HES can alleviate corner-case issue existed in typical threshold-based method.
Information entropy
We established an undirected graph for a conformer, with vertexes representing all atoms with individual atomic energy (i.e. Ei) larger than a pre-defined value Ec (Ec = 10 kcal/mol in this work). Two vertexes are linked by an edge if the distance of the two atoms is less than 2 Å. For each component which contains at least two linked vertexes, its sub-region energy is calculated by summing all the atomic energies of the vertexes in it. The component with the highest sub-region energy undergoes an information entropy calculation, as described in Eqs. (3) and (4), and its information entropy is then used to represent the information entropy of the entire conformer.
Here, H(X) represents the normalized information entropy for component X which is composed of atoms xi satisfying Ei > Ec. N denotes the total number of atoms that satisfy this condition in component X.
Classification criteria for ligand conformation validity
For a given conformation, it is considered invalid if it meets any of the following two criteria. First, it contains any atom with HES larger than 0 (i.e., the atomic energy larger than the atom’s element-wise energy threshold). Second, for the conformation passing the first criteria, if its information entropy is larger than 0.8, it is considered as invalid (Supplementary Algorithm 1).
Evaluation of ligand-protein interaction validity
HEAD can also evaluate the validity of ligand-protein binding interactions. For a given ligand-protein complex, the protein pocket is defined as all residues with atoms within a 20 Å radius of the ligand. This radius is chosen to enable the evaluation of multiple ligands binding to the same protein pocket efficiently. Specifically, defining a large pocket region (20 Å) allows a single pocket definition to be reused across different ligands, thereby reducing the computational overhead associated with repeated pocket extraction and energy calculation for pocket atoms. In contrast, using a smaller radius (e.g., 4 Å) would require redefining the pocket for each ligand individually, increasing processing time. The HEAD model then independently predicts atomic energies for the ligand and pocket in both their isolated state and their bound state. Ligand-protein interactions are deemed invalid under two conditions: (1) local instability, where the number of invalid atoms (as defined in above section) increases in either the ligand or the pocket upon binding, or (2) global energy increase, where the total energy of the bound complex (\({{{{\rm{E}}}}}_{{{{\rm{complex}}}}}^{{{{\rm{bound}}}}}\)) exceeds the sum of the energy of the isolated ligand (\({{{{\rm{E}}}}}_{{{{\rm{ligand}}}}}^{{{{\rm{isolated}}}}}\)) and the isolated pocket (\({{{{\rm{E}}}}}_{{{{\rm{pocket}}}}}^{{{{\rm{isolated}}}}}\)) by a threshold of 20 kcal/mol.
TED development
Torsion energy label preparation
Data preparation commenced with the fragmentation of molecules. This process isolates fragments containing the rotatable bond of interest, while preserving a minimal yet suitable chemical environment around it. The fragmentation process was conducted following the methodology established by Rai (2019)65. It began with identifying a quartet of adjacent atoms that define the dihedral angle for the rotatable bond. This initial quartet was then expanded to include any atoms directly bonded to it. If these additional atoms belonged to predefined structural groups—such as rings or amides—the entire group was included to maintain structural integrity. For cyclic structures, substituents at the ortho position of the quartet were preserved due to their potential interactions that could influence torsion energy. Terminal carbons with unfulfilled valence electrons were capped with hydrogen atoms, while terminal heteroatoms were capped with methyl groups. The segmentation process, illustrated in Supplementary Fig. 8a–c, decomposes a molecule into a set of smaller molecules (hereafter referred to as torsion fragments), each representing a fragment of the original structure containing one rotatable bond of interest (hereafter referred to as the target rotatable bond) and its local chemical environment. This method was applied to our in-house molecular database, which contains over 2 billion molecules. After decomposition, we identified more than 100 million torsion fragments, which were then sorted by their frequency of occurrence. From this sorted list, the top 100,000 torsion fragments were selected for torsion energy surface calculations of its target rotatable bond.
To characterize the torsion energy surface associated with the target rotatable bond in a torsion fragment, it is essential to identify the lowest energy among the conformers that share the same dihedral angle. This ensures that the surface accurately reflects the influence of torsion angles on energy, without being affected by high-energy conformations of other groups in the torsion fragment. This objective was achieved through a three-step sampling approach, as illustrated by the green arrows in Supplementary Fig. 6a. Initially, no more than 20 initial conformations were randomly generated for each torsion fragment using ConfGenX45, providing a diverse set of initial conformations to minimize the risk of trapping in local minima during subsequent steps. The target rotatable bond in each initial conformation was then repeatedly rotated in increments of 15 degrees, resulting in 24 conformations per initial conformation. However, this procedure treats the moving parts of the molecule as rigid, which may lead to steric clashes. To address this issue, the conformations underwent MMFF94 force field optimization with the target torsion angle fixed. Subsequently, to ensure that high torsion energy was influenced solely by the improper torsion angle and not by the high-energy conformations of other groups within the same torsion fragment, molecular dynamics simulations were conducted with the target torsion angle held constant. These simulations, powered by GFN-FF66 at 400 K and conducted using xTB67 program, generated up to 100 conformers per input. As a result, we accumulated no more than 48,000 conformations per torsion fragment (i.e., 20 × 24 x 100). The conformers were subsequently grouped into 24 categories based on the dihedral angle values of the target rotatable bond. For each group, GFN1-xTB optimizations were performed, and the optimized conformations with the lowest GFN2-xTB single-point energy was selected as the representative conformation for that dihedral angle. Further refinement of these representative conformers was conducted using B3LYP-D3(BJ)/def2-SVP optimizations using ORCA68, with the dihedral angles maintained as fixed. This was followed by single-point energy calculations performed at the revDSD-PBEP86-D3(BJ)/def2-TZVPP level. All calculations utilized the SMD water solvent model. The resulting double hybrid DFT level single-point energies were used to plot the torsion energy surface for the rotatable bond.
In addition to the aforementioned procedure of torsion energy surface calculation, we also developed a streamlined version to produce a large amount of data for model pretraining. It retained the key elements of the original approach but excluded the molecular dynamics and DFT components, providing GFN2-xTB level torsion energy surfaces. We applied the streamlined method for 6 million molecules of our 100 million torsion fragments library. These GFN2-xTB level data were used as complementary of the DFT-level data during model training.
We have presented two approaches for calculating the torsion energy of a target rotatable bond in a torsion fragment. From a model-training perspective, both approaches generate labels.
Torsion fragment feature extraction
For torsion fragment conformational feature extraction, our methodology involves characterizing the target atom quartet and the local chemical environment surrounding the target rotatable bond.
We used five key features to characterize a quartet of atoms. For an atom quartet represented as abcd, where a, b, c, and d denote individual atoms and bc denotes the rotatable bond of interest, the features are defined as follows: (1) the dihedral angle between plane abc and bcd; (2) the distance between atoms a and d; (3) the distance between atoms b and c; (4) the product of the atomic numbers of atoms a and d; (5) the product of the atomic numbers of atoms b and c. This results in a 5-dimensional feature for atom quartets.
To extract features of the local chemical environment of a rotatable bond, we adapted atomic environment vectors (AEVs)22,56, which are designed to encode the local chemical environment of a target atom. Given our focus on rotatable bonds, we modified the AEV approach by incorporating distance, angular, and dihedral symmetry functions (SFs) to encode the local chemical environment of rotatable bonds.
Distance symmetry function encodes the distance between the center of bond with any other atoms of the conformer. The formula for distance feature extraction is Eq. (5).
Here, we denote the center of the rotatable bond of interest as point i and any arbitrary atom in the conformer as point j. Rc is a threshold that takes values from the set {1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 10.0}. Rijα denotes the distance between atoms i and j, with the superscript α indicating the atom types: H, C, N, O, F, S, Cl, along with two formal charges, resulting in a total of nine element types. The distance symmetry function \({G}_{r}^{\alpha }\) for each type is computed using Eq. (6).
Here, Zb and Zc represent the atomic numbers of the atoms at both ends of the bond of interest, while η and Rs are set to 10-4 and 0, respectively. This results in a 63-dimension distance feature (9 element types × 7 Rc values).
The angular symmetry function, as shown in Eq. (7), encodes the angles formed by the center of the rotatable bond and any two other atoms in the conformer.
Here, Zα and Zβ represent the atomic numbers of the atoms at both ends of the bond of interest. The subscript i represents the center point of the bond in the dihedral angle, while j and k are any two atoms in the conformation. Rik, Rij, Rjk represent the distances between subscript specified atoms, and ϕijk denotes the angle formed by the edges Rij and Rik. The parameters δ, λ, η are set to 0.5, 0.5, and 10-4, respectively, with Rc valued at 4.0. Given nine types of elements, there are a total of 45 combinations of atom pairs (CC, CN, CO,…, CS, CH), resulting in a 45-dimension angle feature (1 Rc value × 45 combinations of atom pairs = 45).
The dihedral symmetry function, as shown in Eq. (8), encodes the torsion angles of the rotatable bond of interest involving any two other atoms in the conformer.
Here, we denote atoms k and j as the two terminal atoms of the bond on interest. For any other two atoms, i and l, atoms i, k, j, and l form quadruplets, with Rik, Rjl, Ril represent the distances between subscript specified atoms, and ϕikjl representing the dihedral angle of planes ikj and kjl. Rc takes values from the set {2.5, 3.5, 5.0, 10.0}. The parameters δ, λ, η are set to 0.5, 0.5, and 10-4, respectively. Given nine types of elements, there are a total of 45 combinations of atom pairs. This results in a 180-dimension dihedral angle feature (4 Rc values × 45 combinations of atom pairs = 180).
The hyperparameters Rc, δ, λ,and η were configured in accordance with the parameters established in previous AEV studies22,56.
The 63-dimensional distance, 45-dimensional angle, and 180-dimensional dihedral angle features for rotatable bonds and 5-dimensional feature for atom quartets are concatenated to yield a 293-dimensional feature, which is used as input for the model.
Torsion energy prediction model training
Our torsion energy prediction model (i.e. TED-Model) is designed to predict energy values for an input series of 24 conformations with target torsion angles at intervals of 15°. The input conformations should be obtained using inexpensive methods, such as MMFF94 force field, while the output torsion energy must exhibit a high level of accuracy, ideally at DFT level. Otherwise, the model would be unnecessary, as an inexpensive torsion energy surface can be generated simultaneously with the generation of input conformations. The schematic flow indicated with red and green arrows in Supplementary Fig. 6a summarizes the feature extraction and torsion energy labeling process, respectively. This process results in a series of 24 conformations with torsion angle at 15° intervals and a series of 24 energy values as labels.
The data supporting model training consists of two datasets. The first dataset is a double hybrid DFT-level torsion energy dataset involving 100,000 torsion fragments, while the second dataset is a GFN2-xTB level torsion energy dataset involving 6 million torsion fragments.
The training process was divided into two phases: pretraining and fine-tuning. The complete set of GFN2-xTB level data, along with 10% of the double hybrid DFT data, was utilized for pretraining, while the remaining 90% of the double hybrid DFT data was reserved for the fine-tuning phase. In each phase, the data was split into training and test sets using an 8:2 ratio. To prevent information leakage, no molecules in the test set shared more than 0.5 Tanimoto similarity with any molecules in the training set, based on ECFP4 fingerprints69.
The input for the model consists of a series of 24 conformations, with each conformation represented as a 293-dimension vector. This represents the input as a two-dimensional matrix of size 24 × 293. To effectively capture the relationships among the input conformations, we employ a self-attention framework. The output of self-attention module is then flattened and input into a linear-layer module which generate predictions for each conformation, resulting in a final output of size 24 × 1. The model architecture is shown in Supplementary Fig. 6b.
The input matrix M is of size 24 × 293, where each row represents a conformation, and the rows are sequenced in ascending order according to dihedral angle values of specified atom quartet. M undergoes an attention70 transformation using Eq. (9).
This process generates a new matrix representation of size 24 × 293 that encapsulates the internal relationships among the 24 conformations. Thereafter, we apply a flatten operation to compress this matrix into a 7032-dimension vector. Then the 7032-dimension vector is put into linear layers module which consists of seven layers with number of neurons arranged as follows: 3516, 1758, 879, 293, 146, 73, and 24. Each linear layer uses ReLU as active function and employs batch normalization strategy, with a dropout rate of 10% applied during training. The output of the linear layers is a 24-dimension vector. The model’s loss function is defined as the Mean Absolute Error (MAE) of the energy values, expressed as Eq. (10).
Here, \({\hat{y}}_{i}\) and \({y}_{i}\) denote the predicted energy value and energy value label for conformer i, respectively.
Adam was used as optimizer with initial learning rate set as 0.001. Early stop strategy was used to avoid overfitting.
Classification criteria for conformation rationality
This process is illustrated in Supplementary Fig. 6c. For a given conformation, it is first decomposed into several torsion fragments using the approach described in Methods (Torsion Energy Label Preparation). Subsequently, for each torsion fragment, a series of 24 torsion energy values corresponding to 24 torsion angles at 15° intervals is predicted (Supplementary Algorithm 2). The torsion energy surface is then generated by smoothing the 24 discrete points using linear interpolation in SciPy package71. This method ensures that the interpolated values remain within a reasonable range. It helps avoid the extreme values or oscillations that can arise from cubic interpolation, which may lead to unrealistic dips below zero or spikes in energy. The torsion energy for each fragment is determined by checking the corresponding energy value for the torsion angle adopted in that fragment against the torsion energy surface. If any of the torsion fragments derived from the given conformation has a torsion energy exceeding 2 kcal/mol, the conformation is classified as irrational (Supplementary Algorithm 3). This threshold of 2 kcal/mol is based on findings reported in a previous study65.
Datasets
GM-5K
The GM-5K dataset contains 5000 molecules randomly selected from AI-generated molecules produced by five models: Lingo3DMolv2, Pocket2Mol, PocketFlow, TargetDiff, and PMDM. Each model was set to generate 1000 molecules for each of the 102 targets in the DUD-E dataset. We randomly select 1000 molecules from each model, resulting in the final GM-5K dataset of 5000 molecules.
For each conformation in the GM-5K dataset, geometry optimization using the MMFF94 force field was performed. This is followed by QM-level single-point energy calculations (revDSD-PBEP86-D3(BJ)/def2-TZVPP) for both the pre-optimized and post-optimized conformations. The energy differences, calculated as ΔE = EoriDFT - EoptDFT, serve as indicators of conformation quality.
For each ligand-protein complex in GM-5K, the binding free energy of the ligand was calculated using MM/GBSA31,32, where the Amber ff14SB force field72 was used for protein and the General Amber Force Field 2 (GAFF2)73 for organic molecules. Specifically, the MM part describes the bonded (including bond, angle and dihedral), electrostatic, and van der Waals interactions, while the solvation free energies and the non-polar term are approximated by using the generalized Born model (the GB part) and a linear relation to the solvent accessible surface area (SA part), respectively.
GM-1K
The GM-1K dataset is created using the same methodology as GM-5K, but it includes only 1000 molecules that do not share similar counterparts with GM-5K. Similarity is determined using Tanimoto similarity for ECFP4 fingerprints; molecules are considered similar if they have a Tanimoto similarity greater than 0.5.
DFT-5K
The approach described in Methods section (Torsion Energy Label Preparation) and illustrated in Supplementary Fig. 8 was initially applied to our in-house molecular database, which contains over 2 billion molecules, resulting in the identification of more than 100 million torsion fragments. These fragments were then sorted by their frequency of occurrence. From this sorted list, the most frequently occurred 100,000 torsion fragments were selected for torsion energy surface calculations at the DFT level and used for TED-Model training. The DFT-5K dataset consists of 5000 unique torsion fragments selected from the previously mentioned 100-million torsion fragment library, ensuring that no DFT-5K torsion fragment has its Bemis–Murcko scaffolds appearing in the DFT level training set for TED-Model.
Molecule generative models undertest
Five recently reported AI generative models were evaluated in this study. Lingo3DMolv2, Pocket2Mol, and PocketFlow were selected as representatives of autoregressive models, whereas PMDM and TargetDiff were chosen as representatives of diffusion-based models. Lingo3DMolv2 represents an evolution of the original Lingo3DMol, incorporating several key modifications. Firstly, the binding pocket representation has been redesigned in Lingo3DMolv2 to integrate residue-specific information, providing a more comprehensive description of the binding site environment. Secondly, the training paradigm of Lingo3DMolv2 has been adapted to accommodate diverse modeling scenarios, including those with and without NCI (Non-Covalent Interaction) data. Finally, the number of atomic token types in Lingo3DMolv2 has been expanded from 76 to 256 by improving the structural representation of fused ring systems.
Data availability
The GM-5K, GM-1K, and DFT-5K datasets generated in this study have been deposited in the Figshare database74 under accession link https://doi.org/10.6084/m9.figshare.27826488.v6. Source data for figures are provided with this paper. Source data are provided with this paper.
Code availability
Our source code for the HEAD and TED models is publicly available on our GitHub repository at https://github.com/stonewiseAIDrugDesign/HEAD_TED. (The code is also included in the Code Ocean capsule21). The code is distributed under MIT license. We obtained the models for Pocket2Mol, TargetDiff, PocketFlow, and PMDM from their official GitHub repositories and used them for molecule generation. For Lingo3DMolv2, we utilized the online service available at https://sw3dmg.stonewise.cn to generate molecules. The service is freely accessible to academic users, and an academic email address is required to receive the generated results.
References
Zeng, X. et al. Deep generative molecular design reshapes drug discovery. Cell Rep. Med. 3, 100794 (2022).
Jiang, Y. et al. PocketFlow is a data-and-knowledge-driven structure-based molecular generative model. Nat. Mach. Intell. 6, 326–337 (2024).
Feng, W. et al. Generation of 3D molecules in pockets via a language model. Nat. Mach. Intell. 6, 62–73 (2024).
Huang, L. et al. A dual diffusion model enables 3D molecule generation and lead optimization based on target pockets. Nat. Commun. 15, 2657 (2024).
Peng, X. et al. Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets. arXiv:2205.07249 https://ui.adsabs.harvard.edu/abs/2022arXiv220507249P (2022).
Guan, J. et al. 3D Equivariant Diffusion for Target-Aware Molecule Generation and Affinity Prediction. arXiv:2303.03543 https://ui.adsabs.harvard.edu/abs/2023arXiv230303543G (2023).
Wang, L. et al. A pocket-based 3D molecule generative model fueled by experimental electron density. Sci. Rep. 12, 15100 (2022).
Buttenschoen, M., Morris, G. M. & Deane, C. M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 15, 3130–3139 (2024).
Harris, C. et al. Benchmarking Generated Poses: How Rational is Structure-based Drug Design with Generative Models? https://ui.adsabs.harvard.edu/abs/2023arXiv230807413H (2023). arXiv:2308.07413.
Hekkelman, M. L., de Vries, I., Joosten, R. P. & Perrakis, A. J. N. M. AlphaFill. enriching AlphaFold models ligands cofactors. 20, 205–213 (2023).
Ramachandran, S., Kota, P., Ding, F., Dokholyan, N. V. J. P. S., Function, & Bioinformatics. Automated minimization of steric clashes in protein structures. 79, 261-270 (2011).
Kohn, W. & Sham, L. J. J. P. r. Self-consistent equations including exchange and correlation effects. A 140, 1133 (1965).
Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Allinger, N. L., Yuh, Y. H. & Lii, J. H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 111, 8551–8566 (1989).
Halgren, T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519 (1996).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Brooks, B. R. et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 (1983).
Sellers, B. D., James, N. C. & Gobbi, A. A comparison of quantum and molecular mechanical methods to estimate strain energy in druglike fragments. J. Chem. Inf. Model 57, 1265–1275 (2017).
Wang, Y., Walker, B. D., Liu, C. & Ren, P. An efficient approach to large-scale ab initio conformational energy profiles of small molecules. Molecules https://doi.org/10.3390/molecules27238567 (2022).
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (dud-e): better ligands and decoys for better benchmarking. J. Medicinal Chem. 55, 6582–6594 (2012).
Fan, F. et al. Assessing conformation validity and rationality of deep learning-generated 3d molecules [source code]. Code Ocean https://doi.org/10.24433/CO.3893415.v2 (2025).
Devereux, C. et al. Extending the applicability of the ani deep learning molecular potential to sulfur and halogens. J. Chem. Theory Comput. 16, 4192–4202 (2020).
Landrum, G. e. a. RDKit: open-source cheminformatics software. GitHub https://github.com/rdkit/rdkit (2016).
Tong, J. & Zhao, S. Large-scale analysis of bioactive ligand conformational strain energy by ab initio calculation. J. Chem. Inf. Model 61, 1180–1192 (2021).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Roos, K. et al. OPLS3e: extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 15, 1863–1874 (2019).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The cambridge structural database. Acta Crystallogr B Struct. Sci. Cryst. Eng. Mater. 72, 171–179 (2016).
Santra, G., Sylvetsky, N. & Martin, J. M. J. T. J. o. P. C. A. Minimally empirical double-hybrid functionals trained against the GMTKN55 database: revDSD-PBEP86-D4, revDOD-PBE-D4, and DOD-SCAN-D4. 123, 5129–5143 (2019).
Hellweg, A. & Rappoport, D. J. P. C. C. P. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. 17, 1010–1017 (2015).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Computational Chem. 32, 1456–1465 (2011).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Miller, B. R. et al. MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput 8, 3314–3321 (2012).
Cieplinski, T., Danel, T., Podlewska, S. & Jastrzebski, S. Generative models should at least be able to design molecules that dock well: a new benchmark. J. Chem. Inf. Model 63, 3238–3247 (2023).
Jocys, Z., Grundy, J. & Farrahi, K. DrugPose: benchmarking 3D generative methods for early stage drug discovery. Digital Discov. 3, 1308–1318 (2024).
Koes, D. R., Baumgartner, M. P. & Camacho, C. J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model 53, 1893–1904 (2013).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98 (2012).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform 1, 8 (2009).
Rai, B. K. et al. TorsionNet: a deep neural network to rapidly predict small-molecule torsional energy profiles with the accuracy of quantum mechanics. J. Chem. Inf. Modeling 62, 785–800 (2022).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46, D1074–D1082 (2018).
Clark, C. G. et al. Structure based design of macrocyclic factor XIa inhibitors: Discovery of cyclic P1 linker moieties with improved oral bioavailability. Bioorg. Med Chem. Lett. 29, 126604 (2019).
Kovács, D. P. et al. MACE-OFF: short-range transferable machine learning force fields for organic molecules. J. Am. Chem. Soc. 147, 17598–17611 (2025).
Eastman, P. et al. SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials. Sci. Data 10, 11 (2023).
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput Aided Mol. Des. 27, 221–234 (2013).
Wallace, E. R. S., Frey, N. C. & Rackers, J. A. Strain problems got you in a twist? try strainrelief: a quantum-accurate tool for ligand strain calculations. J. Chem. Inf. Model 65, 6613–6620 (2025).
Watts, K. S. et al. ConfGen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Modeling 50, 534–546 (2010).
Peach, M. L., Cachau, R. E. & Nicklaus, M. C. Conformational energy range of ligands in protein crystal structures: The difficult quest for accurate understanding. J. Mol. Recognit. https://doi.org/10.1002/jmr.2618 (2017).
Brameld, K. A., Kuhn, B., Reuter, D. C. & Stahl, M. Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J. Chem. Inf. Model 48, 1–24 (2008).
Zhao, L., Pu, M., Wang, H., Ma, X. & Zhang, Y. J. Modified electrostatic complementary score function and its application boundary exploration in drug design. J. Chem. Inf. Model 62, 4420–4426 (2022).
Ding, K. et al. Observing noncovalent interactions in experimental electron density for macromolecular systems: a novel perspective for protein-ligand interaction research. J. Chem. Inf. Model 62, 1734–1743 (2022).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Kocer, E., Ko, T. W. & Behler, J. Neural network potentials: a concise overview of methods. Annu Rev. Phys. Chem. 73, 163–186 (2022).
Wu, S. et al. Applications and advances in machine learning force fields. J. Chem. Inf. Model 63, 6972–6985 (2023).
Zhang, L. et al. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Schutt, K. T., Sauceda, H. E., Kindermans, P. J., Tkatchenko, A. & Muller, K. R. SchNet - A deep learning architecture for molecules and materials. J. Chem. Phys. 148, 241722 (2018).
Fu, W. et al. Enhancing molecular energy predictions with physically constrained modifications to the neural network potential. J. Chem. Theory Comput 20, 4533–4544 (2024).
Smith, J. S. et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 10, 2903 (2019).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Behler, J. Representing potential energy surfaces by high-dimensional neural network potentials. J. Phys. Condens Matter 26, 183001 (2014).
Zubatyuk, R., Smith, J. S., Leszczynski, J. & Isayev, O. Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network. Sci. Adv. 5, eaav6490 (2019).
Gasteiger, J., Groß, J. & Günnemann, S. J. a. e.-p. Directional Message Passing for Molecular Graphs. arXiv:2003.03123. https://ui.adsabs.harvard.edu/abs/2020arXiv200303123G (2020).
Batatia, I. et al. The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials. arXiv:2205.06643. https://ui.adsabs.harvard.edu/abs/2022arXiv220506643B (2022).
Ramakrishnan, R., Dral, P. O., Rupp, M. & von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 140022 (2014).
Qiao, Z., Welborn, M., Anandkumar, A., Manby, F. R. & Miller, T. F. 3rd. OrbNet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features. J. Chem. Phys. 153, 124111 (2020).
Isert, C., Atz, K., Jimenez-Luna, J. & Schneider, G. QMugs, quantum mechanical properties of drug-like molecules. Sci. Data 9, 273 (2022).
Rai, B. K. et al. Comprehensive assessment of torsional strain in crystal structures of small molecules and protein-ligand complexes using ab initio calculations. J. Chem. Inf. Model 59, 4195–4208 (2019).
Gale, J. D., LeBlanc, L. M., Spackman, P. R., Silvestri, A. & Raiteri, P. A universal force field for materials, periodic gfn-ff: implementation and examination. J. Chem. Theory Comput. 17, 7827–7849 (2021).
Spicher, S. & Grimme, S. Robust atomistic modeling of materials, organometallic, and biochemical systems. Angew. Chem. Int Ed. Engl. 59, 15665–15673 (2020).
Neese, F., Wennmohs, F., Becker, U. & Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 152, 224108 (2020).
Bajusz, D., Racz, A. & Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform 7, 20 (2015).
Vaswani, A. et al. Attention Is All You Need. arXiv:1706.03762. https://ui.adsabs.harvard.edu/abs/2017arXiv170603762V (2017).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph Model 25, 247–260 (2006).
Fan, F. et al. Data for HEAD_TED. Figshare https://doi.org/10.6084/m9.figshare.27826488.v6 (2024).
Acknowledgements
This study was funded by the National Key R&D Program of China (grant no. 2022YFF1203004 received by B.H.). This work was also supported by the Beijing Municipal Science and Technology Commission (grant no. Z241100007724005 received by B.H.).
Author information
Authors and Affiliations
Contributions
B.H. conceived the study and supervised the design of all the experiments. B.X. developed HEAD. F.F. and X.M. developed TED. W.Z. provided instructions for artificial intelligence modeling. F.Z. provided instructions on QM calculations. H.Z. provided instructions for the construction of the torsion fragment. H.W., B.Z., Q.X., W.F., X.W., and W.G. supported the evaluation of AI models. Z.L. and Y.W. supported forced field-based optimization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Charlotte Deane and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, F., Xi, B., Meng, X. et al. Assessing conformation validity and rationality of deep learning-generated 3D molecules. Nat Commun 17, 2481 (2026). https://doi.org/10.1038/s41467-026-69303-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69303-5
