
Abstract
A ‘gut-brain axis’ is an intricate bidirectional connection between the gut and the central nervous system, serving as a key pathway for signal exchange. However, current in vitro models do not fully capture these dynamic interactions, limiting mechanistic insight and therapeutic testing. Here, we show a 3D human gut-brain-vascular microphysiological platform that integrates lumenized villus-like intestinal barrier, blood vascular-astrocyte interactions, and brain tissue to model circulation-mediated crosstalk between the gut and brain. Using this system, we demonstrate gut-to-brain signaling by delivering bacterial-derived toxins to the gut compartment, which traverse the gut and neurovascular barriers and trigger neuroinflammatory responses and tau-associated pathology in the brain tissue. Conversely, we show that Alzheimer’s- and Parkinson’s-relevant stimuli applied to the brain compartment elicit neuroinflammation and disrupt both vascular and intestinal barrier integrity, indicating brain-to-gut feedback. Together, our platform provides a human-relevant tool to dissect mechanisms of bidirectional gut-brain communication and to evaluate therapeutic strategies for neurogastrointestinal disease.
Similar content being viewed by others
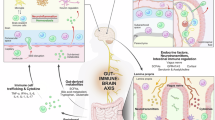
Introduction
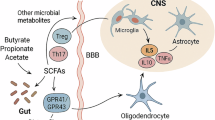
The gut-brain axis (GBA), a bidirectional communication network between the gastrointestinal (GI) tract and the central nervous system (CNS), plays a crucial role in maintaining neurological homeostasis1,2,3,4. This interaction is mediated through neuronal (vagus nerve), immune system (immune cells/cytokine signaling), and circulatory (vasculature) pathways1,3,5,6. These pathways facilitate bidirectional communication, primarily mediated by neuroactive substances, microbial metabolites, and hormones that regulate neurological function and systemic homeostasis. Among these, the vasculature serves as a critical interface, regulating immune surveillance, molecular exchange, and inflammatory responses between the gut, the periphery, and the brain3,4. Mounting evidence underscores the microbiota–gut–brain axis as a key contributor to neurodegeneration, with early alterations in gut microbiota identified in Alzheimer’s disease (AD) and Parkinson’s disease (PD) patients, suggesting a potential causal link between gut dysbiosis and neurodegeneration7,8,9,10,11. Animal studies further support this hypothesis, demonstrating that microbiota-driven neuroinflammation and glial cell dysfunction contribute to disease progression through systemic immune activation, inflammatory cascades, and blood-brain barrier (BBB) disruption12,13,14. Microbial-derived toxins and inflammatory mediators have been implicated in BBB permeability loss, neuronal dysfunction, and protein aggregation, reinforcing the notion that gut dysbiosis is a key driver of neurodegenerative pathology.
Despite these insights, a significant limitation in the field is the lack of physiologically relevant in vitro models that accurately replicate the complex, dynamic interactions among the gut, vasculature, and brain. Most studies depend on animal models, which, while informative, do not capture the human-specific mechanisms underlying gut-brain communication15,16,17. Existing in vitro systems typically concentrate on single-organ models, such as gut-on-chip, BBB-on-chip, or brain-on-chip platforms; however, these models fail to recapitulate the multi-organ interplay that underlies gut-brain dysfunction in neurodegeneration18,19,20. Additionally, traditional Transwell assays and organoid cultures do not integrate circulatory flow, barrier dynamics, and cellular interactions necessary to mimic the in vivo microenvironment21,22,23. Consequently, critical questions regarding the mechanistic underpinnings of gut-brain communication remain unresolved, hindering the development of targeted therapeutic interventions. To tackle these challenges, we developed a 3D microfluidic Gut-Brain-Vascular (GBV) platform that models bidirectional crosstalk in gut-neuropathogenesis. Our system features a 3D lumenized gut barrier with villus-like architecture to replicate the intestinal epithelium, a microvascular structure compartment containing brain endothelial cells, and a 3D brain tissue compartment containing neurons and astrocytes that simulates endothelial-astrocyte interactions, facilitating dynamic molecular exchange.
Our system enables real-time investigation of bidirectional inflammatory crosstalk by modeling gut-to-brain signaling and examining brain-to-gut interactions. With the gut-to-brain axis, bacterial-derived toxins (e.g., lipopolysaccharides, E. Coli-derived toxins) affect the gut compartment, causing gut barrier dysfunction and increased vascular permeability, allowing toxins and pro-inflammatory factors from the gut and vascular organs to infiltrate the brain. This ultimately leads to the accumulation of phosphorylated tau, which is a hallmark of neurodegeneration. In the brain-to-gut axis, AD-like (NPC-derived AD models) and PD-like (PFF α-synuclein aggregates) environments in the brain compartment induce vascular barrier breakdown and subsequent gut epithelial dysfunction. By integrating gut, vascular, and brain compartments within a physiologically relevant microfluidic system, this model serves as a sophisticated microphysiological tool for studying gut-brain interactions in neurodegeneration. Our findings offer critical insights into the role of gut-origin inflammation in neurodegeneration, identifying mechanisms of diseases such as Alzheimer’s and Parkinson’s and laying the groundwork for screening novel therapeutic interventions targeting the gut-brain-vascular axis.
Results
Engineering a physiologically relevant human gut-brain-vascular (hGBV) model
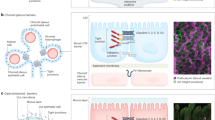
The gut-brain axis (GBA) is a complex bidirectional communication network regulated by neuronal, immune, and vascular interactions5,6. However, current in vitro models lack the physiologically relevant microenvironment needed to study gut-brain-vascular crosstalk. To address this issue, we created a 3D microfluidic Human Gut-Brain-Vascular (hGBV) model that combines lumenized gut epithelium, perfusable vasculature, and a neurovascular brain compartment in a dynamic microfluidic system (Fig. 1a, b, Supplementary Fig. 1). The hGBV system consists of three interconnected compartments: Gut Compartment (GC), Vascular Compartment (VC), and Brain Compartment (BC), designed to recapitulate key features of gut-brain interactions (Fig. 1b). Each compartment was engineered using human-derived cell types to closely mimic native tissue architectures. The gut compartment was developed using Caco-2 cells, forming a 3D villus-like epithelium to replicate intestinal barrier functions. The vascular compartment consisted of induced brain microvascular endothelial cells (iBMECs), forming a microvascular-like endothelium that enables selective molecular transport. The brain compartment incorporated neural progenitor cell (NPC)-derived neurons and astrocytes, which interact with vascular endothelial cells via astrocytic end-feet, representing the neurovascular unit (NVU). The stepwise assembly of the hGBV model (Fig. 1c, Supplementary Fig. 1f) involved the sequential differentiation of brain tissue, gut epithelium formation, and vascular compartment development, ensuring the formation of interconnected and physiologically relevant microenvironments.

a Schematic representation of the Gut-Brain Axis via circulation vessel system. b Gut-Brain-Vascular-on-a-chip platform. c Stepwise assembly of the hGBV tri-organs model. d Immunofluorescence of the integrated model, showing left: large-scale view, gut epithelium-red capillary-like vascular structures-blue, and brain tissue-cyan; right: 3D confocal zoom-in of Gut-VIL1, vascular-CD31, and astrocyte-GFAP. e-i Cross-sectional view of the gut epithelium (GC). e-ii Top view of the gut epithelium (VIL1; DAPI). f Zoom-in 3D of gut villus-like structures (VIL1; DAPI) in GC. g Quantification of VIL1 enrichment on the apical side relative to the nuclear plane (DAPI), supports apical VIL1 polarity in GC. h Immunofluorescence of gut-to-brain axis showing OCLN in GC, ZO-1 in VC, astrocyte-GFAP and neuron-Tuj1 in BC. i Quantification of OCLN localization, ZO-1 localization, and GFAP expression in gut-to-brain axis. j Immunofluorescence of brain-to-gut axis showing pTau in BC, ZO-1 in VC, OCLN in GC. k Quantification of pTau expression, ZO-1 localization, OCLN localization, in brain-to-gut axis. l–n RT-qPCR validation of barrier-related genes OCLN in GC, TJP1 in VC, and astrocytic GFAP in BC, confirming changes quantified in (i). o–q RT-qPCR validation of disease- and barrier-related genes APP in BC, TJP1 in VC, OCLN in GC, confirming the changes observed in (k). Scale bars, 500 μm (d), 100 μm (d, f), 20 μm (e, h, j). Representative images are shown from one experiment; similar results are obtained in three independent experiments with comparable outcomes (d–f, h, j). Quantification is based on n = 10 independent chips per condition (g, i, k), n = 5 in independent chips per condition (l–q). Data represent Mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Statistical analysis: two-sided tests selected per dataset (Student’s t-test/Welch’s t-test or Mann-Whitney U for two-group comparisons; one-way ANOVA/Welch’s ANOVA with Tukey or Kruskal-Wallis with Dunn for multi-group comparisons), with multiple-comparisons adjustments where applicable; exact tests and full statistics (effect sizes, 95% CIs, exact P values) are reported in Supplementary Table S2. CT represents control; E.CCM+, E. Coli-conditioned media, WT wide-type, AD Alzheimer’s disease, GC gut compartment, VC vascular compartment, BC brain compartment, NPC neural progenitor cell. Created in BioRender. Vu, T. A. (2026) https://BioRender.com/702y808 (a) & https://BioRender.com/702y808 (b).
To confirm the physiological relevance of the hGBV model, we assessed the formation of 3D gut villus-like structures using immunofluorescence imaging. The gut compartment exhibited well-organized epithelial cell layers with tight junction integrity, closely resembling native intestinal architecture (Fig. 1d–g, Supplementary Fig. 2c, g). Immunostaining of the vascular and brain compartments further confirmed the presence of iBMECs in the vascular compartment and neurons with astrocyte support in the brain compartment, forming a multicellular microenvironment capable of bidirectional signaling (Fig. 1d, e, Supplementary Fig. 2a–d). Additionally, marker expression analysis confirmed ViL1 in the gut epithelium, Tuj1 in neurons, and GFAP in astrocytes, validating the structural integrity of the system (Fig. 1d, e, Supplementary Fig. 2c). To evaluate barrier integrity and compartmental media separation, we performed FITC-Dextran permeability assays across all compartments. Under physiological conditions, the gut and vascular barriers demonstrated compartment-specific permeability, with approximately 15% FITC-Dextran leakage into the vascular compartment and <5% leakage into the brain compartment (Supplementary Fig. 1g). This selective permeability indicates the presence of intact barrier functions, effectively preventing uncontrolled diffusion of across compartments.
To validate the bidirectional crosstalk of the system, we performed two distinct experiments modeling gut-to-brain and brain-to-gut signaling. To investigate how bacterial-derived toxins from the gut contribute to neurodegeneration, we exposed the gut compartment to E.Coli-conditioned media (E.CCM), mimicking gut dysbiosis-induced inflammation. Exposure to E.CCM led to the disruption of barrier tight junctions and glial activation. Specifically, gut OCLN localization was reduced by ~45% compared to control (CT: 1.00 ± 0.10 AU; E. Coli: 0.55 ± 0.11 AU), while vascular ZO-1 expression decreased by ~50% (CT: 1.00 ± 0.09 AU; E. Coli: 0.54 ± 0.11 AU). In contrast, astrocytic GFAP expression in the brain compartment increased nearly 3-fold (CT: 0.99 ± 0.14 AU; E. Coli: 3.58 ± 1.12 AU), confirming that gut-derived bacterial toxins impair epithelial and vascular barriers and activate neuroinflammatory signaling (Fig. 1h, i). Consistent with these protein-level changes, RT-qPCR showed concordant transcript shifts with a reduction in gut OCLN (Fig. 1i, Supplementary Fig. 3c, d), a decrease in vascular TJP1/ZO-1 (Fig. 1m, Supplementary Fig. 3e, f), and an elevation of brain GFAP (Fig. 1n, Supplementary Fig. 3g, h) after E.CCM relative to CT, supporting E.CCM-driven junction loss and astrocytic activation.
To investigate brain-to-gut signaling, we modeled Alzheimer’s disease (AD)-like conditions in the brain compartment by differentiating NPCs that were transfected with the APP gene for six weeks. Compared with wild-type (WT), AD conditions led to significant pTau accumulation, with a ~2.1-fold increase (WT: 1.00 ± 0.17 AU; AD: 2.01 ± 0.31 AU). This was accompanied by vascular barrier breakdown, as indicated by a ~40% reduction in ZO-1 localization (WT: 1.00 ± 0.03 AU; AD: 0.57 ± 0.07 AU). Similarly, gut epithelial barrier integrity was impaired, with OCLN expression reduced by ~25% (WT: 1.00 ± 0.04 AU; AD: 0.75 ± 0.07 AU), confirming that neuroinflammatory factors contribute to systemic vascular and gut dysfunction (Fig. 1j, k). Consistent with these protein-level changes, RT-qPCR showed concordant transcript shifts with an elevation in brain APP (Fig. 1o, Supplementary Fig. 3i, j), a decrease in vascular TJP1/ZO-1 (Fig. 1p, Supplementary Fig. 3k, l), and a reduction in gut OCLN (Fig. 1q, Supplementary Fig. 3o, p) in AD relative to WT, supporting brain-derived neuroinflammatory signals that weaken vascular and epithelial junctions. Together, these findings illustrate that the hGBV model effectively replicates bidirectional gut-brain-vascular interactions, facilitating the study of gut-to-brain and brain-to-gut signaling in both physiological and disease conditions. The capacity to model gut-derived inflammation, neurovascular dysfunction, and loss of systemic barrier integrity emphasizes the translational potential of this system for investigating neurodegenerative disorders, including Alzheimer’s and Parkinson’s disease.
Physiological properties of the hGBV model
To functionally validate physiological properties of the hGBV model, compared cell culture under static (ST, open well plate) and dynamic (DM, hGBV chip) conditions (Fig. 2, Supplementary Fig. 2e–g). Immunofluorescence confirmed that the hGBV system forms a compartmentalized gut–vascular–brain interface with distinct epithelial, endothelial, and neural identities (Fig. 2a, b, Supplementary Fig. 2a–c). The gut compartment developed villus-like epithelium (VIL1) with tight junction protein OCLN, the vascular endothelium expressed CD31 and ZO-1, and the brain compartment exhibited astrocytic endfeet (AQP4, GFAP) wrapping vascular endothelium (ZO-1) adjacent to neurons, resembling a neurovascular unit.

a 3D confocal images of the gut-vascular (GC-VC), top: epithelium tight junctions (ZO-1 in GC), endothelium (CD31) and astrocyte endfeet contact (GFAP; AQP4) in VC; bottom: polarized VIL1 in GC, endothelium (ZO-1) and astrocyte (GFAP) contacts in VC. b 3D confocal images of the vascular–brain interface (VC–BC), left: endothelium (CD31) contact with astrocytic endfeet (GFAP, AQP4); right: cross-sectional view showing astrocytic endfeet wrapping around vascular endothelium. c Immunofluorescence of gut epithelium showing OCLN and villi (VIL1) in GC. d–g Quantification of epithelial thickness (g), villin expression (e), OCLN expression (f), and paracellular permeability (Papp) measured by 3 kDa and 70 kDa dextran tracers under ST vs DM conditions in GC. h Immunofluorescence of vascular endothelium (CD31; ZO-1) under ST and DM conditions in VC. Quantification of endothelial cell (EC) ZO-1 expression (i) morphology (j) and vascular permeability (Papp) using 3 kDa and 70 kDa dextran tracers (k) in VC. (l) Immunofluorescence of brain tissues showing neurons (Tuj1), astrocytes (GFAP), synaptic marker (SYN1) in BC, and heat maps of Ca²⁺ dynamics (low vs high activity) over time. m Representative Ca²⁺ oscillation trace (ΔF/F₀) averaged from 100 cells. n Quantification of AQP4 immunoreactivity (ICC) in BC and VC. Scale bars, 100 μm (a, b), 20 μm (c, h), 50 μm (l). Representative images are shown from one experiment; similar results are obtained in three independent experiments with comparable outcomes (a–c, h, l). Quantification is based on n = 10 independent chips per condition (d–f, i), n = 5 independent chips per condition (g, k), 30 individual cells per condition (j, n). Box plots show the median (center line), 25th–75th percentiles (box), and minimum to maximum values (whiskers), points indicate individual measurements; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Statistical analysis: two-sided tests selected per dataset (Student’s t-test/Welch’s t-test or Mann-Whitney U for two-group comparisons; one-way ANOVA/Welch’s ANOVA with Tukey or Kruskal-Wallis with Dunn for multi-group comparisons), with multiple-comparisons adjustments where applicable; exact tests and full statistics (effect sizes, 95% CIs, exact P values) are reported in Supplementary Table S2. GC gut compartment, VC vascular compartment, BC brain compartment, ST static cell culture in open well plate, DM dynamic cell culture in hGBV chip.
We assessed the structural, barrier, and transport properties of the gut, vascular, and brain compartments, comparing static and dynamic conditions. Morphometric analysis demonstrated that DM culture in the GBV chip enhanced gut architecture compared with ST conditions in open well plates. 3D reconstruction of the gut compartment revealed markedly taller villus-like structures under DM, with epithelial thickness nearly doubling (ST: 20.4 ± 4.8 μm; DM: 44.3 ± 7.5 μm; ~2.2-fold increase, Fig. 2d). Villin (VIL1) expression was also significantly elevated (ST: 0.98 ± 0.16 AU; DM: 1.74 ± 0.20 AU; ~1.8-fold increase; Fig. 2e, Supplementary Fig. 2g). Tight junction integrity improved under DM, as reflected by enhanced OCLN localization (ST: 0.95 ± 0.27 AU; DM: 2.92 ± 0.44 AU; ~3.1-fold increase, Fig. 2f, Supplementary Fig. 2e-f). Consistent with these structural improvements, barrier tightness was strengthened, as shown by lower Papp values for both 3 kDa dextran (ST: 23.1 ± 8.9 ×10⁻⁶ cm/s; DM: 4.5 ± 2.1 ×10⁻⁶ cm/s; ~5-fold tighter) and 70 kDa dextran (ST: 25.7 ± 8.0 ×10⁻⁶ cm/s; DM: 10.7 ± 2.3 ×10⁻⁶ cm/s; ~2.4-fold tighter; Fig. 2g). These findings indicate that DM culture in the GBV chip enhances epithelial morphology and tight junction function, establishing a physiologically relevant gut barrier. Next, we evaluated vascular barrier properties, particularly endothelial tight junction integrity and dynamic-induced morphological adaptations in the vascular compartment. The vascular compartment also exhibited chip-dependent adaptations. Immunofluorescence staining of ZO-1 showed stronger endothelial tight junctions in DM compared with ST, with expression increasing by ~1.4-fold (ST: 0.99 ± 0.22 AU; DM: 1.38 ± 0.24 AU; Fig. 2h, i). Endothelial cell (EC) morphology analysis confirmed a shear-adapted phenotype in DM, characterized by elongated cell shape and higher Feret ratios (ST: 0.93 ± 0.19; DM: 4.23 ± 1.4, n = 30 representative individual cells), consistent with physiological flow adaptation (Fig. 2j). This shift from static cuboidal morphology to an elongated form is characteristic of physiological endothelial responses to shear stress, confirming that the hGBV vascular compartment successfully recapitulates in vivo-like BBB characteristics. Barrier tightness was further improved under DM, as shown by lower Papp values for both 3 kDa dextran (ST: 2.4 ± 0.6 × 10⁻⁶ cm/s; DM: 0.73 ± 0.29 ×10⁻⁶ cm/s; ~3.3-fold tighter) and 70 kDa dextran (ST: 2.5 ± 0.6 × 10⁻⁶ cm/s; DM: 1.06 ± 0.28 × 10⁻⁶ cm/s; ~2.4-fold tighter; Fig. 2k). Finally, functional imaging confirmed neuronal activity within the brain compartment. Neurons (Tuj1) and astrocytes (GFAP) displayed robust synaptic marker expression (SYN1) and calcium oscillations visualized with Rhod-2 (ΔF/F₀), with time-lapse analysis revealing rhythmic low-frequency Ca²⁺ dynamics (Fig. 2l, m). In addition, aquaporin-4 expression in astrocytic endfeet was significantly upregulated in hGBV vascular compartment (VC) compared with brain compartment (BC), further supporting strengthened vascular–astrocyte coupling (Fig. 2n).
Together, these results demonstrate that our hGBV chip enhances the physiological properties compared with static culture in open well plates, promoting villus-like gut morphology, improving gut barrier function, vascular endothelial integrity, and supporting neurovascular interactions. The integration of flow-adapted villus-like gut epithelium, endothelial cells, and functional neurovascular interfaces establishes hGBV as a biomimetic platform for studying gut-brain-vascular dynamics.
Modeling gut-to-brain inflammation and microbiota-driven neuroinflammation
Dysbiosis and bacterial-derived inflammation are increasingly recognized as key contributors to neurodegenerative disease progression13. To model gut-to-brain inflammation and microbiota-driven neuroinflammation, we exposed the gut compartment (GC) to either E. Coli-conditioned media (E.CCM, 10%) or pure lipopolysaccharide (LPS, 1 μg/mL) to mimic microbial-triggered inflammatory responses (Fig. 3a). we observed progressive barrier leakage and the accumulation of neurotoxic proteins across the interconnected gut–vascular–brain axis (Fig. 3b, Supplementary Fig. 4). Gut paracellular permeability, measured by Papp using 70 kDa dextran, was significantly elevated compared with control (CT: 12.6 ± 2.5 × 10⁻⁶ cm/s; E. Coli: 21.9 ± 4.2 × 10⁻⁶ cm/s; LPS: 20.9 ± 3.1 × 10⁻⁶ cm/s), indicating increased epithelial leakage (Fig. 3c). Endotoxin quantification confirmed systemic leakage, with markedly higher levels in the vascular and brain compartments compared to control (CT: ~0.2–0.1–0.1 EU/mL vs. E. Coli: 5.9–2.5–2.5 EU/mL; LPS: 5.2–2.5–1.1 EU/mL; Fig. 3d). Consistent with this spread, amyloid-β (Aβ) and α-synuclein (α-Syn) accumulated across compartments (Fig. 3b, e–h Supplementary Fig. 4). In E. Coli conditions, Aβ was increased ~3.1-fold in GC, ~2.5-fold in VC, and ~1.6-fold in BC (Fig. 3e), while α-Syn rose ~2.4-fold in GC and ~1.9-fold in BC (Fig. 3g) relative to control. By contrast, phosphorylated tau (pTau) and phosphorylated α-synuclein (pα-Syn) remained unchanged (Fig. 3h). These findings suggest that bacterial stimuli selectively promote Aβ and α-Syn propagation along the gut-to-brain pathway rather than broadly elevating all neurodegenerative markers.

a Schematic of E. Coli and LPS exposure in GC leading to barrier leakage and inflammatory signaling to VC and BC compartments. b Immunofluorescence of neurotoxins across GC, VC, and BC, showing Aβ, pTau, α-Syn, and pα-Syn. Quantification of gut epithelial permeability (Epi Papp, c), endotoxin levels (d), Aβ (e), pTau (f), α-Syn (g), and pα-Syn (h) across compartments. Immunofluorescence of VIL1, CD13 in GC (i), endothelium-astrocytic endfeet contact (AQP4, GFAP) and tight junction ZO-1 in VC (j), and brain SYN1, Tuj1, and C3 in BC (k) under CT, E. Coli, and LPS conditions. Quantification of compartment-specific markers in GC (l): VIL1 (i), CD13 (ii)); in VC (m) AQP4 (i), GFAP (ii), ZO-1 (iii); and in BC (n): SYN1 (i), C3 (ii). o Quantification of pro-inflammatory cytokines in BC: IL-6 (i), IL-8 (ii), CXCL10 (iii), SDF1α (iv), CCL2 (v), CCL3 (vi), and CCL5 (vii) under CT, E. Coli, and LPS conditions. Scale bars: 100 μm (b), 50 μm (i, j, k). Representative images are shown from one experiment; similar results are obtained in three independent experiments with comparable outcomes (b, j, k). Quantification is based on n = 5 independent chips per condition (c, o), 10 independent chips per condition (d–h, l–n). Data represent presented as box plots (a, l–o), showing the median (center line), 25th–75th percentiles (box), and minimum to maximum values (whiskers); Data in (d–h) are presented as Mean ± SEM (d–h); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Statistical analysis: two-sided tests selected per dataset (Student’s t-test/Welch’s t-test or Mann-Whitney U for two-group comparisons; one-way ANOVA/Welch’s ANOVA with Tukey or Kruskal-Wallis with Dunn for multi-group comparisons), with multiple-comparisons adjustments where applicable; exact tests and full statistics (effect sizes, 95% CIs, exact P values) are reported in Supplementary Table S2. CT represents control; E.CCM, E. Coli-conditioned media; and LPS, lipopolysaccharide, GC gut compartment, VC vascular compartment, BC brain compartment. Created in BioRender. Vu, T. A. (2026) https://BioRender.com/702y808 (a).
To assess gut epithelial integrity in response to bacterial-derived toxins, we examined tight junction expression, epithelial permeability, and inflammatory factors release. E.CCM and LPS treatment significantly increased gut permeability index (Papp) in Fig. 3c confirmed barrier dysfunction and indicated by higher endotoxin levels in the vascular (VC) and brain (BC) compartments in Fig. 3d, confirming barrier dysfunction, suggesting that bacterial-derived components translocated across compartments. Structural integrity of the gut barrier was assessed via immunofluorescence and RT-qPCR analysis, Villin-1 expression was significantly reduced under inflammatory conditions (CT: 1.00 ± 0.08 AU; E. Coli: 0.68 ± 0.09 AU; LPS: 0.76 ± 0.11 AU; ~30% reduction; Fig. 3i, l-i), while CD13 expression was strongly upregulated (CT: 1.00 ± 0.10 AU; E. Coli: 3.13 ± 0.34 AU; LPS: 2.73 ± 0.32 AU; ~3-fold increase, Fig. 3i,l-ii), consitent the barrier disruption and epithelial activation in Fig. 1h. Consistent with these protein-level changes, RT-qPCR showed concordant transcript shifts with a reduction in gut OCLN (Fig. 1i, Supplementary Fig. 3c, d), a decrease in vascular TJP1/ZO-1 (Fig. 1m, Supplementary Fig. 3e, f), and an elevation of brain GFAP (Fig. 1n, Supplementary Fig. 3g-h) after E.CCM and LPS relative to CT, supporting E.CCM- and LPS-driven junction loss and astrocytic activation.
To determine whether gut-derived inflammation impacted vascular integrity, we assessed endothelial barrier properties and astrocytic interactions in the vascular compartment (VC). Both E. Coli and LPS exposure significantly damaged vascular tightness, endothelial tight junctions were markedly disrupted with ZO-1 expression reduced by ~40–50% (CT: 0.99 ± 0.03 AU; E. Coli: 0.56 ± 0.05 AU; LPS: 0.63 ± 0.06 AU; Fig. 3m-iii). Astrocytic endfeet exhibited a reactive phenotype: aquaporin-4 (AQP4) expression decreased (CT: 0.99 ± 0.04 AU; E. Coli: 0.68 ± 0.08 AU; LPS: 0.78 ± 0.07 AU; Fig. 3j, m-I, Supplementary Fig. 6), while GFAP intensity was strongly upregulated (~3-fold increase; CT: 0.98 ± 0.09 AU; E. Coli: 2.85 ± 0.42 AU; LPS: 2.70 ± 0.36 AU; Fig. 3j,m-ii). These results indicate that bacterial-derived factors destabilize vascular tight junctions while inducing astrocytic reactivity, thereby disrupting neurovascular unit homeostasis.
To examine the impact of gut-origin inflammation on the brain compartment (BC), we evaluated complement activation (C3), synaptic integrity (Synapsin-1), and inflammatory factors release. In the brain compartment (BC), E. Coli and LPS induced pronounced neuroinflammation and synaptic damage. Complement activation (C3) increased by ~2.5-fold (CT: 0.98 ± 0.06 AU; E. Coli: 2.39 ± 0.42 AU; LPS: 2.43 ± 0.37 AU; Fig. 3k, n-ii), while Synapsin-1 expression was reduced by ~40% (CT: 0.96 ± 0.05 AU; E. Coli: 0.57 ± 0.09 AU; LPS: 0.58 ± 0.08 AU; Fig. 3k,n-i). Cytokine profiling revealed a robust pro-inflammatory signature, with marked elevations in IL-6 (CT: 15.6 ± 2.1 pg/mL; E. Coli: 82.3 ± 7.5 pg/mL; LPS: 75.3 ± 6.8 pg/mL), IL-8 (CT: 2.9 ± 0.4 pg/mL; E. Coli: 7.8 ± 1.0 pg/mL; LPS: 8.2 ± 1.1 pg/mL), and CXCL10/IP-10 (CT: 169.7 ± 15.4 pg/mL; E. Coli: 451.2 ± 34.7 pg/mL; LPS: 896.5 ± 52.1 pg/mL), along with chemokines MCP-1, CCL3, CCL4, CCL5, and SDF-1α (Fig. 3o, Supplementary Fig. 7). Th2-associated cytokines such as IL-4 and IL-13 showed no significant changes, underscoring a primarily pro-inflammatory and chemotactic response. Consistent with these protein-level changes, RT-qPCR showed concordant transcript shifts with an elevation in brain APP (Fig. 1o, Supplementary Fig. 3, j), an increase in brain SNCA (Supplementary Fig. 3m, n), a decrease in vascular TJP1/ZO-1 (Fig. 1p, Supplementary Fig. 3k, l), and a reduction in gut OCLN (Fig. 1q, Supplementary Fig. 3o, p) in AD and PD relative to WT, supporting brain-derived neuroinflammatory signals that weaken vascular and epithelial junctions.
Collectively, these findings confirm that gut-origin risk factors, triggered by bacterial-derived stimuli (E.CCM and LPS), induce a cascade of gut-to-brain inflammatory signaling, leading to neurovascular dysfunction and neurodegeneration. The sequential progression from gut barrier breakdown to vascular, synaptic loss, and tau pathology highlights the critical role of microbiota-driven neuroinflammation in neurodegenerative diseases. This validates the hGBV platform as a translational tool for studying gut-brain interactions in neurodegenerative diseases.
Modeling brain-to-gut crosstalk in neurodegenerative disease
Neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), are increasingly linked to systemic inflammation and gut dysregulation24. To investigate brain-to-gut signaling and its effects on vascular and gut integrity, we modeled AD- and PD-like conditions in the hGBV system. AD-like conditions were induced by transfecting the APP gene into neural progenitor cells (NPCs) and differentiating them for six weeks, while PD was modeled by differentiating NPCs for three weeks, followed by treatment with preformed fibrillar (PFF) α-synuclein (1 µM) and assessment two weeks post-treatment (Fig. 4a–d). We evaluated brain inflammatory responses, vascular dysfunction, and gut structural alterations across compartments to determine the impact of brain-derived neuroinflammation on peripheral systems.

a Schematic of neurotoxins accumulation in BC driving leakage of Aβ, tau, and α-synuclein into VC and GC. Immunofluorescence of WT (b), AD (c), and PD (d) models showing distribution of neurotoxic proteins across compartments: AD markers (Aβ, pTau) and PD markers (α-Syn and pα-Syn). Quantification of AD-related neurotoxin transfer across compartments: soluble Aβ42 (e), Aβ expression (f), and pTau expression (g). Quantification of PD-related neurotoxin transfer across compartments: soluble α-Syn (h), α-Syn expression (i) and pα-Syn expression (j). Immunofluorescence of Tuj1, astrocytic C3, and SYN1 in BC (k); astrocytic endfeet-endothelium contact (GFAP, AQP4) and tight junction ZO-1 in VC (l); and ViL1, CD13 in GC (m). Quantification of compartment-specific markers expression in BC (n): C3 (i), SYN1 (ii); in VC (o): AQP4 (i), GFAP (ii), ZO-1 (iii); and in GC (p): VIL1 (i), CD13 (ii). q Quantification of pro-inflammatory cytokines in BC: IL-6 (i), IL-8 (ii), CXCL10 (iii), CCL2 (iv), CCL3 (v), CCL5 (vi), and SDF1α (vii) under WT, AD, and PD conditions in BC. Scale bars: 100 μm (b–d), 50 μm (k–m). Representative images are shown from one experiment; similar results are obtained in three independent experiments with comparable outcomes (b–d, k–m). Quantification is based on n = 10 independent chips per condition (f, g, i, j, n–p), 5 independent chips per condition (e, h, q). Data in (e–j) are presented as mean ± SD; Data in (n–q) are presented as box plots, showing the median (center line), 25th–75th percentiles (box), and minimum to maximum values (whiskers); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Statistical analysis: two-sided tests selected per dataset (Student’s t-test/Welch’s t-test or Mann-Whitney U for two-group comparisons; one-way ANOVA/Welch’s ANOVA with Tukey or Kruskal-Wallis with Dunn for multi-group comparisons), with multiple-comparisons adjustments where applicable; exact tests and full statistics (effect sizes, 95% CIs, exact P values) are reported in Supplementary Table S2. WT wild-type, AD alzheimer’s disease, PD parkinson’s disease, GC gut compartment, VC vascular compartment, BC brain compartment. Created in BioRender (a). Vu, T. A. (2026) https://BioRender.com/702y808 (a).
To confirm the induction of AD- and PD-like pathology, we quantified the secretion and distribution of neurotoxic protein aggregates (Aβ42 and α-Synuclein) across all compartments. In the AD-like model, ELISA revealed a marked increase in Aβ42 levels in the brain compartment (BC; WT: ~5 pg/mL vs. AD: ~300 pg/mL; Fig. 4e), with detectable leakage into the vascular (VC, ~41.2 pg/mL) and gut (GC, ~10 pg/mL) compartments. Consistently, intensity analysis showed ~5.5-fold higher Aβ immunoreactivity in BC, accompanied by moderate increases in VC (~2.7-fold) and GC (~1.5-fold) compared with WT controls (Fig. 4f, Supplementary Fig. 4). By contrast, phosphorylated tau (pTau) exhibited only mild increases (~3.2-fold in BC, ~1.3-fold in VC, and 1.0-fold in GC; Fig. 4g), suggesting that Aβ42 accumulation was the dominant feature of the AD-like model. In the PD-like model, α-synuclein (α-Syn) concentrations were substantially elevated in the brain (WT: ~200 pg/mL vs. PD: ~900–1250 pg/mL; Fig. 4h), with increased levels also detected in VC (~650 pg/mL) and GC (~350 pg/mL). Intensity analysis further confirmed ~2.0-fold increases in BC, with sustained elevations in VC (~1.5-fold) and GC (~1.1-fold) (Fig. 4i). Notably, phosphorylated α-synuclein (pα-Syn) did not differ significantly from WT (Fig. 4j), indicating that aggregate accumulation rather than phosphorylation was the predominant phenotype in PD-like conditions. Together, these results confirm that AD-like pathology is driven primarily by Aβ42 accumulation and trans-compartmental leakage, whereas PD-like pathology is characterized by robust α-Syn aggregation and dissemination across the gut–vascular–brain axis. This suggests that neurotoxic aggregates can permeate through the vasculature, potentially contributing to systemic neuroinflammation.
In the brain compartment, both AD- and PD-like conditions exhibited pronounced neuroinflammatory and degenerative features compared with WT. Complement activation was significantly elevated, with C3 expression increasing by ~1.6-fold in AD and ~1.5-fold in PD relative to WT, indicating robust inflammatory signaling (Fig. 4k, n-i). Synaptic integrity was impaired, as Synapsin-1 (SYN1) intensity was reduced by ~35–45% in AD and ~20–30% in PD compared with WT, consistent with synaptic loss and diminished neuronal connectivity (Fig. 4k, n-ii). These findings were further supported by decreased Tuj1+ neuronal networks, reflecting neuronal density loss under both disease conditions. Cytokine profiling further highlighted a distinct pro-inflammatory signature in the brain environment of AD- and PD-like models. In the AD model, average levels of several cytokines were strongly elevated, including Eotaxin (CCL11: ~15 pg/mL vs. ~0 in WT), GRO-α/CXCL1 (~26 pg/mL vs. 3 pg/mL), IL-6 (~3351 pg/mL vs. ~15 pg/mL), IL-8 (~1712 pg/mL vs. ~14 pg/mL), IP-10/CXCL10 (~3780 pg/mL vs. 170 pg/mL), and MCP-1/CCL2 (~8100 pg/mL vs. ~1058 pg/mL). Chemokines such as RANTES (CCL5: ~88 pg/mL vs. ~6 pg/mL) and SDF-1α (~646 pg/mL vs. ~45 pg/mL) were also increased, confirming broad inflammatory and chemotactic activation (Fig. 4q; Supplementary Fig. 7). In contrast, the PD model displayed a more moderate cytokine profile. Average levels of IL-6 (~40 pg/mL), IL-8 ( ~4.8 pg/mL), IP-10/CXCL10 (~527 pg/mL), and MCP-1/CCL2 (~1662 pg/mL) were consistently higher than WT but substantially lower than AD. Other cytokines, including Eotaxin, GM-CSF, and GRO-α, remained near baseline levels (Fig. 4q; Supplementary Fig. 7). PD brains displayed a similar but less pronounced pattern, with modest increases in GRO-α (4.2 pg/mL vs. ~1.7 pg/mL in WT), IL-6 (~38–40 pg/mL), and IL-8 (~4–5 pg/mL), alongside consistently low Eotaxin and GM-CSF. Other cytokines, including IFN-γ, IL-1β, IL-2, IL-4, and IL-5, showed only minor or inconsistent changes. Together, these results demonstrate that AD-like pathology drives a highly pro-inflammatory cytokine and chemokine milieu in the brain, while PD-like pathology induces a more moderate inflammatory response. Both models converge on complement activation, synaptic degeneration, and neuronal loss, confirming that brain-derived neuroinflammation underlies the pathological signaling captured in the hGBV platform.
Given the role of neuroinflammation in vascular dysfunction, we assessed endothelial inflammation, vascular barrier integrity, and permeability changes in the vascular compartment (VC). Both AD- and PD-like conditions triggered pronounced astrocytic remodeling. AQP4 expression in astrocytic endfeet was significantly reduced (WT: 1.00 ± 0.05 AU; AD: 0.60 ± 0.06 AU; PD: 0.97 ± 0.08 AU; Fig. 4l, o-i, Supplementary Fig. 6), while GFAP intensity was markedly elevated (WT: 1.00 ± 0.08 AU; AD: 2.3 ± 0.4 AU; PD: 2.1 ± 0.3 AU; Fig. 4l, o-ii, Supplementary Fig. 6), consistent with reactive astrogliosis and neurovascular unit activation. In parallel, endothelial junctional integrity was damaged as evidenced by decreased ZO-1 expression (WT: 1.00 ± 0.03 AU; AD: 0.56 ± 0.07 AU; PD: 0.79 ± 0.08 AU; Fig. 4l, o-iii, Supplementary Fig. 6), confirming tight junction loss (Fig. 4o-iii). Together, these findings indicate that brain-derived inflammatory signals initiate astrocytic endfeet reactivity, followed by endothelial junction disruption and vascular leakage, driving neurovascular dysfunction in both AD- and PD-like models.
To determine whether brain-to-gut signaling disrupts gut homeostasis, we examined gut morphology, epithelial barrier integrity, and inflammatory activation in the gut compartment (GC). Unlike the pronounced vascular inflammation and endothelial barrier dysfunction observed in the VC, the gut compartment exhibited only mild structural alterations. Villus marker ViL1 expression was reduced in both disease models (WT: 1.00 ± 0.06 AU; AD: 0.71 ± 0.09 AU; PD: 0.82 ± 0.08 AU; Fig. 4m, p-i), consistent with villus disruption. In contrast, the activation marker CD13 was markedly upregulated (WT: 1.00 ± 0.08 AU; AD: 1.67 ± 0.22 AU; PD: 1.28 ± 0.17 AU; Fig. 4m, p-ii), confirming epithelial remodeling and inflammatory activation. However, ZO-1 expression remained relatively preserved across conditions (WT: 1.00 ± 0.05 AU; AD: 0.90 ± 0.08 AU; PD: 0.93 ± 0.07 AU; n = 10), suggesting that tight junctions were not severely compromised. Together, these findings suggest that while brain-derived inflammatory signals propagate to the gut epithelium, driving villus loss and epithelial activation, accompanied by increased slightly permeability, they primarily induce structural remodeling rather than direct gut barrier disruption.
These findings demonstrate that brain-origin inflammation in AD- and PD-like conditions leads to significant vascular dysfunction while only mildly affecting the gut barrier. The vascular compartment exhibited pronounced endothelial inflammation, tight junction disruption, and increased permeability, confirming brain-to-vascular inflammatory signaling. In contrast, the gut compartment showed only mild structural alterations, with reduced villus integrity and increased CD13 expression, but no significant tight junction disruption or epithelial loss. This study supports the hypothesis that brain-derived neuroinflammation propagates inflammatory signals to peripheral compartments, with strong effects on the vasculature but limited gut barrier breakdown. These results establish the hGBV platform as a valuable model for studying brain-to-gut crosstalk in neurodegenerative diseases.
Discussion
The gut-brain axis (GBA) is increasingly recognized as a key regulator of systemic homeostasis, with accumulating evidence implicating bidirectional communication between the gut, vasculature, and brain in the progression of neurodegenerative diseases1,5. Despite this recognition, current in vitro models fail to fully capture the complex, multi-compartmental interactions that underlie gut-brain inflammatory signaling. In this study, we developed and validated a 3D human Gut-Brain-Vascular (hGBV) model, successfully integrating a physiologically relevant, villus-like gut epithelium, perfusable vasculature, and a neurovascular brain compartment. Using this platform, we demonstrate that gut-origin inflammation (gut-to-brain axis) can drive neurovascular dysfunction and neurodegeneration, while brain-origin neuroinflammation (brain-to-gut axis) predominantly affects vascular integrity, resulting in only mild alterations to the gut barrier. These findings provide critical mechanistic insights into microbiota-driven neuroinflammation and brain-to-gut crosstalk, establishing the hGBV system as a valuable translational model for neurodegenerative disease research.
Our results confirm that gut-derived inflammatory stimuli, including E. Coli-conditioned media (E.CCM) and lipopolysaccharide (LPS), trigger systemic inflammation that spreads to the vasculature and brain. The observed gut barrier disruption, characterized by tight junction loss (OCLN, ZO-1), villus marker loss (ViL1), and epithelium activation(CD13 upregulation), further increased permeability and the release of inflammatory factors, aligns with previous reports linking gut dysbiosis to intestinal barrier breakdown in neurodegenerative diseases25,26. Importantly, bacterial-derived endotoxins translocate into the vascular and brain compartments, where they induce endothelial leakage and neurodegenerative phenotypes. Notably, we observed selective neurotoxic protein accumulation: amyloid-β and α-synuclein were significantly increased across compartments, whereas phosphorylated tau and phosphorylated α-synuclein remained unchanged. This indicates a specific, rather than generalized, mechanism of aggregate propagation. These findings are consistent with preclinical and clinical studies reporting circulating endotoxins and inflammatory cytokines in Alzheimer’s disease (AD) and Parkinson’s disease (PD) patients27,28,29. At the vascular interface, gut-derived inflammatory signals also triggered astrocytic endfeet remodeling, marked by decreased aquaporin-4 (AQP4) expression and upregulated GFAP intensity. This reactive astrocytic response, together with endothelial junction loss, reflects neurovascular unit dysfunction as an early mediator of gut-to-brain inflammatory signaling. At the neurovascular interface, prolonged exposure to gut-derived inflammatory stimuli results in synaptic dysfunction, complement C3 activation, and neuroinflammation. The neuroinflammation was confirmed by a distinct pro-inflammatory cytokine signature. Both E.CCM and LPS exposure markedly increased IL-6, IL-8, and IP-10/CXCL10, along with chemokines MCP-1/CCL2, CCL3, CCL4, and RANTES, while Th2-associated cytokines (IL-4, IL-13) remained largely unchanged. This indicates a primarily pro-inflammatory and chemotactic milieu, reinforcing the link between gut-derived signals and brain immune activation. Consistent with prior work from our group demonstrating OMV/genotoxin-driven microglial NLRP3–IL-18 activation (Tran et al., 202530) and toxin-mediated neuroinflammation (Tran et al., 202531), we posit that conserved Gram-negative OMV/LPS ( ± flagellin) rather than species-specific toxins underlie the cytokine responses observed with E. Coli–CM. This progression supports the hypothesis that chronic gut inflammation contributes to neurodegenerative pathology through systemic immune activation, changes in BBB permeability, and direct neuronal damage11,13,32,33. Notably, our model recapitulates the sequential progression observed in vivo, where gut inflammation precedes vascular impairment and neuronal loss, reinforcing the critical role of the gut-brain axis in neurodegeneration.
In contrast to the gut-driven model, brain-origin neuroinflammation (brain-to-gut axis) in AD- and PD-like conditions primarily induced vascular dysfunction, with limited gut barrier disruption. APP-transfected NPCs (AD model) and α-synuclein-treated NPCs (PD model) exhibited neurotoxic protein accumulation (Aβ42, α-Syn), synaptic loss, complement activation, and pTau accumulation, consistent with postmortem brain analyses of AD and PD patients34,35,36,37. In parallel, cytokine profiling revealed distinct inflammatory signatures: AD brains displayed a strongly pro-inflammatory and chemotactic milieu, with markedly elevated IL-6, IL-8, IP-10/CXCL10, MCP-1/CCL2, GRO-α, Eotaxin, and RANTES, whereas PD brains exhibited a more moderate profile dominated by IL-6, IL-8, and IP-10. This highlights disease-specific inflammatory patterns, with AD driving a more aggressive neuroimmune cascade compared with PD. At the vascular interface, brain-derived signals first triggered astrocytic endfeet remodeling, characterized by reduced AQP4 expression and marked GFAP upregulation, consistent with reactive astrogliosis. These changes were followed by endothelial dysfunction, with ZO-1 disruption and significantly increased vascular permeability, confirming breakdown of barrier integrity. In contrast, the gut compartment exhibited only modest remodeling. Villus marker ViL1 expression was moderately reduced, and CD13was upregulated, consistent with partial epithelial activation. However, ZO-1 expression remained preserved, and paracellular permeability changes were limited, indicating that tight junction integrity was largely intact. This suggests that brain-to-gut inflammatory signaling is primarily mediated through vascular dysfunction rather than direct gut barrier breakdown. These findings challenge previous hypotheses suggesting severe gut barrier dysfunction in neurodegenerative disease models, instead proposing that gut remodeling in response to brain inflammation is subtler38,39,40. This distinction underscores the need for physiologically relevant multicompartment models like hGBV, which allow for dynamic gut-brain-vascular interactions that are difficult to capture in traditional 2D cultures or in vivo models.
By utilizing our hGBV platform, our findings have several translational implications. First, it provides a human-specific model to study gut-brain-vascular interactions in neurodegeneration, reducing reliance on animal models that often fail to replicate human pathophysiology. Second, our findings suggest that gut-to-brain signaling plays a more direct role in neurodegeneration, while brain-to-gut signaling primarily affects vascular integrity. This highlights the potential for gut-targeted therapeutics to prevent or slow neurodegenerative progression. In this case, several avenues for future research emerge from these findings. First, integrating patient-derived induced pluripotent stem cells (iPSCs) into the hGBV model could enable personalized disease modeling and drug screening. Second, investigating specific microbial metabolites (e.g., short-chain fatty acids) and their effects on neurovascular integrity could help elucidate mechanistic pathways linking gut dysbiosis to neurodegeneration. Finally, leveraging this platform for high-throughput drug screening could accelerate the identification of gut-targeted therapeutics for AD and PD.
While our hGBV platform successfully recapitulates gut-brain-vascular interactions, several limitations remained. First, the physical distance between the gut and brain compartments in our chip is shorter than in vivo, potentially accelerating inflammatory signaling and molecular transport. Second, the model lacks immune cells (e.g., monocytes, T-cells, microglia), which play a crucial role in neuroinflammation and blood-brain barrier (BBB) regulation. Although the presence of endothelial cells and astrocytes provides a neurovascular interface, the addition of circulating peripheral immune cells could further enhance the physiological relevance of the platform. Third, gut microbiota interactions are simplified in this system, as only microbial-derived toxins (e.g., E.CCM, LPS) were introduced. A system integrating live commensal and pathogenic microbes in a gut-microbiome-on-a-chip setup could provide deeper insights into host-microbe interactions in neurodegeneration. Lastly, while this study models AD and PD phenotypes, the system could be expanded to investigate other neurodegenerative conditions, such as ALS or multiple sclerosis, where gut-brain interactions are increasingly implicated. Incorporating genetic risk factors related AD and PD would enable disease modeling and therapeutic screening. While our compartment-resolved readouts implicate gut-vascular-brain crosstalk, pinpointing the cytokine-producing cell types will require single-cell resolution; future work will apply scRNA-seq ( ± spatial transcriptomics) to map ligand-receptor signaling and attribute cytokine transcripts to specific epithelial, endothelial, microglial, and astrocytic subpopulations. Despite these limitations, our hGBV system represents a major advancement over traditional in vitro models, integrating gut, vascular, and brain compartments to create a more physiologically relevant platform. Addressing these limitations in future versions will further enhance its translational potential in neurodegenerative research and therapeutic development.
This study establishes the hGBV system as a physiologically relevant human model for investigating gut-brain-vascular interactions in neurodegeneration. Our findings highlight the differing effects of gut-origin and brain-origin inflammation, showing that gut-derived inflammation drives neurovascular dysfunction and neurodegeneration, while brain-derived neuroinflammation primarily leads to vascular dysfunction with minimal gut remodeling. These results provide critical insights into microbiota-driven neuroinflammation and brain-to-gut communication, supporting gut-targeted therapeutic strategies for AD and PD. By capturing the dynamic interplay between the gut, vasculature, and brain, the hGBV platform serves as a powerful translational tool for elucidating disease mechanisms, identifying biomarkers, and accelerating the development of targeted therapeutics for neurodegenerative diseases.
Method
Ethics statement
All research was performed in accordance with relevant ethical guidelines and regulations and in alignment with the 2021 ISSCR Guidelines for Stem Cell Research and Clinical Translation. Human embryonic stem cell–derived endothelial cells were obtained from WiCell under authorized agreements (Agreement Nos. 22-W0724) and used in accordance with supplier documentation and institutional guidelines. No human participants or identifiable human samples were involved; therefore, no additional institutional review board (IRB) or stem-cell oversight approval was required at Sungkyunkwan University. Donor-informed consent and ethical procurement were obtained and documented by the supplier in accordance with applicable regulations. Human participant information (sex/gender, age, consent, compensation) is not applicable, as this study did not involve human participants or the collection of individual-level human data.
To systematically investigate the role of gut-brain-vascular interactions in neuroinflammation, we developed a 3D microfluidic platform that recapitulates the physiological architecture of the gut, vasculature, and brain. Below, we detail the fabrication of the microfluidic system, cellular components, and experimental conditions used to simulate gut-origin and brain-derived inflammatory processes.
Microfluidic chip fabrication
The microfluidic chip was fabricated using soft lithography techniques, where a silicon substrate served as the base for device molding. The photolithography process sequentially patterned an SU-8 negative photoresist (MicroChem, Round Rock, TX) onto the silicon wafer, forming the microfluidic channel mold. To replicate the microfluidic structures, a polydimethylsiloxane (PDMS) mixture was prepared by combining PDMS elastomer with a curing agent (Sylgard 184 A/B, Dow Corning, Midland, MI) at a 9:1 ratio. The mixture was poured onto the SU-8 mold and degassed under vacuum to remove air bubbles. The PDMS was then thermally cured at 65 °C for 2 hours to ensure complete polymerization. Once cured, the PDMS replica was carefully removed from the mold, and fluid reservoirs were created by punching inlet and outlet holes using a 2 mm biopsy punch. Additional plastic media reservoirs were fabricated using a computer-controlled laser cutter (Epilog Laser, Golden, CO) on a 5 mm thick acrylic plate to enhance experimental throughput and facilitate media handling. These acrylic reservoirs were bonded to the PDMS chip using an additional layer of liquid PDMS to ensure a stable, leak-free connection. For final assembly, the PDMS chip was bonded to a custom glass-bottomed uni-plate (MatTek, Ashland, MA) via oxygen plasma treatment (Plasma Etch, Carson City, NV), ensuring strong adhesion and optical transparency for high-resolution imaging (Supplementary Fig. 1). After assembly, the microfluidic chip was sterilized under UV light for 30 minutes before cell seeding and experimental use.
Functionalization of microfluidic devices before cell seeding
To promote cell adhesion and extracellular matrix (ECM) compatibility, the microfluidic compartments were functionalized with Poly-L-lysine (PDL) and collagen type I (Col I) following a well-established protocol [ref]. This functionalization step enhances cell attachment, barrier formation, and tissue-specific microenvironment stability within the microfluidic system. The entire microfluidic network, including all compartments and channels, was infused with 1 mg/mL Poly-L-lysine (Sigma Aldrich, St. Louis, MO) and incubated for 30 minutes at room temperature (RT) to facilitate surface charge modification and ECM adhesion. After incubation, the device was rinsed thoroughly with cold 1× PBS (phosphate-buffered saline) three times to remove excess PDL. Following PDL treatment, the gut compartment (GC) and vascular compartment (VC) were infused with 1 mg/mL collagen type I (Corning, Corning, NY), adjusted to pH 7.5. The functionalized device was incubated at 37 °C for 1 hour to allow proper ECM polymerization and surface coating. After functionalization, all compartments were washed with fresh 1× PBS to remove unbound proteins. The microfluidic devices were then immediately seeded with cells to ensure optimal cell adhesion and tissue integration.
3D culture of brain tissue in the hgbv microfluidic device
To establish a functional 3D neural microenvironment within the brain compartment (BC) of the hGBV model, human neural progenitor cells (ReN cell VM NPCs) were differentiated into neurons and astrocytes over a three-week period. This approach promoted cellular maturation, synaptic formation, and astrocyte-neuron interactions, ensuring a physiologically relevant brain tissue model. Human ReN cell VM neural progenitor cells (NPCs) were obtained from Millipore Sigma (Billerica, MA, USA) and maintained at passages <20 to preserve their differentiation capacity. Cells were expanded in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12, Life Technologies, Grand Island, NY, USA) supplemented with 2 mg/mL Heparin (StemCell Technologies, Vancouver, Canada), 2% (v/v) B27 neural supplement (Life Technologies, Grand Island, NY, USA), 20 ng/mL Epidermal Growth Factor (EGF) (Sigma-Aldrich, St. Louis, MO, USA), 20 ng/mL Basic Fibroblast Growth Factor (bFGF) (Stemgent, Cambridge, MA, USA), and 1% (v/v) Penicillin/Streptomycin (Sigma-Aldrich, St. Louis, MO, USA). NPCs were plated in T25 flasks coated with 1% Matrigel (BD Biosciences, San Jose, CA, USA) and cultured at 37 °C with 5% CO₂ in a humidified incubator. The culture medium was refreshed every three days until confluence was reached.
To generate a 3D mini-brain tissue model, ReN NPCs were suspended at 2 × 104 cells/device in a 10% (v/v) Matrigel-DMEM/F12 differentiation medium mixture (Supplementary Fig. 3). 10 µL of the NPC-Matrigel suspension was injected into the brain compartment. The device was incubated at 37 °C for 1 hour to facilitate gel polymerization and cell attachment. After polymerization, 100 µL of differentiation medium was added to both the brain and vascular compartments. The devices were placed in a 37 °C, 5% CO₂ incubator for long-term culture. Over 21 days, the NPCs progressively differentiated into neurons and astrocytes. Culture medium was replaced every two days to maintain optimal differentiation conditions.
3D gut epithelium barrier generation
To establish a functional gut epithelium within the gut compartment (GC) of the hGBV model, the Caco-2 epithelial cell line was cultured and differentiated under dynamic perfusion conditions to mimic the intestinal barrier. Human Caco-2 cells were obtained from the Cell Lines Service (CLS, Eppelheim, Germany) and maintained between passages 40–70 to ensure stable barrier formation properties. Cells were cultured in 75 cm² tissue culture flasks using high-glucsoe Dulbecco’s Modified Eagle’s Medium (high glucose DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) Fetal Bovine Serum (Sigma-Aldrich, St. Louis, MO, USA), 1% (v/v) Penicillin/Streptomycin (Sigma-Aldrich, St. Louis, MO, USA). Cells were maintained at 37 °C in a 5% CO₂/95% air atmosphere with constant humidity, and the culture medium was refreshed every two days until the cells reached 80–90% confluence.
At day 7 of neuronal and astrocyte differentiation, Caco-2 cells were seeded into the gut compartment (GC) of the microfluidic chip to allow for co-culture with the neurovascular system. The cells were introduced at a density of 106 cells/mm2 and allowed to attach for 4 hours before adding fresh media. The loaded cells adhered to the upper surface of the compartments when the platforms were inverted for 4 hours in a 37 °C incubator with 5% CO2. After the cells adhered to the upper surface, newly cultured Caco-2 cells were gently reintroduced to the bottom surface in their normal orientation for an additional two hours to ensure proper adherence. Once both surfaces were covered with adherent cells, we added an extra 100 μL of fresh medium to the reservoirs. To enhance intestinal barrier function, dynamic perfusion conditions were introduced using a rocker integrated into the incubator. The culture medium was replaced daily to maintain optimal differentiation conditions.
3D vascular endothelium barrier generation
To establish a functional blood-brain barrier (BBB) within the vascular compartment (VC) of the hGBV model, human embryonic stem cell-derived endothelial cells (hECs) were differentiated into induced brain microvascular endothelial cells (iBMECs) and cultured under dynamic perfusion conditions to mimic physiological endothelial barrier function.
Human embryonic stem cells (hEC, H9) were obtained from Wi-Cell (USA) and differentiated into iBMECs following a previously established protocol (ref). Cells were grown in mTeSR Plus medium (StemCell Technologies, Vancouver, Canada) supplemented with 10 μM Y-27632 dihydrochloride (Tocris #1254) to enhance survival and proliferation. Upon reaching ~70% confluency, differentiation into iBMECs was initiated. Cells were switched to an unconditioned medium (Thermo Fisher Scientific #11330057) containing 20% KnockOut Serum Replacement (Thermo Fisher Scientific #10828010), 1% Non-Essential Amino Acids (NEAA, Thermo Fisher Scientific #11140050), 0.5% Glutamax (Thermo Fisher Scientific #35050079), 0.1 mM β-Mercaptoethanol (Sigma-Aldrich #M6250, St. Louis, MO, USA). This differentiation medium was maintained for 6 days to promote iBMEC-like characteristics. Cells were transitioned to EC++ medium, consisting of human Endothelial Serum-Free Medium (SFM, Thermo Fisher Scientific #11111044), 1% Platelet-Poor Plasma-Derived Serum, Bovine (Alfa Aesar #15406419, Haverhill, MA, USA), 20 ng/mL Basic Fibroblast Growth Factor (bFGF), 10 μM Retinoic Acid (Sigma-Aldrich #R2625). After 2 days, the medium was replaced with maturation medium (SFM without bFGF and retinoic acid) to stabilize endothelial barrier function. iBMECs were harvested using Accutase (Corning #25-058-CI, Corning, NY, USA) and prepared for seeding onto the vascular compartment (VC) of the hGBV microfluidic device.
At day 18 of neuronal and astrocyte differentiation with Caco-2, a suspension of iBMECs (5 × 106 cell/mm²) was loaded into the vascular compartment (VC). The microfluidic chip was inverted for 4 hours at 37 °C to facilitate cell attachment to the upper surface. After 4 hours, the chip was returned to its normal orientation, and a second suspension of iBMECs was gently reintroduced to the bottom surface for 2 additional hours to ensure bilayer coverage. Once both surfaces were fully covered with adherent iBMECs, 100 μL of fresh differentiation medium was added to one reservoir, allowing flow through the vascular compartment. To maintain a constant supply of fresh nutrients, the medium was replenished daily by adding 100 μL of fresh medium to one reservoir while removing the same volume from the opposite reservoir. To enhance endothelial barrier integrity, dynamic perfusion was introduced using a rocker integrated within the incubator.
Barrier integrity assessment
To evaluate the permeability of the 3D barrier model, including the gut (GC) and vascular (VC) compartments, we performed a FITC-dextran permeability assay. This method assessed barrier integrity by quantifying the diffusion of fluorescently labeled dextran molecules across the epithelial and endothelial layers. FITC-conjugated dextran molecules of different molecular weights were used to evaluate paracellular permeability, including low molecular weight FITC-dextran (3–5 kDa) to assess small molecule permeability and high molecular weight FITC-dextran (70 kDa) to assess macromolecule leakage. Both dextran solutions were obtained from Sigma-Aldrich (St. Louis, MO, USA) and prepared in sterile 1× PBS at a final concentration of 10 µg/mL. FITC-dextran was introduced into the luminal side of either the gut compartment (GC) or vascular compartment (VC) of the microfluidic device. Time-lapse imaging was performed to monitor fluorescence intensity changes during the perfusion of FITC-dextran across the barrier layer. Imaging was conducted using a Nikon Eclipse Ti2-E microscope (Nikon Corporation, Tokyo, Japan) equipped with a filter cube for FITC detection, 10× objective lens, and frames were captured at 1 second per frame to track dextran diffusion dynamics. Raw fluorescence intensity values were extracted using Nikon NIS-Elements software. The fluorescence intensity ratio between the perfused FITC-dextran and the initial luminal concentration was used to calculate the perfusion index. The apparent permeability coefficient (Papp, cm/s) was determined using the following equation:
Where: Papp is permeability coefficient (cm/s), dCi/dt is rate of change in FITC-dextran concentration in the receiving compartment over time (t), Co is initial FITC-dextran concentration in the luminal compartment (10 µg/mL), V is volume of the receiving compartment, A is surface area of microfluidic channel (0.013 cm²). Lower Papp values indicate an intact and selectively permeable barrier. Higher Papp values suggest barrier leakage.
Brain calcium signaling
To assess neuronal activity within the brain compartment (BC), we monitored intracellular calcium dynamics using Oregon-Green-BAPTA-1 (OGB-1), a Ca²⁺ indicator (Life Technologies, Thermo Fisher Scientific). Calcium transients were analyzed using real-time fluorescence imaging, providing insights into neuronal and astrocyte interactions. Brain compartments were incubated in artificial cerebrospinal fluid (aCSF) at 37 °C for 30 minutes to mimic physiological conditions. The aCSF composition included 25 mM NaHCO₃, 25 mM D-glucose, 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH₂PO₄, 1 mM MgCl₂, 2 mM CaCl₂. During incubation, Pluronic F-127 (20% w/v in DMSO, Sigma-Aldrich) was added to facilitate OGB-1 dye loading into the cytosol. After 30 minutes of dye loading, samples were rinsed twice with fresh aCSF to remove unbound dye. Time-lapse fluorescence imaging was performed using a Nikon Eclipse Ti2-E microscope (Nikon Corporation, Tokyo, Japan) equipped with a Texas red filter cube (for Rhod-2 detection), 20× objective lens, and frames were captured at 1 frame per second to monitor dynamic changes in intracellular Ca²⁺ levels. Raw fluorescence intensity values were extracted using Nikon NIS-Elements software.
E. Coli Bacteria Conditioned Media Collection
Escherichia coli (E. Coli; ATCC 25922) colonies were grown on Luria–Bertani (LB) agar plates at 37 °C for 18–24 hours. To prepare the bacterial suspension, colonies were collected by gently scraping the plate surface with sterile phosphate-buffered saline (PBS). The bacterial suspension was adjusted to an approximate optical density (OD₆₀₀ ≈ 0.5–0.6) to reflect mid-log phase concentration. The suspension was then centrifuged at 4000 × g for 10 minutes to pellet the bacteria. The supernatant was collected and passed through a 0.22 µm sterile filter to remove any remaining bacterial cells. The resulting E. Coli–conditioned media (E.CCM) was aliquoted and stored at –80 °C until further use in the gut compartment of the GBV-on-a-chip. For positive control experiments, lipopolysaccharide (LPS) from E. Coli O111:B4 (Sigma-Aldrich) was used at 1 µg/mL to induce inflammatory responses across compartments.
Alzheimer’s Disease (AD) model construction in the hGBV platform
To generate Alzheimer’s disease (AD) human neural progenitor cells (hNPCs), ReN hNPCs were transduced with a lentiviral vector encoding the APPSL mutation, resulting in the overexpression of a familial Alzheimer’s disease (FAD) variant of the amyloid precursor protein (APP). This model enabled the production of high levels of Aβ peptides, mimicking AD-like pathophysiology in the brain compartment (BC) of the hGBV system.
ReN hNPCs were cultured in six-well plates and transduced using a commercially available APPSL-GFP Alzheimer’s lentivirus (cat#SCR526, EMD Millipore). The APPSL lentiviral vector encodes a mutated form of human APP containing two familial Alzheimer’s disease (FAD) mutations, K670N/M671L (Swedish mutation) for increasing β-secretase cleavage, leading to higher Aβ42 productio,n and V717I (London mutation) for altering γ-secretase cleavage, promoting Aβ42 over Aβ40 production. ReN hNPCs were incubated with 5 µL of APPSL-GFP lentivirus (1 × 10⁹ IFU/mL) in the presence of 2 mg/mL polybrene (EMD Millipore) to enhance transduction efficiency. Cells were incubated for 24 hours at 37 °C with 5% CO₂. GFP fluorescence was monitored to confirm initial viral expression. After three washes with PBS, a second round of viral infection was performed by adding fresh APPSL-GFP lentivirus to the culture medium. Cells were incubated for another 24 hours. After 48 hours, cells were washed three times with PBS and cultured in fresh medium to allow transgene expression stabilization. Fluorescence microscopy was used to confirm GFP-positive transgene expression. Transgene-positive cells were enriched using fluorescence-activated cell sorting (FACS, BD FACS Aria II, BD Biosciences) to ensure a homogeneous AD hNPC population. All plasmids and viral vectors used in this study are commercially sourced and are listed in Supplementary Table S1 (Key Resources Table) with supplier information and catalog/identifier numbers.
Parkinson’s Disease (PD) model construction in the hGBV platform
To establish a Parkinson’s disease (PD) model within the hGBV platform, preformed fibril (PFF) α-synuclein aggregates were introduced into the brain compartment (BC), where they induced pathological α-synuclein aggregation, neuroinflammation, and neuronal dysfunction. This model enabled the study of brain-to-gut crosstalk in PD pathology. ReN-derived NPCs were differentiated into neurons and astrocytes for 3 weeks in the brain compartment, following the differentiation protocol described in Section 1.3. PFF α-Synuclein (PFF α-Syn, 1 μM, StressMarq Bioscience) was added to the brain compartment (BC) for 6 hours to allow for fibril uptake by neurons and astrocytes. Cells were then washed and replaced by fresh medium to remove excess extracellular fibrils. The cultures were maintained for 7-14 additional days to allow for α-synuclein aggregation and pathology development.
Chromogenic endotoxin measurement assay
To quantify endotoxin levels in conditioned media and microfluidic compartments, we performed a chromogenic endotoxin measurement assay using the Pierce™ Chromogenic Endotoxin Quant Kit (Thermo Fisher Scientific, Catalog #A39552S). This assay was conducted following the manufacturer’s user guide and is based on the Limulus Amebocyte Lysate (LAL) reaction. Samples were collected from the gut compartment (GC), vascular compartment (VC), and brain compartment (BC). Samples were diluted 1:10 in endotoxin-free water to ensure that measurements fell within the linear range of the standard curve. The optical density (OD) at 405 nm was measured using a microplate reader (Biotek, USA). Sample endotoxin levels were calculated using the standard curve and expressed in endotoxin units per milliliter (EU/mL). Endotoxin levels were compared between control (CT), E. Coli–conditioned media (E.CCM), and LPS-treated conditions to assess gut-to-brain endotoxin translocation. Each condition was prepared in triplicate to ensure data reliability and reproducibility.
Amyloid beta 42 ELISA assay
Quantifying amyloid beta 42 (Aβ42) levels was performed an enzyme-linked immunosorbent assay (ELISA) using the Human Aβ42 ELISA Kit (Abcam, Catalog #ab289832). The assay was conducted following the manufacturer’s user guide, which employs a sandwich ELISA method for the sensitive and specific detection of Aβ42 in conditioned media. Conditioned media were collected from the brain compartment (BC), vascular compartment (VC), and gut compartment (GC) after 6 weeks of neuronal differentiation in wide-type (WT) and AD-like conditions (APP-overexpressing NPCs, Section 1.9). Sample concentrations were interpolated from the standard curve and expressed as pg/mL Aβ42. Total Aβ42 levels were compared between control (CT) and AD conditions to assess the release and potential translocation of Aβ42 across the brain-gut axis. All samples were analyzed in triplicate to ensure data reproducibility.
Alpha-Synuclein ELISA assay
To quantify alpha-synuclein (α-Syn) levels in the brain compartment (BC) and assess its potential translocation into the vascular compartment (VC) and gut compartment (GC), we performed an enzyme-linked immunosorbent assay (ELISA) using the Human Alpha-Synuclein ELISA Kit (Abcam, Catalog #ab260052). The assay was conducted according to the manufacturer’s user guide and utilized a sandwich ELISA format for the specific and sensitive detection of α-Syn in conditioned media. Conditioned media were collected from the brain compartment (BC), vascular compartment (VC), and gut compartment (GC) following 7 days of PFF α-Synuclein treatment in the PD model (Section 1.10). Measured α-Syn levels were compared between wild type (WT) and PD-like conditions to assess neurotoxic α-Syn secretion and potential translocation across compartments. All samples were analyzed in triplicate to ensure data reproducibility.
Multicytokine profiling assay
To quantify inflammatory cytokine levels in different compartments, we used the ProcartaPlex™ Human Th1/Th2 Cytokine and Chemokine Panel 1, 20plex (Thermo Fisher Scientific, USA), following the manufacturer’s instructions. Conditioned media were collected from the gut, vascular, and brain compartments under various treatment conditions and stored at −80 °C until analysis. Samples were incubated with magnetic beads conjugated to capture antibodies for 20-targeted human cytokines and chemokines. After washing and biotinylated detection antibody incubation, streptavidin-PE was added for signal detection. Plates were read using the Luminex 200™ system (Luminex Corporation, USA), and data were analyzed using xPONENT software. Concentration values were determined using standard curves generated from known concentrations of recombinant cytokines. All conditions were tested in technical duplicates across five biological replicates (n = 5), and inter-assay variability was minimized through parallel processing. This approach allowed sensitive, multiplexed cytokine quantification to assess compartment-specific inflammatory responses. Cytokine expression levels were compared across control (CT), E.Coli-conditioned media (E.CCM)-treated, LPS-treated, and neuroinflammatory conditions (AD-like, PD-like environment). In addition, a heatmap of log₂ fold-change cytokine expression was generated to visualize differential secretion patterns across experimental conditions.
Immunostaining assay
To visualize cellular markers and structural integrity within the 3D hGBV microfluidic platform, an immunostaining assay was performed following a standard fixation, permeabilization, blocking, and antibody incubation protocol.
First, cells cultured within the microfluidic device were rinsed twice with 1× PBS and fixed at room temperature for 30 minutes using 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA). After fixation, cells were washed three times with 1× PBS at 10-minute intervals. To enhance antibody penetration, cells were permeabilized for 30 minutes at room temperature with 1× PBS supplemented with 0.1% (v/v) Triton X-100 and 0.1% (v/v) Tween-20. Subsequently, cells underwent three additional 10-minute washes with 1× PBS before blocking to minimize non-specific antibody binding. For blocking, cells were incubated for 2 hours at 4 °C in a solution of 1× PBS supplemented with 0.1% (v/v) Tween-20 and 4% (v/v) human serum albumin (HSA). Following blocking, cells were rinsed three times with 1× PBS containing 0.1% (v/v) Tween-20 at 10-minute intervals. Cells were then incubated with the primary antibody diluted in 1% (v/v) blocking solution for 6 hours at 4 °C. The dilution ratios of all primary and secondary antibodies are provided in Table S1 (Supplementary Information). After the primary antibody incubation, cells were washed three times with 1× PBS containing 0.1% (v/v) Tween-20 at 10-minute intervals before the addition of the secondary antibody. Secondary antibodies were diluted in 1× PBS supplemented with 0.1% (v/v) Tween-20 and 4% (v/v) HSA and incubated for 2 hours at 4 °C. Following secondary antibody binding, cells were washed three times with 1× PBS supplemented with 0.1% (v/v) Tween-20 at 10-minute intervals to remove excess antibodies.
Immunostained samples were visualized using a Nikon Eclipse Ti2-E fluorescence microscope (Nikon Corporation, Tokyo, Japan) and a K1-Fluo Confocal Laser Microscope (Nanoscope, Korea). For high-resolution 3D imaging, especially of the villus structure and barrier interfaces, we additionally employed the Zeiss LSM980 with Airyscan 2 (Carl Zeiss AG, Germany), enabling 2.5D and volumetric reconstructions for thickness and surface area analysis. Regions of interest (ROIs) were manually selected for each compartment (gut, vascular, and brain) to quantify the mean fluorescence intensity (MFI). Image acquisition and MFI quantification were performed using NIS-Elements software (Nikon, Japan) and Fiji/ImageJ (NIH, Wayne Rasband, USA). To account for variations in cell density, MFI values were normalized to the counts of DAPI-stained nuclei. Cell morphology and tight junction integrity were assessed by measuring ZO-1 and OCLN fluorescence intensity at cell junctions, which were processed in Fiji/ImageJ using segmentation-based analysis to determine tight junction localization and disruption. For 3D reconstruction and polarity evaluation, orthogonal views and surface renderings were generated to visualize villin-1 distribution and luminal architecture across compartments.
RT-qPCR validations
Total RNA was extracted from each compartment of the GBV-on-a-chip (gut, vascular, and brain) using the PureLink™ RNA Mini Kit (Thermo Fisher Scientific, USA), followed by on-column DNase treatment using the PureLink™ DNase Set to eliminate genomic DNA contamination. RNA quantity and purity were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized from 500 ng of total RNA using the SuperScript™ IV VILO Master Mix (Thermo Fisher Scientific), according to the manufacturer’s protocol. RT-qPCR was performed using TaqMan™ Fast Advanced Master Mix and TaqMan™ Gene Expression Assays (Thermo Fisher Scientific) specific to human GFAP, ZO-1 (TJP1), OCLN, SYN1, NOS2 (iNOS), and GAPDH (as the housekeeping control). Amplification reactions were conducted on a QuantStudio™ 5 Real-Time PCR System (Thermo Fisher Scientific), with relative gene expression levels calculated using the ΔΔCt method. All reactions were performed in triplicate and normalized to GAPDH expression. Data are presented as fold changes relative to control conditions. All oligonucleotides (including qPCR primers/probes) used in this study are commercially sourced and are listed in Supplementary Table S1 (Key Resources Table) with supplier information and catalog/identifier numbers. For commercial TaqMan assays, the manufacturer’s assay IDs are provided (primer/probe sequences are proprietary). No new plasmids, oligonucleotides, or viral constructs were generated in this study.
Raw fluorescence (Rn) was baseline-corrected by the instrument to yield ΔRn for each cycle. A single quantification threshold was set within the exponential phase for each gene and applied to all samples on the plate; the Ct was the first cycle where ΔRn/ARn crossed this threshold. For each sample, ΔCt = Cttarget − CtGAPDH. The indicated comparator group (CT for gut-to-brain, WT for brain-to-gut) served as the calibrator, and ΔΔCt = ΔCtsample − mean(ΔCtcalibrator). Fold change relative to the calibrator was computed as 2^-ΔΔCt; for presentation as normalized arbitrary units (AU), values were scaled so that the calibrator mean equals 1. When reporting on a log scale, log₂ fold-change = −ΔΔCt. Biological replicate numbers (n = 5) are provided in the figure legends.
Quantification and statistical analysis
All statistical analyses were conducted using SPSS (IBM SPSS Statistics, Armonk, NY, USA) and GraphPad Prism (GraphPad Software, San Diego, CA, USA). For each dataset, we first assessed the normality of distribution using the Shapiro–Wilk test and evaluated the homogeneity of variances using Levene’s test. Based on these assessments, parametric tests such as the independent Student’s t-test (for two-group comparisons) or one-way ANOVA with Tukey’s post hoc test (for three or more groups) were used when data were normally distributed with equal variances. If variances were unequal, Welch’s t-test or Welch’s ANOVA was applied. For non-normally distributed data, non-parametric tests, including the Mann–Whitney U test for two groups or the Kruskal–Wallis test with Dunn’s post hoc correction for multiple groups, were performed. Data are presented as mean ± SEM for plots with error bars; where indicated, 95% confidence intervals (CIs) are reported. Box plots show the median (center line), 25th–75th percentiles (box), and minimum to maximum values (whiskers). Statistical significance was assessed at α = 0.05 (two-tailed); symbols denote p < 0.05, 0.01, 0.001, or 0.0001. Exact statistical tests, sample sizes, and post hoc corrections are described in the respective figure legends and supplementary Table S2. No statistical methods were utilized to pre-determine sample sizes; however, experiments were repeated to improve reproducibility, with n = 5 for biochemical assays and n = 10 for imaging-based quantifications. The data collection and analysis were not blinded to the experimental conditions, and randomization and exclusions were not implemented.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Source Data file and the Supplementary Information. Large raw image datasets are available under restricted access due to file-size constraints; access can be obtained by contacting the corresponding author at h.cho@g.skku.edu with a brief description of the requested dataset and intended use. Requests will be processed within 15 business days, and approved data will be shared for non-commercial research purposes. Source data are provided with this paper.
References
Loh, J. S. et al. Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct. Target. Ther. 9, 37 (2024).
Parker, A., Fonseca, S. & Carding, S. R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 11, 135–157 (2020).
Yadav, H. et al. Unveiling the role of gut-brain axis in regulating neurodegenerative diseases: A comprehensive review. Life Sci. 330, 122022 (2023).
Knox, E. G. et al. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 27, 2659–2673 (2022).
Agirman, G., Yu, K. B. & Hsiao, E. Y. Signaling inflammation across the gut-brain axis. Science 374, 1087–1092 (2021).
Yang, D., Almanzar, N. & Chiu, I. M. The role of cellular and molecular neuroimmune crosstalk in gut immunity. Cell. Mol. Immunol. 20, 1259–1269 (2023).
Ahmed, H. et al. Microbiota-derived metabolites as drivers of gut-brain communication. Gut Microbes 14, 2102878 (2022).
Vogt, N. M. et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 7, 13537 (2017).
Jung, J. H. et al. Gut microbiome alterations in preclinical Alzheimer’s disease. PLoS One 17, e0278276 (2022).
Zhang, F. et al. Altered gut microbiota in Parkinson’s disease patients/healthy spouses and its association with clinical features. Park. Relat. Disord. 81, 84–88 (2020).
Romano, S. et al. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. npj Park. Dis. 7, 27 (2021).
Mou, Y. et al. Gut Microbiota interact with the brain through systemic chronic inflammation: implications on neuroinflammation, neurodegeneration, and aging. Front. Immunol. 13, 796288 (2022).
Bairamian, D. et al. Microbiota in neuroinflammation and synaptic dysfunction: a focus on Alzheimer’s disease. Mol. Neurodegener. 17, 19 (2022).
Warren, A., Nyavor, Y., Zarabian, N., Mahoney, A. & Frame, L. A. The microbiota-gut-brain-immune interface in the pathogenesis of neuroinflammatory diseases: a narrative review of the emerging literature. Front. Immunol. 15, 1365673 (2024).
Nguyen, T. L., Vieira-Silva, S., Liston, A. & Raes, J. How informative is the mouse for human gut microbiota research? DMM Dis. Model. Mech. 8, 1–16 (2015).
Oriá, R. B. et al. Revisiting inbred mouse models to study the developing brain: The potential role of intestinal microbiota. Front. Hum. Neurosci. 12, 358 (2018).
Kim, M. H., van Noort, D., Sung, J. H. & Park, S. Organ-on-a-chip for studying gut-brain interaction mediated by extracellular vesicles in the gut microenvironment. Int. J. Mol. Sci. 22, 13513 (2021).
Ashammakhi, N. et al. Gut-on-a-chip: Current progress and future opportunities. Biomaterials 255, 120196 (2020).
Taavitsainen, S. et al. Gut-on-chip devices as intestinal inflammation models and their future for studying multifactorial diseases. Front. Lab a Chip Technol. 2, 1337945 (2024).
Raimondi, I. et al. Organ-On-A-Chip in vitro models of the brain and the blood-brain barrier and their value to study the microbiota-gut-brain axis in neurodegeneration. Front Bioeng Biotechnol 7, 435 (2020).
Leung, C. M. et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Prim. 2, 33 (2022).
Porter, R. J., Murray, G. I. & McLean, M. H. Current concepts in tumour-derived organoids. Br. J. Cancer 123, 1209–1218 (2020).
Urzì, O. et al. Three-dimensional cell cultures: the bridge between in vitro and in vivo models. Int. J. Mol. Sci. 24, 12046 (2023).
Denman, C. R., Park, S. M. & Jo, J. Gut-brain axis: gut dysbiosis and psychiatric disorders in Alzheimer’s and Parkinson’s disease. Front. Neurosci. 17, 1268419 (2023).
Park, A. M. & Tsunoda, I. Helicobacter pylori infection in the stomach induces neuroinflammation: the potential roles of bacterial outer membrane vesicles in an animal model of Alzheimer’s disease. Inflamm. Regen. 42, 39 (2022).
Guo, S., Al-Sadi, R., Said, H. M. & Ma, T. Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 182, 375–387 (2013).
Jaworowicz, D. J., Korytko, P. J., Lakhman, S. S. & Boje, K. M. K. Nitric oxide and prostaglandin E2 formation parallels blood-brain barrier disruption in an experimental rat model of bacterial meningitis. Brain Res. Bull. 46, 541–546 (1998).
Brown, G. C., Camacho, M. & Williams-Gray, C. H. The Endotoxin Hypothesis of Parkinson’s Disease. Mov. Disord. 38, 1143–1155 (2023).
Brown, G. C. & Heneka, M. T. The endotoxin hypothesis of Alzheimer’s disease. Mol. Neurodegener. 19, 30 (2024).
Tran, V. T. A. et al. Helicobacter pylori-Vacuolating Cytotoxin A Induces Neurotoxic Neuroinflammation in Human Multicellular Models. Biochip J. 19, 556–576 (2025).
Tran, V. T. A. et al. Escherichia Coli K1-colibactin meningitis induces microglial NLRP3/IL-18 exacerbating H3K4me3-synucleinopathy in human inflammatory gut-brain axis. Commun. Biol. 8, 382 (2025).
Mulak, A. & Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 21, 10609–10620 (2015).
Zhang, X., Tang, B. & Guo, J. Parkinson’s disease and gut microbiota: from clinical to mechanistic and therapeutic studies. Transl. Neurodegener. 12, 59 (2023).
Zhang, W., Xiao, D., Mao, Q. & Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 8, 267 (2023).
Janelidze, S. et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer's disease. Neurology 91, e867–e877 (2018).
Gilbert, M. A. G. et al. CryoET of β-amyloid and tau within postmortem Alzheimer’s disease brain. Nature 631, 913–919 (2024).
Irwin, D. J., Lee, V. M. Y. & Trojanowski, J. Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636 (2013).
Pei, Y., Lu, Y., Li, H. Z., Jiang, C. Y. & Wang, L. Gut microbiota and intestinal barrier function in subjects with cognitive impairments: a cross-sectional study. Front. Aging Neurosci. 15, 1174599 (2023).
Kowalski, K. & Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 25, 48–60 (2019).
Guan, J. et al. Vascular degeneration in Parkinsons disease. Brain Pathol 23, 154–164 (2013).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024-00336758); the Korea Dementia Research Project through the Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and the Ministry of Science and ICT, Republic of Korea (RS-2022-KH126808); the Technology Development Program funded by the Ministry of SMEs and Startups (MSS, Republic of Korea) (RS-2023-00224842); and the Fourth Stage of the Brain Korea 21 Project in the Department of Intelligent Precision Healthcare, Sungkyunkwan University (SKKU), awarded to H.C. We also acknowledge MicroTAS 2024 for supporting this project through the CBMS Student/Young Researcher Grant and the Biomicrofluidics Best Paper Award (M.T.).
Author information
Authors and Affiliations
Contributions
M.T. developed the microfluidic device, designed and performed the experiments, analyzed the data, and wrote the manuscript. H.W.J. designed and developed the microfluidic device. C.B. conducted experiments using the fabricated microfluidic devices, while A.J. performed experiments related to Alzheimer’s disease (AD) modeling, and M.J.A. conducted experiments on Parkinson’s disease (PD) modeling. S.M.K. fabricated SU-8 molds for the microfluidic platforms. L.P.L. provided insightful feedback and contributed to the overall supervision of the project. H.C. conceived, initiated, provided insightful feedback, and supervised the entire study.
Corresponding authors
Ethics declarations
Competing interests
M.T. and H.C. are inventors on a patent application related to the gut–brain–vascular microphysiological platform described in this study (filed by Sungkyunkwan University). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Marco Campisi, Inhee Mook-Jung, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tran, M., Jeong, H.W., An, M. et al. A 3D gut-brain-vascular platform for bidirectional crosstalk in gut-neuropathogenesis. Nat Commun 17, 2504 (2026). https://doi.org/10.1038/s41467-026-69318-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69318-y
