
Abstract
Sequencing-based spatial transcriptomics (sST) enables transcriptome-wide gene expression mapping but falls short of reaching the optical resolution (200–300 nm) of imaging-based methods. Here, we present Seq-Scope-X (Seq-Scope-eXpanded), which empowers submicrometer-resolution Seq-Scope with tissue expansion to surpass this limitation. By physically enlarging tissues, Seq-Scope-X minimizes transcript diffusion effects and increases spatial feature density by an additional order of magnitude. In liver tissue, this approach resolves nuclear and cytoplasmic compartments in nearly every single cell, uncovering widespread differences between nuclear and cytoplasmic transcriptome patterns. Independently confirmed by imaging-based methods, these results suggest that individual hepatocytes can dynamically switch their metabolic roles. Seq-Scope-X also works in brain and colon, and can be adapted for spatial proteomics, profiling hundreds of barcode-tagged antibody stains at microscopic resolutions in mouse spleens and human tonsils. Together, these findings establish Seq-Scope-X as a powerful platform for ultra-high-resolution whole-transcriptome and proteome profiling, expanding the spatial precision achievable for studying cellular architecture, function, and disease mechanisms.
Similar content being viewed by others
Introduction
Spatial transcriptomics (ST) has revolutionized our ability to interrogate gene expression within intact tissues, enabling studies of cellular organization, function, and interactions in situ1. By mapping the transcriptome directly onto tissue sections, ST reveals intricate spatial patterns critical for understanding complex processes such as development, homeostasis, and disease pathology1,2,3,4,5.
Among various ST techniques, sequencing-based approaches (sST)4 combine spatially coordinated nucleotide barcodes with Next-Generation Sequencing (NGS) to resolve transcript locations. These techniques, including the original ST6, 10x Visium7, Slide-Seq8,9, DBiT-seq10, and HDST11, offer genome-wide coverage and have progressively achieved higher spatial resolution over time. However, even the latest sST platforms—such as Seq-Scope12, Stereo-Seq13, Pixel-Seq14, as well as Seq-Scope’s recent variations (e.g., including Open-ST and Nova-ST)15,16,17—struggle to reach the resolution of optical microscopy (~ 200 nm). The spatial barcode spacing is inherently constrained to ~ 0.5 µm by the procedures underlying NGS-based spatial barcode sequencing. In addition, RNA diffusion during tissue permeabilization and capture further exacerbates this limitation, reducing the effective resolution to approximately 1–3 µm1,2,3.
In contrast, imaging-based ST (iST)4 methods—such as MERFISH18, CosMx19, and Xenium20—achieve resolutions as fine as ~ 200 nm by optically detecting fluorescently labeled RNAs through sequential hybridization or in situ sequencing. Tissue expansion techniques have further advanced iST resolution into the nanoscale regime21,22. However, with the exception of ExSeq21, which supports both unbiased and targeted analyses, iST methods are generally constrained to predefined gene sets, limiting their ability to comprehensively capture global transcriptomic complexity, including splicing variants and somatic mutations. Scaling iST methods to encompass global transcriptomic complexity becomes prohibitively expensive due to the need for unique imaging probes. Moreover, while sST methods are time-efficient, completing experiments in a single measurement, iST methods are time-intensive, requiring repeated imaging–erasing cycles to detect multiple targets. These iterative cycles not only limit scalability and practicality4,5, but also introduce cumulative errors—such as photobleaching, incomplete probe removal, and imaging artifacts—that compromise precision and diminish the resolution advantage4,5. Consequently, a critical gap persists: no existing platform currently offers whole-transcriptome coverage at a reasonable time and cost while achieving spatial resolution at or beyond the diffraction limit of optical microscopy.
Here, we introduce Seq-Scope-X (Seq-Scope-eXpanded), a platform that delivers super-resolution spatial analysis—surpassing the diffraction limit of optical microscopy—while preserving the whole-transcriptome coverage and cost-effectiveness of the original Seq-Scope. Inspired by tissue expansion–based approaches in iST21,22 and their integration into lower-resolution sST workflows23, Seq-Scope-X synergistically incorporates tissue expansion into Seq-Scope’s submicrometer-resolution capabilities. This innovation increases spatial feature density by an additional order of magnitude, generating tens of millions of hexagonally arranged coordinate points per square millimeter. Furthermore, proportional tissue expansion reduces distortion and limits RNA diffusion, enhancing spatial precision beyond what was achieved by existing variations of Seq-Scope15,16,17.
In addition to providing improved resolution, Seq-Scope-X facilitates high-resolution multi-omics profiling by decoupling molecular probe staining from spatial capture. We demonstrate that Seq-Scope-X supports massively multiplexed proteomics analysis using hundreds of barcode-tagged antibodies, while maintaining the same resolution and throughput as its transcriptomic assays. By bridging the gap between comprehensive NGS-based profiling and the nanoscale precision of imaging-based methods, this integrated approach paves the way for deeper insights into cellular architecture, subcellular heterogeneity, and disease mechanisms across diverse tissues.
Results
Seq-Scope-X Workflow
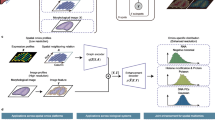
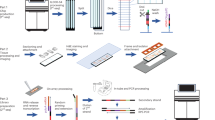
The Seq-Scope-X protocol builds on two recent advances in spatial transcriptomics: the tissue expansion method for enhanced resolution24 and the submicrometer-resolution Seq-Scope platform12,15. By integrating these approaches, Seq-Scope-X achieves sub-200 nm spatial transcriptomics, surpassing the diffraction limit of optical microscopy. Frozen tissue sections were fixed (Fig. 1a), permeabilized, and hybridized with locked nucleic acid (LNA)-containing oligo-dT probes labeled with a red fluorescent dye and an acrydite motif (Fig. 1b). The hybridized tissue was embedded in a polyacrylate-containing expandable hydrogel, incorporating the acrydite-bound oligo-dT and associated mRNA species into the polymer matrix (Fig. 1c). After embedding, the tissue was digested and cleared with proteinase K, and the gel was expanded by placing it in a hypotonic buffer. The expanded gel, now containing spatially preserved mRNAs, was overlaid onto the Seq-Scope’s spatially barcoded array (Chip), which was produced using a NovaSeq 6000 S4 flow cell15. To facilitate efficient transcript transfer, the gel-array assembly was incubated at 45 °C. At this temperature, the 15-mer LNA-modified oligo-dT probes in the gel, with a melting temperature of about 39 °C, release their hybridized mRNAs. In contrast, the 30-mer oligo-dT probes on the array, with a melting temperature above 50 °C, capture the released transcripts and maintain stable hybridization23. This temperature differential enables directional release of transcripts from the gel and their efficient capture by the array probes (Fig. 1d). Finally, the Seq-Scope Chip, with mRNAs transferred from the gel, was processed into spatially labeled cDNA libraries using the conventional Seq-Scope library prep method12,15.

a–d Schematic diagram depicting the Seq-Scope-X procedure. The tissue (a) was hybridized with ATTO590-labeled (*) oligo-dT acrydite probes (b). Then it was embedded in a polyacrylate-containing expandable hydrogel (c), which was expanded after tissue digestion. (d) The mRNAs were then transferred to Seq-Scope through a heat-mediated hybridization partner switching. e, f Comparison of spatial performance across technologies. The x-axis indicates release or publication date, and the y-axis shows resolution (e) or spatial pixel density (f). Red trendlines indicate overall improvement trends. g–j Comparison of conventional Seq-Scope (g, i) and Seq-Scope-X (h, j) data visualized at the same scale of Seq-Scope–analyzed (g, h) or original (i, j) tissue. Magenta and green dots indicate spliced and unspliced transcripts, respectively. Yellow boxes were magnified in insets. White dotted boxes were magnified in (i, j). k–m Digitally-enhanced images of spliced (magenta) and unspliced (green) transcripts (k, m) are overlaid with nuclei (blue; l, m) identified by CellProfiler-based segmentation of DAPI-stained images. n, o Side-by-side comparison of Seq-Scope-X (n) and conventional Seq-Scope (o) data in Glul + Slc1a2 expression, which mark centrilobular (CL) hepatocytes. Cell boundaries were estimated using an accompanying histology image (conventional Seq-Scope) or RNA aggregation patterns (Seq-Scope-X), as described in Supplementary Fig. 2l–o. p For each segmented cell, the area was stratified into 8 radial bins from the nuclear center. Relative enrichment of unspliced RNA over the whole-cell baseline (set to 1) is shown as box plots. Conventional Seq-Scope data (red, n = 1694) and Seq-Scope-X data (blue, n = 6570), which have ≥ 5 unspliced RNAs in each bin, were compared side by side. A two-sided two-way ANOVA with method and radial interval as factors showed a significant interaction (F(7, 66,096) = 572.14, P < 2.2 × 10−16). (q) Comparison of different technologies on their transcript capture efficiency. UMI, a unique molecular identifier. Scale bars represent the scale corresponding to the original tissue. Box plots show median (center line), interquartile range (box), and whiskers extending to 1.5 × IQR; outliers are not shown. Source data are provided as a Source Data file.
Seq-Scope-X Tissue Expansion Maintains Structural Integrity and Uniformity
We first verified that tissue expansion in our workflow preserves overall structural integrity, which is essential for accurate spatial transcriptomics. Across experimental batches, the expansion factor consistently ranged between 2 and 3 and was reproducibly determined by measuring gel dimensions before and after expansion. DAPI-labeled nuclei remained stably anchored within the hydrogel, indicating minimal diffusion of nuclear material. The tissue expanded uniformly across the section, maintaining nuclear positions and proportional nuclear areas (Supplementary Fig. 1a, b). Computational dilation of pre-expansion images showed that nuclear positions before and after expansion were nearly identical across most regions (Supplementary Fig. 1a, b).
Quantitative analyses, including displacement vector maps, anisotropy heatmaps, and root mean square (RMS) distortion measurements across tissues (Supplementary Fig. 1c–e), further showed that expansion was largely isotropic. In the liver and the brain, RMS distortion was less than 1% of the distance, corresponding to greater than 99% spatial fidelity. In the colon and spleen, distortion remained below 5% of the distance, corresponding to greater than 95% fidelity, although somewhat higher than in the liver and brain. This modest increase may reflect their dense and compositionally heterogeneous tissue layers, which may impose uneven in-plane resistance during expansion and may also affect polymer penetration or tissue clearing. Additional optimization may further reduce distortion in these tissues.
Finally, red fluorescence signals from the oligo-dT acrydite probe remained enriched near DAPI-labeled nuclei after expansion (Supplementary Fig. 1f), consistent with the high concentration of mRNA in perinuclear regions where translation is active.
Seq-Scope-X achieves super-resolution spatial transcriptomics
The latest version of Seq-Scope and its variations achieve a resolution of 0.6 µm in a hexagonally arranged pixel array15,16,17, with a pixel density of approximately 3 million per mm². However, when combined with the 3-fold tissue expansion procedure in Seq-Scope-X, the resolution is effectively improved to 0.2 µm, and the pixel density is increased to approximately 27 million per mm². This represents a substantial enhancement over existing sequencing-based spatial transcriptomics (sST) technologies in both resolution (Fig. 1e) and spatial feature density (Fig. 1f), enabling a much more detailed view of the tissue transcriptome, particularly in densely packed or subcellular regions.
To evaluate whether Seq-Scope-X maintains its high-resolution utility in practical tissue analyses, we applied it to liver tissue—a model well-suited for cross-dataset comparisons due to its repeating parenchymal units and well-defined metabolic zonation layer structure. Liver tissue minimizes common challenges in benchmarking procedures such as variability in tissue depth, orientation, and cell type composition. We directly compared Seq-Scope-X liver data with datasets generated using the original Seq-Scope15. Seq-Scope-X consistently produced the expected spatial readout with markedly improved resolution and throughput. In the original Seq-Scope data, spliced (magenta) and unspliced (green) transcripts allowed visualization of single cell-level hepatocellular textures, but cell boundaries were often diffuse, and subcellular details were ambiguous (Fig. 1g). Seq-Scope-X, in contrast, clearly resolved individual cells, including transcript-depleted extracellular domains, and identified nuclear-like clusters of unspliced transcripts centered within hepatocytes (Fig. 1h). When both datasets were viewed at the same scale corresponding to the original tissue, conventional Seq-Scope exhibited blurred cell boundaries and only occasionally captured nuclear-cytoplasmic features in large cells (Fig. 1i). Seq-Scope-X, however, identified nearly every nucleus as a compact cluster of unspliced transcripts surrounded by the corresponding cytoplasmic spliced transcripts (Fig. 1j). These unspliced transcript clusters showed strong concordance with DAPI-marked nuclei (Fig. 1k–m), demonstrating the microscopic precision of Seq-Scope-X.
Such high resolution and precision also enabled clear cell segmentation based on RNA aggregates distributed around each nucleus. Spatial mapping of Glul and Slc1a2, which mark centrilobular hepatocytes and are confined to only one or two hepatocytes adjacent to the central vein, showed sharply defined cell boundaries: Glul/Slc1a2-high centrilobular hepatocytes were distinctly separated from neighboring hepatocytes (Fig. 1n). These sharp boundaries were not observed in conventional Seq-Scope data, where Glul/Slc1a2 signals appeared diffused across histology-defined cell borders15 (Fig. 1o). These findings indicate that the high resolution and precision of Seq-Scope-X is valuable not only for subcellular analyses but also for improving cell segmentation accuracy.
We also performed a quantitative analysis of subcellular transcript distribution. For each cell, radial distance bins from nuclear centers were defined, and the relative concentration of unspliced transcripts was measured. In both datasets, regions within 2.5 µm of the nuclear center were enriched for unspliced transcripts, consistent with the expected nuclear area. However, the magnitude of enrichment was substantially higher in Seq-Scope-X compared to conventional Seq-Scope (Fig. 1p). These findings confirm that Seq-Scope-X achieves its advanced resolution in experimental applications, enabling the visualization of cellular and subcellular transcriptomic structures with precision across the tissue section.
Seq-Scope-X retains high RNA capture efficiency
While Seq-Scope-X’s resolution was clearly enhanced, another important consideration is whether it provides sufficient transcriptome coverage comparable to that of the original Seq-Scope and other ST methodologies. To address this, we analyzed seven publicly available ST datasets, all prepared from C57BL/6 WT mouse liver, including those from the original ST25, 10x Visium26, Slide-Seq8, xDBiT (an improved version of DBiT-Seq)27, Seq-ScopeMISEQ (conventional Seq-Scope implemented with Illumina MISEQ)12, Stereo-Seq28, and Seq-ScopeNOVASEQ (conventional Seq-Scope implemented with Illumina NOVASEQ600015, similar to Open-ST16 and Nova-ST17), alongside the Seq-Scope-X data. Because these datasets differ markedly in spatial resolution (Fig. 1e, f), we processed the Seq-ScopeNOVASEQ and Seq-Scope-X datasets by generating hexagonal or square spatial grids whose effective spatial unit sizes matched those of the comparison datasets (Supplementary Fig. 2a). In an area-normalized evaluation of transcript capture efficiency, Seq-Scope-X showed strong performance per µm² (Fig. 1q), comparable to most other technologies and only slightly less than the values observed for Stereo-Seq and Seq-ScopeNOVASEQ (Fig. 1q and Supplementary Fig. 2b–k).
We also compared transcript capture efficiency on a per-cell basis using Seq-ScopeNOVASEQ data15, the only other dataset with available cell segmentation and with procedures identical to Seq-Scope-X except for the expansion step. The segmented cell areas, adjusted for XY expansion, were similar between the two datasets (Supplementary Fig. 2l–n). Consistent with the area-normalized results (Fig. 1q and Supplementary Fig. 2b), Seq-Scope-X showed lower per-cell capture efficiency (1489 ± 953.6 UMIs per cell) than Seq-ScopeNOVASEQ (Supplementary Fig. 2o; 3110 ± 3281 UMIs per cell). Because expansion increases both XY and Z dimensions, a 3-fold expansion would be expected to reduce per-cell capture to one-third due to transcript dilution, yet we observed only about a one-half reduction, indicating a modest dilution effect. These results indicate that Seq-Scope-X maintains high capture efficiency while operating at much higher spatial resolution.
Seq-Scope-X matches bulk RNA-seq accuracy
The transcriptome content of Seq-Scope-X was highly similar to bulk RNA-seq29, conventional Seq-Scope12,15, and other ST techniques8,26,27,28, with correlation coefficients of approximately 0.9 in most pairwise comparisons (Supplementary Fig. 3a, b). High concordance with bulk RNA-seq and with most other ST datasets was retained regardless of whether the intersection or union of genes were used to evaluate the correlation between the datasets (Supplementary Fig. 3c–g; correlation over 0.7). These results indicate that Seq-Scope-X preserves transcriptome accuracy comparable to both its predecessor and other spatial transcriptomics platforms.
Seq-Scope-X reveals nuclear-cytoplasmic transcriptome disparities
Using Seq-Scope-X, we performed subcellular-level analysis by leveraging the distribution of spliced and unspliced transcripts, which represent cell boundary and nuclear positions, respectively (Fig. 1j–m). Using this image, the Watershed cell segmentation algorithm divided the spatial area into single cells and labeled the local maxima of unspliced transcripts in each segment. This information was then used to define nuclei and cytoplasm (Fig. 2a). Each nuclear and cytoplasmic area pair constituted a single cell area (Fig. 2b), and was subjected to a cell type clustering analysis (Fig. 2c, d).

a–h Segmentation-based nuclear-cytoplasmic analysis. Number of segments analyzed: n = 18,448. Boxed regions are progressively magnified. a Gaussian-filtered spliced (red), unspliced (green), and mitochondrial (blue) transcripts segmented into nuclear and cytoplasmic regions using a Watershed algorithm. b Nuclear (gray) and cytoplasmic (white) segments. c Segments colored by hepatocyte zonation type: centrilobular (CL), pericentral (PC), central-side midzone (CM), portal-side midzone (PM), and periportal (PP). d–f UMAP of nuclear and cytoplasmic segments colored by cell type (d) compartment (e) or transcript count (f). g Nuclear–cytoplasmic pairs from single cells connected in UMAP space. h Heatmap showing mismatches between nuclear and cytoplasmic cell types across neighboring zonation clusters. i Segmentation-free FICTURE analysis of Seq-Scope-X liver data showing pixel-level cell type factors overlaid with nuclei. Boxed areas are magnified on the right. Arrows indicate nuclear–cytoplasmic mismatches. j FICTURE analysis of MERSCOPE liver data31 showing similar nuclear–cytoplasmic differences. Boxed areas are magnified on the right. Arrows indicate nuclear–cytoplasmic mismatches. k, l smFISH images of zonation markers with F-actin–defined cell boundaries, illustrating nuclear or cytoplasmic localization. Boxed areas are magnified on the bottom. Reproduced with permission from © 2017 Springer Nature32. All rights reserved. Scale bars represent the scale corresponding to the original tissue.
Both nuclear and cytoplasmic transcriptomes are grouped into five well-established hepatocellular zonation types: periportal (PP), portal-side midzone (PM), central-side midzone (CM), pericentral (PC), and centrilobular (CL) hepatocytes (Fig. 2d). These assignments reflect known portal–central zonation patterns supported by canonical marker gradients. Nuclear and cytoplasmic profiles were largely concordant within each zonal group (Fig. 2e), although they differed in nuclear- and mitochondrial-enriched transcripts (Supplementary Fig. 4a), which produced a consistent relative shift within the UMAP visualization (Fig. 2e). In addition, cytoplasmic clusters generally exhibited a higher number of unique transcripts compared to nuclear clusters (Fig. 2f and Supplementary Fig. 4b).
In the histological space, five hepatocellular types were arranged nicely, corresponding to the portal-central axis of liver metabolic zonation, forming a clearly layered structure (Fig. 2c and Supplementary Fig. 4c). Interestingly, while nuclear and cytoplasmic clusters were concordant in most cases, about one third of cells exhibited differing transcriptomic phenotypes between the subcellular compartments (Fig. 2c and Supplementary Fig. 4c; magnified images). These disparities were predominantly observed between clusters of two neighboring zones (Fig. 2c, g, h and Supplementary Fig. 4c), which represented relatively similar clusters.
From these results, we speculate that the cytoplasmic transcriptome reflects the more immediate functional state of each cell, whereas the nuclear transcriptome may capture transcripts undergoing processing or awaiting export. This distinction suggests that nuclear and cytoplasmic compartments highlight different facets of hepatocyte biology, including potential temporal aspects of zonal function. Although the study was performed during daytime, when hepatocyte metabolism is generally stable, we nonetheless observed heterogeneous nuclear-cytoplasmic distributions of several established zonation markers, indicating substantial subcellular diversity within and across hepatocyte layers.
Segmentation-Free Analysis Aligns with Nuclear-Cytoplasmic Transcriptome Differences
To validate that our observed subcellular differences were not artifacts of imperfect segmentation or contamination from neighboring cells, we employed FICTURE, a segmentation-free, pixel-level cell type inference method30. Using FICTURE, we projected the five hepatocellular clusters (PP, PM, CM, PC, and CL) into pixel-level histological space. Although the analysis revealed a clearly layered structure along the portal-central axis as seen from the segmentation analysis, it also presented rough boundaries and scattered fragments between layers (Fig. 2i, left). When we overlaid the FICTURE factor map with DAPI-marked nuclei, we found that these fragmented clusters aligned closely with nuclear regions where the transcriptome profile differed from that of the surrounding cytoplasm (Fig. 2i, right). Both our subcellular segmentation analysis (Fig. 2a–h) and segmentation-free FICTURE analysis (Fig. 2i) of the Seq-Scope-X dataset congruently confirm the presence of subcellular transcriptome heterogeneity, specifically demonstrating distinct nuclear and cytoplasmic transcriptome characteristics within individual cells.
In situ imaging confirms subcellular transcriptome heterogeneity
To independently validate the observed subcellular transcriptome heterogeneity, particularly the disparity between nuclear and cytoplasmic phenotypes, we analyzed publicly available datasets from imaging-based spatial transcriptomics (iST) techniques with comparable resolution to Seq-Scope-X (~ 200 nm). Using MERFISH mouse liver data, an iST dataset with an optical resolution (200–300 nm)31, we applied segmentation-free FICTURE analysis30 to evaluate whether this method could detect similar disparities. Consistent with our Seq-Scope-X findings, MERFISH data revealed distinct hepatocellular phenotypes of metabolic zonation between nuclear and cytoplasmic compartments within the same cell, again observed widely across the dataset (Fig. 2j).
To further assess subcellular differences, we re-examined single-molecule RNA fluorescence in situ hybridization (smFISH) data reported in the literature32. Markers for the central-most CL (Glul, Fig. 2k) and portal-most PP (Ass1, Fig. 2l) demonstrated distinct subcellular transcript localization in single cells. In many cases, transcripts were observed exclusively in either the nucleus (lower left panels) or the cytoplasm (lower right panels), confirming pronounced subcellular differences at the single-transcript level. These findings not only corroborate the observations from Seq-Scope-X but also suggest that hepatocellular phenotypes could dynamically change, with nuclear and cytoplasmic transcriptomes reflecting different functional states. This underscores the robustness of subcellular transcriptome heterogeneity revealed through Seq-Scope-X and its relevance across diverse orthogonal techniques.
Seq-scope-X delivers spatial precision for cell- and tissue-level analysis
In addition to enabling subcellular analysis, the higher precision of Seq-Scope-X compared to conventional Seq-Scope could also enhance the spatial precision of cell- and tissue-level analysis. Using FICTURE, which we previously applied to detect nuclear–cytoplasmic transcriptome disparities, we evaluated how Seq-Scope-X data and conventional Seq-Scope data (the Seq-ScopeNOVASEQ data described above) perform in resolving the overall tissue organization of metabolic zonation. Even though Seq-Scope-X and conventional Seq-Scope data both were able to reveal five major hepatocellular clusters (PP, PM, CM, PC, and CL) and non-parenchymal (NP) cell clusters in hexagonal binning analysis (Fig. 3a, d), and generally produced similar clustering patterns with similar size of hexagonal bins (Supplementary Fig. 5), FICTURE-mediated projection of these cell types into pixel-level histological space produced far clearer qualitative differences in spatial details (Fig. 3b, c, e, f). For instance, conventional Seq-Scope data showed overlapping or discontinuous layer structures (Fig. 3e, f), which were much more clearly defined in the Seq-Scope-X data (Fig. 3b, c). Subcellular level details shown in Seq-Scope-X data (Figs. 2i, 3c) were also not attainable by conventional Seq-Scope data (Fig. 3f). Gene plotting analysis also clearly demonstrated distinct domains of cluster-specific marker expression in the Seq-Scope-X data (Fig. 3g), whereas the conventional Seq-Scope data exhibited a continuously diffused pattern that obscured the detailed domain architecture (Fig. 3h). These findings underscore Seq-Scope-X’s superior resolution and spatial precision, enabling a detailed and accurate understanding of tissue architecture.

a, d Cell type clustering results from Seq-Scope-X (a) and conventional Seq-Scope (d) datasets. Both datasets were processed through 14 μm-sided hexagons (corresponding to the array scale, not accounting for expansion). Both results identified cell types corresponding to Periportal (PP), Portal- and Central-Midzones (PM and CM), Pericentral (PC), Centrilobular (CL) hepatocytes, and non-parenchymal (NP) cell types. The results are presented in UMAP manifolds. For analyses using additional hexagonal sizes, refer to Supplementary Fig. 5. Number of hexagons analyzed: n = 55,191 (a), 23,564 (d). b, c, e–h Spatial plot of cell type factors (b, c, e, f) and individual gene expressions (g, h) using the Seq-Scope-X (b, c, g) and conventional Seq-Scope (e, f, h) datasets. The spatial cell type factor maps (b, c, e, f) were constructed using cell type clusters (a, d) through segmentation-free FICTURE projection. Individual factors were projected in a spatial heat map, indicating the probability of each factor (b, e). Individual dots in the spatial gene expression plot (g, h) represent the digital gene expression of the indicated genes in the same color. Scale bars represent the scale corresponding to the original tissue.
Because both conventional Seq-Scope and Seq-Scope-X datasets were generated from mouse liver under closely matched experimental conditions, we examined whether they remained congruent when analyzed within an identical analytical framework. To do this, we binned both datasets using hexagonal grids corresponding to 12 to 24 µm hexagonal flat-to-flat width (according to the original tissue dimension), which produced stable cell type mapping in both platforms (Supplementary Fig. 5). We then integrated the two datasets for combined analysis. Across all comparisons, conventional Seq-Scope and Seq-Scope-X yielded similar UMAP structures (Supplementary Fig. 6a, e, i) and cluster geometries (Supplementary Fig. 6b, f, j), as well as comparable cell type arrangements (Supplementary Fig. 6c, g, k) and compositions (Supplementary Fig. 6d, h, l). Despite these broad similarities, Seq-Scope-X consistently produced spatial maps with clearer layer organization and reduced noise, even at these lower resolution analyses (insets in Supplementary Fig. 6c, g, k). Together, these results demonstrate that Seq-Scope-X retains full compatibility with conventional Seq-Scope while providing substantially improved spatial precision.
Seq-Scope-X performs high-resolution spatial mapping of brain and colon transcriptomes
To evaluate the broad applicability of Seq-Scope-X, we analyzed brain and colon tissues, which are frequently studied using ST technologies. In brain tissue, Seq-Scope-X effectively captured cellular heterogeneity while maintaining high spatial resolution. The spatial distribution of spliced and unspliced transcripts aligned well with DAPI-marked nuclei from the same section (Fig. 4a), validating the spatial precision and subcellular capability of the technique. Cell type clustering successfully identified multiple neuronal populations, including inhibitory GABAergic neurons (Gad1, Gad2, Gabra1, Sst), excitatory neurons (Camk2a, Ppp3ca, Slc1a2), and glutamatergic neurons (Slc17a6, Rora, Zic1; Supplementary Fig. 7a, b). Additional populations such as Foxp2⁺ neurons, granular cells, pyramidal neurons, and non-neuronal cells (astrocytes, oligodendrocytes, choroid plexus cells) were also clearly distinguished. Segmentation-free FICTURE analysis provided high-resolution spatial mapping of these cell types (Supplementary Fig. 7c, d).

a Spatial plotting of spliced (red), unspliced (green), and mitochondrial (blue) transcripts processed with a Gaussian filter in mouse brain tissue profiled using Seq-Scope-X. The transcript signals are shown together with nuclei (white, right) identified by Cellpose-based segmentation of DAPI-stained images from the same expanded tissue section. Insets show magnified views of the regions indicated by colored boxes. b, c Spatial gene expression plots comparing cell type- or region-specific gene expression in conventional Seq-Scope (left) and Seq-Scope-X (right) datasets. Mouse brain samples are shown in (b), and colon samples in (c). For a more detailed analysis of the Seq-Scope-X data, refer to Supplementary Fig. 7. For the list of genes used for the plotting, see the Methods section. Scale bars represent the scale corresponding to the original tissue.
Similarly, Seq-Scope-X analysis of colon tissue revealed detailed cellular organization along the surface-crypt axis (Supplementary Fig. 7e–i). The technique distinguished between crypt-localized (type I, deep crypt secretory cells or DCSC) and surface-localized (type II) goblet cell populations, marked by differential expression of Zg16/Fcgbp and Muc2/Cryab/Agr2, respectively. Other identified populations included colonocytes, Paneth/stem cells, immune cells, smooth muscle cells, fibroblasts, and macrophages. Nuclear transcriptome signatures characterized by Malat1 and Neat1 expression were also detected (Supplementary Fig. 7f), again confirming the capabilities of detecting subcellular transcriptome structures.
Side-by-side comparison of cell type- and region-specific markers between Seq-Scope-X and conventional Seq-Scope demonstrated that Seq-Scope-X data are consistent with previous datasets while enabling massively magnified spatial analysis (Fig. 4b, c). To further confirm that conventional Seq-Scope and Seq-Scope-X datasets are congruent, we combined both datasets using grids of identical size (24 µm hexagonal flat-to-flat width, based on the original tissue dimensions) and performed an integrated analysis. In both brain (Supplementary Fig. 8a–d) and colon (Supplementary Fig. 8e–h), the two platforms produced highly similar UMAP structures (Supplementary Fig. 8a, e), cluster geometries (Supplementary Fig. 8b, f), spatial arrangements (Supplementary Fig. 8c, g), and cell type compositions (Supplementary Fig. 8d, h), confirming strong concordance between the datasets. These results establish Seq-Scope-X as a reliable tool for high-resolution spatial transcriptomics across diverse tissue types.
Seq-Scope-X profiles mouse splenic proteome using barcode-tagged antibodies
Previously, we attempted to capture spatial proteomic information in conventional Seq-Scope using poly-A–labeled barcoded antibodies, a technique originally developed for single-cell CITE-seq33,34 and recently implemented in several low-resolution sST platforms35,36,37. The workflow involved attaching tissue to the Seq-Scope Chip, performing barcoded antibody staining, and then releasing the antibody-attached barcode oligonucleotides for poly-A–based capture. However, despite extensive optimization efforts, generating high-resolution proteomic maps proved difficult due to multiple technical challenges. The primary obstacle was the difficulty in optimizing tissue permeabilization conditions to efficiently release antibody-tagged barcode sequences while maintaining spatial tissue integrity. Moreover, the method suffered from high background noise and non-specific signals, likely caused by premature capture of antibody-tagged poly-A tails by the Seq-Scope Chip during the staining process.
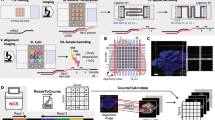
Given Seq-Scope-X’s success in transcriptomic applications, we hypothesized it could overcome the proteomic challenges faced by the conventional Seq-Scope. The key advantages of Seq-Scope-X were twofold: it separated the antibody staining step from poly-A capture, and it eliminated the need for optimizing tissue permeabilization by stably anchoring poly-A to the expansion hydrogel during tissue digestion. To test this approach, we used a mouse spleen tissue and a panel of 119 antibodies targeting distinct mouse cell-surface antigens associated with various immune lineages. Each antibody was labeled with an oligonucleotide containing a unique antibody-derived barcode tag (ADT) and a poly(dA) tail38. After staining mouse spleen tissue sections with this antibody cocktail, we subjected the stained slides to the Seq-Scope-X procedure (Fig. 5a).

a Schematic of the Seq-Scope-X procedure modified for proteome analysis. Tissues were stained with oligonucleotide barcode-tagged antibodies (AbTags) before undergoing the standard Seq-Scope-X procedure (Fig. 1a). b–f Seq-Scope-X analysis of mouse spleen stained with an AbTag cocktail targeting 119 unique cell surface antigens, including principal lineage markers, plus 9 isotype controls (total 128 AbTags). b Fluorescence images of DAPI (blue) and oligo-dT probe (red) post-tissue expansion. The magnified pink boxed area reveals preserved nuclear-membrane structures. The image spatially corresponds to the digitally reconstructed Seq-Scope-X image shown in (c). c Spatial plot of antibody tags (red) detected in Seq-Scope-X data. The magnified pink boxed area demonstrates structural similarities with (b) but appears more diffused. Boxed areas in white, yellow, and pink are further analyzed in (d, e), and (c, right panel), respectively. d Representative spatial expression plot of antibody tags targeting specific proteins within the white boxed region in (c). For spatial expression plots of all 128 antibody tags, refer to Supplementary Fig. 9a. e, f FICTURE projection of spatial factors identified via LDA analysis of protein expression results. Twelve distinct factors are annotated and color-coded. The yellow box in (c) is magnified in (e). The blue box in (e) is further magnified in (f). In (f), the positional probability of selected LDA factors was presented in spatial heat maps. For spatial heat maps of all 12 LDA factors for a wider area, refer to Supplementary Fig. 9b. g, h Seq-Scope-X analysis of human tonsil stained with an AbTag cocktail targeting 154 unique cell surface antigens, including principal lineage markers, plus 9 isotype controls. g FICTURE projection of spatial factors identified via LDA analysis of protein expression results (left). Twelve factors are annotated and color-coded (right). h Spatial heat maps for the top two factors (CD4 + T cells and Germinal Center B cells) demonstrate the positional probability of each factor. For spatial heat maps of all 12 LDA factors, refer to Supplementary Fig. 10b. Scale bars represent the scale corresponding to the original tissue.
Fluorescence imaging of the expanded gel revealed that oligo-dT probes—which hybridize with poly(dA) and thus highlight ADT locations—were mostly localized around the cell membranes surrounding DAPI-stained nuclei (Fig. 5b). Notably, although the nuclear pattern was clearly visible in fluorescence imaging (Fig. 5b, right panel), it was less distinct in the Seq-Scope-X data (Fig. 5c, right panel). This discrepancy likely results from physical differences between the fluorescence imaging plane (in the middle of the gel) and the ADT capture interface (at the gel–array contact). Subtle subcellular details may have been obscured by diffusion of ADT molecules and their subsequent capture by spatial barcode molecules on the Seq-Scope Chip.
Nevertheless, mapping individual ADTs revealed highly localized expression patterns (Fig. 5d and Supplementary Fig. 9a), indicating that Seq-Scope-X successfully captured spatial proteomic information with intact positional fidelity and a broad dynamic range. Using Latent Dirichlet Allocation (LDA)-based FICTURE analysis, we identified 12 distinct spatial factors representing diverse immune cell types and states (Fig. 5e and Supplementary Fig. 9b). These factors included multiple T- and B-lymphocyte populations and myeloid-lineage cells (Fig. 5f and Supplementary Fig. 9c). T and B cell zones were clearly separated, and within the T cell areas, CD4⁺ and CD8⁺ T cells were distinguishable, mirroring their tight intermingling observed in multispectral imaging39. While single-cell CITE-seq (scCITE-seq)33,34,38 achieves similar molecular resolution in distinguishing these populations, it lacks spatial context. Conversely, current spatial proteomics platforms such as SM-Omics35, SPOTS36 and spatial CITE-seq37 provide coarse spatial information but lack the resolution to reliably distinguish closely associated cell populations at the tissue level. We also observed rich diversity in the B cell compartment, with distinct cell populations expressing IgM, CD20, CD21, and CD79b, as well as antibody staining patterns indicating myeloid cells such as dendritic cells and macrophages (Supplementary Fig. 9c).
These results establish that Seq-Scope-X exhibits both the single cell-level molecular precision of scCITE-seq and substantially higher spatial resolution than existing spatial proteomics technologies.
Antibody detection in Seq-Scope-X proteomics workflow is sensitive and Precise
To verify the precision of Seq-Scope-X proteome analysis, we performed additional validation by subjecting serial sections of the same mouse spleen to Seq-Scope-X proteomic profiling and to immunofluorescence (IF) confocal microscopy targeting a subset of antigens included in the proteomic assay. IF-confocal microscopy is considered the gold standard for spatial protein detection. IF-confocal imaging of CD4 and CD8b, which mark helper and cytotoxic T cell populations, produced spatial patterns that were nearly identical to Seq-Scope-X digital detection of CD4 and CD8b antibody barcodes from the nearby section (Supplementary Fig. 9d). Similarly, IF-confocal imaging of CD21-35, representing mature B cell populations, closely matched the Seq-Scope-X spatial detection of CD21-35 antibody barcodes (Supplementary Fig. 9e). These results demonstrate the accuracy and sensitivity of Seq-Scope-X in detecting individual antibodies: its detection performance is comparable to IF-confocal microscopy while enabling simultaneous profiling of more than 100 antibodies in a single experiment.
Seq-Scope-X Maps human tonsil spatial proteome
We next evaluated the applicability of Seq-Scope-X spatial proteomics for human tissues by analyzing tonsil sections from a donor with chronic tonsillitis. Using an antibody cocktail targeting 154 human cell surface antigens, Seq-Scope-X successfully identified a broad range of immune and stromal cell types, including diverse T- and B-lymphocyte populations, myeloid cells, endothelial cells, and several less-characterized subsets such as CD9/CD24-high and CD49f/CD49b-high populations/areas (Fig. 5g, h and Supplementary Fig. 10a–c). Spatially resolved expression patterns revealed the organization of distinct B cell subtypes within follicular and marginal zone regions, as well as CD4⁺ and CD8⁺ T cells in extrafollicular T cell zones (Fig. 5g and Supplementary Fig. 10b). Plasma cells and other immune populations also exhibited distinct localization patterns (Supplementary Fig. 10b). These results confirm that Seq-Scope-X is capable of high-resolution spatial proteomic mapping in complex human tissues.
DMAA Chemistry enables greater expansion for seq-scope-X proteomic workflow
All Seq-Scope-X data in Figs. 1–5 were generated with standard expansion gels composed of acrylamide and acrylate, which could allow for up to fourfold tissue expansion in conditions allowing for RNA capture. This translates to a maximum effective resolution of 150–300 nm (Fig. 6a). To push the resolution further, we incorporated X10 expansion chemistry, which replaces acrylamide with dimethylacrylamide (DMAA) to achieve up to tenfold expansion40,41,42. In the X10 chemistry, DMAA’s unique polymerization properties produce a softer, more swellable hydrogel, enabling a maximum effective resolution of 60–180 nm (Fig. 6a), which is considered as a true nanoscale regime.

a Schematic diagram comparing expansion chemistry using standard (left) and DMAA (right) gels. While not a precise representation of the final polymer arrangement, this schematic provides a conceptual visual guide to illustrate the differences between the two gels. The standard gel uses a separate backbone (acrylamide-acrylate) and crosslinker (methylenebisacrylamide), forming a more defined net structure. In contrast, the X10 gel employs dimethylacrylamide, which can function as both a backbone (with acrylate) and a crosslinker, resulting in a more flexible structure capable of gross expansion. b–j Seq-Scope-X analysis of the spatial proteome in mouse spleen, enhanced by DMAA gel expansion, using an antibody tag cocktail targeting 119 unique cell surface antigens, including principal lineage markers, plus 9 isotype controls. b Comparison of the spleen tissue used in the analysis, before and after tissue expansion. The spleen section was imaged for oligo-dT-acrydite fluorescence, which reflects the presence of Poly-A tags in the tissue. The boxed area is digitally magnified (left inset), and compared with the expanded image (center inset) and the digitally reconstructed image from the Seq-Scope-X dataset (right inset). The dimensions and geometry of salient features (white and yellow arrows) are concordant across representations. c–j FICTURE projection of spatial factors identified via LDA analysis of protein expression results. Twenty-four factors are annotated and color-coded as shown in (c). The pink box in (c) is magnified in (g). Blue boxes in (d–g) are further magnified in (h–j, left). Yellow boxes in (h–j, left) are subsequently magnified in (h–j, right). Different B cell (d), T cell (e), and myeloid cell (f) subtypes are represented with reddish, greenish, and bluish colors, respectively. Scale bars represent the scale corresponding to the original tissue.
To assess the compatibility of Seq-Scope-X with the DMAA expansion chemistry, we repeated the mouse spleen analysis (Fig. 5a–f) using DMAA instead of acrylamide. This resulted in an approximately 3.3-fold expansion while preserving the salient spatial features of the original tissue (Fig. 6b, first inset; indicated in arrows), as validated in both the expanded gel (Fig. 6b, second inset) and the Seq-Scope-X array’s digital output (Fig. 6b, third inset). Quantitative image distortion analysis, performed on a separate spleen section, showed that the DMAA expansion is largely isotropic with over 95% of fidelity (Supplementary Fig. 1d, e). FICTURE analysis of the Seq-Scope-X dataset produced from the DMAA gel recapitulated all major cell types and histological regions identified in the standard Seq-Scope-X procedure, while providing enhanced spatial detail (Fig. 6c). In particular, it revealed a wider diversity of B-cell (Fig. 6d), T-cell (Fig. 6e), and myeloid cell (Fig. 6f) subtypes, along with their specific marker expression (Supplementary Fig. 11a) and spatial distributions (Supplementary Fig. 11b). Furthermore, digital magnification of the spatial factor map demonstrated clear, cellular-level organization of these immune populations (Fig. 6g–j), confirming that Seq-Scope-X could be successfully combined with the DMAA expansion chemistry, which could open a way of further increasing the resolution to enable nanoscale spatial analysis.
Seq-Scope-X detects extracellular and intracellular proteins and post-translational modifications
All the Seq-Scope-X proteome analyses shown above in Figs. 5, 6 and S9–S11 utilized barcoded antibody cocktails originally developed for single-cell CITE-seq studies. These panels primarily target cell membrane proteins, which are the main detectable targets in the single-cell CITE-seq workflow. However, because Seq-Scope-X profiles both extracellular and intracellular regions, its capabilities are not restricted to cell surface proteins, and in principle, any antigen accessible to antibody staining can be detected with this workflow. To evaluate this, we tested whether Seq-Scope-X could be adapted to detect extracellular and intracellular proteins, as well as post-translational modifications, in mouse skeletal muscle tissue engineered to exhibit strong mechanistic target of rapamycin complex 1 (mTORC1) signaling43. This tissue displays extracellular collagen deposition, intracellular actin bundles, and high mTORC1 activity leading to marked phosphorylation of downstream targets such as 4E-BP1 in restricted cytosolic domains. Both Seq-Scope-X–based multiplexed digital detection (Supplementary Fig. 12a, b) and classical immunohistochemistry (Supplementary Fig. 12c–h) visualized these features: collagen antibodies labeled extracellular matrix structures (Supplementary Fig. 12a–d), skeletal muscle actin antibodies marked intracellular fibers (Supplementary Fig. 12a, b, e, f), and phospho-4E-BP1 antibodies highlighted localized cytosolic activation domains (Supplementary Fig. 12a, b, g, h). Together, these results show that Seq-Scope-X can be readily extended beyond cell surface markers to detect extracellular matrix components, intracellular structural proteins, and post-translationally modified targets, demonstrating broad versatility in spatial proteomic profiling.
Discussion
The development of Seq-Scope-X represents a significant advance in sequencing-based spatial omics by closing the resolution gap between sST and iST methodologies. By integrating tissue expansion with Seq-Scope, Seq-Scope-X enhances spatial precision, transcriptomic coverage, and compatibility with multiplexed proteomic analyses, enabling detailed subcellular and cellular investigations across various tissues.
In this study, we demonstrated that Seq-Scope-X could achieve a resolution up to ~ 180 nm—higher than most conventional light microscopes. In terms of pixel density, this translates to an order-of-magnitude improvement over standard Seq-Scope (from ~ 3 million pixels per mm² to ~ 30 million pixels per mm²). Theoretically, DMAA-based expansion chemistry is capable of reaching approximately 60 nm resolution, which would represent another order-of-magnitude improvement. However, such high expansion levels are more likely susceptible to biochemical, physical, and practical challenges. Maximal DMAA swelling requires very low ionic strength, which weakens poly(T)–poly(A) anchoring interactions, increasing the likelihood of target dissociation. Expansion also occurs in three dimensions, so large linear expansion factors create extreme volumetric dilution that reduces effective transcript concentration and makes downstream library preparation difficult. In addition, highly swollen DMAA gels become fragile, increasing the risk of distortion or mechanical damage during handling. Together, these constraints limit the practical expansion range for transcriptome-quality data, even though improved experimental strategies may make larger, more stable expansion achievable in the future.
Despite these limitations, the 3–3.3 fold expansion demonstrated here enabled precise mapping of subcellular transcriptomic heterogeneity. The ability to resolve such features brings Seq-Scope-X’s resolution closer to that of iST technologies like Xenium, MERFISH, and CosMx, while maintaining the key advantages of sequencing-based approaches: comprehensive whole-transcriptome coverage and superior scalability in both time and cost. In addition, because mRNA transcripts are captured and immobilized prior to tissue expansion, the effective lateral diffusion is diminished in Seq-Scope-X relative to standard Seq-Scope, scaling inversely with the expansion factor. Indeed, our analyses of liver, brain, and colon tissues confirm Seq-Scope-X’s superior spatial resolution and precision, which are critical for understanding subcellular structures, organelle distributions, zonation patterns, and other fine-scale cellular and tissue-level properties.
Looking ahead, several technical developments may support larger and more stable expansion. These include stronger anchoring chemistries that remain stable under low-salt conditions, or anchoring systems that can be conditionally released by light or enzyme triggers at the transfer step to allow full swelling without loss of RNA or antibody tags. In addition, in situ amplification strategies could potentially increase effective molecular abundance after large volumetric dilution, improving capture efficiency and making downstream library preparation feasible. Mechanical approaches such as iterative expansion or scaffold-supported gels may also help maintain structural stability at higher swelling. Together, these directions may enable Seq-Scope-X to achieve larger expansion factors and operate closer to the theoretical resolution limits of DMAA-based hydrogels.
One inherent limitation of Seq-Scope-X is that tissue expansion requires a larger capture area or multiple capture arrays to assay the same physical tissue region compared with non-expanded methods. As a result, when using the same array size, Seq-Scope-X interrogates proportionally smaller tissue areas, and larger coverage demands proportionally greater array usage. Although the expansion process itself adds minimal cost, the required capture area scales with the square of the linear expansion factor, leading to an increase in chip-related costs when broad tissue coverage is desired. Even with this consideration, Seq-Scope-X maintains a favorable balance of resolution, throughput, and cost relative to imaging-based spatial transcriptomics, making it a practical choice for high-resolution whole-transcriptome analysis.
Another limitation is that tissue expansion introduces geometric distortion, which could become more pronounced as the expansion factor increases. However, in the expansion range used in this study (2–3.3 fold), quantitative distortion analyses showed that structural fidelity was high, with RMS errors less than 1 percent in liver and brain and less than 5 percent in colon and spleen. Distortion was usually restricted to the outermost edges of the expanded gel, while tissue boundaries and internal voids such as blood vessels showed relatively little deformation. Although these residual distortions did not alter biological interpretation at the cellular or subcellular scales examined here, they can make analysis of very large contiguous regions more difficult unless local registration is performed. As expected from the physics of hydrogel swelling, higher expansion factors, such as those produced with DMAA chemistry, are more prone to geometric distortion. These issues highlight a trade-off between achieving higher resolution and maintaining uniform structural fidelity, and they set a practical ceiling for the expansion factors that can be used for biologically useful analyses at present.
The power of Seq-Scope-X allowed us to reveal previously inaccessible biological insights in liver transcriptome. While recent single-cell and spatial studies have established that hepatocytes express distinct gene sets based on their histological niches25,32, effectively dividing the liver’s metabolic labor, the flexibility of these metabolic roles remained unknown. Through systematic analysis of the Seq-Scope-X dataset, using both segmentation-based nuclear/cytoplasmic analyses and segmentation-free FICTURE approaches, we uncovered widespread mismatches between nuclear and cytoplasmic transcriptomic profiles within individual cells. These observations were independently validated through orthogonal imaging-based methods, including MERFISH and smFISH. Our findings suggest that hepatocellular characteristics are not fixed, but rather that hepatocytes can dynamically switch their metabolic roles over time.
Seq-Scope-X’s capacity to separate nuclear and cytoplasmic transcriptomes offers opportunities to improve cell trajectory analyses. Traditional approaches frequently rely on splicing states, where spliced transcripts represent a cell’s immediate function and unspliced transcripts suggest preparatory regulatory directions44,45. However, splicing-based methods can be confounded by substantial gene-specific differences in splicing dynamics, raising concerns about their reliability in depicting temporal changes45,46,47. In contrast, Seq-Scope-X, as well as other high-resolution methods like MERFISH48, can delineate transcriptomic properties into nuclear (future) and cytoplasmic (current) compartments, thereby sidestepping the biases introduced by splicing kinetics. Our observations show that while unspliced and spliced reads from the same cell often diverge in clustering space (Supplementary Fig. 4d–g), nuclear and cytoplasmic reads from the same cell integrate well (Fig. 2e), differentiating only in transcripts known to be nuclear- or mitochondria-specific (e.g., Malat1, Neat1, and mitochondria-encoded genes).
This nuclear-cytoplasmic segmentation is most effective in larger, regularly shaped cells like hepatocytes, which have clear nuclear and cytoplasmic boundaries. Smaller or irregular cells (e.g., immune cells, cholangiocytes) pose additional challenges, as do inherently complex tissues such as the brain and colon, where precise subcellular segmentation remains more difficult—even though nuclear-specific factors can still be detected by unbiased clustering. Furthermore, our current liver data reflect a stable metabolic state in conventionally reared mice; the semi-random mismatches we observed between nuclear and cytoplasmic phenotypes may represent fine-grained fluctuations rather than broader shifts driven by metabolic cues. Future work could examine how physiological or experimental perturbations influence nuclear and cytoplasmic transcriptome relationships in hepatocytes. Approaches such as controlled feeding or fasting regimens, endocrine stimulation, or liver injury models may help clarify whether the nuclear-cytoplasmic differences reflect regulated transitions in hepatocellular states. In addition, improvements in expansion chemistry may further increase spatial resolution, which could broaden the applicability of nuclear-cytoplasmic segmentation to a wider range of tissues and cell types.
Beyond transcriptomics, Seq-Scope-X can detect any poly-A–labeled molecule, allowing single-cell–level proteomic analyses using nucleotide-tagged antibodies. Current commercial nucleotide-tagged antibody cocktails primarily target cell surface proteins because they were originally developed for single-cell CITE-seq. However, recent advances have enabled CITE-seq-based detection of intracellular proteins and selected post-translational modifications49. As additional oligonucleotide-tagged antibodies for intracellular or matrix-associated targets become available, Seq-Scope-X should accommodate a broader range of protein classes and post-translational modifications. In this study, we also demonstrate that laboratories can generate custom oligonucleotide-conjugated antibodies through a straightforward workflow.
Seq-Scope-X’s strategy of separating tissue staining from spatial array capture confers several advantages over existing array-based spatial proteomics platforms (e.g., SM-OMICs35 and SPOTs36), which rely on low-resolution arrays and apply antibodies directly to the capture surface. This can result in non-specific capture of antibody barcodes leading to undesirable noises. While microfluidic-based techniques, such as spatial CITE-seq/DBiT-Seq37, or slide maneuvering-based techniques, such as 10x Visium CytAssist20, mitigate nonspecific capture, they still operate at lower resolution (> 2–20 µm). In contrast, Seq-Scope-X capitalizes on the high resolution of Seq-Scope augmented by tissue expansion, achieving nanoscale omics exploration with greater precision.
Separating tissue preparation from spatial capture further broadens Seq-Scope-X’s versatility. Various poly-A–labeled probes—such as wheat germ agglutinin, phalloidin, aptamers, or RNA in situ probes—can be applied without risking damage to the capture array. It is even conceivable to integrate genomic assays (e.g., as demonstrated in spatial genomics50, spatial-ATAC51,52 and spatial-Cut&Tag53,54) by labeling fragmented DNA with poly-A or other capture sequences, thereby extending high-resolution mapping to epigenomic states and transcription factor binding sites on a solid-phase platform.
Despite its capabilities, Seq-Scope-X proteomic workflow has limitations requiring further refinement. One prominent challenge is the suboptimal performance of the current antibody panel for spatial applications. The antibody cocktail used was originally optimized for CITE-seq in suspension cells, which may not preserve epitope integrity or accessibility in fixed tissue contexts, leading to false negative results (i.e., failure to detect proteins that are present). Background signals from nonspecific antibody binding (false positives) also remain an issue, potentially leading to diffuse or misleading spatial patterns. To address these problems, protocol optimization for histological staining, including fixation conditions, antigen retrieval, and antibody validation in tissue contexts, is necessary. Similarly, the reliance on temperature-based melting for poly-A release imposes potential problems with efficiency and diffusion artifacts; alternatives like photocleavable linkers or non–poly-A sequences may offer greater flexibility.
In summary, Seq-Scope-X integrates tissue expansion with sequencing-based spatial profiling to increase spatial resolution and enable proteomic measurements. Although several technical aspects require further development and optimization, the approach broadens the range of biological questions that can be addressed with sequencing-based spatial omics. Continued methodological improvements will determine the extent to which the platform can be extended to additional tissues, molecular targets, and experimental contexts.
Methods
Experimental procedures
Tissue samples
Mouse tissues, including liver, colon, and spleen, were collected from 8-week-old male Mus musculus C57BL/6 J wild-type mice (The Jackson Laboratory), and brain tissue was obtained from a 6-month-old male C57BL/6 J wild-type mouse. Liver from 8-week-old Alb-Cre/Tsc1F/F/Depdc5F/F female mice55 and gastrocnemius (GTN) skeletal muscle from 10-week-old CKM-Cre/Tsc1F/F/Depdc5F/F male mice43, both in the C57BL/6 J background, were also used. Mice were housed under specific pathogen-free conditions with a 12 h light and 12 h dark cycle, ambient temperature maintained at 22 ± 2 °C, and relative humidity maintained at 40–60%, with food and water provided ad libitum. Human tonsil tissue was obtained from the UCLA Pathology Biobank as a discarded surgical specimen from an adult patient diagnosed with chronic tonsillitis.
Tissue Sectioning, Attachment, Fixation, and Permeabilization
Tissue expansion techniques, previously used to enhance the resolution of iST21,48,56 and low-resolution sST23 approaches were adapted for Seq-Scope-X analysis. Fresh frozen tissues embedded in Optimal Cutting Temperature compound (OCT) were sectioned using a cryostat (Epredia, 957250 L CryoStar NX50) at − 17 °C, with a 5° cutting angle and a thickness of 10–16 µm. The tissue sections were mounted on charged microscope slides and fixed for 10 minutes at room temperature in 4% formaldehyde solution prepared by diluting 16% formaldehyde (28908, Thermo Scientific) in 1X PBS (14190, GIBCO). After fixation, tissue sections were washed three times with 1X PBS to remove residual formaldehyde. The tissues were then permeabilized with ice-cold methanol at − 20 °C for 20–30 min, followed by three washes in 2X saline sodium citrate (SSC), prepared by diluting 20X SSC stock solution (BP1325, Fisher Bioreagents) with UltraPure water (10977, Invitrogen).
Probe hybridization
Tissues were incubated at room temperature in 30% formamide (AM9342, Invitrogen) diluted in 2X SSC for 30 min, then blow-dried using nitrogen gas. The anchor probe hybridization solution was prepared with 2X SSC, 30% formamide, 1 mg/mL Yeast tRNA (15401011, Invitrogen), 10% (w/v) dextran sulfate (J63606, Thermo Scientific Chemicals), 1% (v/v) SUPERase-In (AM2696, Invitrogen), and 2 mM Anchor-590ST (/5Acryd/TT + T T + TT + TT + T T + TT + TT + T /3ATTO590N/ as an HPLC-purified oligonucleotide; Integrated DNA Technologies) in UltraPure water (10977, Invitrogen). Dried tissues were submerged in the hybridization solution, covered with an RNase Zap (AM9780, Invitrogen)-treated coverslip, and incubated in a pre-warmed humidification chamber at 37 °C for 48 h.
Tissue embedding
The tissue embedding procedure was adapted from a previously described expansion MERFISH protocol22. The monomer solution was prepared as 2 M NaCl, 12% (w/v) sodium acrylate (408220, Sigma-Aldrich), 3% (v/v) 19:1 acrylamide/bis-acrylamide solution (BP1406, Fisher Chemical), and 60 mM Tris-HCl (15568, Invitrogen) in UltraPure water, followed by sonication at 40 kHz for 10–20 min (Branson, model no.1800). After disassembling the humidified chamber, hybridized samples were washed three times with 2X SSC and then incubated at 4 °C for 30–45 min in pre-chilled monomer solution supplemented with 0.2% tetramethylethylenediamine (TEMED; 17919, Thermo Scientific). After removing the initial monomer solution, a polymerization chamber was constructed using a spacer (SLF-0601, Bio-Rad) with open ends for solution entry and a Gel Slick (50640, Lonza)-treated coverslip. Polymerization was initiated in a separate pre-chilled aliquot of monomer solution by adding 0.2% ammonium persulfate (0486, VWR) and 0.2% TEMED, without 4-hydroxy-TEMPO delay, and the pre-polymerized solution was introduced onto the tissue, where capillary forces between the slide and coverslip maintained uniform coverage. Polymerization proceeded at room temperature in the polymerization chamber, with a damp paper towel to maintain humidity and protection from light under foil, minimizing thermal and chemical stress on RNA while preserving gel integrity, and was completed within 1–2 h.
Tissue clearing
The tissue digestion buffer consisted of 2X SSC, 2% (w/v) sodium dodecyl sulfate (SDS; 428015, Millipore), and 0.5% Triton X-100 (M143, AMRESCO) prepared in UltraPure water. After polymerization, the gel, which remained attached to the charged slide, was retrieved by disassembling the polymerization chamber, and the slide was trimmed using a glass cutter (FU-200, MSE Supplies) to fit within a 60 mm petri dish. The trimmed gel-slide complex was placed in the dish, briefly washed three times with digestion buffer, and then incubated with 3–4.5 mL of the same buffer supplemented with 1% Proteinase K (P8107S, NEB) to facilitate tissue clearing. The petri dish was covered with foil and incubated in a humidified chamber at 37 °C for 24 h.
Tissue expansion
The digestion buffer was removed from the petri dish, and the gel was washed with 1X SSC. Tissue expansion was performed by incubating the gels in 0.1X SSC. The solution was refreshed after 15 min and replaced again after another 15 minutes. Following this, the gels were incubated for an additional ~ 30 min or until a plateau was achieved.
The extent of expansion is governed primarily by the ionic strength of the medium. Bath volume and exchange frequency influence how rapidly the gel reaches its equilibrium state by accelerating the effective reduction in salt concentration. In our workflow, we cannot use very low-salt conditions such as pure water because reduced ionic strength can destabilize the hybridization between mRNA and the anchor probe, resulting in molecular loss. For this reason, we carried out expansion in 0.1X SSC with two buffer exchanges. During the final incubation, we monitored the gel until the desired scale factor was reached in a plateau; if additional expansion was required, we added more 0.1X SSC to allow the gel to reach the target expansion factor.
During the final 30 min of incubation, DAPI (D21490, Invitrogen) was added to assess tissue integrity and assist with tissue alignment.
Imaging and Probe Release
Expanded tissue gels were trimmed with a razor to fit the ~ 7 mm × 7 mm dimensions of the Seq-Scope Chip, produced as described in the previous Seq-Scope protocol15. DAPI and poly-dT probe (ATTO590) images of the expanded tissue samples were captured in a digital darkroom (Keyence BZ-X810). To minimize gel drying, distortion, and RNA damage, imaging was often skipped or performed at low magnification (e.g., using a 4X objective lens). When captured, the DAPI and poly-dT images were used for downstream alignment and segmentation. Following imaging, the gels were incubated in a humidity-maintained environment, achieved by a wet paper towel placed inside a RapidFISH slide hybridizer (240200, Boekel Scientific™), at 45 °C for 30 min. The heat facilitated the release of RNAs or antibody tags from the anchor probes, which were subsequently captured by the HDMI-oligo-dT probes on the surface of the Seq-Scope Chip23.
Standard procedure and library preparation
After incubation, the gel was carefully removed from the sandwich to retrieve the Seq-Scope Chip, which contained spatially captured RNAs. The established Seq-Scope protocol15 was then followed, starting from the reverse transcription step. All subsequent steps described in the Seq-Scope protocol15 were strictly adhered to, except for the tissue digestion step (Proteinase K treatment), which was omitted as no real tissue was attached to the Seq-Scope Chip.
Tissue processing and antibody staining for Seq-Scope-X proteome analysis
Tissues were permeabilized with ice-cold methanol at − 20 °C for 20 minutes. Following permeabilization, tissues were washed 3 times for 5 min each, with PBS containing 0.1% Triton-X (Ab-wash solution), and subsequently incubated with a blocking solution consisting of 1% BSA (BP1605, Fisher Scientific) and 1% RNase inhibitor (30281, Lucigen) in Ab-wash solution for 1 hour at room temperature. For the proteomic analysis of mouse spleen and human tonsil, TotalSeq™-A Human (399907, BioLegend) and Mouse (199901, BioLegend) Universal Cocktail were reconstituted according to the manufacturer’s recommendation and diluted 1:3 in SignalStain® Antibody Diluent (8112, Cell Signaling). For the proteomic profiling of mouse skeletal muscle, oYo-Link® Oligo Custom antibody (AT1002-25ss, Alpha Thera) conjugation was performed with Alpha Thera LED lamp (ZS-L09, Alpha Thera), following the manufacturer’s instructions. The oligonucleotide sequences have the following structure: [TruSeq Read 2 sequence: TCA GAC GTG TGC TCT TCC GAT CT] [10-base antibody barcode: NNN NNN NNN N] [10-base UMI: NNN NNN NNN B] [32-base poly-A: AAA AAA AAA AAA AAA AAA AAA AAA AAA AAA*A*A]. Oligo conjugation was performed for the following antibodies: COL1A1 (72026, Cell Signaling), ACTA1 (17521-1-AP, Proteintech), and p-4E-BP1 (2855, Cell Signaling), and diluted in blocking solution. The tissues were incubated with the diluted antibody cocktail overnight at 4 °C. After incubation, the antibody solution was removed, and the tissues were washed three times with the Ab-wash solution for 10 minutes each at 37 °C. Finally, the tissues were subjected to post-fixation with 4% formaldehyde in PBS for 10 min to stabilize the staining.
Tissue Expansion and Library Preparation for Seq-Scope-X Proteome Analysis
After post-fixation, the antibody-stained slides were processed following the same steps as the Seq-Scope-X transcriptome analysis, including “Probe Hybridization,” “Tissue Embedding,” “Tissue Clearing,” “Tissue Expansion,” and “Imaging & Probe Release.” However, for Seq-Scope-X proteome analysis, specific modifications were made to capture the DNA oligonucleotides tagging TotalSeq™-A antibodies. The antibody-tagging DNA oligonucleotides encode small RNA Read 2 (SR2) PCR adapter, antibody tag (AT) barcode, a random nucleotide (used as a unique molecular identifier (UMI) in our analysis), and poly-dA sequences. Following the “Imaging & Probe Release” step, reverse transcription was performed as in the Seq-Scope-X transcriptome analysis. During this step, reverse transcriptase, with its DNA polymerase activity, extended the antibody-tagging DNA oligonucleotides from its poly-dA tail using the Seq-Scope Chip’s DNA template encoding oligo-dT, HDMI, and Truseq Read 1 (TR1) adapter sequences. Consequently, the primary antibody tag (AT) library, containing SR2, Ab-barcode, a unique molecular identifier (UMI), poly-A, HDMI, and TR1 sequences, was produced on the Seq-Scope Chip. The reverse transcription solution was removed from the Chip, and the primary AT library was eluted using 30 μL of 0.1 N NaOH for 5 min. This elution step was repeated twice to collect a total of 60 μL of elution solution, which was neutralized with 30 μL of 3 M potassium acetate, pH 5.5 (AM9610, Invitrogen). The neutralized primary AT library was purified using the Oligo Clean & Concentrator kit (D4060, Zymo Research) per the manufacturer’s instructions, yielding a 40 μL final elution volume. The primary AT library was then amplified using an AT-PCR reaction comprising 100 μL of 2X Kapa HiFi Hotstart ReadyMix (KK2602, KAPA Biosystems), 1 μL each of 100 mM RPEPCR*F (TCT TTC CCT ACA CGA CGC*T*C) and SR2-RPEPCR*R primers (CCT TGG CAC CCG AGA ATT C*C*A), 40 μL of the purified primary library, and 58 μL of Ultrapure water. The AT-PCR cycle condition is: 95 °C for 3 min, followed by 15 cycles of 95 °C for 30 s, 60 °C for 1 min, 72 °C for 1 min, and a final extension at 72 °C for 2 min. The AT-PCR product was purified using AMPure XP beads (A63881, Beckman Colter) with a 1.8X bead/sample ratio and eluted in 30 μL. The sequencing library was prepared in an AT-Indexing-PCR reaction using 20 μL of 2X Kapa HiFi Hotstart ReadyMix, 4 μL each of 10 mM P5TR1* (AAT GAT ACG GCG ACC ACC GAG ATC TAC AC [10-base index barcode] ACA CTC TTT CCC TAC ACG ACG CTC TT*C) and P7SR1* (CAA GCA GAA GAC GGC ATA CGA GAT [10-base index barcode] GTG ACT GGA GTT CCT TGG CAC CCG AGA ATT CC*A) primers, 3 μL of the purified AT-PCR product, and 9 μL of Ultrapure water. The AT-Indexing-PCR cycle condition is: 95 °C for 3 min, followed by 6–8 cycles of 95 °C for 30 s (8 cycles for > 0.2 ng/μL, 7 cycles for > 0.5 ng/μL, and 6 cycles for > 0.8 ng/μL), 60 °C for 1 min, 72 °C for 1 minute, and a final extension at 72 °C for 2 min. The AT-Indexing-PCR products were purified using AMPure XP beads with a 1X bead/sample ratio and eluted in 30 μL. The purified AT-Indexing-PCR products underwent size selection via agarose gel elution to isolate the 250 bp band using the Zymoclean Gel DNA Recovery Kit (D4001, Zymo Research) according to the manufacturer’s instructions. The final library was sequenced using an Illumina NovaSeq-X sequencer. Seq-Scope-X proteome analysis using custom oYo-Link®–conjugated antibodies followed the same procedure, except that the custom antibody oligos contained TruSeq Read 2 (TR2) PCR adapter sequences; therefore, TR2-targeting primers, as used in the transcriptome library construction15, were used in place of SR2 primers.
Immunohistochemistry
Tissue sections were mounted on charged microscope slides and fixed for 5 min at room temperature in 4% formaldehyde. After fixation, the sections were washed three times with 1X PBS to remove residual formaldehyde. A slide adapter (SLF-0601, Bio-Rad) was then mounted around the tissues to prevent liquid spillage. The tissues were immersed in − 20 °C methanol, placed on a petri dish, and incubated at − 20 °C for 10 min. Samples were then washed three times with 1X PBS and subsequently incubated with 3% hydrogen peroxide (H1009, Sigma-Aldrich) at room temperature for 10 min to block endogenous peroxidase activity for DAB-based detection. Excess reagents were removed by washing the samples three times with Ab-wash solution, 5 min each, after which the tissues were incubated for 1 h at room temperature in blocking solution.
For DAB-based immunostaining, the tissues were incubated overnight at 4 °C with primary antibodies diluted in blocking solution, using anti-COL1A1 (72026, Cell Signaling, 1:300), anti-ACTA1 (17521-1-AP, Proteintech, 1:200), and anti-p-4E-BP1 (2855, Cell Signaling, 1:200). After primary incubation, the sections were washed three times with Ab-wash solution for 5 minutes at room temperature and then incubated with biotinylated anti-rabbit secondary antibody (BA-1000, Vector laboratories, 1:200) for 40 min at room temperature. Following the same washing procedure, the tissues were incubated with Streptavidin-Horseradish Peroxidase (554066, BD Biosciences, 1:300) for 35 min at room temperature. After washing, DAB substrate (D4293, Sigma) was applied according to the manufacturer’s instructions. Because the DAB reaction develops rapidly, color formation was closely monitored to avoid overdevelopment and high background; the reaction was stopped by washing with 1X PBS, and the tissues were counterstained with hematoxylin.
For fluorescence-based staining, the tissues were incubated overnight at 4 °C with FITC-labeled CD21-CD35 (123407, BioLegend, 1:100), Alexa488-labeled CD4 (100532, BioLegend, 1:100), and Alexa594-labeled CD8b (126635, BioLegend, 1:33) antibodies, as appropriate. After incubation, the tissues were washed three times with Ab-wash solution for 5 min at 37 °C. The samples were then dried and mounted in ProLong Gold Antifade Mountant (P36934, Invitrogen) for confocal imaging.
Confocal images of antibody-stained spleen tissue were acquired using a Zeiss LSM 980 microscope equipped with an Airyscan 2 detector at 10X magnification. For large tissue areas, the microscope’s tile-scanning function was used, and the resulting images were automatically stitched using Zen software. Fluorescent signals were visualized using the 488 nm (green) and 561 nm (red) channels.
Tissue expansion with DMAA gels
Tissue expansion using the X10 system was achieved with Dimethyl Acrylamide (DMAA) gels, as previously described for expansion microscopy41. In this method, the Acryl/bis-acrylamide used in the standard monomer solution was replaced with DMAA (274135, Sigma Aldrich) to allow for larger expansion. The DMAA monomer solution was prepared following the X10 expansion microscopy protocol41, using a defined composition of 1.335 g DMAA, 0.32 g sodium acrylate, and 2.85 mL of buffer containing 1.75 M NaCl and 100 mM Tris-Cl (pH 8.0), corresponding to an approximately 4:1 molar ratio of DMAA to sodium acrylate (that is, ~ 80:20 within the monomer fraction). Polymerization was conducted with a KPS/TEMED redox initiation pair, and oxygen was removed by nitrogen bubbling to prevent free-radical inhibition, consistent with the requirements of the X10 procedure. These conditions supported robust gelation around thin (~ 10 µm) tissue sections and ensured stable incorporation of acrydite-bound probes into the polymer matrix.
The extent of expansion achieved with DMAA gels depended primarily on the ionic strength and equilibration of the expansion medium. Therefore, larger bath volumes of 0.1X SSC supported more complete equilibration and produced higher expansion factors, whereas smaller volumes limited swelling. However, very large expansions reduced gel stability and complicated physical handling. To achieve a practical balance, we used a volume of 5-10 mL 0.1X SSC, yielding a consistent expansion factor of approximately 3.3 ×.
Data analysis
Seq-Scope-X transcriptome data processing
Seq-Scope-X data analysis was performed using the same pipeline as the Seq-Scope pipeline15 with standard parameters and adjustments for the experimentally determined scale factor. The analysis followed the steps outlined in the NovaScope and NovaScope Exemplary Downstream Analysis (NEDA) documentation, which include the generation of spatial barcode maps, alignment of sequencing reads to reference genomes, creation of spatially resolved gene expression (SGE) matrices, hexagonal binning, Seurat-based clustering57, Latent Dirichlet Allocation (LDA)-based spatial factor discovery, pixel-level spatial factor projection using FICTURE30, alignment of transcriptome data with microscopic imaging data, and image-based segmentation analyses12. The fluorescence images, such as DAPI and oligo-dT fluorophore images, did not have fiducial marks, so we aligned them to the Seq-Scope-X transcriptome image with salient features, such as blood vessel locations and nuclear centers estimated by unspliced RNAs, using the georeferencing function of QGIS. The aligned image was further processed as described in the Image Handling section and figure legends. The Seurat v5 package57 was used to analyze the hexagonally binned data through PCA and clustering analyses, as well as differential expression analyses and data integration analyses based on harmony58. FICTURE was also used to project seurat clusters into Seq-Scope raw pixel space, in the NEDA workflow. Additional analyses not performed on this pipeline and packages are outlined separately below.
Resolution and pixel density benchmarking
For resolution benchmarking in Fig. 1e, Seq-Scope-X was compared with 11 prior technologies. The center-to-center resolution of each method was as follows: ST (200 μm)6, 10x Visium (100 μm)7, Slide-Seq (10 μm)8,9, HDST (4 μm)11, DBiT-Seq (20 μm)27, Seq-Scope (0.5 μm)12, Stereo-Seq (0.5 μm)13, Pixel-Seq (1 μm)14, Expansion ST (40 μm)23, 10x Visium HD (2 μm)20, and Open-ST (0.6 μm; this resolution point also represents Seq-ScopeNOVASEQ and Nova-ST)15,16,17. These values were obtained as nominal resolutions from their respective publications. Seq-Scope-X demonstrated a resolution of 0.2 μm, achieved through a 3 × expansion factor, as validated using the mouse liver dataset. Even though it is not included in Fig. 1e, incorporation of the DMAA chemistry into Seq-Scope-X achieved 3.3 × expansion factor and 180 nm resolution and can potentially achieve up to a 6–10 × expansion factor and 60–100 nm. These reflect an expansion-mediated enhancement from the 0.6 μm resolution of NovaSeq-based Seq-Scope. Please note that the feature size of 10x Visium and Stereo-Seq is 55 μm and 0.2 μm; however, to perform the fair comparison between technologies, we only considered center-to-center distances for this benchmarking comparison.
For pixel density benchmarking in Fig. 1f, the following values in barcode/mm² were used: ST (25), Visium (154), Slide-Seq (2,210), HDST (94,100), DBiT-Seq (2,500), Seq-Scope (1,000,000), Stereo-Seq (4,000,000), Pixel-Seq (1,000,000), Expansion ST (625), Visium HD (250,000), Open-ST (2,961,000), and Seq-Scope-X (26,649,000). The method for calculating pixel density is done corresponding to our previous study12. In brief, for the technologies that have a defined pixel area (ST, Visium, HDST, DBiT-Seq, Stereo-Seq, Expansion ST, Visium HD, Open-ST, and Seq-Scope-X), pixel density was calculated as the inverse of the pixel area, adjusted for the expansion factor (Expansion ST & Seq-Scope-X). Note that HDST, Stereo-Seq, Open-ST, and Seq-Scope-X technologies employed patterned flow cells with a defined resolution and pixel density; however, due to sequencing errors, effective pixel density could be lower than the nominal density. For Slide-Seq, Slide-SeqV2 and Seq-Scope, pixel density was calculated in 150 μm grids (Slide-Seq and Slide-SeqV2) and 10 μm grids (original Seq-Scope) of the final dataset. Pixel-Seq pixel density was estimated to be the same as the original Seq-Scope as they are based on the similar sequencing chemistry.
Also note that Open-ST, Nova-ST, and NovaSeq-version of Seq-Scope were all published in 2024 with largely similar methodologies with minor differences in experimental and data processing procedures15,16,17. Their performances are similar to each other; so for simplicity, we only indicated Open-ST, which first appeared in the literature, in the graph.
For technologies developed from academic space, the date corresponds to either their peer-reviewed publication date or the preprint release date if not yet published before the current paper. For commercial technologies, the date reflects when the technology became publicly and commercially available.
RNA Capture Benchmarking and Transcriptome Comparison
To objectively and fairly benchmark transcriptome capture efficiency and precision, we compared Seq-Scope-X liver datasets with publicly available datasets derived from the same type of tissue, specifically normal young (2–4 months of age) wild-type male C57BL/6 mouse liver, ensuring consistency in biological context. These datasets included 10x Visium26, xDBiT-Seq27, Stereo-Seq28, Seq-ScopeMISEQ12, and Seq-ScopeNOVASEQ15. Additional datasets, including ST25, which analyzed normal young (2-3 months of age) wild-type female C57BL/6 mouse liver, and Slide-Seq (v1)8, which analyzed normal mouse liver of an unspecified origin, were also analyzed. For ST, Visium, Slide-Seq, and xDBiT-Seq, transcriptome capture efficiency per spatial feature (UMI/feature) was calculated and multiplied by spatial feature density (feature/μm²). For higher-resolution technologies such as Stereo-Seq and Seq-Scope, transcriptome capture efficiency was calculated using publicly available binned datasets (50 µm square grid for Stereo-Seq, and 10 µm square grid for Seq-ScopeMISEQ) as UMI/bin divided by bin area. Seq-ScopeNOVASEQ and Seq-Scope-X data was binned using 14 μm-sided hexagonal grid (24 μm flat-to-flat width, according to the original tissue scale) or additionally indicated grids and processed similarly. Outliers or non-tissue regions were removed using custom thresholds, such as minimum or maximum feature counts. Summary statistics for UMI counts were computed, and violin plots of UMI per unit area (UMI/μm²) were generated for comparison across datasets. To visualize spatial distribution and data quality, XY scatterplots stratified by raw (low-resolution ST) or binned (high-resolution ST) dataset were generated, with each spatial coordinate point colored by UMI count (nCount), allowing comparison of transcriptome capture across spatial features and datasets.
To assess transcriptomic similarity across datasets, we performed a comprehensive comparison using pseudo-bulk gene expression profiles aggregated from each ST dataset, along with genuine bulk RNA-seq results (six biological replicates) previously generated from the same type of tissue (normal wild-type C57BL/6 male mouse liver)29. Sum counts of gene expression from Seq-Scope-X and other datasets (ST, Visium, Slide-Seq, xDBiT, Stereo-Seq, Seq-ScopeMISEQ, Seq-ScopeNOVASEQ, and bulk RNA-seq samples) were merged based on shared genes. Transcriptome-wide similarity was visualized through scatterplots and heat maps of pairwise comparisons (Supplementary Fig. 3). These analyses underscore the consistency and reliability of Seq-Scope-X’s transcriptome data relative to prior methods.
Comparison of spatial gene expression, presented in Figs. 3g, h, 4b, c, were conducted by plotting each individual digital gene expression as colored dots. Following gene list, retrieved from clustering or factoring results as well as area-specific differential expression analyses, were used for spatial plotting. Figures 3g, h: Mup20 (blue), Hamp (green), Mup17 (orange) and Glul (red). Figure 4b: C1ql3, Cntnap5a, Dpyd, Glis3, Htr4, Il1rap, Itga8, Jph1, Lrrtm4, Maml2, Ppfia2, Prkd1, Prox1, Rasal2, Rfx3, Sema5a, Stxbp6, Tafa2 and Trpc6 (DG Granular Cells, red), Atp2b1, Cacnb2, Epha6, Galntl6, Hs6st3, Kcnh7, Man1a, Nr3c2, Ppm1e, Ryr3, Slc24a3 and Slc8a1 (CA1, green), Cacna2d3, Cpne4, Kcnq5, Nrip3, Tafa1, Wipf3, Zfp804a, Cntnap5c, Hpca, Khdrbs3, Neto1, Sv2b, Tafa1 and Ywhah (CA2/3, yellow), Chgb, Epha3, Lmo4, Neurod6, Nptxr, Nrp1, Rnf182, Slit2, Trhde and Trps1 (CA3, orange), Adarb1, Amotl1, Kcnc2, Lhfp, Nefh, Nell1, Nexn, Ntng1, Pde7b, Pdp1, Prkcd, Rora, Shox2, Slc17a6, Synpo2, Tcf7l2 and Zfhx3 (Excitatory, lightblue), Adam18, Adam32, Aip, Akr1c14, Ankrd52, Arrdc3, Atad5, Bcar3, Bdh1, C2cd4c, Cacna1i, Cct3, Cdh18, Cntnap4, Col12a1, Commd6, Csrnp2, Ctso, Dcbld1, Ddt, Dhrs3, Dnaaf11, Dpp10, Dzip1l, ENSMUSG00000120124, Efhd1, Fam124b, Ggh, Gm10033, Gm13322, Gm5468, Gm57059, Gpn3, Gpr176, Hdac11, Hey2, Ipp, Josd1, Kcnj16, Kirrel, Lrrc61, Ltbp1, Lzts1, Maip1, Med12, Mtmr2, Nrg3os, Nxt2, Pdzrn4, Peli3, Phkg2, Pik3r3, Pms1, Ppargc1b, Ppil2, Ppp2r1b, Prdm15, Prox1os, Ptpn20, Rnd3, Rnf121, Sdad1, Sema5b, Septin9, Sh3yl1, Syt2, Tent4a, Tmem132a, Tmem196, Tmem268, Tox, Tspan31, Unk, Vwc2l, Zbtb25, Zfp68, Zfp703, Zfp715, Zfp763, Zfp982 and Zxdb (Astrocyte, violet), Erbb4, Pcdh9, Pde4b, Plp1, Qki, Tmeff2 and Tspan2 (Oligodendrocyte, blue), Ttr and Enpp1 (Choroid Plexus, white). Figure 4c: Acta2, Actg2, Cnn1, Csrp1, Des, Flna, Hspb1, Myh11, Myl9, Mylk, Tagln, Tpm1 and Tpm2 (Smooth Muscle, white), Agr2, Cryab and Muc2 (Goblet I (DCSC), red), Zg16 and Fcgbp (Goblet II, orange), Mptx1 (Paneth, purple), Igha, Jchain and Ly6e (Immune, yellow), S100a6 (Fibroblast, blue), Saa1 and Aqp8 (Colonocyte I, cyan), Krt20, Cldn4 and Fabp2 (Colonocyte II, green).
Image-based single cell and subcellular segmentation
Image-based segmentation for single cells was performed as described in NEDA documentation and our previous protocol15, using the Watershed algorithm implemented in the NIH ImageJ and NEDA pipeline, with the following modifications. Instead of using the hematoxylin-eosin image that was exemplified in our previous protocol15, we used the spliced (red) and unspliced (green) RNA density image, processed through Gaussian filtering (as in Figs. 1k and 2a), for image-based cell segmentation. When this image was converted to grayscale, green unspliced signal is more pronounced and suitable to identify local maxima, which is the nuclear center, while the dimmer red spliced signal will be still effectively used to estimate the cell boundary as spliced transcripts are scarcely found in extracellular areas. Each Watershed segments are then considered as a single cell, and a circular area centered at the local maxima with a radius of 5 µm (expansion-adjusted unit) is considered as a nuclear compartment, while the remainder of the single cell area was considered as a cytoplasmic compartment. Performance of this segmentation method is generally concordant with visual inspection of the image (as in Fig. 2a). For downstream analysis, each compartment was separately collapsed as an independent aggregate, even though the data are labeled so that the aggregate pair from a single cell can be tracked for the cytoplasmic-nuclear mismatch analysis (as in Fig. 2g, h). The spliced and unspliced gene counts were retrieved using velocyto function in STARsolo, implemented in the NovaScope pipeline, and whole cell segment was used to aggregate single cell spliced and unspliced transcriptomes used for analysis in Supplementary Fig. 4d–g. Cell segmentation for the conventional Seq-Scope dataset was taken from our previously published results15.
Analysis of in situ imaging data
MERFISH mouse liver data was retrieved from the Vizgen website31 and subjected to FICTURE analysis as described previously30 and analyzed with the DAPI staining data after image alignment. Mouse smFISH liver data was retrieved from a previous publication32, and reproduced with permission from Springer Nature32. These data demonstrate the discrepancies between nuclear and cytoplasmic hepatic gene expression related to metabolic zonation, an observation consistent with Seq-Scope-X findings.
Seq-Scope-X proteome analysis
To enable proteome analysis, the NovaScope pipeline was modified to ingest the Read2 sequencing reads encoding antibody tag (AT) barcode, UMI, and poly-dA sequences. Instead of running STARsolo for transcriptome alignment, a custom software (spatula match-tag, available at https://github.com/seqscope/spatula) was used to match the FASTQ reads with the antibody barcodes. For the TotalSeq™-A Human Universal Cocktail, V1.0 (399907, BioLegend) and the TotalSeq™-A Mouse Universal Cocktail, V1.0 (199901, BioLegend), the first 15 bases of Read 2 were used as antibody tags, and the next base was used as the UMI. For custom antibody cocktails conjugated in-house using oYo-Link® technology, the first 10 bases of Read 2 were used as antibody tags, and the following 10 bases were used as the UMI. Sequence reads that do not match with any antibody barcodes are discarded, and the HDMI counts having the same AT barcode and UMI were deduplicated. The spatial digital gene expression (sDGE) output was constructed in a format similar to sDGE produced from transcriptome analysis, except that it only has one expression count and does not have intron-containing gene counts (GeneFull) or spliced/unspliced gene counts. The sDGE was used for downstream analysis in the same way as what was described in NEDA, such as gene plotting, LDA and FICTURE analyses15.
Image registration and displacement analysis of expanded tissue
First, each image pair was aligned by manual rigid registration (scaling, rotation, and translation). The unexpanded image was used as the fixed reference, and the expanded image was used as the moving image. All later steps were performed in MATLAB (MathWorks) using the Image Processing Toolbox and Image Registration Toolbox. We applied non-linear registration with the function imregdeform to deform the expanded image onto the unexpanded image (10 pyramid levels, GridRegularization = 0.6). This produced a dense 2D displacement field at each pixel in x and y. To restrict the analysis to tissue, we generated a binary mask from the unexpanded image. We smoothed the image with a Gaussian filter (σ = 8 pixels), thresholded the blurred image (Otsu threshold scaled to one-third), and removed small objects with area < 150 pixels using bwareaopen. From the displacement field, we calculated the displacement magnitude at each pixel in micrometers and applied the tissue mask. We visualized these values as displacement heatmaps and overlaid down sampled displacement vectors (one every 186.4 µm for brain, colon, and liver datasets, and 178.5 µm for the spleen dataset) using quiver for direction.
To visualize displacement error in expanded tissue, we plotted the Root Mean Square Error (RMSE) of the displacement error over different measurement lengths. Each displacement vector consists of (i) the measurement location and (ii) the displacement computed by the image alignment method described above. We first down-sampled the displacement field as described above and then generated all pairwise combinations of vectors from this down-sampled set. For each pair, we computed the measurement distance and displacement error. The measurement distance is the Euclidean distance between the two measurement locations. The displacement error is the Euclidean distance between the two displacement vectors (that is, the magnitude of their difference). For each unique measurement distance present in the down-sampled set, we calculated the RMSE of the displacement errors as sqrt(mean(error2)), and plotted RMSE versus measurement distance.
Image handling
All images within the same comparison were processed identically. When displayed at low magnification, data were downsampled to match the displayed image size, and contrast was enhanced using the same linear adjustment across corresponding panels. Due to the high dynamic range of the DAPI signal and substantial spatial variation in background intensity in expanded tissues, nuclei were identified using nuclear detection algorithms implemented in CellProfiler59 (Fig. 1l, m) and Cellpose60 (Fig. 4a). Accordingly, nuclei were used solely for anatomical reference and not for quantitative comparison. Transcriptomic and nuclear images were aligned using image registration procedures to account for distortion introduced during tissue expansion. Finally, a magenta and green pseudocolor scheme was applied to enhance accessibility for color-blind readers. QGIS, Adobe Photoshop and NIH ImageJ were used for image alignment and processing.
Statistics & reproducibility
No statistical method was used to predetermine sample size. Sample sizes were chosen based on practical considerations and prior experience with the experimental system. No data were excluded from the analyses except through standard quality control procedures for single-cell and spatial data. The experiments were not randomized. The investigators were not blinded to allocation during experiments or outcome assessment. Unless otherwise indicated, experiments were performed once for each application to demonstrate the technical feasibility of the method; where indicated, experiments were repeated independently with similar results and representative data are shown.
Use of large language models
ChatGPT (OpenAI) was used to refine grammar, improve clarity, and edit text during manuscript preparation. The authors reviewed all LLM-assisted edits and take full responsibility for the content.
Ethical statement
All mouse experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan.
This study involved analysis of leftover human specimens obtained during routine clinical care (e.g., surgery or biopsy). Specimens were fully and irreversibly de-identified prior to receipt by the investigators, with no codes, keys, or mechanisms available to permit re-identification. Investigators had no interaction or intervention with human subjects and received no clinical or demographic data that could enable identification of individuals, either directly or indirectly.
Under UCLA Policy 991 (Protection of Human Subjects in Research), which implements the federal Common Rule (45 CFR 46), research that does not involve interaction with living individuals and does not use identifiable private information or identifiable biospecimens does not constitute human subjects research. Because investigators could not reasonably ascertain the identity of any individual, this activity was classified by UCLA as Not Human Subjects Research, and IRB review or exemption was not required.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Seq-Scope-X datasets generated from this study were deposited to GEO under accession code GSE316811 and to the Deep Blue Data repository61 as raw sequences and spatially annotated digital gene expression (sDGE) matrix. Publicly available datasets used in this study include GEO accession codes GSE230402 (Bulk), GSE165141 (ST), GSE192741 (Visium), GSE169706 (Seq-ScopeMISEQ), and GSE207843 (xDBiT), as well as data available at Deep Blue Data (Seq-ScopeNOVASEQ: https://doi.org/10.7302/tw62-4f97), LISTA database (Stereo-Seq: https://db.cngb.org/stomics/lista/download/), Broad Single Cell Portal (Slide-Seq: https://singlecell.broadinstitute.org/single_cell/study/SCP354/slide-seq-study), and MERFISH Mouse Liver Map31. The source image files corresponding to all microscopy figures are available in Figshare62. The datasets used for benchmarking and distortion analyses are provided as Source Data. Source data are provided in this paper.
Code availability
The NovaScope pipeline (https://github.com/seqscope/NovaScope)63, spatula tookit (https://github.com/seqscope/spatula)64and NEDA package (https://github.com/seqscope/NovaScope-exemplary-downstream-analysis)65 to process these datasets are available at Github and zenodo repositories.
References
Rao, A., Barkley, D., Franca, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Liu, L. et al. Spatiotemporal omics for biology and medicine. Cell 187, 4488–4519 (2024).
Kang, H. M. & Lee, J. H. Spatial single-cell technologies for exploring gastrointestinal tissue transcriptome. Compr. Physiol. 13, 4709–4718 (2023).
Tian, L., Chen, F. & Macosko, E. Z. The expanding vistas of spatial transcriptomics. Nat. Biotechnol. 41, 773–782 (2023).
Moffitt, J. R., Lundberg, E. & Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 23, 741–759 (2022).
Stahl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Rodriques, S. G. et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39, 313–319 (2021).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681 (2020).
Vickovic, S. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019).
Cho, C. S. et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559–3572 (2021).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792 (2022).
Fu, X. et al. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell 185, 4621–4633 (2022).
Kim, Y. et al. Seq-Scope: repurposing Illumina sequencing flow cells for high-resolution spatial transcriptomics. Nat. Protoc. (2024).
Schott, M. et al. Open-ST: High-resolution spatial transcriptomics in 3D. Cell (2024).
Poovathingal, S. et al. Nova-ST: Nano-patterned ultra-dense platform for spatial transcriptomics. Cell Rep. Methods 4, 100831 (2024).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
He, S. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 40, 1794–1806 (2022).
Oliveira, M. F. et al. High-definition spatial transcriptomic profiling of immune cell populations in colorectal cancer. Nat. Genet. 57, 1512–1523 (2025).
Alon, S. et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science 371, https://doi.org/10.1126/science.aax2656 (2021).
Wang, G., Moffitt, J. R. & Zhuang, X. Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci. Rep. 8, 4847 (2018).
Fan, Y. et al. Expansion spatial transcriptomics. Nat. Methods 20, 1179–1182 (2023).
Chen, F., Tillberg, P. W. & Boyden, E. S. Expansion microscopy. Science 347, 543–548 (2015).
Hildebrandt, F. et al. Spatial Transcriptomics to define transcriptional patterns of zonation and structural components in the mouse liver. Nat. Commun. 12, 7046 (2021).
Nikopoulou, C. et al. Spatial and single-cell profiling of the metabolome, transcriptome and epigenome of the aging mouse liver. Nat. Aging 3, 1430–1445 (2023).
Wirth, J. et al. Spatial transcriptomics using multiplexed deterministic barcoding in tissue. Nat. Commun. 14, 1523 (2023).
Xu, J. et al. A spatiotemporal atlas of mouse liver homeostasis and regeneration. Nat. Genet. 56, 953–969 (2024).
Suchacki, K. J. et al. The effects of caloric restriction on adipose tissue and metabolic health are sex- and age-dependent. Elife 12, https://doi.org/10.7554/elife.88080 (2023).
Si, Y. et al. FICTURE: scalable segmentation-free analysis of submicron-resolution spatial transcriptomics. Nat. Methods 21, 1843–1854 (2024).
Vizgen MERFISH Mouse Liver Map. https://info.vizgen.com/mouse-liver-data (2022).
Halpern, K. B. et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017).
Vickovic, S. et al. SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 13, 795 (2022).
Ben-Chetrit, N. et al. Integration of whole transcriptome spatial profiling with protein markers. Nat. Biotechnol. 41, 788–793 (2023).
Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol. 41, 1405–1409 (2023).
Gayoso, A. et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021).
Feng, Z. et al. Multispectral Imaging of T and B Cells in Murine Spleen and Tumor. J. Immunol. 196, 3943–3950 (2016).
Truckenbrodt, S. et al. X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO Rep. 19, https://doi.org/10.15252/embr.201845836 (2018).
Truckenbrodt, S., Sommer, C., Rizzoli, S. O. & Danzl, J. G. A practical guide to optimization in X10 expansion microscopy. Nat. Protoc. 14, 832–863 (2019).
Sheard, T. M. D. & Jayasinghe, I. Enhanced expansion microscopy to measure nanoscale structural and biochemical remodeling in single cells. Methods Cell Biol. 161, 147–180 (2021).
Cho, C. S. et al. Simultaneous loss of TSC1 and DEPDC5 in skeletal and cardiac muscles produces early-onset myopathy and cardiac dysfunction associated with oxidative damage and SQSTM1/p62 accumulation. Autophagy 18, 2303–2322 (2022).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Soldatov, R. A., Kharchenko, P. V. & Theis, F. J. RNA velocity-current challenges and future perspectives. Mol. Syst. Biol. 17, e10282 (2021).
Zheng, S. C., Stein-O’Brien, G., Boukas, L., Goff, L. A. & Hansen, K. D. Pumping the brakes on RNA velocity by understanding and interpreting RNA velocity estimates. Genome Biol. 24, 246 (2023).
Gorin, G., Fang, M., Chari, T. & Pachter, L. RNA velocity unraveled. PLoS Comput. Biol. 18, e1010492 (2022).
Xia, C., Fan, J., Emanuel, G., Hao, J. & Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. USA 116, 19490–19499 (2019).
Ariss, M. M. et al. InTraSeq: a multimodal assay that uncovers new single-cell biology and regulatory mechanisms. Preprint at https://doi.org/10.21203/rs.3.rs-5284652/v1 (2024).
Zhao, T. et al. Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature 601, 85–91 (2022).
Deng, Y. et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 609, 375–383 (2022).
Llorens-Bobadilla, E. et al. Solid-phase capture and profiling of open chromatin by spatial ATAC. Nat. Biotechnol. 41, 1085–1088 (2023).
Deng, Y. et al. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 375, 681–686 (2022).
Zhang, D. et al. Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature 616, 113–122 (2023).
Cho, C. S. et al. Concurrent activation of growth factor and nutrient arms of mTORC1 induces oxidative liver injury. Cell Discov. 5, 60 (2019).
Allen, W. E., Blosser, T. R., Sullivan, Z. A., Dulac, C. & Zhuang, X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 186, 194–208 (2023).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinform. 22, 433 (2021).
Stringer, C., Wang, T., Michaelos, M. & Pachitariu, M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021).
Anacleto, A., Cheng, W., Kang, H. M. & Lee, J. H. Seq-Scope-X datasets [Data set]. University of Michigan - Deep Blue Data., https://doi.org/10.7302/kj26-7j44 (2025).
Anacleto, A., Zhao, Q., Park, A. & Lee, J. H. Raw images for “Seq-Scope-eXpanded: Spatial Omics Beyond Optical Resolution”. figshare, https://doi.org/10.6084/m9.figshare.31093639 (2026).
Cheng, W. & Kang, H. M. NovaScope (v1.1.0). Zenodo, https://doi.org/10.5281/zenodo.18292675 (2026).
Kang, H. M. spatula: A C++ toolkit for spatial transcriptomics (v1.1.0). Zenodo, https://doi.org/10.5281/zenodo.18327578 (2026).
Cheng, W. & Kang, H. M. NovaScope Exemplary Downstream Analysis (NEDA) (v1.0.0). Zenodo, https://doi.org/10.5281/zenodo.18291721 (2026).
Acknowledgements
The authors thank the U-M Advanced Genomics Core (AGC) for their cooperation and help in performing Seq-Scope and sequencing analysis. We thank Lee, Han and Kang lab members for their help in experiments and analysis. The work was supported by the Taubman Institute (to H.M.K. and J.H.L.), NIH (F31AG094300 to A.A., T32AG000114 to A.A., C.S.C. and Y.S.K., UH3CA268091 and R01DK133448 to J.H.L., R01AG079163 to M.K. and J.H.L., R35GM147420 to H.-S.H., and R01HG011031 to Y.S. and H.M.K.), Chan Zuckerberg Initiative (to H.M.K.), and Glenn Foundation (to J.H.L.) grants.
Author information
Authors and Affiliations
Contributions
A.A. conceived and designed the study, performed experiments, and developed the Seq-Scope-X methodology with guidance and mentoring from H.-S.H., H.M.K., and J.H.L. A.A., W.C., Y.H., Y.S., A.W.S., and S.Y. performed computational and statistical analyses. Q.F., A.P., C.S.C., Y.K., J.E.H., Q.Z., X.Z., D.K., and M.S. assisted with experiments. M.K. and C.H.K. contributed to data interpretation. R.T., R.L.M., O.P., J.E.G., and M.K. provided tissue samples. A.A., H.-S.H., H.M.K., and J.H.L. drafted the manuscript. All authors reviewed, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.H.L. is an inventor on U.S. Patent No. 12,319,955, filed by The Regents of the University of Michigan, which covers the Seq-Scope method for localized detection of nucleic acids in tissue samples as described in this manuscript. Y.H. is currently an employee of Samsung Semiconductor. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhangsheng Yu, who co-reviewed with Zhouhui Qi and Xin Yuan, Christoph Ziegenhain, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Anacleto, A., Cheng, W., Feng, Q. et al. Seq-Scope-eXpanded: spatial omics beyond optical resolution. Nat Commun 17, 2564 (2026). https://doi.org/10.1038/s41467-026-69346-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69346-8
