
Abstract
CD8+ T cell abundance within the tumor microenvironment is a critical determinant of immunotherapy efficacy. Here we show that CD8⁺ T cells lacking STING or TOX display markedly improved antitumor activity, with enhanced tumor infiltration and elevated IFN-γ and granzyme B production. These STING or TOX deficient cells exhibit a stem-like transcriptional state and resist ferroptosis by suppressing lipid peroxidation pathways while promoting mitochondrial biogenesis. Mechanistically, STING and TOX form a positive regulatory loop that represses HO-1 expression, leading to iron accumulation, mitochondrial oxidative stress, and ferroptosis in tumor-infiltrating CD8⁺ T cells. We further identify lactate as a microenvironmental trigger of STING–TOX–HO-1-mediated CD8+ T-cell ferroptosis. In mouse tumor models, engineered STING/TOX-deficient CD8⁺ T cells synergize with immune checkpoint blockade, chemotherapy, or STING agonist to enhance tumor control. These findings reveal a central pathway governing CD8⁺ T-cell ferroptosis in tumors and suggest therapeutic strategies to overcome immunotherapy resistance.
Similar content being viewed by others

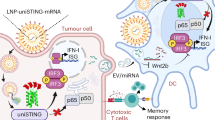

Introduction
Dysfunction of CD8+ cytotoxic T cells within the tumor microenvironment (TME) is a key driver for tumor immune escape and a significant barrier to the clinical efficacy of immunotherapies, such as immune checkpoint blockade (ICB) and adoptive cell therapy (ACT). Anti–PD-1/PD-L1 antibodies have shown substantial objective response rates (ORR) in several malignancies, including non–small cell lung cancer (NSCLC)1,2,3. However, due to the complexity and heterogeneity of the TME, blockade of alternative inhibitory receptors such as TIM-3 has yielded limited clinical benefit, and more than 60% of initial responders eventually develop acquired resistance4,5,6. Consequently, enhancing the efficacy and durability of ICB while mitigating resistance has become a major focus in cancer immunotherapy. Current efforts often involve combining immune checkpoint inhibitors (ICIs) with chemoradiotherapy, targeted therapies, or cancer vaccines7,8. Despite these advances, there remains an urgent need to design combination strategies that are both broadly effective and minimally toxic.
STING (stimulator of interferon genes), a sensor of cytosolic DNA activated via cyclic dinucleotides, regulates innate immune responses by inducing production of interferons and cytokines9. Beyond its role in innate immunity, STING also influences CD8+ T cell-mediated responses to DNA damage10. Although STING is broadly expressed in a wide variety of cell types throughout normal tissues, its activation produces tissue-specific effects that can either support protective immunity or contribute to pathological outcomes11,12. Current evidence indicates that STING activation in tumor cells enhances type I interferon signaling, thereby stimulating the activation of antigen-presenting cells and anti-tumor T cell responses13. However, clinical trials evaluating the STING agonist MIW815 (ADU-S100) in advanced cancers have demonstrated limited clinical efficiency14,15. Notably, STING expression is critical for immune cell development and functionality, human peripheral T cells exhibit low basal levels of STING under physiological conditions in contrast to their murine counterparts; However, STING expression is significantly upregulated upon T cell activation in contexts such as infection and cancer16,17. Recently, emerging research shows that STING activation within human and murine T cells suppresses proliferation and triggers their death by apoptosis18. Moreover, STING signaling has been implicated in mitochondrial-dependent ferroptosis in tumor cells19.
Tumor-infiltrating CD8+ T cells undergo functional remodeling within the TME in response to both intercellular and intracellular signals20. The nuclear factor thymocyte selection-associated high mobility group box (TOX) has emerged as a master regulator of tumor-specific T cell differentiation21. Under conditions of chronic antigen stimulation, such as in cancer or persistent viral infections, TOX expression is upregulated, promoting T cell exhaustion and dysfunction. TIM-3 and PD-1 double-positive exhausted T cells usually are co-expressed with TOX within TME22. Murine studies demonstrate that TOX or TOX2 knockout enhances tumor control by augmenting interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) secretion in tumor-specific T cells23. Despite these advances, the regulatory mechanisms linking TOX to CD8+ T cell dysfunction within the TME remain poorly understood.
Here, we show that the mice harboring CD8⁺ T cells deficient in either STING or TOX exhibit markedly enhanced antitumor activity. STING signaling synergizes with TOX to suppress heme oxygenase-1 (HO-1) expression, thereby promoting ferrous-ion-accumulation-induced mitochondrial ferroptosis in CD8+ T cells. Moreover, the lactate accumulation in the TME serves as a critical upstream driver of STING-TOX-HO-1-mediated ferroptosis. These findings provide mechanistic insight into the regulation of CD8⁺ T-cell ferroptosis in the TME and highlight potential therapeutic strategies to overcome immunotherapy resistance.
Results
STING- or TOX-deficient CD8+ T cells display enhanced tumor clearance
To investigate the regulatory role of intracellular STING and TOX signaling in CD8+ T cells during antitumor immunity, the Cd8creSting1flox/wt (Sting1-cKD), Cd8creSting1flox/flox (Sting1-cKO), Cd8creToxflox/flox (Tox-cKO) and double-knockdown (Sting1/Tox-D-cKD) mice were generated by crossing Sting1flox/flox and/or Toxflox/flox CD45.2+ C57BL/6 J mice with Cd8cre CD45.2 C57BL/6 J mice, and Sting1flox/flox, Toxflox/flox and wild-type (WT, CD45.1+ and CD45.2+) C57BL/6 J mice were included as controls. The Cd8creSting1flox/wt (Sting1-cKD), Cd8creSting1flox/flox (Sting1-cKO), Cd8creToxflox/flox (Tox-cKO) and double-knockdown (Sting1/Tox-D-cKD) mice exhibited phenotypes comparable to Sting1flox/flox, Toxflox/flox, Cd8cre and WT mice (Supplementary Fig. S1a–c). Notably, Sting1-cKD, Tox-cKO, and Sting1/Tox-D-cKD mice displayed enhanced tumor clearness across multiple models (TC-1, MC38, and B16F10) compared with WT mice (Fig. 1a and Supplementary Fig. S1d–e). These cKO mice displayed increased frequencies of CD3+CD8+ tumor-infiltrating lymphocytes (TILs), CD8+PD1+TIM3-CXCR5+ stem-like and CD8+TCF-1+Ly108+CD69+ precursor of exhausted TILs, as well as CD8+ TILs producing interferon-gamma (IFN-γ) and Granzyme B (GrB); additionally, an increased CD8+Ki67+ proliferating TIL subset was observed in tumor-bearing Tox-cKO and Sting1/Tox-D-cKD mice but not in Sting1-cKD mice related to WT mice. In contrast, the proportion of CD8+PD-1+TIM-3+CXCR5- exhausted TILs was reduced in these cKO mice; and the number of CD3+CD4+ TILs remained equal in these three cKO and WT mice (Supplementary Fig. S2a and Fig. 1b–d). Accordingly, peripheral T cell homeostasis remained unaffected in tumor-bearing Sting1-cKD and Tox-cKO mice compared with WT controls (Supplementary Fig. S2b). In ACT models, TC-1-bearing NSG mice receiving preactivated CD45.1⁺ WT-CD3+ T cells combined with preactivated CD45.2+CD3+ T cells from WT, Sting1-cKD, Tox-cKO, or Sting1/Tox-D-cKD donors exhibited greater tumor regression than PBS-treated controls (Fig. 1e, f). Donor-derived CD45.2⁺CD8⁺ TILs from Sting1-cKD, Tox-cKO, and Sting1/Tox-D-cKD groups showed higher infiltration and greater production of IFN-γ and GrB compared with CD45.1+ WT counterparts. In contrast, CD45.2+ WT-derived CD8+ TILs did not significantly differ from CD45.1+ WT TILs in terms of infiltration or cytokine production (Supplementary Fig. S2c and Fig. 1g, h). Furthermore, in TC-1-bearing CD45.1+ WT mice, adoptive transfer of pre-activated CD45.2+ WT CD8+ T cells treated with the STING inhibitor H151 led to enhanced tumor clearance compared with untreated CD45.2+ WT CD8+ T cell and PBS control groups, accompanied by increased lymphocyte infiltration and elevated IFN-γ production (Supplementary Fig. S3).

a–d Xenograft model was set up in WT (n = 4 mice, black), Sting1-cKD (n = 3 mice, blue), Tox-cKO (n = 4 mice, red), and Sting1/Tox-D-cKD (n = 4 mice, yellow) mice by subcutaneous injection of 4 × 10⁵ TC-1 tumor cells, and the lymphocytes were isolated from tumor tissues of each group after the tumor was harvested. Tumor growth kinetics and endpoint weights (a), frequencies of CD3⁺CD8⁺ and CD3⁺CD4⁺ TILs (b), Ki67⁺ proliferating, PD1⁺TIM3⁻CXCR5⁺ stem-like, TCF-1⁺Ly108⁺CD69⁺ precursor-exhausted, and PD1⁺TIM3⁺CXCR5⁻ terminally exhausted CD8⁺ TIL subsets (c), and IFN-γ⁺ and GrB⁺ CD8⁺ TILs (d). e, f TC-1 xenograft model was set up in NSG mice receiving adoptive transfer of CD45.1⁺ WT CD3⁺ T cells together with CD45.2⁺ WT (black), Sting1-cKD (blue), Tox-cKO (red), or Sting1/Tox-D-cKD (yellow) CD3⁺ T cells or PBS control (gray). Schematic of therapeutic protocol was generated in Generic Diagraming Platform. Zhu, Q. (2025) https://www.BioGDP.com/. e and tumor growth curves were shown (f) (n = 5 NSG mice/group). g, h Donor-derived CD45.2⁺ and CD45.1⁺ TIL subsets (g) and their IFN-γ and GrB producing (h) in the indicated groups (n = 4 NSG mice/group). Each dot represents an individual mouse; data are shown as mean ± SD. Statistical significance was determined by a two-sided unpaired Student’s t test and one-way ANOVA.
Transcriptomic features of tumor-infiltrating CD8+ T cells deficient in STING or TOX show resistance to ferroptosis
We performed bulk RNA sequencing of tumor-infiltrating CD8+ T cells from WT, Sting1-cKO, and Tox-cKO tumor-bearing mice. Differential gene expression analysis revealed 1,860 downregulated and 723 upregulated genes in Sting1-cKO CD8+ TILs, and 362 downregulated and 286 upregulated genes in Tox-cKO CD8+ TILs, compared to WT CD8+ TILs ( | log2FC | > 1, FDR < 0.05; Supplementary Fig. S4). Notably, transcripts encoding Sting1, the immune inhibitory receptors (Tox, Havcr2, Pdcd1, Lag3, Ctla4, and Entpd1), chemokine receptors (Cx3cr1, Ccr5 and Cxcr6) related to CD8+ T cell homing and ferroptosis drivers (Trim21, Pparg, Taz, Ptpn6, Flt3, and Ncoa4) were significantly downregulated. In contrast, genes associated with proliferative and stem-like properties (Mki67, Pcna, Top2a, Sell, Tcf7, and Cxcr5), along with the ferroptosis suppressor genes (Hmox1, Slc7a11, Acsl3, Trib2, Slc40a1, and Lcn2), were upregulated in both Sting1-cKO- and Tox-cKO-CD8+ TILs relative to WT CD8+ TILs (Supplementary Fig. S4 and Fig. 2a, b). We further demonstrated reductions in STING, phosphorylated TBK1 (p-TBK1) and IRF3 (p-IRF3), TOX, TIM-3 and PD-1, and ACSL4 and cathepsin B (CTSB). Conversely, expression of ferroptosis suppressors HO-1 and GPX4 was elevated in Sting1-cKO and Tox-cKO CD8+ TILs compared to WT CD8+ TILs (Fig. 2c). Multicolor immunofluorescence (IF) staining of tumor tissues revealed reduced frequencies of CD8+TOX+, CD8+PD-1+, CD8+TIM-3+, and CD8+CTSB+ cells, alongside increased CD8+HO-1+ and CD8+GPX4+ populations in Sting1-cKO and Tox-cKO mice (Fig. 2d, e). Pathway enrichment analysis identified shared alterations in Sting1-cKO- and Tox-cKO-CD8+ TILs, including pathways involved in iron ion transport regulation, lipid metabolic process and granzyme-mediated cell death (Fig. 2f). Specifically, intracellular iron homeostasis pathway was upregulated in both Sting1-cKO- and Tox-cKO-CD8+ TILs, while fatty acid metabolism pathway was downregulated in Sting1-cKO-CD8+ TILs; and ferroptosis-associated gene pathway was downregulated in both Sting1-cKO and Tox-cKO CD8+ TILs compared to WT CD8+ TILs (Fig. 2g). Consistently, Sting1-cKO- and Tox-cKO- CD8+ TILs exhibited decreased total cellular iron levels and reduced lipid peroxidation relative to WT-CD8+ TILs (Figs. 2h, i).

a CD8+ T cells were isolated from tumor tissues of TC-1-bearing WT, Sting1-cKD and Tox-cKO mice, and then for RNA sequencing. Heatmap of differential gene expression (DEGs) of CD8+ TILs. b, c Splenic CD8+ T cells from Sting1-cKO (blue), Tox-cKO (red) and Floxed controls (black) were co-cultured with TC-1 cells plus OKT-3; Quantitative PCR analysis of Sting1, Isg15 (STING signal pathway), Tox, Havcr2, Pdcd1 (T cell exhaustion markers), Hmox1, Gpx4, Acsl4, and Ctsb (ferroptosis-related genes) is shown (b) (n = 3 biologically independent experiments); Western blot analysis of STING, p-TBK1, p-IRF3, TOX, TIM-3, PD-1, HO-1, GPX4, ACSL4, and CTSB is shown (c). Immunoblot analysis was performed as three biologically independent experiments, and representative image from one experiment is shown in (c). d Representative fluorescence images of TOX (yellow), PD-1 (red), TIM-3 (green) expression in intratumoral CD8+ T cells (CD8, white), with quantification of CD8+TOX+, CD8⁺PD-1⁺, and CD8⁺TIM-3⁺ frequencies across tumor sections from WT (black), Sting1-cKO (blue), and Tox-cKO (red) mice (n = 3 mice/group). e Representative fluorescence images of ferroptosis-related marker HO-1 (pink), GPX4 (green), and CTSB (orange) expression in intratumoral CD8+ T cells (CD8, white), with quantification of CD8⁺HO-1⁺, CD8⁺GPX4⁺, and CD8⁺CTSB⁺ frequencies in tumor sections from the indicated mice (n = 3 mice/group). Scale bars, 100 μm. The scale bar denotes 100 μm in (d, e). Original magnification × 20. f Gene ontology (GO) analysis of pathways enriched in the common DEGs in both Sting1-cKO-CD8+ TIL and Tox-cKO-CD8+ TIL versus WT-CD8+ TIL. g GSEA on the Sting1-cKO-CD8+ TIL and Tox-cKO-CD8+ TIL versus WT-CD8+ TIL, the GSEA plots of intracellular iron ion homeostasis and lipid metabolic process were displayed. h, i Representative histogram and quantification of total iron accumulation (Phen Green SK, h) and lipid peroxidation (oxidized BODIPY-FITC, i) in intratumoral CD8⁺ T cells from TC-1-bearing WT (black), Sting1-cKD (blue), and Tox-cKO (red) mice (n = 4 mice/group for WT and Tox-cKO; n = 3 mice for Sting1-cKD). Numerical data was presented mean ± SD. Statistical analyses were performed using one-way ANOVA.
STING- and TOX-deficient tumor-infiltrating CD8+ T cells exhibit reduced susceptibility to mitochondria-dependent ferroptosis
To investigate enhanced tumor infiltration observed in CD8+ T cell in Sting1-cKO and Tox-cKO mice, we evaluated the proliferation and cell death rates of STING- and TOX-deficient CD8+ T cells compared to WT CD8+ T cells in vitro and found out that the proliferation remained comparable across genotypes (Supplementary Fig. S5a), but the STING- and TOX-deficient CD8+ T cells exhibited significantly reduced mortality following co-culture with TC-1 tumor cells and the STING agonist DMXAA (Supplementary Fig. S5b). Notably, the treatment of ferroptosis inhibitor Ferrostatin-1 (Fer-1), but not necrosis inhibitor Necrostatin-1 (Nec-1) or apoptosis inhibitor Z-VAD-FMK (Z-VAD), extremely significant rescued cell death of WT CD8+ T cells upon TC-1-stimulation (Supplementary Fig. S5c). Furthermore, the STING- and TOX-deficient CD8+ T cells displayed dose-dependent resistance to ferroptosis induced by Erastin, an SLC7A11 inhibitor (Supplementary Fig. S5d). Consistent with these results, WT CD8+ T cells showed increased nuclear CTSB expression and lipid peroxidation following TC-1/DMXAA stimulation, relative to their STING- and TOX-deficient counterparts (Supplementary Figs. S5e, f). Single-cell transcriptomic analysis revealed that ferroptosis is the predominant mode of cell death in tumor-infiltrating CD8+ and CD4+ T cells in contrast to their peripheral counterparts from patients with cervical cancer (CC, n = 2) or nasopharyngeal carcinoma (NPC, n = 10), accompanied by upregulation of STING and lipid metabolic pathways (Supplementary Fig. S6a, b). Consistently, pharmacological inhibition with Fer-1, but not with Nec-1 or Z-VAD, prevented tumor-induced human CD8+ T cell death in vitro (Supplementary Fig. S6c).
Subcellular localization analysis revealed mitochondrial enrichment of STING and ferroptosis-associated proteins, including STING, IRF3, HO-1, and ACSL4 in all CD8+ T cells upon TC-1 stimulation; whereas the CD8+ T cells deficient in STING or TOX exhibited reduced STING and ACSL4 expression but elevated HO-1 expression following TC-1 stimulation (Fig. 3a). Transcriptomic clustering using MSigDB (https://www.gsea-msigdb.org/gsea/msigdb) identified upregulated survival pathways (Cluster1) and downregulated death/lipid metabolism genes (Cluster3) in both STING- and TOX-deficient CD8+ TILs. Notably, TOX-deficient CD8+ TILs uniquely showed enrichment in iron ion homeostasis and cytokine production pathways (Cluster 2) (Fig. 3b). Functional assessments demonstrated preserved mitochondrial integrity in STING- and TOX-deficient CD8+ T cells, evidenced by reduced mitochondrial ROS (Mito-ROS) and mitochondrial lipid peroxidation (Mito-lipid peroxidation) along with increased mitochondrial mass (Mito-mass), and sustained ATP production and mitochondrial (mt)DNA copy number following TC-1 stimulation (Fig. 3c–g). Using a transwell co-culture system with murine tumor cell lines (CT26, B16F10 and TC-1), human tumor cells (SiHa and C666), and viral antigens peptide (HPV-E6/E7 and EBV-LMP1), we observed activation of STING signaling in murine and human CD8+ T cells. This was characterized by elevated levels of Ifit1, Isg15, and Isg56, as well as increased p-TBK1, p-IRF3, alongside TOX upregulation and HO-1 downregulation (Supplementary Fig. S7a, b), as well as the metabolic collapse in WT murine and human CD8+ T cells following tumor cell or viral antigen stimulation in vitro (Supplementary Fig. S7c–g). Collectively, these data suggest that STING/TOX signaling promotes ferroptosis of tumor-derived CD8+ T cells through mitochondrial metabolic reprogramming within the TME.

a Western blot analysis of STING, p-TBK1, IRF3, p-IRF3, and ferroptosis regulators (HO-1, GPX4, and ACSL4) in cytoplasm and mitochondria fractions of peripheral CD8⁺ T cells gotten from Sting1-cKO, Tox-cKO and Floxed control mice, and co-cultured with OKT-3 plus TC-1 cells for 48 h. Immunoblot analysis was performed as three biologically independent experiments, and a representative image was shown. b Heatmap and Gene Oncology analysis showing the DEGs associated with mitochondrial metabolism and morphology in intratumoral CD8⁺ T cells from TC-1-bearing WT, Sting1-cKO, and Tox-cKO mice. c–e Representative histograms and quantification of mitochondrial ROS (Mito-ROS) (c), mitochondrial lipid peroxidation (Mito-lipid peroxidation) (d), and mitochondrial mass (Mito-mass) (e) in CD8⁺ TILs from TC-1-bearing WT, Sting1-cKD, and Tox-cKO mice (n = 4 mice/group for WT and Tox-cKO; n = 3 mice for Sting1-cKD). f, g Quantification of ATP production (f) and mtDNA content (mt-Co2 normalized to nuclear App, g) in CD8⁺ T cells under co-culture transwell conditions as in (a). Numerical data was presented as mean ± SD. Statistical analyses were performed using one-way ANOVA.
STING-TOX-HO-1 axis drives tumor-primed ferroptosis in CD8+ T cells through iron-overloaded mitochondrial metabolic reprogramming
Transcriptional analysis revealed reciprocal regulation between STING and TOX in CD8+ T cells, with HO-1 upregulated in both STING- and TOX-deficient CD8+ T cells (Fig. 2a, b). Knockout of TOX or STING reduced the sensitivity of SiHa-stimulated human CD8+ T cells to ferroptosis, as indicated by decreased cell death, diminished ferrous ion accumulation, and reduced lipid peroxidation (Supplementary Fig. S8a–c). These deficiencies also restored mitochondrial metabolism, reflected by increased Mito-mass and reduced Mito-ROS (Supplementary Fig. S8d, e), and reversed the induction of TOX, STING, p-TBK1, p-IRF3, and HO-1 (Supplementary Fig. S8f). Knockdown of IRF3, a downstream mediator of the STING signaling pathway, in WT CD8+ T cells led to decreased TOX expression and increased HO-1 expression, accompanied by decreased total iron accumulation and increased Mito-mass (Supplementary Fig. S9a–c). Ectopic expression of TOX or STING in TC-1-stimulated STING- and TOX-deficient CD8+ T cells, respectively, restored sensitivity to ferroptosis, as evidenced by elevated cell death, increased ferrous ion accumulation, and enhanced lipid peroxidation (Fig. 4a–c). Simultaneously, ectopic expression of TOX or STING induced mitochondrial depolarization, evidenced by reduced Mito-mass and elevated Mito-ROS (Fig. 4d, e), while reactivating the STING-TOX-HO-1 signaling axis in the corresponding deficient cells (Fig. 4f).

Murine peripheral CD8+ T cells were gotten from Sting1-cKO, Tox-cKO, or Floxed control mice. STING- or TOX-deficient CD8+ T cells were transfected with lentivirus vectors encoding TOX (Lenti-TOX-OE) or STING (Lenti-STING-OE), respectively, or with shRNA targeting Hmox1 (Lenti-shHmox1); Floxed CD8⁺ T cells transduced with empty vectors (Lenti-PCDH or Lenti-pLKO) served as controls. All cells were co-cultured with OKT-3 plus TC-1 cells for 48 h before analysis. a–e Quantification of cell death (a) cytoplasmic iron levels (b) lipid peroxidation (c) mitochondrial mass (Mito-mass) (d) and mitochondrial ROS (Mito-ROS) (e) (n = 3 biologically independent experiments). f Immunoblot analysis of TOX, STING, p-TBK1, p-IRF3, and HO-1 expression in CD8+ T cells under the indicated conditions. Immunoblot analysis was performed as three biologically independent experiments. Representative image was shown. g, k Quantification of cell death (g) cytoplasmic iron levels (h) lipid peroxidation (i) Mito-mass (j) and Mito-ROS (k) in CD8⁺ T cells as indicated conditions (n = 3 biologically independent experiments). l Schematic model illustrating the STING–TOX–HO-1–dependent ferroptosis pathway in CD8⁺ T cells within the tumor microenvironment was generated in the Generic Diagraming Platform. Zhu, Q. (2025) https://www.BioGDP.com/. Numerical data was presented mean ± SD. Statistical analyses were performed using one-way ANOVA.
Previous studies have shown that HO-1 suppression can induce ferrous ion overload24,25. However, HO-1 knockdown paradoxically increased ferroptosis susceptibility in TC-1-stimulated STING- and TOX-deficient CD8+ T cells, as evidenced by significantly elevated cell death and ferrous ion levels without affecting lipid peroxidation. However, this intervention also exacerbated mitochondrial depolarization, as indicated by reduced Mito-mass and increased Mito-ROS (Fig. 4g–k). Iron chelation experiments using Deferoxamine (DFO) persevered mitochondrial integrity, reflected by increased Mito-mass and modulated mitochondrial dynamics by reducing DRP1 expression while increasing levels of MFN1 and HO-1 (Supplementary Fig. S9d–f). HO-1 knockdown in TOX-deficient CD8+ T cells impaired tumor clearance in TC-1-bearing CD45.1 WT mice, which was associated with reduced lymphocyte infiltration and diminished IFN-γ production (Supplementary Fig. S10). Collectively, these data identify the STING-TOX-HO-1 axis as a key ferroptosis checkpoint that regulates mitochondrial iron homeostasis in CD8+ T cells (Fig. 4l).
Tumor microenvironment induces STING-TOX-HO-1-mediated ferroptosis of tumor-infiltrating CD8+ T cells through lactate accumulation
In vitro and in vivo data demonstrated that ferroptosis was the predominant form of cell death of in both murine and human tumor-infiltrating CD8+ T cells (Supplementary Figs. S5, S6). Given prior evidence linking lactate to mitochondrial metabolic dysfunction and ferroptosis26, we observed that the lactate release in human (HNE-1, EBV+-TW03, SiHa) and murine (TC-1, CT26, B16F10) tumor cells were largely exceeded those in human and murine peripheral blood mononuclear cells (PBMCs, Supplementary Fig. S11a). Notably, WT CD8⁺ T cells exhibited greater sensitivity to lactate-induced cell death in a dose-dependent manner compared to WT CD4⁺ T cells and TC-1 cells (Supplementary Fig. S11b). Lactate treatment also induced iron accumulation, reduced Mito-mass, and decreased mtDNA copy number in WT CD8⁺ T cells (Supplementary Fig. S11c–1e). Further analysis revealed dose-dependent increases in the mitochondrial fission regulator DRP1, reductions in the mitochondrial fusion protein MFN1 and ferroptosis suppressor HO-1, along with elevated STING signaling (p-TBK1 and p-IRF3) and TOX expression in lactate-stimulated WT CD8+ T cells (Fig. S11f, g). Notably, STING- or TOX-deficient CD8+ T cells were resistant to lactate-induced cell death compared to their WT counterparts (Fig. 5a). Lactate specifically induced nuclear CTSB expression, lipid peroxidation, and mtDNA loss in WT-CD8+ T cells but not in STING- or TOX-deficient CD8+ T cells (Figs. 5b–f). Strikingly, treatment with lactate, TC-1 cells, or STING agonist DMXAA increased p-TBK1, p-IRF3 and TOX levels while suppressing HO-1 expression in WT CD8+ T cells while these effects were absent in STING- or TOX-deficient CD8+ T cells (Fig. 5g). However, these lactate-induced alterations in WT CD8⁺ T cells were reversed by the MCT1 inhibitor AZD3965, and this effect was markedly attenuated in Sting1-cKO or Tox-cKO CD8⁺ T cells, consistent with impaired activation of STING signaling and ferroptosis pathways (Fig. 5h). The Transcriptional profiling further revealed a pronounced reduction in several solute carrier family 16 (SLC16) members which was involved in lactate transport27, including Slc16a2, Slc16a3, Slc16a4, Slc16a6, Slc16a7, Slc16a11, and Slc16a13, in CD8⁺ TILs from Sting1-cKO or Tox-cKO mice compared to WT controls in vivo. In vitro, co-culture with TC-1 tumor cells resulted in reduced MCT11 protein abundance in Sting1-cKO CD8+ T cells, but not changed in Tox-cKO CD8+ T cells, whereas MCT1 levels remained unchanged (Fig. S11h, i). Collectively, these findings identify that the SLC16 transporters may be involved in lactate-driven ferroptosis in T cells mediated by STING-TOX-HO-1 signals. Consistent with this, AZD3965 inhibition of MCT1 markedly impaired B16F10 tumor growth in WT mice compared with PBS-treated controls, yielding a phenotype comparable to that observed in Sting1-cKO mice (Fig. S12a b). AZD3965 treatment increased the frequencies of CD8⁺ TILs, CD4⁺ TILs, TCF-1⁺Ly108⁺CD69⁺ precursor of exhausted CD8⁺ T cells, and IFN-γ–producing CD8⁺ TILs, but not Ki67⁺CD8⁺ proliferating TILs in tumor-bearing WT mice (Fig. S12c–f). Furthermore, AZD3965 reduced apoptosis and lipid peroxidation in CD8⁺ TILs compared to WT control (Fig. S12g, h). These results suggest that lactate enrichment in the TME promotes CD8+ T cell ferroptosis by inducing mitochondrial damage and activating the STING-TOX-HO-1 signaling axis.

Peripheral CD8+ T cells were gotten from Sting1-cKO (blue), Tox-cKO (red), or Floxed control (black) mice and stimulated under three conditions: lactate (2 mM), TC-1 tumor cell supernatant, or STING agonist DMXAA (1 μg/mL). a Quantification of cell death in CD8⁺ T cells of the indicated genotypes under the specified treatments (n = 3 biologically independent experiments). b–e Representative fluorescence images and quantification of CTSB expression (b, c) and lipid peroxidation (d, e) in CD8⁺ T cells under the indicated conditions (n = 3 biologically independent experiments). Scale bars, 30 μm. Immunofluorescence analysis was performed as three biologically independent experiments, and a representative image was shown in (b, d). f mtDNA levels (mt-Co2 normalized to nuclear App) in CD8⁺ T cells stimulated with OKT3 in the presence or absence of lactate (n = 3 biologically independent experiments). g Immunoblot analysis of STING, p-TBK1, p-IRF3, TOX, and HO-1 in CD8⁺ T cells treated with OKT3 plus lactate, TC-1 (transwell), or DMXAA. h Immunoblot analysis of these indicative proteins in CD8⁺ T cells stimulated with lactate in the presence or absence of the MCT1 inhibitor AZD3965 (500 nM). Immunoblot analysis in (g, h) were performed as three biologically independent experiments, and a representative image was shown. Numerical data was presented mean ± SD. Statistical analyses were performed using an unpaired two-sided Student’s t test and one-way ANOVA.
Neoadjuvant combined therapy based on ACT using STING- or TOX-deficient engineered T cells together with ICIs, STING agonist or cisplatin enhances tumor clearance
ACT using Sting1-cKD or Tox-cKO CD8+ T cells, in combination with low-dose PD-1 or TIM-3 neutralizing antibodies, significantly enhanced antitumor efficacy compared to monotherapy with low-dose ICIs alone or with WT CD8+ T cells combined with ICIs (Figs. 6a–c and Supplementary Fig. S13). To further explore combinatorial strategies, we evaluated DDP, a widely used broad-spectrum chemotherapeutic agent that induced DNA damage, and the STING agonist ADU-S10014,28. We found that DDP effectively suppressed TC-1 tumor growth in NSG mice, but ADU-S100 alone showed no significant antitumor activity in the murine tumor model (Fig. 6d, e). To assess potential synergy, we reconstituted CD8+ T cell-depleted CD45.1 WT mice (via CD8-neutralizing antibody) with CD45.2 Sting1-cKD, Tox-cKO, or WT CD8+ T cells and established TC-1 xenografts. Mice reconstituted with Sting1-cKD or Tox-cKO CD8+ T cells exhibited significantly reduced tumor growth rate and tumor weight compared to those receiving WT CD8+ T cells, regardless of DDP or ADU-S100 treatment (Figs. 6f–h). Strikingly, combining Sting1-cKD- or Tox-cKO CD8+ T cells with DDP or ADU-S100 further enhanced tumor suppression, underscoring the therapeutic potential of ACT using genetic engineering T cells by targeting T cell-intrinsic STING/TOX axis in combination with conventional chemotherapy or immunomodulatory agents.

a Schematic of TC-1-bearing CD45.2 WT mice accepted with ACT combined with checkpoint blockade, cisplatin (DDP), ADU-S100 or isotype IgG. In briefly, the TC-1-bearing mice received intravenous injection (i.v.) infusion of pre-activated WT-(black), Sting1-cKD- (blue), or Tox-cKO-CD8⁺ T cells (red) on day 4, followed by intraperitoneal injection (i.p.) injection of anti-PD-1, anti-TIM-3 antibodies or IgG (50 μg per mouse) on days 4, 7, 10, and 13. b, c Tumor growth curves and endpoint tumor weights in TC-1-bearing CD45.2 WT mice treated with ACT plus anti-TIM-3 (b) (n = 4 mice/group) or anti-PD-1 (c) (n = 4 mice/group). d, e Experimental design (d) and tumor growth curves (e) of TC-1-bearing NSG mice (n = 5 mice/group) injected with DDP (50 μg per mouse, i.p.) or intratumorally (i.t.) injected with ADU-S100 (25 μg per mouse) on days 6, 9, 12, and 15. f Schematic of immune reconstitution: the CD45.1 WT mice were depleted of endogenous CD8⁺ T cells, reconstituted with WT- (black), Sting1-cKD- (blue), or Tox-cKO-CD8⁺ T cells (red), and implanted with TC-1 tumors, followed by DDP (50 μg per mouse, i.v.) on days 8, 11, 14, 17, and 20 or ADU-S100 (25 μg per mouse, i.t.) on days 9, 12, 15, and 18. g-h, Tumor growth curves and endpoint tumor weights in reconstituted CD45.1 WT mice (n = 4 mice/group) treated with DDP (g) or ADU-S100 (h). Numerical data was presented mean ± SD. Statistical analyses were performed using an unpaired two-sided Student’s t test. Schematics of therapeutic protocol in (a, d, f) were generated in the Generic Diagraming Platform. Zhu, Q. (2025) https://www.BioGDP.com/.
Prognostic significance of TOX and HO-1 in tumor specimens from patients with cervical cancer
Single-cell RNA-seq of peripheral blood and tumor-infiltrating immune cells from patients with CC (n = 2), together with flow cytometry analysis (n = 7), revealed preferential TOX expression in tumor-derived CD8⁺ and CD4⁺ T-cell clusters relative to peripheral counterparts, STING transcripts were broadly expressed across CD4⁺ and CD8⁺ T-cell subsets, with relative enrichment in intratumoral CD4⁺ T cells, and HO-1 mRNA was lower in peripheral and tumor-associated T-cell clusters relative to peripheral and tumor-associated B-cell clusters (Fig. 7a, b). Furthermore, transcriptional levels of TOX and STING exhibited a positively correlated in tumor-infiltrating CD8+ T cells (Fig. 7c). In our previous study, we demonstrated that a high abundance of STING-positive lymphocytes within tumor tissues was associated with poor prognosis in patients with cervical cancer29. Here, immunohistochemical (IHC) analysis of tumor specimens from 123 patients with CC revealed markedly higher levels of TOX and HO-1 in tumor-associated lymphocytes compared with adjacent non-tumor tissues (n = 14 paired samples; Fig. 7d). Strikingly, increased TOX expression or decreased HO-1 expression in tumor-infiltrating lymphocytes correlated with shorter disease-free survival (DFS) and overall survival (OS) in the CC cohort (n = 123; Fig. 7e, f). Together, these findings establish the STING-TOX–HO-1 axis as a prognostic determinant in cervical cancer progression.

Single-cell transcriptomic profiling was performed on CD45⁺ immune cells from peripheral blood and tumor-infiltrating lymphocytes (TILs) of patients with cervical cancer (CC, n = 2 patients, HRA003358). a Bubble plot showing expression of TOX, TMEN173, and HMOX1 across CD8⁺ T cells, CD4⁺ T cells, NK cells, and B cells from PBMCs and TILs. b Flow cytometric quantification of TOX⁺CD4⁺ and TOX⁺CD8⁺ T cells in PBMCs (green) and TILs (orange) from CC patients (n = 7 patients). c Violin plots of Sting1 expression in TOXlow and TOXhigh CD8⁺ TILs from 2 CC patients (two-sided Wilcoxon rank-sum test). d Representative Immunohistochemistry (IHC) images and quantification of TOX and HO-1 in tumor and matched adjacent tissues from CC patients (n = 14 patients). Original magnification × 400. Insets highlight TOX or HO-1 expression in tumor-infiltrating immune cells. Scale bars, 100 μm. e, f Kaplan–Meier analysis of disease-free survival (DFS, e) and overall survival (OS, f) in CC patients (n = 123 patients) stratified by TOX or HO-1 expression in TILs. Survival cutoffs were determined using the survminer R package; p-values were calculated by the log-rank test. g Schematic model summarizing the clinical association of lactate-induced CD8⁺ T cell dysfunction with poor survival and a preclinical framework for neoadjuvant combination immunotherapy was generated in the Generic Diagraming Platform. Zhu, Q. (2025) https://www.BioGDP.com/. b–d Statistical analyses were performed using an unpaired two-sided Student’s t test.
Discussion
The abundance of cytotoxic CD8+ T cells within the TME is a critical determinant of clinical outcomes in cancer patients undergoing immunotherapy, including ICB and ACT3. While activation-induced cell death (AICD) plays a key role in regulating T cell homeostasis during responses to allogeneic antigens, such as those encountered in viral infections or tumors, we demonstrate that STING- or TOX-deficient CD8+ T cells are resistance to ferroptosis, exhibiting enhanced tumor clearance, elevated intratumoral infiltration, increased cytotoxic cytokine production, a greater proportion of stem-like phenotypes, and reduced expression of exhaustion markers. These functional improvements are associated with upregulation of ferroptosis-inhibitory pathways and downregulation of genes involved in lipid peroxidation. Mechanistically, STING signaling acts in concert with TOX to repress HO-1 expression, leading to iron-overloaded mitochondrial dysfunction, characterized by reduced mitochondrial mass, diminished mtDNA copy numbers, impaired ATP synthesis, and elevated mitochondrial ROS and lipid peroxidation, culminating in CD8+ T cell ferroptosis. Tumor-derived lactate further amplifies this process through activation of STING and TOX pathways. Therapeutically, combining adoptive transfer of STING- or TOX-knockout engineered T cells with immune checkpoint inhibitors (anti-TIM-3/PD-1), STING agonizts (ADU-S100), or DDP significantly improved tumor clearance in murine models. Clinically, elevated TOX and reduced HO-1 levels in intratumoral T cells correlated with poor survival in patients with cervical cancer.
As an endoplasmic reticulum (ER)-resident adapter, STING is best known for mediating cytosolic DNA sensing through the cGAS-STING axis, thereby coordinating antimicrobial immunity and inducing type I interferons and proinflammatory cytokines30. Beyond its canonical role, accumulating evidence implicates STING in diverse contexts, including age-associated cellular senescence and ROS/DNA damage–driven tumor sensitization31,32. In tumor cells and myeloid populations, STING activation promotes antitumor immunity through type I IFN production19,29,30. By contrast, TCR engagement in T cells can also trigger STING signaling via ER stress and the unfolded protein response (UPR), leading to apoptosis and impaired memory T cell differentiation through induction of C/EBP homologous protein (CHOP) and indoleamine 2,3-dioxygenase (IDO) activity18,33. Moreover, T cell–intrinsic STING signaling has been shown to paradoxically promote immune tolerance by enhancing regulatory T cell expansion and driving calcium/ER stress–dependent apoptosis18,29. Similarly, TOX, a key regulator of thymic development, has been shown to promote exhaustion in tumor-specific CD8+ T cell and serves as a predictive biomarker for immunotherapy response21,34. In this study, we found that conditional knockout of STING or TOX in CD8+ T cells resulted in enhanced antitumor immunity, characterized by improved tumor clearance, increased intratumoral infiltration, a stem-like phenotype, and robust cytotoxic cytokine production. Furthermore, transcriptomic profiling of tumor-infiltrating CD8+ T cells revealed that STING or TOX deficiency reprograms pathways involved in iron transport, lipid metabolism, and granzyme-mediated apoptosis. These cells exhibited decreased expression of PD-1, TIM-3, ACSL4, and CTSB, but increased levels of HO-1 and GPX4. Functional assays confirmed reduced ferric ion accumulation and lipid peroxidation, alongside preserved mitochondrial integrity and viability under tumor challenge. This resistance to ferroptosis contrasts with established paradigms in which cGAS-STING activation promotes mtDNA release and ferroptotic cell death in tumor and myeloid cells, underscoring the cell type-specific regulation of this death pathway. STING activation has been reported to promote the formation of mitochondria-associated endoplasmic reticulum membranes (MAMs), thereby facilitating its trafficking from the ER to mitochondria35,36. Intriguingly, in both in vivo and in vitro settings, we observed mitochondrial localization of key regulators of type I interferon signaling and ferroptosis, including STING, phosphorylated TBK1, IRF3, HO-1, and ACSL4, in CD8⁺ T cells activated by tumor cells. A comparable mechanism, whereby STING activation drives mitochondrial ferroptosis, has also been reported in cardiomyocytes subjected to ischemia–reperfusion injury37. While prior studies have shown that viral activation of STING recruits the E3 ubiquitin ligase RNF5 for mitochondrial ubiquitination and degradation in myeloid cells38, our data reveal that tumor-induced STING signaling instead upregulates TOX expression and elevates Mito-ROS and lipid peroxidation in murine and human CD8+ T cells (Fig. S7). In addition, we identified a STING-TOX transcriptional positive feedback loop at the mRNA level, with IRF3 knockdown (shIrf3) in human and murine CD8+ T cells markedly reduced TOX protein expression (Supplementary Figs. S8f and S9a). Mechanistically, CD8+ T cells exhibited reduced HO-1 expression, increased ferric ion accumulation and low mitochondrial mass upon tumor cell or viral antigen stimulation, alongside the phenotypes reversible upon inhibition of STING signaling or iron chelation. HO-1, a rate-limiting heme oxygenase, liberates redox-active iron, a recognized ferroptosis driver in sickle cell disease39. However, HO-1 has been reported as a ferroptosis suppressor in liver cancer and renal proximal tubule cells40,41. Our findings are consistent with this suppressive role: HO-1 attenuated ferroptosis by mitigating iron overload, Mito-ROS, and mitochondrial lipid peroxidation. Importantly, ectopic expression of TOX or STING, or pharmacological inhibition of HO-1, restored both iron accumulation and ferroptosis susceptibility in TC-1-stimulated STING- and TOX-deficient CD8⁺ T cells in vitro. Therapeutically, adoptive transfer of Lenti-shHmox1-transduced TOX-deficient CD8⁺ T cells abrogated the enhanced tumor control conferred by TOX deficiency in vivo (Supplementary Fig. S10). Collectively, these results establish the STING-TOX-HO-1 axis as a critical regulator of ferroptosis in CD8+ T cells within TME.
Ferroptosis, an iron-dependent form of metabolic cell death, plays a dual role in tumor biology42; while its induction in tumor cells enhances antigen presentation and T cell cytotoxicity to suppress malignancies, it paradoxically undermines antitumor immunity by targeting infiltrating immune cells in the TME43. In the present study, we further demonstrated that tumor-infiltrating CD8⁺ T cells from patients with CC and NPC displayed heightened ferroptotic signatures, including increased STING activation and lipid peroxidation, relative to circulating counterparts (Supplementary Fig. S6). In vitro, tumor cell engagement induced STING and TOX signaling, suppressed HO-1 expression, and provoked mitochondrial dysfunction in CD8⁺ T cells (Supplementary Fig. S7); While specific inhibition of HO-1 in T cells declined the enhanced anti-tumor immunity of TOX-deficient T cells in the murine tumor model with ACT (SupplementaryFig. S10). Effector T cell functionality in the TME is regulated not only by canonical TCR (signal 1), co-stimulatory (signal 2), and cytokine (signal 3) pathways but also by metabolic cues (signal 4), including nutrient availability, ion homeostasis, and local pH44. Notably, TME-enriched factors such as labile iron, cholesterol, and fatty acids have been shown to drive ferroptosis in immune cells43,45. For instance, elevated fatty acids promote CD8+ T cell ferroptosis via CD36-mediated lipid uptake, thereby impairing their antitumor capacity43 Furthermore, the Warburg effect in tumor cells exacerbates lactate accumulation in the TME, a metabolite linked to metastatic progression and poor clinical outcomes46. Lactate has been shown to suppresses T and NK cell survival, facilitating immune evasion47,48,49. Lactate, abundantly secreted by tumors, selectively sensitizes murine CD8+ T cells but not tumor cells to mitochondrial reprogramming and ferroptosis (Supplementary Fig. S11a–g). Strikingly, STING- or TOX-deficient CD8+ T cells exhibited resistance to lactate-driven ferroptosis and mitochondrial dysregulation, a phenotype potentially linked to altered expression of Slc16a family genes (SupplementaryFig. S11h, i). WT-CD8+ T cells showed upregulation of STING and TOX signals and suppression of HO-1 expression under lactate stress, all of which were reversed by the MCT1 inhibitor AZD3965. In contrast, this reversal was markedly diminished in Sting1-cKO or Tox-cKO CD8⁺ T cells, consistent with defective activation of STING signaling and ferroptosis pathways. These findings identify lactate as a TME-derived mediator of CD8+ T cell ferroptosis through activation of the STING-TOX-HO-1 axis.
The clinical efficacy of ICB (e.g., anti-TIM-3), T cell-based adoptive cell therapy, and targeted agents such as STING agonizts in advanced cancers is substantially limited by the heterogeneity, complexity, and spatiotemporal dynamics of the tumor immune microenvironment4,14,50. This limitation persists despite the robust antitumor immunity elicited by STING agonizts in preclinical tumor models51,52,53. Drug resistance remains a major therapeutic barrier, promoting a shift toward neoadjuvant combinatorial strategies aimed at mitigating toxicity and limited efficacy associated with monotherapies54. Preclinical studies have demonstrated that combining TIL-based ACT with PD-1 blockade can overcome ICB resistance in solid tumors, while synergizing ADU-S100 or CAR-T-based ACT with PD-1 inhibitors enhances therapeutic outcomes15,55,56. In this study, we show that STING- or TOX-deficient engineered T cells not only augment the efficacy of ACT but also improve tumor clearance in neoadjuvant regimens combining ACT with TIM-3/PD-1 blockade, DDP, or ADU-S100 in murine models. Notably, ADU-S100 monotherapy lacked efficacy in NSG mice, underscoring its dependent on immune activation rather than direct tumor cytotoxicity. In contrast, DDP, a DNA-damaging agent, amplifies STING signaling across multiple cellular TME compartments57. Our findings suggest that inhibition of intracellular STING/TOX pathways enhances CD8+ T cell survival and effector functions (e.g., cytokine secretion), thereby potentiating ICB, chemotherapy, and targeted therapies in neoadjuvant settings. Clinically, elevated STING, TOX and reduced HO-1 expression in CC patients were associated with poor prognosis, reinforcing their respective roles in promoting immune tolerance. Collectively, we demonstrate that lactate accumulation in the TME induces iron-overloaded mitochondrial reprogramming and ferroptosis in CD8+ T cells via the STING-TOX-HO-1 axis, contributing to immune escape and adverse clinical outcomes. Importantly, the use of STING- or TOX-deficient engineered T cells significantly enhances tumor clearance in preclinical models when combined with TIM-3/PD-1 blockade, cisplatin, or ADU-S100 in neoadjuvant ACT regimens (Fig. 7f). These findings reveal a potential strategy to enhance anti-tumor immunity through modulation of T cell-intrinsic STING–TOX signaling and provide a rationale for optimizing neoadjuvant combination therapies integrating ICB, chemotherapy, targeted agents, and engineered adoptive T-cell therapy.
Methods
Generation of CD8 conditional STING or TOX knockout mice
Toxflox/flox and Cd8cre CD45.2 C57BL/6 J mice were purchased from Gempharmatech Co., Ltd (JiangSu, China). Sting1flox/flox CD45.2 C57BL/6 J mice were a gift from XJ. Xia (Sun Yat-sen University). Sting1flox/flox or/and Toxflox/flox CD45.2 C57BL/6 J mice were crossed with Cd8cre CD45.2 C57BL/6 J mice to generate STING or TOX conditional knockout (cKO) or knockdown (cKD) mice: Cd8creSting1flox/wt (Sting1-cKD), Cd8creSting1flox/flox (Sting1-cKO), Cd8creToxflox/flox (Tox-cKO) and Cd8creSting1flox/wtToxflox/wt (Sting1/Tox-D-cKD) mice. Wild type (WT) CD45.2 and CD45.1 C57BL/6 J mice, Sting1flox/flox and Toxflox/flox CD45.2 C57BL/6 J mice were included as controls. Mice were bred and maintained under specific pathogen-free conditions in the Animal Facility of the Sun Yat-sen University Cancer Center, and all experiments were performed according to the regulations of Sun Yat-sen University (L102012022001Z). For all experiments, 6 to 8-week-old mice were used unless otherwise indicated, with a randomized gender. Mice were kept under standard housing conditions with a 12:12 h light-dark cycle, an ambient temperature between 20–24 °C and humidity between 45–65%.
Human and murine cell lines culture
Human HEK-293T (ATCC, #CRL-3216), human cervical cancer cell line SiHa (ATCC, #HTB-35), human NPC cell lines HNE-1, C666 (provided from Dr. Mu-sheng Zeng, Sun Yat-sen University Cancer Center), and EBV+-TW03 (provided from Maria G Masucci, Karolinska Institutet), murine TC-1 (Jining BIO, #JN26363), B16F10 (ATCC, #CRL-6475), MC38 (Procell, #CL--0972) and CT26 (ATCC, #CRL-2638) cell lines were cultured in Dulbecco’s Modified Eagle Medium (Gibco) or RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum (ExcellBio, China). All of the cell lines were subjected to the mycoplasma test and were found to be free of contamination.
Human samples
Paraffin-embedded tumor tissues (n = 123) were collected from patients diagnosed with primary CC patients at the Sun Yat-Sen University Cancer (Guangzhou, China) from 2011 to 2015. The detailed clinical data are described in Supplementary Table 1. Peripheral blood from healthy donors was also included in this study.
Isogenous xenograft tumor models and therapy models
Isogenous xenograft tumor models
Sting1-cKO, Sting1-cKD, Tox-cKO, Sting1/Tox-D-cKD and WT mice (4 mice/group) were injected subcutaneously in the right rear flank with 4 × 105 TC-1 cells, 1 × 105 MC38 cells, and 2 × 105 B16F10 cells. In ACT models, TC-1-bearing NSG mice (5 mice/group) were administered preactivated 5 × 105 CD45.1⁺ WT-CD3+ T cells in combination with 5 × 105 CD45.2⁺ CD3⁺ T cells derived from WT, Sting1-cKD, Tox-cKO, or Sting1/Tox-D-cKD mice. Tumor growth was monitored every 3 days, and the tumor volume was calculated using the following formula: V = 0.5 × L × W2, where L is the length, and W is the width (in mm). The mice were sacrificed on the indicated days. The spleen, lymph nodes, and partial tumor were acquired from tumor-bearing mice and used for T cell isolation, and the partial tumor was fixed by formalin (Biosharp, China) for IHC or IF staining analysis.
Adoptive cell therapy using T cells with STING or HO-1 inhibition
For H151 (STING inhibitor)-treated-CD8+ T cell adoptive therapy, CD8+ T cells isolated from TC-1-preimmune CD45.2 WT mice were stimulated with 5 μg/mL H151 (MCE, USA) for 2 days in vitro. For Hmox1-knockdown (Lentivirus containing shHmox1 vector)-CD8+ T cells adoptive therapy, CD8+ T cells were isolated from the spleens and lymph nodes of TC-1-preimmune WT and Tox-cKO mice; TC-1-activated-WT-CD8+ T cells were treated with Lenti-pLKO (pLKO, empty vector), and TC-1-activated-Tox-cKO CD8+ T cells were transfected with Lenti-shHmox1 for 48 hours. CD45.1 WT mice (4 mice/group) were injected subcutaneously in the right rear flank with 4 × 105 TC-1 cells. TC-1-bearing CD45.1 WT mice were intravenously injected with 1 × 106 H151-treated-WT CD8+ T cells or shHmox1-transfected-Tox-cKO CD8+ T cells. After the mice were sacrificed, the tumor tissues were excised and weighed, and the T cells were isolated from the tumor tissues and used for subsequent experiments.
Combined therapy with STING- or TOX-deficient CD8+ T cells and immune checkpoint inhibitors, STING agonist or cisplatin in murine tumor model
For the ICB therapy group, WT mice (5 mice/group) were injected subcutaneously in the right rear flank with 2 × 105 TC-1 cells, and the TC-1-bearing WT mice were injected with anti-PD1 and anti-TIM3 antibodies (200 μg per mouse) on days 19, 21, 24, and 27. After these mice were sacrificed on day 37, the tumor tissues were excised and weighed. For combination therapy groups receiving ACT with engineered STING-, TOX-deficient or WT CD8+ T cells and low-dose PD-1 or TIM-3 neutralizing antibodies (50 μg per mouse), the TC-1-bearing CD45.2 WT mice were intravenously injected with 1 × 106 TC-1-preactivated CD8+ T cells from Sting1-cKD-, Tox-cKO-, or WT mice (4 mice/group) and intraperitoneal injected with anti-PD-1 and anti-TIM-3 antibodies (50 μg per mouse, BIOXCELL, USA) on days 4, 7, 10, and 13. After the mice were sacrificed on day 22, the tumor tissues were excised and weighed.
For the ADU-S100 and DDP monotherapy group: NSG mice (5 mice/group) were injected subcutaneously in the right rear flank with 4 × 105 TC-1 cells. TC-1-bearing NSG mice were intraperitoneal injected with DDP (50 μg per mouse) or intratumoral injected with ADU-S100 (25 μg per mouse) on days 6, 9, 12, and 15. The tumor was measured every 3 days, and weighted after the mice were sacrificed. Immune reconstitution: the endogenous CD8+ T cells of CD45.1 WT mice were depleted by intraperitoneal injected with mouse CD8-neutralizing antibody (200 μg per mouse, BIOXCELL, USA), and then intravenously injected with 1 × 106 CD45.2 Sting1-cKD-, Tox-cKO-, or WT-CD8+ T cells. The immune reconstituted CD45.1 mice were injected subcutaneously in the right rear flank with 4 × 105 TC-1 cells (4 per group). TC-1-bearing immune reconstituted CD45.1 WT mice were treated with DDP (50 μg per mouse, intraperitoneal injection, i.v.) on days 8, 11, 14, 17, and 20 or ADU-S100 (25 μg per mouse, intratumoral administration, i.t.) on days 9, 12, 15, and 18. The tumor was measured every 3 days, and weighted after the mice were sacrificed.
T cell isolation, transwell co-culture and transfection system
Lymphocytes were separated from health human donors or mice using human or murine Ficoll (Dakewe, China) and isodensity centrifugation, and cultured in OKT-3-coated 24-well plates (Corning, USA) in X-VIVO medium (Lonza, USA) with IL-2. CD8+ T cells isolated from lymphocytes using the CD4-positive selection kit and cocultured with tumor cells (Transwell, 0.4 μm pore size), antigens peptide (0.05 mg/mL E6/E7 or 0.05 mg/mL LMP1), cell death inhibitors, indicated concentrations of ferroptosis-inducer Erastin, 1 μg/mL DMXAA (STING agonist), TC-1 cells in the presence of increasing concentrations of the iron chelator DFO, indicated concentrations of lactate, or 500 nM AZD3965 (MCT1 inhibitor) for 48 h in OKT-3-coated 24-well plates in vitro.
Lentivirus was produced containing mouse TOX and STING full-length protein expression vector PCDH-TOX (TOX-OE) and PCDH-STING (STING-OE) or mouse Hmox1 and Irf3 knockdown vector pLKO-shHmox1 (shHmox1) and pLKO-shIrf3 (shIrf3), or human STING1- and TOX-sgRNAs recombinant plasmid. Lentivirus was added to 1 × 106 enriched CD8+ T cells supplemented with polybrene (10 μg/mL, Sigma-Aldrich, USA) and centrifuged at 800 × g at 37 °C for 1 h. The lentivirus-treated cells were cocultured with TC-1 in OKT-3-coated 24-well plates with a transwell co-culture system for 48 h in vitro. The primers of TOX and STING overexpression and shHmox1 and shIrf3 sequences are listed in Supplementary Dataset 1.
Bulk and single-cell transcriptome profile and quantitative RT-PCR analysis
Bulk RNA sequencing
CD8+ T cells from tumor tissues from tumor-bearing Sting1-cKO, Tox-cKO and WT mice were sorted using the mouse CD8a-positive selection kit (Stemcell, Canada) and used for bulk RNA-seq. RNA-seq libraries were sequenced by Illumina (Illumina, USA) with 150-bp paired-ends at Ribobio Co., Ltd (Guangzhou, China). Differential expression analysis was performed using the edgeR R package58 with the read counts as input. Gene set enrichment analysis (GSEA) was performed by the clusterProfiler R package59. The raw sequence data were deposited in the GSA database (Accession number: CRA011572) and publicly accessible at https://ngdc.cncb.ac.cn/.
Single-cell RNA sequencing
A single-cell library was constructed using Chromium Single Cell 3′ Reagent Kits following the manufacturer’s instructions (10 × Genomics) at Guangzhou Saliai Stem Cell Science and Technology Co., Ltd (Guangzhou, China). The gene expression data were aligned to the human genome reference sequence (GRCh38) using the Cellranger pipelines (version 3.0.2, USA). The DoubletFinder package was used to remove doublets, with an expected doublet rate of 0.0446. The data were analyzed by the Seurat R package60. DEGs were identified using the ‘FindMarkers’ function in the Seurat R package, with a significance threshold of p < 0.05 and |log fold change | > 0.25. Gene signatures were calculated using the ‘AddModuleScore’ function in the Seurat R package. A list of cell death-related genes can be found in Supplementary Dataset 1. GSEA was performed by the clusterProfiler R package61. The raw sequence data have been deposited in the Genome Sequence Archive58 in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA003358) and publicly accessible at https://ngdc.cncb.ac.cn/gsa-human. Single-cell RNA-seq data for nasopharyngeal carcinoma (GEO: GSE162025) was publicly accessible at the GEO database (http://www.ncbi.nlm.nih.gov/geo/).
Real-time quantitative PCR analysis
According to the manufacturer’s instructions, total RNA was extracted from cells using the EZ-press RNA Purification Kit (EZBioscience, USA). Quantitative real-time polymerase chain reaction (qPCR) was performed using a RevertAid First-Strand cDNA Synthesis Kit (Invitrogen, USA) and qPCR SYBR Master Mix (Vazyme, China). The primers are synthesized by Rui Biotech (Beijing, China) and primer sequences are shown in Supplementary Dataset 1.
Flow cytometry, immunofluorescence, immunoblot and Immunohistochemistry
Flow cytometry
Cells were washed twice using phosphate buffered saline (PBS) (Biosharp, China), labeled with fixable viability dye (eBioscience, USA), and stained for biomarkers of CD3, CD8, CD4, CD19, CD16, Ki67, PD1, TIM-3, CXCR5, TCF-1, Ly108, CD69, PI, and Annexin Ⅴ using fluorochrome-conjugated antibodies (see Supplementary Dataset 1) according to the manufacturer’s instructions. Intracellular cytokine IFN-γ and GrB staining was performed using a fixation/permeabilization solution kit (BD, USA) following the manufacturer’s instructions. For the cell proliferation assay, cells were labeled with CellTrace CFSE (CFDA-SE, Invitrogen, USA) according to the manufacturer’s instructions. Data were acquired by a Beckman Colter flow cytometer (CytoFLEX LX, Beckman, Germany) and analyzed by FlowJo software (BD, USA).
Immunofluorescence
Multiplex immunofluorescence staining was performed using the Opal Polaris 7-Color Manual IHC Kit (Akoya Biosciences, USA) according to the manufacturer’s instructions. The following primary antibodies were used with designated fluorophores: anti-mouse CD8 (white), TOX (yellow), PD-1 (red), TIM-3 (green), HO-1 (pink), GPX4 (green), and CTSB (orange) (see Supplementary Dataset 1 for details). Nuclei were counterstained with DAPI. Images were acquired using the Vectra 3.0 imaging system and analyzed with Vectra software (Akoya Biosciences, USA). Quantification of CD8⁺, TOX⁺, PD-1⁺, TIM-3⁺, HO-1⁺, GPX4⁺, and CTSB⁺ cells was performed using HALO software (Indica Labs). The frequencies of CD8⁺TOX⁺, CD8⁺PD-1⁺, CD8⁺TIM-3⁺, CD8⁺HO-1⁺, CD8⁺GPX4⁺, and CD8⁺CTSB⁺ subsets were calculated as a proportion of total CD8⁺ T cells across the entire tissue section.
Immunoblot
Murine or human CD8+ T cells were subjected to lysis in ice-cold lysis buffer (Beyotime, China) supplemented with 5 mg/mL protease inhibitor cocktail (Coolaber, China). Extracts (15–20 μL) were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The antibodies used are available in Supplementary Dataset 1.
Immunohistochemistry
Tumor tissue sections from 123 CC patients were incubated with anti-TOX and anti-HO-1 at 4 °C overnight (see Supplementary Table 1 and 2), and operated using an IHC kit (Zhongshan Jinqiao Biotechnology, China) according to the manufacturer’s instructions. The secondary antibody (Zhongshan Jinqiao Biotechnology, China) was incubated for 30 minutes at room temperature, and 3,3’-diaminobenzidine (DAB, Zhongshan Jinqiao Biotechnology, China) was used for visualization. Finally, the pathological sections were counterstained with hematoxylin (Zhongshan Jinqiao Biotechnology, China) and then dehydrated and sealed with neutral gum (Jieli Biotechnology, China) for optical microscopy (ECLIPSE Ni, NIKON, Japan).
Ferroptosis and metabolism assay
Cell death and viability assay
A total of 2 × 105 pre-treated-CD8+ T cells were seeded into 96-well plates (Corning, USA) and 10 μL of CCK-8 (Mien, China) mixed into each well. The absorbance at 450 nm was measured after 3 h using a microplate reader (Bio-Tek EPOCH2, USA).
Metabolism measurement
Experiments were performed according to the manufacturer’s protocol. Briefly, cells were washed with phosphate-buffered saline (PBS) twice and stained with 10 nM of Phen Green SK (Glpbio, USA) for 15 min at 37 °C, Lipid Peroxidation Sensor (1 μM, Thermo Fisher, USA) for 30 minutes at 37 °C, MitoTracker (25 nM, ThermoFisher, USA) for mitochondrial mass and MitoSOX Red (1 μM, Beyotime, China) for Mito-ROS for 30 minutes at 37 °C, and MitoPePP (0.2 μM, DOJINDO, Japan) for mitochondrial lipid peroxidation for 15 min at 37 °C. Cells were washed with PBS and examined by flow cytometry.
Nuclear cathepsin B and total cellular oxidized lipid peroxides measurement
Cells were stained by CTSB primary antibody by an Opal Polaris 7 color manual immunohistochemistry kit (Akoya Biosciences, USA). For cytosolic lipid peroxidation staining, cells incubated in 1640 cell culture medium with Lipid Peroxidation Sensor (1 μM, Thermo Fisher, USA) in a humidified chamber at 37 °C with 5% CO2 for 30 min. All stained cells were smeared to polylysine-coated slide and confocal laser microscopy LSM880 (Zeiss, Germany), and ImageJ plugin StarDist (NIH, USA) were used to analyze the images.
Quantification of ATP production and mtDNA content
The ATP was determined by use of the ATP determination assay (Beyotime Biotechnology, China). Mitochondrial DNA (mtDNA) was quantified with real-time PCR. Total DNA was isolated from CD8+ T cells using the Phenol-chloroform extraction method, and mtDNA was quantified using primers specific for the mouse mtCo2 gene and normalized using primers specific for the murine reference gene App. The primer sequences are shown in Supplementary Dataset 1.
Statistical analysis
All statistical analyses were performed using R software (version 4.0.3), GraphPad Prism 9 (La Jolla, CA, USA), and SPSS software (version 18.0; Chicago, IL, USA). Continuous variables are presented as the mean ± standard deviation (SD) and were compared using the unpaired Student’s t test or one-way analysis of variance (ANOVA), as appropriate. Nonparametric data were analyzed using the two-sided Wilcoxon test. Patient survival was assessed using the Kaplan–Meier method, and differences between groups were evaluated with the log-rank test. Optimal cut-off values were determined using the survival and survminer packages in R. A P-value < 0.05 was considered statistically significant; ns indicates no statistical significance (P > 0.05).
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of Sun Yat-sen University Cancer Center (G2022-133-01), and all patients and healthy donors signed a consent form. All experiments involved mice were performed according to the regulations of Sun Yat-sen University (L102012022001Z), and the maximum tumor volume is limited to 2000 mm³, or when the tumor metastasizes or grows rapidly to ulcerate, causing infection or necrosis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authenticity of this article has been validated by uploading the key raw data to the Research Data Deposit (RDD) public platform [https://www.researchdata.org.cn/] under RDD approval number RDDB2025700905. The single-cell RNA sequencing data generated in this study have been deposited in the GSA-Human database under accession code HRA003358 [https://ngdc.cncb.ac.cn/gsa-human/]. The bulk RNA sequencing data generated in this study have been deposited in the GSA database under accession code CRA011572 [https://ngdc.cncb.ac.cn/]. Single-cell RNA-seq data for nasopharyngeal carcinoma (GEO: GSE162025) was publicly accessible at the GEO database [http://www.ncbi.nlm.nih.gov/geo/]. All other data are available in the article and Supplementary files or from the corresponding author, Jiang Li (lijiang2@mail.sysu.edu.cn) upon request. Source data are provided in this paper.
References
Zebley, C. C., Zehn, D., Gottschalk, S. & Chi, H. T cell dysfunction and therapeutic intervention in cancer. Nat. Immunol. 25, 1344–1354 (2024).
Wang, T. W. et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611, 358–364 (2022).
Philip, M. & Schietinger, A. CD8(+) T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223 (2022).
Memon, D. et al. Clinical and molecular features of acquired resistance to immunotherapy in non-small cell lung cancer. Cancer Cell 42, 209–224 (2024).
Harding, J. J. et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: a phase Ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin. Cancer Res. 27, 2168–2178 (2021).
Schoenfeld, A. J. & Hellmann, M. D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 37, 443–455 (2020).
Oladejo, M., Paulishak, W. & Wood, L. Synergistic potential of immune checkpoint inhibitors and therapeutic cancer vaccines. Semin. Cancer Biol. 88, 81–95 (2023).
Lorusso, D. et al. Pembrolizumab or placebo with chemoradiotherapy followed by pembrolizumab or placebo for newly diagnosed, high-risk, locally advanced cervical cancer (ENGOT-cx11/GOG-3047/KEYNOTE-A18): a randomised, double-blind, phase 3 clinical trial. Lancet 403, 1341–1350 (2024).
Chen, C. & Xu, P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. 33, 630–648 (2023).
Li, W. et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci. Transl. Med. 12, eaay9013(2020).
Rai, P. et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 22, 312–321 (2021).
Wang, Q. et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014).
Somatilaka, B. N. et al. STING activation reprograms the microenvironment to sensitize NF1-related malignant peripheral nerve sheath tumors for immunotherapy. J. Clin. Invest. 134, e176748 (2024).
Meric-Bernstam, F. et al. Phase I dose-escalation trial of MIW815 (ADU-S100), an intratumoral STING agonist, in patients with advanced/metastatic solid tumors or lymphomas. Clin. Cancer Res. 28, 677–688 (2022).
Meric-Bernstam, F. et al. Combination of the STING agonist MIW815 (ADU-S100) and PD-1 inhibitor spartalizumab in advanced/metastatic solid tumors or lymphomas: an open-label, multicenter, phase Ib study. Clin. Cancer Res. 29, 110–121 (2023).
Knox, K. et al. Dynamic STING repression orchestrates immune cell development and function. Sci. Immunol. 10, eado9933 (2025).
Larkin, B. et al. Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 199, 397–402 (2017).
Wu, J. et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 216, 867–883 (2019).
Zhang, S. et al. Manganese induces tumor cell ferroptosis through type-I IFN dependent inhibition of mitochondrial dihydroorotate dehydrogenase. Free Radic. Biol. Med. 193, 202–212 (2022).
Galassi, C., Chan, T. A., Vitale, I. & Galluzzi, L. The hallmarks of cancer immune evasion. Cancer Cell 42, 1825–1863 (2024).
Scott, A. C. et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019).
Khan, O. et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218 (2019).
Seo, H. et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 116, 12410–12415 (2019).
Poss, K. D. & Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 94, 10919–10924 (1997).
Kovtunovych, G., Eckhaus, M. A., Ghosh, M. C., Ollivierre-Wilson, H. & Rouault, T. A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116, 6054–6062 (2010).
Deng, H. et al. Ubiquinol-mediated suppression of mitochondria-associated ferroptosis is a targetable function of lactate dehydrogenase B in cancer. Nat. Commun. 16, 2597 (2025).
Felmlee, M. A., Jones, R. S., Rodriguez-Cruz, V., Follman, K. E. & Morris, M. E. Monocarboxylate transporters (SLC16): function, regulation, and role in health and disease. Pharmacol. Rev. 72, 466–485 (2020).
Kowalczyk, J. T. et al. Exploiting the immunogenic potential of standard of care radiation or cisplatin therapy in preclinical models of HPV-associated malignancies. J. Immunother. Cancer 10, 142503 https://doi.org/10.1136/jitc-2022-005752 (2022).
Ni, H. et al. T cell-intrinsic STING signaling promotes regulatory T cell induction and immunosuppression by upregulating FOXP3 transcription in cervical cancer. J. Immunother. Cancer 10, https://doi.org/10.1136/jitc-2022-005151 (2022).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Glück, S. et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061 (2017).
Dou, Z. X. et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017).
Quaney, M. J. et al. STING controls T cell memory fitness during infection through T cell-intrinsic and IDO-dependent mechanisms. Proc. Natl. Acad. Sci. USA 120, e2205049120 (2023).
Sekine, T. et al. TOX is expressed by exhausted and polyfunctional human effector memory CD8(+) T cells. Sci. Immunol. 5, https://doi.org/10.1126/sciimmunol.aba7918 (2020).
Padhiar, A. A. et al. MAM-STAT3-driven mitochondrial Ca(+2) upregulation contributes to immunosenescence in type a mandibuloacral dysplasia patients. Adv. Sci. 12, e2407398 (2025).
Liu, J. et al. cGAS-STING, inflammasomes and pyroptosis: an overview of crosstalk mechanism of activation and regulation. Cell Commun. Signal 22, 22 (2024).
Wang, X. et al. STING aggravates ferroptosis-dependent myocardial ischemia-reperfusion injury by targeting GPX4 for autophagic degradation. Signal Transduct. Target. Ther. 10, 136 (2025).
Zhong, B. et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 (2009).
Menon, A. V. et al. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 139, 936–941 (2022).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Adedoyin, O. et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Renal. Physiol. 314, F702–F714 (2018).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Ma, X. et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 33, 1001–1012.e1005 (2021).
Raynor, J. L. & Chi, H. B. Nutrients: Signal 4 in T cell immunity. J. Exp. Med. 221, https://doi.org/10.1084/jem.20221839 (2024).
Xu, S. et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577 (2021).
Dai, E., Wang, W., Li, Y., Ye, D. & Li, Y. Lactate and lactylation: Behind the development of tumors. Cancer Lett. 591, 216896 (2024).
Quinn, W. J. et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 33, https://doi.org/10.1016/j.celrep.2020.108500 (2020).
Lopez, E. et al. Inhibition of lactate transport by MCT-1 blockade improves chimeric antigen receptor T-cell therapy against B-cell malignancies. J. Immunother. Cancer 11, https://doi.org/10.1136/jitc-2022-006287 (2023).
Brand, A. et al. LDHA-Associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 24, 657–671 (2016).
Hsu, J. M. et al. MTAP deficiency confers resistance to cytosolic nucleic acid sensing and STING agonists. Science 390, eadl4089 (2025).
Zhang, H. et al. Endothelial STING-JAK1 interaction promotes tumor vasculature normalization and antitumor immunity. J. Clin. Invest. 135, https://doi.org/10.1172/jci180622 (2025).
Girard, M. et al. STING agonists drive recruitment and intrinsic type I interferon responses in monocytic lineage cells for optimal anti-tumor immunity. J. Immunol. 214, 3634–3646 (2025).
Ramanjulu, J. M. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443 (2018).
Chi, H., Pepper, M. & Thomas, P. G. Principles and therapeutic applications of adaptive immunity. Cell 187, 2052–2078 (2024).
Hall, M. S. et al. Combination nivolumab, CD137 agonism, and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Clin. Cancer Res. 28, 5317–5329 (2022).
L’Orphelin, J. M. et al. NIVO-TIL: combination anti-PD-1 therapy and adoptive T-cell transfer in untreated metastatic melanoma: an exploratory open-label phase I trial. Acta Oncol. 63, 867–877 (2024).
Cohen, S. M. & Lippard, S. J. Cisplatin: from DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 67, 93–130 (2001).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Stuart, T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 (2019).
Xu, C. et al. Comprehensive multi-omics single-cell data integration reveals greater heterogeneity in the human immune system. iScience 25, 105123 (2022).
Acknowledgements
This work was supported by the National Key Research and Development Program (2025YFC3409800) and the National Natural Science Foundation of China (grant no. 82272780 and 81972874 to J.L., and 8216110322 to H.H.).
Author information
Authors and Affiliations
Contributions
J.L., Q.Z., J.B.Z., C.P.N., and X.F.L. were responsible for the study conceptualization. Q.Z., J.B.Z., C.P.N., L.P.Z., M.L., X.L.Z., and J.H. conducted experiments. X.F.L. contributed to the bioinformatics analysis. H.H., X.J.X, S.G., and X.S.Z. provided gene-edited mice and reagents. J.L., Q.Z., and X.F.L. were involved in the writing and revision of the manuscript. All authors have read and approved the manuscript. The order of the co-first authors was determined on the basis of their efforts and contributions to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Peer review
Peer review information
Nature Communications thanks anonymous reviewers for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Q., Zhang, Jb., Nie, Cp. et al. STING synergizes with TOX suppressing HO-1 expression to trigger ferroptosis in tumor-infiltrating CD8+ T cell and immunotherapy resistance. Nat Commun 17, 2543 (2026). https://doi.org/10.1038/s41467-026-69350-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69350-y
