
Abstract
Inflammatory bowel disease (IBD) is characterized by chronic inflammation and impaired immune tolerance, for which current therapies provide only partial and transient relief. Here, we introduce PrEXO-a23, a biomimetic nanotherapeutic engineered by fusing regulatory T cell (Treg)-derived exosomes with platelet membrane vesicles and conjugating interleukin-23 (IL-23) antibodies via a matrix metalloproteinase (MMP)-cleavable linker. This design exploits the inherent homing ability of platelets and Tregs, enabling PrEXO-a23 to preferentially accumulate in inflamed colonic tissues in murine IBD models. At the disease site, elevated MMP activity triggers antibody release to inhibit IL-23-mediated inflammation, while exosomal cargo reprograms dendritic cells and promotes Treg expansion, thereby restoring immune tolerance. This dual-action strategy significantly alleviates IBD, prevents complications like intestinal fibrosis and colitis-associated colorectal cancer, and shows p53-dependent efficacy in carcinogenesis prevention. These findings highlight PrEXO-a23 as a promising nanotherapeutic platform for durable immune reprogramming and long-term IBD management.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic relapsing gastrointestinal disorder characterized by debilitating symptoms such as diarrhea, abdominal pain, and rectal bleeding, which often progress to life-threatening complications1. A key driver of IBD pathogenesis is sustained intestinal inflammation coupled with impaired immune tolerance, leading to a vicious cycle of immune dysregulation2,3. Although current treatments, including probiotics, small-molecule medicines, immunosuppressants, and biologics, can reduce disease symptoms, their efficacy in reversing immune dysregulation remains limited4,5,6. Critically, unresolved immune imbalance increases the risk of disease relapse and contributes to progressive complications such as intestinal fibrosis and colitis-associated colorectal cancer (CAC). Intestinal fibrosis is characterized by thickened intestinal walls and reduced peristalsis, eventually leading to intestinal stenosis and life-threatening obstruction. The cumulative incidence of intestinal fibrosis is reported as >50% in CD and 2–11% in UC7,8. In parallel, chronic immune dysregulation promotes somatic mutations in intestinal epithelial cells, including key tumor suppressor genes such as TP53, driving the progression to CAC. Patients with IBD have an ~2–3-fold increased risk of developing CAC compared to the general population9,10,11. Despite this, there are currently no FDA-approved treatments for intestinal fibrosis8, and CAC remains associated with poor clinical outcomes11. Therefore, there is an urgent need for therapeutic strategies that not only suppress inflammation but also restore immune tolerance to correct immune dysregulation, with the goal of reducing disease severity and long-term complications.
The proinflammatory cytokine interleukin-23 (IL-23) plays a central role in the pathogenesis of IBD by promoting the differentiation, expansion, and effector function of T-helper 17 (Th17) cells12. Th17 cells exacerbate IBD by secreting proinflammatory cytokines (e.g., IL-17A), which drive epithelial barrier dysfunction and chronic tissue damage, thereby perpetuating the inflammatory cascade13,14. Therapeutic antibodies targeting IL-23 specifically neutralize these cytokines, effectively disrupting the inflammatory cascade and reducing disease symptoms. Clinical studies have shown that IL-23 antibodies achieve remission rates of ~40%, substantially outperforming conventional therapies, which typically yield remission rates below 20%15,16. However, systemic administration of IL-23 antibodies may lead to off-target immunosuppression, increasing the risk of opportunistic infections17. Consequently, the development of targeted delivery systems is essential to enhance therapeutic safety and improve treatment efficacy.
Regulatory T cells (Tregs) are crucial for maintaining immune tolerance18,19, making Treg-based therapies promising candidates for IBD treatment, as supported by ongoing clinical and preclinical studies20,21,22. However, the direct application of Tregs is challenged by complex isolation and expansion protocols, as well as their reduced stability within the inflammatory microenvironment14,23. As an alternative, exosomes derived from Tregs have emerged as potent immunomodulatory nanovesicles. These nanoscale vesicles carry diverse functional biomolecules, including cytokines, microRNAs, and proteins, that facilitate immune regulation by targeting and reprogramming recipient immune cells24,25. Importantly, rEXO therapy bypasses the stability issues associated with cell-based Treg therapy, making it a compelling strategy for modulating immune tolerance in IBD.
The pathological features of IBD include an inflammatory environment and damaged intestinal vasculature26,27. High-affinity integrins that bind inflammation-upregulated endothelial adhesion molecules are expressed on the Treg cell surface, enabling the targeting of inflamed lesions28,29. Platelets, which possess unique surface molecules (e.g., GPIbα), can preferentially localize to damaged vasculature through robust adhesion to collagen exposed on the injured vascular endothelium30,31. The platelet-Treg hybrid engineering strategy offers an approach for enabling lesion-specific drug delivery through a dual-targeting mechanism for IBD therapy.
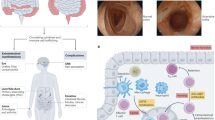
Inspired by these findings, we engineered a platelet-Treg hybrid nanovesicle system (PrEXO-a23) for lesion-specific delivery of anti-inflammatory and immune tolerance agents to address immune dysregulation for IBD treatment. rEXO was fused with platelet membrane vesicles (PMV) to generate hybrid nanovesicles (PrEXO), followed by the conjugation of IL-23 antibodies (a23) via a matrix metalloproteinase (MMP)-cleavable peptide linker. MMPs are overexpressed and released by activated stromal cells and immune cells infiltrating the inflamed intestinal mucosa in IBD32. The introduction of an MMP9-cleavable linker can exploit the pathological microenvironment to control drug release, thereby maximizing local therapeutic efficacy while minimizing systemic exposure and off-target immunosuppression24,33. Thus, after intravenous administration of PrEXO-a23, it selectively accumulated at sites of damaged and inflamed colonic lesions due to platelet-mediated damage adhesion and Treg-derived inflammation homing. Cleavage by MMPs promoted the release of a23 and PrEXO within MMP-enriched IBD lesions. The release of a23 neutralized the cytokine IL-23, which suppressed inflammation, while the PrEXO, enriched with immunomodulatory molecules from Tregs, reprogrammed local immune cells into regulatory phenotypes, such as tolerogenic dendritic cells (tDCs) and Tregs, promoting immune tolerance. As a result, in murine IBD models, PrEXO-a23 treatment effectively modulated colonic immune dysregulation, leading to comprehensive IBD remission, including symptom relief, intestinal barrier repair, and microbiota normalization. Furthermore, PrEXO-a23 significantly decreased the risk of complications, such as intestinal fibrosis and CAC. Notably, the action of PrEXO-a23 to prevent CAC progression is partially dependent on p53 (Fig. 1).

The preparation of PrEXO-a23 and its experimental design for addressing immune dysregulation to ameliorate IBD and decrease the risk of complications, such as intestinal fibrosis and colitis-associated colorectal cancer (CAC). The action of PrEXO-a23 in preventing IBD-to-CAC progression is partially dependent on p53. Treg: regulatory T cells, rEXO: Treg-derived exosomes, PMV: platelet membrane vesicles, a23: interleukin-23 antibodies, MMP: matrix metalloproteinase, mDC: mature dendritic cells, tDC: tolerogenic dendritic cells, Th17: T-helper 17 cells.
Results
Engineering biomimetic exosome-based nanovesicle (PrEXO-a23) for targeted delivery of the immunomodulatory IL-23 antibody
Naïve CD4⁺ T cells isolated from mouse spleens successfully differentiated into Tregs, as confirmed by flow cytometry analysis showing CD25 and FOXP3 expression on day 7 (Supplementary Fig. 1). Subsequently, Treg-derived exosomes (rEXO) were isolated from the cellular supernatant by ultracentrifugation, and verified by Western blotting to retain Treg immunosuppressive proteins, such as IL-10 and CTLA-4 (Supplementary Fig. 2). Platelet membrane vesicles (PMV) were prepared from whole blood using repeated freeze-thaw cycles followed by serial extrusion. rEXO and PMV were then fused through additional coextrusion to generate hybrid vesicles (PrEXO). Nanoflow cytometry analysis revealed that a 1:1 mass ratio of PMV to rEXO achieved the highest fusion efficiency (Supplementary Fig. 3), and this ratio was used for all subsequent experiments. To equip these hybrid vesicles for controlled antibody delivery, we employed an MMP-cleavable peptide linker (Arg-Val-Gly-Leu-Pro) bearing N-hydroxysuccinimide (NHS) and maleimide (Mal) termini. The NHS end reacted with primary amines on the antibody to yield linker-a23 (Supplementary Fig. 4). When the molar ratio of a23 to linker was 1:50, the conjugation efficiency of linker to a23 was the highest (Supplementary Fig. 5a). Subsequently, the surface thiols of PrEXO were activated by mild reduction and reacted with linker-a23 to form stable thioether bonds through the maleimide group to generate PrEXO-a23. Enzyme-linked immunosorbent assay (ELISA) determined the conjugation efficiency of a23 to PrEXO, revealing that a PrEXO-to-a23 mass ratio of 1:0.5 achieved the optimal conjugation efficiency (Supplementary Fig. 5b). Therefore, throughout this study, a a23-to-linker molar ratio of 1:50 and a PrEXO-to-a23 mass ratio of 1:0.5 were used to prepare the PrEXO-a23 formulations.
Transmission electron microscopy (TEM) analysis demonstrated that PrEXO-a23 maintained the typical cup-shaped morphology of exosomes (~100 nm), and immunogold labeling confirmed surface conjugation of a23 (Fig. 2a). Compared with PrEXO, PrEXO-a23 exhibited a slight increase in particle size and a reduction in zeta potential (Fig. 2b, c and Supplementary Fig. 6). Confocal laser scanning microscopy (CLSM) results revealed high colocalization of DiI-labeled rEXO, DiD-labeled PMV, and Alexa Fluor 488-labeled a23 signals in PrEXO-a23 (Fig. 2d), confirming successful construction of PrEXO-a23. Western blot analysis verified the presence of exosomal proteins (ALIX, TSG101), chemokine receptor (CCR6), platelet markers (GPIbα and CD62P), and a23 on PrEXO-a23 (Fig. 2e and Supplementary Fig. 7), indicating the retention of functional components. MMP9 expression was markedly upregulated in inflamed colonic tissues of IBD mice compared with healthy controls (Supplementary Fig. 8a, b) and it was significantly higher than that in the serum of the same mice (Supplementary Fig. 8c). This enzyme facilitated responsive release of a23 from PrEXO-a23, which was suppressed by the MMP9 inhibitor GM6001 (Fig. 2f). The released a23 preserved its IL-23 neutralization activity (Fig. 2g). Notably, PrEXO-a23 remained stable over 7 days in PBS at 4 °C (Fig. 2h) and in plasma at 37 °C (Supplementary Fig. 9), with no detectable changes in size or zeta potential, supporting its potential for in vivo application.

a Representative TEM images of rEXO, PMV, PrEXO, and PrEXO-a23. PrEXO-a23 was bound to secondary antibody-linked gold nanoparticles. Scale bar, 50 nm. b, c Particle size (b) and zeta potential (c) of rEXO before and after engineering with PMV and a23. d CLSM images of PrEXO-a23. Orange: rEXO; Red: PMV; Green: a23. Scale bar, 10 μm. Insets show images of the outlined regions in the main image. Scale bar, 100 nm. e Western blot analysis of protein markers in rEXO, PMV, a23, and PrEXO-a23. Full scans are provided in Supplementary Fig. 7. f ELISA quantification of a23 release under different treatments. g ELISA-based binding analysis of released a23 and free a23. h Size and zeta potential of PrEXO-a23 in PBS over 7 days. For (b, c, f–h), data were presented as mean ± SD (n = 3 independent experiments). For (f), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Anti-inflammatory and epithelial cell protective effects of PrEXO-a23
To investigate the regulatory effects of PrEXO-a23 on Th17 cell differentiation and its potential to protect intestinal epithelial cells, we established a coculture model. Naïve CD4+ T cells were seeded in the upper chamber and treated with IL-6 and IL-23 to induce Th17 polarization. Cells were then treated with PBS (Th17 group), rEXO (rEXO group), PMV (PMV group), PrEXO (PrEXO group), a23 (a23 group), or PrEXO-a23 (PrEXO-a23 group). Untreated cells served as the negative control (Control group). After 72 h of treatment, colonic epithelial NCM460 cells were seeded into the lower chamber and cocultured with the pretreated T cells for an additional 24 h to evaluate the epithelial response (Fig. 3a).

a Experimental schema of the coculture system between naïve T cells and NCM460 cells. Created in BioRender. Zhang, F. (2026) https://BioRender.com/1wcbfv8. b Flow cytometric quantification of Th17 cells (CD4+ IL-17A+) in CD4+ T cells. c, d ELISA analysis of IL-17A (c) and TNF-α (d) levels in CD4+ T cell culture supernatants. e Flow cytometric quantification of cell death rate in NCM460 cells. f Experimental schema of the shift of mDCs into tDCs. Created in BioRender. Zhang, F. (2026) https://BioRender.com/sp3rrne. g Flow cytometric analysis of the expression of CD80/CD86 and MHC II in CD11c+ cells for DCs. h ELISA analysis of IL-6, TNF-α, IL-10, and TGF-β, in supernatant of DCs. i Experimental schema of T cell differentiation assay in naïve T cells cocultured with pretreated DCs, analyzed using flow cytometry. Created in BioRender. Zhang, F. (2026) https://BioRender.com/sp3rrne. j, k Polarization of Tregs (CD25+ FOXP3+) (j) and Th17 (CD4+ IL-17A+) cells (k) in CD4+ T cells. For (b–e, g, h, j, k), data were presented as mean ± SD (n = 4 independent experiments in b–e, n = 5 independent experiments in g, h, j, k). For (b–e, g, h, j, k), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Flow cytometry analysis revealed that IL-6 and IL-23 successfully induced the differentiation of naïve T cells into Th17 cells in the Th17 group, as evidenced by an increased proportion of CD4+IL-17A+ cells compared to the control (Fig. 3b and Supplementary Fig. 10a). Compared with the Th17 group, PMV treatment showed no inhibitory effects on Th17 cell differentiation. Treatment with the IL-23 neutralizing antibody a23 significantly reduced Th17 differentiation (P = 0.0001), confirming its functional blocking activity. rEXO and PrEXO treatments showed effective inhibitory effects, while PrEXO-a23 exhibited the most pronounced suppression, reducing Th17 cell differentiation to levels comparable to untreated controls. Meanwhile, ELISA analysis of supernatants revealed elevated levels of proinflammatory cytokines IL-17A and TNF-α in the Th17 group, consistent with Th17 activation. There was no significant difference in the secretion of these inflammatory cytokines in the PMV group (Fig. 3c, d). In contrast, rEXO and PrEXO treatments reduced the levels of these cytokines, with PrEXO-a23 displaying a more pronounced reduction and bringing them to levels comparable to the control.
We next examined the effects of Th17 polarization on epithelial cell viability. The results showed that apoptosis and necrosis of NCM460 cells were significantly increased when cocultured with Th17 cells in the Th17 group (P < 0.0001), indicating epithelial cell damage. Similarly, PMV alone exerted minimal protective effect against Th17-induced epithelial damage. Notably, PrEXO-a23 treatment significantly alleviated this damage, reducing both apoptosis and necrosis and demonstrating a robust epithelial protective effect (P < 0.0001, Fig. 3e and Supplementary Fig. 10b, c). These findings clearly indicate that PrEXO-a23 protects intestinal epithelial cells from damage by inhibiting Th17 cell differentiation and inflammatory cytokine secretion, thereby exerting anti-inflammatory and epithelial protective effects.
PrEXO-a23 reprograms dendritic cells toward a tolerogenic phenotype and promotes the expansion of Tregs
Dendritic cells (DCs) are key regulators of immune responses, shaping T-cell differentiation through their distinct phenotypes34. Mature DCs (mDCs) drive immune effector functions, characterized by elevated expression of the costimulatory markers (CD80, CD86 and MHC II) and secretion of proinflammatory cytokines (e.g., IL-6 and TNF-α). In contrast, tolerogenic DCs (tDCs) display reduced levels of these markers and increased production of immunosuppressive cytokines, including IL-10 and TGF-β, promoting immune tolerance by facilitating Treg induction and suppressing Th17 cell differentiation35,36,37.
Given the role of Treg-derived exosomes in maintaining immune tolerance38, we evaluated whether PrEXO-a23 could reprogram mDCs toward a tolerogenic phenotype. The immature DCs (imDCs) were stimulated with lipopolysaccharide (LPS) and IFN-γ to induce mature mDCs and subsequently treated with various formulations, including PBS (mDC group), rEXO (rEXO group), PMV (PMV group), PrEXO (PrEXO group), a23 (a23 group), or PrEXO-a23 (PrEXO-a23 group) for 24 h. Untreated immature DCs (imDCs group) served as the negative control (Fig. 3f). The expression of surface markers and cytokine profiles was analyzed using flow cytometry and ELISA, respectively. As expected, mDCs showed increased expression of CD80, CD86, and MHC II compared to imDCs (Fig. 3g and Supplementary Fig. 11a), as well as increased production of IL-6 and TNF-α (Fig. 3h). PMV treatment did not significantly affect these markers or cytokines on mDCs. While treatment with a23 markedly downregulated these maturation markers and reduced the proinflammatory cytokine levels, indicating an anti-inflammatory effect, the levels of IL-10 and TGF-β were comparable to those in mDCs, suggesting that a23 alone was insufficient to fully reprogram DCs into a tolerogenic state. In contrast, both rEXO and PrEXO treatments resulted in downregulation of maturation markers and proinflammatory cytokines while promoting secretion of IL-10 and TGF-β, making a shift toward a tolerogenic phenotype. Notably, PrEXO-a23 exhibited the most potent suppressive effects on inflammatory markers and the strongest promotion of anti-inflammatory cytokine production, suggesting its robust role in inducing DC transformation into tDCs. This enhanced efficacy was likely due to the anti-inflammatory effects of a23, which created a favorable microenvironment for PrEXO to reprogram mDCs into tDCs.
To further explore the potential of PrEXO-a23–induced tDCs in promoting Treg differentiation, we cocultured the pretreated mDCs with naïve T cells for 72 h, and T cell phenotypes were determined via flow cytometry (Fig. 3i). Flow cytometry analysis revealed that PBS-treated mDCs from the mDC group did not significantly induce Treg differentiation compared to imDCs (P = 0.9998, Fig. 3j and Supplementary Fig. 11b). Likewise, PMV-treated and a23-treated mDCs failed to promote Treg differentiation. However, the proportion of Tregs (CD25+ FOXP3+) among naïve T cells was significantly increased in both rEXO- and PrEXO-treated mDCs groups compared to the mDCs group. Remarkably, PrEXO-a23-induced tDCs led to the most significant increase in Treg differentiation (P < 0.0001). Additionally, these tDCs effectively reduced the proportion of Th17 cells (CD4+ IL-17A+) compared to other treatments, with rEXO, PrEXO, and a23 showing moderate effects, and PMV alone showing a negligible effect (Fig. 3k and Supplementary Fig. 11c).
Collectively, while a23 monotherapy effectively inhibited Th17 differentiation and proinflammatory cytokine secretion, it showed limited capacity to promote tDC and Treg generation. PMV did not induce immune tolerance under these conditions. In contrast, both rEXO and PrEXO effectively induced Treg differentiation over Th17 cells by promoting DC polarization toward a tolerogenic phenotype, thereby supporting immune tolerance. Notably, PrEXO-a23 demonstrated a substantially greater ability to induce tDCs and expand Tregs. This enhanced efficacy stems from the environmental regulation of DC and T cell differentiation, where an inflammatory microenvironment-dominated by cytokines like IL-23, drives polarization toward proinflammatory states. Sustained IL-23 signaling suppresses the expression of FOXP3, a transcription factor critical for Treg function, thereby impairing Treg differentiation and exacerbating immune tolerance loss19,39,40. By neutralizing IL-23, a23 mitigated this inhibitory milieu, enabling the intrinsic tolerogenic cargo of PrEXO to fully reprogram DCs and T cells toward a regulatory state.
PrEXO-a23 enables precise targeted delivery and MMP-responsive release of antibody to inflamed colonic lesions
During IBD, inflammatory injury to the intestinal endothelium exposes subendothelial collagen, to which platelets rapidly adhere via GPIbα-von Willebrand factor (vWF) interactions in the bloodstream41. To determine whether PrEXO‑a23 retains this critical adhesive capability, we performed an in vitro blood circulation assay using collagen-coated flow channels that mimic damaged vessels. DiO-labeled formulations, including rEXO, PMV, PrEXO, or PrEXO-a23, were introduced into the flow channel under arterial flow conditions (1000 s−1), and fluorescence imaging was performed using CLSM. While rEXO displayed weak fluorescence in the flow channel, both PrEXO and PrEXO-a23 showed strong fluorescent signals (Supplementary Fig. 12a), indicating strong adherence to the collagen matrix. These results confirmed that platelet membrane hybridization conferred enhanced adhesion under high shear, prolonging retention at lesion sites, and that a23 conjugation did not impair this capability.
Next, to determine its targeting to inflammatory cells, we incubated LPS-activated RAW264.7 macrophages and neutrophils with DiI-labeled formulations. CLSM revealed that all formulations strongly bound to LPS-treated RAW264.7 cells after 2 h, whereas binding to untreated controls was minimal (Supplementary Fig. 12b). Flow cytometry result of LPS-pretreated neutrophils confirmed this trend (Supplementary Fig. 12c). This enhanced targeting ability is likely due to the increased molecular interactions between adhesion receptors overexpressed on inflamed cells and the corresponding ligands on the hybrid membranes. Additionally, in the presence of RAW264.7 cells, PrEXO-a23 efficiently released a23 upon exposure to MMP9 (Supplementary Fig. 12d).
Encouraged by these in vitro findings, we investigated the in vivo targeting performance of PrEXO-a23 in a dextran sulfate sodium (DSS)-induced murine IBD model. DiD-labeled formulations, including rEXO, PMV, rEXO + PMV (physical mixture), PrEXO or PrEXO-a23, were administered intravenously, and biodistribution was monitored via an in vivo imaging system. PrEXO and PrEXO-a23 showed strong accumulation in inflamed intestinal tissues within 2 h after injection, reached a peak at 12 h, and maintained strong retention for up to 48 h. The other formulations showed weaker and less durable intestinal targeting (Fig. 4a and Supplementary Fig. 13a). Pharmacokinetic analysis showed that the blood circulation half-life of PrEXO-a23 was ~1.44 h (Supplementary Fig. 13b). Ex vivo imaging of harvested colons and frozen tissue sections confirmed significantly higher lesion-targeting by PrEXO and PrEXO-a23 compared to controls (P < 0.0001, Fig. 4b–d). PrEXO-a23 exhibited the highest colon-to-liver ratios of fluorescence intensity, further indicating its enhanced disease-site selectivity (Supplementary Fig. 13c–e). Additionally, flow cytometry of intestinal immune cells revealed efficient uptake of PrEXO-a23 by dendritic cells, macrophages, T cells, B cells, and neutrophils (Fig. 4e and Supplementary Fig. 13f). Importantly, PrEXO-a23 outperformed rEXO in lesion-targeting, suggesting that membrane fusion synergistically enhances adhesion to damaged vessels (via PMV) and targeting of inflamed cells (via rEXO). Thus, PrEXO-a23 demonstrates dual-targeting capability, efficiently homing to both damaged vasculature and inflamed colonic tissue, offering a promising targeted delivery strategy for IBD therapy.

a Quantification of fluorescent radiant efficiency in the whole body of IBD mice at the indicated time points after injection of DiD-labeled formulations. b, c Colon tissues were isolated and imaged at 12 h. Ex vivo fluorescence images of isolated colonic tissues (b) and quantification of fluorescent radiant efficiency (c). d CLSM images and quantitative data of DiD-labeled formulations in frozen colonic sections. Red: DiD; Blue: nucleus; Scale bar, 100 μm. e Flow cytometric analysis of DiD-positive immune cells in colonic lamina propria including neutrophils, DCs, macrophages, T cells, and B cells. f Representative in vivo fluorescence images of the whole body and isolated colon of IBD mice at the indicated time points after injection of PrEXO + a23, PrEXO-fL-a23, or PrEXO-a23 (a23 labeled with Alexa Fluor 647; PrEXO labeled with DiR). g Quantification of fluorescence radiant efficiency in the whole body of PrEXO (top) and a23 (bottom) in (f). h Quantification of fluorescence radiant efficiency in isolated colon at 12 h after injection for PrEXO and a23 in (f). i Representative CLSM images (left) and quantitative analysis (right) of colonic tissue sections. Red: PrEXO; Green: a23. Scale bar, 20 μm. For (a, c–e, g, h), data were presented as mean ± SD (n = 5 mice in a, c, g, h; n = 3 mice in d, e). For (c–e, h), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
To further elucidate the mechanism of targeted delivery and MMP-responsive antibody release by PrEXO-a23, we performed dual-label in vivo tracking analysis to independently monitor the antibody (a23) and the carrier (PrEXO). a23 was labeled with Alexa Fluor 647, and the PrEXO was labeled with DiR. Three groups were included for comparison: PrEXO + a23 (a physical mixture of PrEXO and a23), PrEXO-fL-a23 (PrEXO and a23 were conjugated with a non-cleavable peptide, fL), and PrEXO-a23 (PrEXO and a23 were conjugated with the MMP-cleavable peptide). Following intravenous administration, in vivo and ex vivo imaging were performed at predetermined time points to assess distribution and retention in DSS-induced IBD mice and isolated colons. In the PrEXO + a23 group, the DiR-PrEXO signal robustly accumulated in colonic lesions, peaked at 12 h, and persisted for up to 48 h, demonstrating that PrEXO effectively targets inflamed colon tissues (Fig. 4f–h), consistent with the targeting observed in Fig. 4a–c. However, the Alexa Fluor 647-a23 signal in this group was weak and rapidly diminished, becoming almost undetectable after 12 h, indicating poor lesion-targeting and rapid clearance of free antibody. In contrast, both PrEXO-fL-a23 and PrEXO-a23 groups displayed PrEXO signals of similar intensity and duration to the physical mixture, but importantly, their a23 signals were markedly stronger and more persistent (higher than in the physical mixture, Fig. 4h). These results demonstrated that both PrEXO and a23 in PrEXO-a23 were effectively delivered to colonic lesions, which was attributed to the lesion-targeting capability of PrEXO, enabling the co-delivery of a23 via linkage.
Next, we performed CLSM analysis on colon tissue sections from IBD mice to confirm the release of PrEXO and a23 following MMP9-responsive cleavage of PrEXO-a23 at lesions. As shown in Fig. 4i, the non-cleavable PrEXO-fL-a23 group displayed strong spatial colocalization of a23 (green) and PrEXO (red), indicating the simultaneous delivery of the antibody and carrier to lesions. In contrast, the clear spatial separation of the two signals was detected in the PrEXO-a23 group, confirming in situ cleavage of the MMP-cleavable linker and subsequent release of a23 and PrEXO at the colonic lesions.
Taken together, these findings demonstrate that PrEXO-a23 not only achieves targeted co-delivery of both carrier and antibody to the site of inflammation but also undergoes efficient MMP-mediated cleavage to release the therapeutic antibody and PrEXO.
PrEXO-a23 corrects immune dysregulation via dual modulation of inflammation and tolerance in IBD
We evaluated the therapeutic efficacy of PrEXO-a23 in a DSS-induced colitis model, focusing on its ability to modulate immune dysregulation by simultaneously suppressing inflammation and promoting immune tolerance. C57BL/6 mice were administered 3% (w/v) DSS in drinking water for 7 days to induce colitis, and randomly divided into five treatment groups receiving intravenous injections every other day: PBS (DSS group), PrEXO (PrEXO group), IL-23 antibody (a23 group), rEXO-a23 (rEXO-a23 group), or PrEXO-a23 (PrEXO-a23 group). Healthy mice without DSS served as controls (Health group). Colonic tissues were collected on day 8 for downstream analyses.
Persistent inflammation in IBD is characterized by excessive infiltration of innate immune cells and dysregulated cytokine production3,42. Immunofluorescence analysis of colon tissues revealed that PrEXO-a23 treatment significantly reduced infiltration of CD86⁺ dendritic cells (P < 0.0001) and CD68⁺ macrophages (P = 0.0010), with greater reductions than those observed for PrEXO or a23 monotherapies (Fig. 5a–c). Further evaluation of colonic cytokine profiles via immunohistochemistry, ELISA, and RT-qPCR demonstrated that DSS treatment markedly upregulated proinflammatory cytokines IL-17A, IL-6, TNF-α, IL-23, and IFN-γ, while anti-inflammatory IL-10 was suppressed compared to healthy controls (Fig. 5d–f and Supplementary Fig. 14a, b). These cytokines are critical mediators of IBD pathogenesis: IL-23 and IL-17 promote pathogenic Th17 differentiation3, IFN-γ and TNF-α exacerbate chronic inflammation and intestinal tissue damage43,44, and IL-6 correlates with disease activity45, whereas IL-10 promotes immune tolerance and mucosal protection46. While free a23 demonstrated some anti-inflammatory activity in vitro, its in vivo efficacy was limited, only moderately reducing IFN-γ, IL-23, and IL-17A, and showing negligible impact on IL-6, TNF-α, or IL-10. This was attributed to poor lesion-targeting and insufficient drug enrichment. Moreover, a23 monotherapy had insufficient efficacy in regulating immune tolerance. In contrast, PrEXO-a23 broadly suppressed all major proinflammatory cytokines to near-healthy levels, while significantly upregulating IL-10 (P < 0.0001). These therapeutic effects likely result from the hybrid membrane-mediated lesion-targeting, which delivers a23 to locally inhibit the IL-23/Th17 axis, combined with PrEXO’s role as an immune tolerance modulator to regulate the inflammatory microenvironment.

a–c Representative immunofluorescence staining images of CD86/CD68 (a) and quantification of CD86 (b), and CD68 (c) in colonic sections. Green: CD68; Red: CD86; Blue: nucleus; Scale bar, 100 μm. d, e Representative immunohistochemical staining images (d), and quantitative analysis (e) of IL-17A and IFN-γ expression in colon tissues. Scale bars, 100 μm. f Cytokine levels (IL-23, IL-6, IL-10) in colonic homogenates measured by ELISA. g, h Flow cytometric plots (g) and statistical analysis (h) of Tregs (CD4+ FOXP3+), Th1 cells (CD4+ IFN-γ+), and Th17 cells (CD4+ IL-17A+) in colonic lamina propria. i–k RNA sequencing analysis of colonic tissues from mice (n = 5 mice). GSEA of the inflammatory bowel disease pathway for differentially expressed genes (DEGs) between DSS group and PrEXO-a23 group (i). NES: normalized enrichment score. The KEGG pathway analysis of DEGs (log2|fold change|≥1 and adjusted P ≤ 0.05) derived from DSS-, PrEXO-, a23-, rEXO-a23-, and PrEXO-a23-treated groups compared with indicated controls (j). Hierarchical clustering heatmap showing DEGs associated with inflammatory response (k). For (b, c, e, f, h), data were presented as mean ± SD (n = 3 mice in b, c, e; n = 4 mice in f; n = 5 mice in h). For (b, c, e, f, h), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Previous studies have suggested that promoting Treg differentiation and inhibiting Th1/Th17 responses can significantly alleviate IBD by restoring immune tolerance22,47. Immunofluorescence staining of colon tissues showed that PrEXO-a23 treatment substantially decreased CD4⁺ T cell infiltration compared to DSS mice (Supplementary Fig. 14c). Flow cytometry analysis was then performed to quantify the proportions of Tregs (CD4⁺ FOXP3⁺), Th1 cells (CD4⁺ IFN-γ⁺), and Th17 cells (CD4⁺ IL-17A⁺) among CD4⁺ T cells. Compared with healthy mice, DSS mice exhibited reduced Treg frequencies and elevated Th1 and Th17 proportions, confirming immune tolerance breakdown (Fig. 5g, h). Compared to the DSS group, treatment with a23 significantly decreased Th1 (P = 0.0061) and Th17 cell (P = 0.0090) populations but had minimal effects on Treg proportion, consistent with its in vitro anti-inflammatory profile and limited capacity to promote tolerogenic reprogramming. Although PrEXO showed some potential to induce Tregs and suppress Th17 cells in vitro, its Treg-promoting effect was attenuated in vivo, likely due to antagonistic inflammatory signals such as IL-23-mediated Treg instability19,39,40. In contrast, PrEXO-a23 elicited the strongest immunoregulatory response, with significant Treg induction and concurrent suppression of Th1 and Th17 cells (P < 0.0001), restoring an immune profile comparable to healthy mice. This enhanced effect likely results from the synergistic combination of a23, which establishes an anti-inflammatory milieu conducive to tolerance, and PrEXO, which promotes Treg differentiation. Collectively, these data demonstrate that PrEXO-a23 provides a dual therapeutic strategy in the complex IBD microenvironment by: (1) suppressing the IL-23/Th17 inflammatory axis through targeted a23 delivery and (2) promoting immune tolerance via Treg induction and tolerogenic reprogramming.
To further elucidate the molecular basis of PrEXO-a23’s immunoregulatory effects, we performed RNA sequencing (RNA-seq) on colonic tissues, using the DSS group as a control. PrEXO-a23 treatment induced 1,134 differentially expressed genes (DEGs): 199 upregulated and 935 downregulated (Supplementary Fig. 15a). Gene set enrichment analysis (GSEA) showed significant downregulation of IBD-related and Th17-differentiation gene signatures (P < 0.05; Fig. 5i and Supplementary Fig. 15b). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of downregulated DEGs highlighted suppression of cytokine–cytokine receptor interaction, TNF, NF-κB, IL-17, JAK/STAT, Toll-like receptor signaling, and Th17 differentiation pathways (Fig. 5j). Gene Ontology (GO) analysis further indicated significant downregulation of processes involved in immune cell infiltration, inflammatory response, proinflammatory cytokine production, and neutrophil chemotaxis (P < 0.05, Supplementary Fig. 15c). Importantly, PrEXO-a23 markedly downregulated key inflammatory genes (e.g., Ccr2, Ccr5, Il-17a, and Tnf), and upregulated anti-inflammatory or tolerance-associated genes (e.g., Fem1a) (Fig. 5k). Collectively, these results establish PrEXO-a23 as a dual-function therapeutic platform capable of concurrently extinguishing pathogenic IL-23 signaling and regulating immune tolerance, thereby resolving the fundamental immune dysregulation underlying IBD pathogenesis.
Potent therapeutic efficacy of PrEXO-a23 in treating IBD
To evaluate the therapeutic efficacy of PrEXO-a23, we employed a DSS-induced IBD mouse model characterized by hallmark pathological features such as body weight loss, elevated disease activity index (DAI), colon shortening, and splenomegaly. Mice received 3% (w/v) DSS in drinking water for 7 days. Meanwhile, mice were treated on days 2, 4, and 6 by intravenous administration, as outlined in Fig. 6a. DAI, including weight change, stool consistency, and fecal bleeding, was monitored daily. Colon and spleen tissues were harvested for analysis on day 8. Treatment with PrEXO, a23, or rEXO-a23 partially alleviated the DSS-induced weight loss (Fig. 6b) and DAI elevation (Fig. 6c and Supplementary Fig. 16a–c), with PrEXO-a23 demonstrating the most significant therapeutic benefit. Colon length, severely reduced in the DSS group (~4.2 cm), was partially restored by rEXO-a23 (~5.4 cm) and further extended to near-normal levels by PrEXO-a23 (~6.6 cm; Fig. 6d). Spleen index analysis indicated significant splenomegaly in DSS-treated mice (P < 0.0001), which was not observed in the PrEXO-a23 group (Supplementary Fig. 16d), suggesting attenuation of systemic inflammation48. These findings highlight the robust efficacy of PrEXO-a23 compared to monotherapies. Mechanistic analysis suggests that while PrEXO or a23 alone showed limited therapeutic effects due to insufficient immune regulation, PrEXO-a23 combines lesion-targeted delivery with synergistic immunomodulation. rEXO facilitates precise delivery and contributes to immune tolerance through its intrinsic bioactivity, while PMV-fusion-mediated vascular adhesion enhances retention at inflamed sites, amplifying payload accumulation and therapeutic outcomes beyond those of rEXO-a23.

a Experimental design for evaluating the therapeutic effect of PrEXO-a23 in the IBD mouse model. b, c Body weight change (b) and total DAI scores (c) during the experiment. On day 8, mice were euthanized. d Photographs of colon tissues and corresponding colon lengths. e Representative H&E-stained images of colon sections and histopathological damage scores. Scale bars, 100 μm. f Representative ZO-1 and Occludin labeling staining images of colon tissues and relative expression levels. Green: ZO-1; Red: Occludin; Blue: nucleus; Scale bars, 100 μm. g–i Gut microbiota analysis. Circos diagram of relative abundance for gut microbiome at family level (g), relative abundance of dominant bacterial families (h), and distribution histogram based on linear discriminant analysis (LDA) using the LEfSe method (i). LDA score (LDA (log10) >4.0, P < 0.05) cutoff was set at the top 5. For (b–f, h), data were presented as mean ± SD (n = 5 mice in b–d, h; n = 3 mice in e, f). For (d–f, h), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
In IBD, intestinal epithelial cell damage and inflammatory cell infiltration disrupt the integrity of the mucosal barrier49. Hematoxylin and eosin (H&E) staining and histological scoring revealed that PrEXO-a23 treatment preserved colonic epithelial integrity, maintaining intact epithelia, regular crypt architecture, and mucosal layers, along with reduced inflammatory cell infiltration compared to DSS group (Fig. 6e and Supplementary Fig. 16e). Immunofluorescence staining demonstrated restored expression of the tight junction proteins zonula occludens-1 (ZO-1) and Occludin, which were downregulated in DSS-treated mice, indicating improved intestinal barrier repair (Fig. 6f and Supplementary Fig. 16f). These results confirm that PrEXO-a23 substantially prevents physiological damage to colonic tissue and preserves intestinal barrier function.
Beyond barrier integrity, gut microbiota composition is a critical indicator of intestinal health. Dysregulated immune microenvironment in IBD favors pathogenic bacteria overgrowth while suppressing beneficial probiotics, leading to dysbiosis50. We next investigated microbiota modulation by PrEXO-a23 in IBD mice via 16S ribosomal RNA (rRNA) gene sequencing of fecal samples. A community circos diagram illustrated that PrEXO-a23 alleviated DSS-induced perturbations in microbiota composition (Fig. 6g). Specifically, PrEXO-a23 mitigated the reduction of beneficial bacteria, such as Lachnospiraceae and Akkermansiaceae, families known for anti-inflammatory and probiotic properties51,52, while suppressing overgrowth of Erysipelotrichaceae (Fig. 6h), a family associated with IBD progression53,54. Linear discriminant analysis (LDA) effect size (LEfSe) analysis further identified dominant microbial taxa influencing the gut environment from family to species levels. Consistent with previous findings, the abundances of IBD-associated pathogenic bacteria, including Erysipelotrichaceae, Turicibacter, and Bacteroides, were increased in DSS-treated mice (Fig. 6i). PrEXO-a23 treatment reduced these pathogenic bacteria while enriching beneficial microbes such as Lachnospiraceae and Akkermansiaceae, suggesting restoration of gut microbial homeostasis. This significant microbiota-regulating effect of PrEXO-a23 is likely attributed to its dual anti-inflammatory and pro-tolerant actions, which together restore immune homeostasis and repair the epithelial barrier, thereby creating a microenvironment favorable for microbiota recovery.
In clinical practice, IBD is associated with various extraintestinal inflammatory responses, such as skin inflammation. To explore whether PrEXO-a23 can simultaneously treat these inflammatory conditions, we established an IBD-comorbid psoriasis mouse model using DSS and Imiquimod (IMQ). PrEXO-a23 was then administered intravenously to assess its accumulation and therapeutic effects at the site of inflammation. The results showed that PrEXO-a23, through its synergistic “inflammation homing + damage adhesion” strategy, was enriched not only in colonic lesions but also in extraintestinal inflamed skin sites, without accumulating in healthy tissues (Supplementary Fig. 17). In addition, PrEXO-a23 treatment effectively alleviated IBD and psoriasis symptoms, as demonstrated by suppressed body weight loss and colon shortening, and reduced disease activity scores for both diseases (Supplementary Fig. 18).
Finally, the biosafety of PrEXO-a23 was evaluated both in vitro and in vivo. In vitro cytotoxicity assays showed no adverse effects on colonic epithelial cell viability across a range of PrEXO-a23 concentrations (Supplementary Fig. 19a, b). In vivo safety assessment following intravenous administration of PrEXO-a23 in healthy mice revealed no histopathological abnormalities in major organs (Supplementary Fig. 19c). Blood biochemistry (Supplementary Fig. 19d, e) and cytokine analyses confirmed that levels of IFN-γ, IL-6, and TNF-α (Supplementary Fig. 19f) remained within normal physiological ranges, indicating favorable biocompatibility.
PrEXO-a23 effectively decreases the risk of CAC and fibrosis
We further analyzed the RNA-seq data on colonic tissues from DSS-induced IBD mice, comparing gene expression profiles across healthy, DSS-treated, and PrEXO-a23-treated groups. Among the expressed genes, 295 were consistently different in DSS mice compared with healthy controls and the PrEXO-a23 group. Using STRING and Cytoscape, we constructed a protein-protein interaction network and extracted an 89-node, 1044-edge subnetwork. MCODE analysis revealed two highly interconnected modules (module 1, score = 23.5; module 2, score = 8.0), with module 1 containing 49 targets and 564 interactions. Degree-centrality ranking within this module identified Trp53 as the pivotal hub (Fig. 7a and Supplementary Fig. 20). As a tumor suppressor governing cell cycle, DNA repair, and apoptosis, Trp53 dysfunction is an early event in ulcerative colitis‑associated colorectal carcinogenesis10,55. Notably, PrEXO-a23 therapy regulated p53 in DSS-treated colons, suggesting that PrEXO-a23 has the potential to decrease the risk of progression from IBD to CAC.

a Schematic illustration of the protein-protein interaction network analysis for identifying Trp53 in PrEXO-a23 treatment. Created in BioRender. Zhang, F. (2026) https://BioRender.com/rx9bdp9. b Diagram of treatment schedule for PrEXO-a23 in AOM/DSS-induced CAC mouse model. c, d Survival curves (c) and DAI score (d) in CAC mice receiving indicated treatments. e–g, Photos (e), tumor burden (f), and tumor number (g) of colons in CAC mice at week 10. h, i ELISA quantification of TNF-α (h) and IL-6 (i) in colon tissue. j H&E-stained histological images of colon sections in CAC mice. Scale bar, 250 μm. k Representative Ki67-stained images of colon sections. Green: Ki67; Blue: nucleus; Scale bar, 100 μm. l H&E-stained histological images of colon sections in intestinal fibrosis mice. Scale bar, 100 μm. m–o Thickness of colonic wall (m), muscularis propria (n), and muscularis mucosa (o) of colonic section in mice. p, q Representative immunohistochemical staining images (p) and quantification (q) for α-SMA in colon sections. Scale bar, 100 μm. For (j–l, p), experiment was repeated three times independently with similar results. For (c, d, f–i, m–o, q), data were presented as mean ± SD (n = 8 mice in c; n = 4 mice in d, f, g; n = 3 mice in h, i, m–o, q). For (f–i, m–o, q), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. P value for the survival data (c) was analyzed using the log-rank (Mantel–Cox) test. Source data are provided as a Source Data file.
To evaluate the long-term therapeutic potential of PrEXO-a23 in preventing IBD-associated complications such as intestinal fibrosis and CAC, we employed an azoxymethane (AOM)/DSS-induced CAC mouse model. Mice were administered AOM followed by repeated cycles of DSS to induce chronic colitis and tumorigenesis, with untreated healthy mice serving as controls (Health group). During disease progression, mice received intravenous injections of PBS (AOM/DSS group), PrEXO (PrEXO group), a23 (a23 group), rEXO-a23 (rEXO-a23 group), or PrEXO-a23 (PrEXO-a23 group) (Fig. 7b). As expected, AOM/DSS-treated mice exhibited significantly reduced survival (P = 0.0208; Fig. 7c), substantial weight loss (Supplementary Fig. 21a), and significantly elevated DAI scores (Fig. 7d), shortened colon length, and increased tumor burden, both in terms of number and size (Fig. 7e–g and Supplementary Fig. 21b). Among all treatment groups, PrEXO-a23 demonstrated the robust therapeutic efficacy, markedly improving survival, body weight, DAI, and colon morphology to levels comparable with healthy controls. Importantly, while PrEXO, a23, and rEXO-a23 treatments conferred partial protection, none of these monotherapies prevented tumor development. In contrast, PrEXO-a23 treatment completely inhibited tumor formation, highlighting its potent anti-carcinogenic effect and capacity to halt IBD-to-CAC progression.
Given that CAC development involves sustained mucosal immune activation, epithelial barrier disruption, and hyperproliferation11, we further investigated the impact of PrEXO-a23 on these pathological hallmarks. Compared with the AOM/DSS group, PrEXO-a23 significantly decreased proinflammatory cytokine levels (TNF-α and IL-6) in colon tissues (P = 0.0016 and P = 0.0112; Fig. 7h, i) and normalized the spleen index (Supplementary Fig. 21c), suggesting effective suppression of both local and systemic inflammation. Histological analysis by H&E staining confirmed preserved epithelial architecture and reduced inflammatory cell infiltration in the PrEXO-a23 group (Fig. 7j). Furthermore, Ki67 immunofluorescence staining demonstrated suppressed epithelial hyperproliferation, with proliferation indices comparable to those of healthy controls (Fig. 7k and Supplementary Fig. 21d). Together, these data indicate that PrEXO-a23 improves colonic integrity and function, as demonstrated by its ability to modulate immune response, repair colonic barrier and control epithelial proliferation.
To evaluate whether PrEXO-a23 also protects against intestinal fibrosis, a common and severe consequence of chronic IBD, we employed a chronic DSS-induced colitis model with three DSS cycles. Mice in the AOM/DSS group exhibited marked thickening of the colonic wall, muscularis mucosa, and muscularis propria, characteristic features of fibrosis (Fig. 7l–o). Immunohistochemical staining for α-smooth muscle actin (α-SMA), a marker of myofibroblast activation, showed significant upregulation in fibrotic colons (P < 0.0001; Fig. 7p, q). While PrEXO, a23, and rEXO-a23 monotherapies did not inhibit fibrotic progression, PrEXO-a23 treatment preserved normal colon thickness and suppressed α-SMA expression to baseline levels, indicating effective prevention of fibrotic changes. PrEXO-a23 mitigated fibrotic progression through a coordinated dual mechanism. By neutralizing IL-23, it could suppress Th17-driven inflammation, while simultaneously delivering Treg-derived exosomes to promote immune tolerance. This combined anti-inflammatory and pro-tolerance strategy not only corrected the underlying immune dysregulation before pathological fibrosis occurs but also reprogrammed the local microenvironment to favor beneficial immunosuppressive and tolerogenic Treg functions, thereby preemptively blocking the pathological progression toward fibrosis.
We previously demonstrated that PrEXO-a23 affected the Trp53 expression change in DSS-treated colons. To evaluate the contribution of p53 to the therapeutic effect of PrEXO-a23, the mice were co-treated with the p53 inhibitor pifithrin-α (ip53). This intervention markedly diminished the therapeutic efficacy of PrEXO-a23 and negated its anti-tumor effects (Fig. 8a–g), suggesting that p53 pathway activity is associated with the nanotherapy’s ability to prevent CAC. We therefore examined key mediators of p53 signaling in colonic tissues using RT-qPCR and Western blot analysis. RT-qPCR analysis revealed that during CAC progression in the AOM/DSS group, expression of Trp53, Cdkn1a, and Bax was significantly downregulated compared to healthy controls, reflecting pathway dysfunction. Treatment with PrEXO-a23 restored their expression to near-normal levels, an effect that was partially blocked by p53 inhibition (PrEXO‑a23 + ip53) (Fig. 8h). Western blot analysis confirmed these changes at the protein level for p53, p21, and Bax (Fig. 8i, j). Together, these data demonstrate that the capacity of PrEXO-a23 to prevent the progression from chronic inflammation to cancer is partially dependent on functional p53 signaling. Collectively, PrEXO-a23 not only attenuates active inflammation but also provides durable protection against fibrosis and tumorigenesis, highlighting it as a promising therapeutic strategy for the long-term management of IBD and its complications.

a, b During the treatment of AOM/DSS-induced CAC mice with PrEXO-a23 or PrEXO-a23 and the p53 inhibitor (ip53), body weight (a) and DAI score (b) of mice were monitored daily. c spleen index analysis of mice. d–g Colon photographs (d), colon length (e), tumor number (f), tumor burden (g) of CAC mice at week 10. h Relative mRNA expression levels of Trp53, Cdkn1a and Bax in colonic tissues of CAC mice with indicated treatment measured by RT-qPCR assay. i, j Western blot images (i) and relative protein level analysis (j) of p53, p21, and Bax expressed in colonic tissues. For (a–c, e–h, j), data were presented as mean ± SD (n = 4 mice in a–c, e–g; n = 5 mice in h; n = 3 mice in j). For (c, e–h, j), statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Discussion
“How is immune homeostasis maintained and regulated?” was highlighted as one of the 125 most important scientific questions by Science in 2021. IBD, a prototypical disorder of immune dysregulation, remains inadequately managed by conventional therapies, which primarily offer transient symptom relief and often fail to achieve durable immune modulation4,5, leaving patients vulnerable to relapse and progression of complications. Antigen-specific immune tolerance induction is widely regarded as an optimal strategy for treating inflammatory diseases, as it selectively restores tolerance to pathogenic antigens while preserving systemic immune competence37,56. However, this strategy is currently infeasible for IBD, given the incomplete characterization of disease-driving antigens. Tregs, which play a central role in maintaining immune tolerance, have thus emerged as a viable alternative strategy for IBD management20,21,22. Nevertheless, the induction and expansion of Tregs in a disease-specific manner remain technically challenging. IL-23 antibodies have shown promise as clinical therapeutics for IBD15,16. Our findings demonstrate that free IL-23 antibodies effectively inhibit Th17 differentiation and proinflammatory cytokine secretion in vitro. However, it exhibits a limited capacity to induce tDCs or expand Tregs, which are all essential for reestablishing immune tolerance. Moreover, precise in vivo delivery and safety concerns associated with cytokine-based therapies must be carefully addressed to improve therapeutic outcomes.
Treg-derived exosomes (rEXO) are enriched in a variety of immunomodulatory molecules, including proteins and miRNAs, which contribute to their immunomodulatory functions24,25. In this study, we found that Treg-derived exosomes have potent tolerance-inducing capabilities in vitro, effectively promoting the generation of tDCs and Tregs, while suppressing Th17 polarization. Building upon this observation, we developed a dual-functional nanotherapeutic system, PrEXO-a23, that integrates both inflammation suppression and immune tolerance induction. This nanoplatform is engineered by fusing Treg-derived exosomes with platelet membrane vesicles, granting it the ability to target colonic lesions through inflammatory homing and collagen binding. Furthermore, IL-23 antibodies (a23) are conjugated to the hybrid vesicles via MMP-cleavable peptide linkers, which release a23 in response to the inflammatory microenvironment. Locally released a23 neutralizes IL-23 and inhibits Th17-driven inflammation, while the exosomal cargo reprograms dendritic cells to a tolerogenic phenotype and promotes Treg expansion, further suppressing Th17 polarization. This synergistic “anti-inflammatory and pro-tolerant” approach helps effectively modulate immune dysregulation in IBD, resulting in symptom relief, intestinal barrier repair, and gut microbiota normalization. Notably, PrEXO-a23 demonstrated the potential to maintain long-term remission and reduce the risk of intestinal fibrosis and CAC. Additionally, the efficacy of PrEXO-a23 to prevent the progression from IBD to CAC partially depends on p53.
While our approach demonstrates strong therapeutic potential for IBD, several important considerations warrant further investigation. First, existing murine models do not fully recapitulate the complex and heterogeneous immune landscape of human IBD, highlighting the need for rigorous validation of PrEXO-a23 in advanced preclinical models and, ultimately, in clinical settings57,58. Second, given the substantial inter-patient variability in IBD, the therapeutic efficacy of PrEXO-a23 may differ across disease subtypes, such as Crohn’s disease versus ulcerative colitis, and between disease stages (active vs. remission)1,59. Therefore, future studies should incorporate biomarker-based patient stratification, such as IL-23 receptor (IL-23R) expression and Treg/Th17 ratios, to identify responsive populations and support the development of subtype-specific disease models. Encouragingly, the PrEXO platform is designed as a modular and adaptable delivery system, enabling the integration of alternative immunomodulatory agents, such as monoclonal antibodies or small-molecule inhibitors, tailored to an individual’s immune profile.
In summary, the PrEXO-a23 nanomedicine offers a promising strategy for overcoming the immune-regulation limitations of current IBD therapies. Through lesion-specific targeting via platelet-Treg exosome hybrid vesicles, combined with concurrent suppression of inflammation and regulation of immune tolerance, PrEXO-a23 effectively mitigates disease progression and associated complications. This work exemplifies precision immunomodulation via engineered biomimicry and represents a major step toward next-generation, personalized IBD treatments.
Methods
Reagents and materials
Naïve CD4+ T cell isolation kit was purchased from Miltenyi Biotec. The peptide linker (Arg-Val-Gly-Leu-Pro, RVGLP) was designed and synthesized by ChinaPeptides. InVivoMAb IL-23 was obtained from BioXcell. Primary antibodies for immunostaining MMP9, ALIX, TSG101, CCR6, CD68, CD4, ZO-1, Occludin, CD62P, CTLA-4, GAPDH, and Ki67 were provided by Abcam. Anti-mouse GPIbα, IFN-γ, IL-10 and α-SMA were supplied by Affinity. Anti-mouse CD86, IL-17A and p53 were purchased from Proteintech. Anti-mouse p21 and Bax were obtained from Cell Signaling Technology. BV421-anti-CD11c, PE-anti-CD86, APC-anti-CD80, FITC-anti-MHC II, BV421-anti-CD45, FITC-anti-CD3, APC/FireTM 750-anti-CD4, APC-anti-CD25, PE-anti-IL-17A, APC/FireTM 750-anti-CD45, PE-anti-CD19, BV421-anti-F4/80, and PE-Ly6G were purchased from BioLegend. PE-anti-FOXP3 and APC-anti-IFN-γ were provided by eBioscience. 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI), 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate (DiD), and tris(2-carboxyethyl) phosphine (TCEP) were obtained from Invitrogen. IL-6, IL-23, IFN-γ, and IL-1β, were provided by PeproTech. IL-2, TGF-β, MMP9, GM6001, NHS-PEG-MAL linker, and pifithrin-α were purchased from MedChemExpress. Dextran sulfate sodium salt (DSS), calcein-AM/propidium iodide (PI) stain kit, PI/Annexin V apoptosis detection kit, and mouse type I collagen were supplied by Yeasen. Azoxymethane (AOM) and lipopolysaccharide (LPS) were purchased from Sigma-Aldrich. 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) and ELISA kits for IFN-γ, TNF-α, IL-17A, IL-23, IL-6, and IL-10 were purchased from Solarbio. ELISA kits for TGF-β1 and MMP9 were obtained from Elabscience. X-VIVO medium was purchased from Lonza. Dulbecco’s Modified Eagle Medium (DMEM), RPMI 1640 medium, fetal bovine serum (FBS), and penicillin–streptomycin (P/S) were purchased from Gibco.
Animal and cell lines
All C57BL/6 female mice (6−8 weeks old) were obtained from the SPF Biotechnology Co., Ltd (Beijing, China), and housed in groups of six in individually ventilated cages under a 12 h light/dark cycle at 20–22 °C with ad libitum access to standard rodent chow and water. All animal studies were conducted in accordance with the animal experimental protocols approved by the Institutional Animal Care and Use Committee of the Chinese Academy of Medical Sciences &Peking Union Medical College Institute of Radiation Medicine (Approval number: IRM/2-IACUC-2311-002).
RAW264.7 (murine macrophage cell line) and HT-29 cells (human colorectal carcinoma cell line) were obtained from Procell. NCM460 cells (human normal colonic epithelial cell line) were purchased from SUNNCELL. All cell lines were maintained in DMEM supplemented with 10% FBS and 1% P/S. Bone marrow-derived neutrophils and DCs were isolated from 6-week-old C57BL/6 mice. The bone marrow cells were cultured in RPMI 1640 medium containing 10% FBS, 1% P/S, 20 ng/mL recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF; Sino Biological), and 10 ng/mL IL-4 (Sino Biological) for DC differentiation. Half of the culture medium was replaced with fresh medium on day 3, and cells were used on day 6 as immature DCs. For neutrophil isolation, bone marrow cells were added to a Percoll gradient (55, 65, and 78%; v/v), followed by centrifugation at 500×g for 30 min. Neutrophils were collected at the interface between the 65 and 78% Percoll layers, washed three times with PBS, and maintained in RPMI 1640 medium containing 10% FBS and 1% P/S for further analysis. All cells were cultured at 37 °C in a humidified 5% CO2 atmosphere.
Preparation of rEXO and PMV
Naïve CD4+ T cells were isolated from the spleen of healthy C57BL/6 mice using a CD4-positive T cell isolation kit and polarized to regulatory T cells in X-VIVO medium supplemented with anti-CD3 (5 μg/mL), anti-CD28 (2 μg/mL), IL-2 (20 ng/mL), and TGF-β (5 ng/mL) for 7 days. Treg culture supernatant was sequentially centrifuged at 300×g for 10 min, 2000×g for 20 min, and 10,000×g for 30 min to remove debris, followed by ultracentrifugation at 100,000×g for 70 min to obtain rEXO.
Platelets isolated from the whole blood of healthy C57BL/6 mice were subjected to freeze-thaw cycles, and platelet membranes were extracted by centrifugation at 8000×g for 20 min. The processed membranes were sequentially extruded through polycarbonate porous membrane filters with decreasing pore sizes (0.8, 0.4, 0.2, and 0.1 μm) to obtain PMV. Protein concentrations of rEXO and PMV were quantified using a BCA assay kit (Accurate Biotechnology).
Preparation and characterization of PrEXO-a23
Co-extrusion of PMV and rEXO. PrEXO was prepared by mixing PMV and rEXO at protein mass ratios of 1:1, 1:2, or 1:3, followed by sonication (40 kHz, 5 min) and then repeated extrusion as described for the PMV preparation. Fusion efficiency of PrEXO was assessed by NanoFlow Cytometer (CytoFLEX, Beckman).
Synthesis and conjugation of MMP9-cleavable linker to a23. An MMP-cleavable peptide linker (Arg-Val-Gly-Leu-Pro, RVGLP) with an NHS group at the arginine (Arg) terminus and a Mal group at the proline (Pro) terminus was used to conjugate the IL-23 antibody (a23) to the hybrid vesicles (PrEXO). To determine the optimal molar ratio for linker-a23 conjugation, a23 was reacted with a linker at molar ratios (a23:linker) of 1:1, 1:10, 1:50, 1:75, or 1:100 in PBS at 4 °C for 12–18 h. Unreacted linker was removed by centrifugation at 8000×g for 30 min using a centrifugal filter (100 kDa; Jet Biofil). The purified linker-a23 was analyzed using matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS; AXIMA-Performance MA, Shimadzu).
Preparation of PrEXO-a23. PrEXO was prepared by coextrusion of platelet membrane vesicles and Treg-derived exosomes at a 1:1 protein mass ratio, followed by protein quantification using a BCA assay. PrEXO was treated with 1 mM TCEP at 37 °C for 30 min to expose sulfhydryl (SH) groups on its membrane. Pretreated PrEXO was then added to purified linker-a23 at a mass ratio (PrEXO:a23) of 1:0.1, 1:0.25, 1:0.5, 1:1, or 1:2, and incubated at 25 °C for 1 h. Unreacted linker-a23 was removed by centrifugation at 8000×g for 30 min using a centrifugal filter (300 kDa). ELISA was used to determine the conjugation efficiency of a23 to PrEXO. Using the same method, PrEXO was conjugated to a23 using a non-MMP-cleavable linker (fL: NHS-PEG-MAL) to generate PrEXO-fL-a23.
Characterization of PrEXO-a23. Morphological analysis was performed using a TEM instrument (JEM-1400 Flash, JEOL, Japan). Particle sizes and zeta potentials were measured by a Nano ZS Malvern analyzer (Malvern, UK). PrEXO-a23 was stored in PBS (pH 7.4) at 4 °C and in plasma at 37 °C, and particle size and zeta potential were measured daily for 7 days.
Protein analysis of PrEXO-a23
IL-10 (1:50), CTLA-4 (1:50), ALIX (1:50), TSG101 (1:50), CCR6 (1:50), CD62P (1:50), GPIbα (1:50), and a23 were detected using an automated Digital Capillary Electrophoresis (CE)-Western system (JS4167, ProteinSimple, USA) according to the manufacturer’s protocol. Specifically, protein samples were prepared using loading buffer without reducing agents (e.g., DTT) to allow analysis under non-reducing conditions. Protein samples, primary antibody dilution, secondary antibody dilution, and chemiluminescence substrate were sequentially added to the lane plates for protein detection. All analyses were carried out using Compass for Simple Western software (v 6.0.0, ProteinSimple).
PrEXO-a23 colocalization assay
rEXO, PMV, and a23 were labeled with DiI, DiD, and Alexa Fluor 488, respectively. All samples were incubated with 5 μg/mL dye at 37 °C for 30 min. Triple-labeled PrEXO-a23 was observed using confocal laser scanning microscopy (Eclipse Ti2, Nikon, Japan) with a 100× oil-immersion objective.
In vitro assessment of a23 release from PrEXO-a23
To evaluate MMP9-responsive properties of the peptide linker, PrEXO-a23 was incubated with MMP9 with or without the MMP9 inhibitor GM6001 at predetermined time intervals. Released a23 was quantified using an ELISA kit.
Coculture model of CD4+ T cells and intestinal epithelial cells
Naïve CD4+ T cells were polarized to Th17 cells by culturing in RPMI 1640 medium containing TGF-β (1 ng/mL), IL-6 (50 ng/mL), IL-23 (5 ng/mL), IL-1β (10 ng/mL), anti-IFN-γ (10 μg/mL), and anti-IL-4 (10 μg/mL) for 72 h. Cells were treated with rEXO (25 μg/mL), PMV (25 μg/mL), PrEXO (25 μg/mL rEXO and 25 μg/mL PMV), a23 (7.5 μg/mL), or PrEXO-a23 (25 μg/mL rEXO, 25 μg/mL PMV, and 7.5 μg/mL a23), respectively. After 72 h, T cells were collected and stained with APC/FireTM 750-CD4 antibody and PE-IL-17A antibody to detect the percentage of Th17 cells using a flow cytometer (CytoFLEX, Beckman).
For coculture, the pretreated T cells (2 × 105 per well) were seeded in the upper chamber of 12-well Transwell inserts (NEST Biotechnology), while NCM460 cells (5 × 105 cells per well) were plated in the lower chamber. After 24 h, NCM460 cells were collected and stained with PI/Calcein-AM or Annexin V/PI for 15 min according to the manufacturer’s protocol. The stained cells were then analyzed using fluorescence microscopy or flow cytometry.
Induction of tDCs and Tregs
On day 6, immature DCs were stimulated with LPS (1 μg/mL) and IFN-γ (20 ng/mL) to generate mature DCs for 24 h. For the induction of tDCs, the cells were co-treated with rEXO, PMV, PrEXO, a23, or PrEXO-a23, respectively. After 24 h, cells were incubated with CD16/CD32 (BioLegend) for 15 min to block Fcγ receptors, then stained with BV421-CD11c, APC-CD80, PE-CD86, and FITC-MHC II antibodies, and analyzed by flow cytometry. Culture supernatants were collected for cytokine analysis (IL-10, TGF-β, IL-6, and TNF-α) using ELISA kits.
To activate lymphocytes, 5 × 105 CD4+ T cells were cocultured with pretreated 1 × 105 DCs for 72 h. After incubation, CD4+ T cells were stained with antibodies, including APC/FireTM 750-CD4, PE-IL-17A, APC-CD25, and PE-FOXP3, to assess the proportion of Th17 cells and Tregs.
In vitro flow assay
A flow channel was coated with 100 μg/mL Type I collagen by incubating overnight at 37 °C. DiO-labeled rEXO, PMV, PrEXO, or PrEXO-a23 were loaded into a 1 mL syringe for perfusion over the collagen-coated flow channel at a shear rate of 500 s−1 using a syringe pump (Harvard, USA). Then, the flow channel was perfused with PBS at the same shear rate before imaging by CLSM.
In vitro inflammatory targeting assay
RAW264.7 cells or neutrophils were incubated with DiI-labeled rEXO, PMV, PrEXO, or PrEXO-a23 for 2 h, with or without the pre-stimulation of LPS (1 μg/mL). After incubation, cells were washed twice using PBS, then the percentage of DiI-positive cells or mean fluorescence intensity was measured using a flow cytometer or CLSM. PrEXO-a23 was incubated with or without LPS-activated RAW264.7 at 37 °C in the MMP9 condition. The cumulative release of a23 was measured by ELISA at the indicated time points.
In vivo biodistribution of PrEXO-a23 in the IBD model
To generate the IBD mouse model, C57BL/6 mice were fed with 3% (w/v) DSS for 7 days after being pretreated with a fluorescence-free diet. Mice were intravenously injected with DiD-labeled rEXO, PMV, rEXO + PMV, PrEXO, or PrEXO-a23, respectively. At 0, 2, 6, 12, 24, 36, and 48 h after injection, the mice were imaged using an in vivo imaging system (ABL-X5, Tanon, China) to analyze the fluorescence biodistribution of DiD. Mice from each group were euthanized, and their major organs (heart, liver, spleen, lung, kidney, and colon) were collected for further analysis. For localization analysis, frozen colon sections were stained with DAPI, and images were captured using CLSM. For cellular targeting analysis, colon tissues were processed into single-cell suspensions for flow cytometry analysis. The relative DiD fluorescence in DCs (APC/FireTM 750-CD45 and BV421-CD11c), macrophages (APC/FireTM 750-CD45 and BV421-F4/80), T cells (APC/FireTM 750-CD45 and FITC-CD3), B cells (APC/FireTM 750-CD45 and PE-CD19) and neutrophils (APC/FireTM 750-CD45 and PE-Ly6G) were analyzed to determine immune cell uptake.
In vivo pharmacokinetics study
DiD-labeled PrEXO-a23 was intravenously administered to C57BL/6 mice, and blood samples were collected at predetermined time points (0.5, 1, 2, 4, 24, 48, and 72 h). Blood samples were placed in 96-well plates, and fluorescence intensity was measured at an excitation wavelength of 650 nm using an in vivo imaging system.
Dual-label tracking analysis in vivo
a23 was labeled with Alexa Fluor 647, and PrEXO was labeled with DiR. DSS-induced IBD mice were intravenously injected with PrEXO + a23 (a physical mixture of PrEXO and a23), PrEXO-fL-a23 (PrEXO and a23 were conjugated with a non-cleavable peptide, fL), or PrEXO-a23 (PrEXO and a23 were conjugated with the MMP-cleavable peptide), respectively. The biodistribution of PrEXO and a23 was monitored using an in vivo imaging system at 0, 2, 6, 12, 24, 36, and 48 h after injection. Colon tissues were harvested at 12 h after injection for ex vivo imaging analysis. Cryosections were processed for CLSM imaging to examine the colocalization of PrEXO (DiI-labeled) and a23 (Alexa Fluor 647-labeled) signals.
Evaluation of clinical signs in DSS-induced IBD model
Following a 7-day acclimatization period, female C57BL/6 mice (8 weeks old) were given 3% (w/v) DSS in drinking water for 7 days to establish an IBD model. Healthy mice in the normal group were not treated, while DSS-induced mice in the DSS, PrEXO, a23, rEXO-a23, and PrEXO-a23 groups were injected intravenously with PBS, PrEXO, a23, rEXO-a23, and PrEXO-a23 on days 2, 4, and 6 of DSS feeding, respectively. The dose was administered at 5 mg/kg for rEXO and 1.5 mg/kg for a23. Mice were observed daily, and the body weight and disease activity index (DAI) were evaluated. DAI scores were calculated daily based on weight loss (0 to 4), stool consistency (0 to 4), and fecal bleeding (0 to 4) scores. On day 8, mice were euthanized, and their colons were collected for length measurement and subsequent analysis. Spleen index was calculated as follows: spleen index = (spleen weight/body weight) × 100%.
Evaluation of the inhibition effect in the AOM/DSS-induced CAC model
To induce the CAC mouse model, six-week-old female C57BL/6 mice were intraperitoneally injected with AOM (10 mg/kg). After 7 days, mice were subjected to three cycles of DSS treatment, each consisting of 2.5% DSS for 7 days followed by a 14-day recovery period with regular water. During each 7-day DSS treatment period, PBS, PrEXO, a23, rEXO-a23, and PrEXO-a23 were administered intravenously every 2 days, as described for the IBD model. At the end of the 10-week experiment, mice were euthanized, and colons were excised for tumor enumeration and length measurement.
Histological analysis
Colonic tissues were fixed in 10% formalin for 24 h, embedded in paraffin, and sliced into 4 μm sections with a microtome (Thermo Fisher). Subsequently, hematoxylin and eosin (H&E) staining was performed on the tissue sections. Colonic histological damage was blindly scored for epithelial damage (0–6) and inflammatory infiltration (mucosa: 0–3; submucosa: 0–2; muscle/serosa: 0–1) according to established criteria60.
For immunofluorescence, the fixed colon slides were permeabilized with 0.3% Triton X-100 for 10 min, blocked with 5% goat serum, and incubated overnight at 4 °C with primary antibodies against ZO-1 (1:100), Occludin (1:100), CD86 (1:400), CD68 (1:1000), CD4 (1:500), or Ki67 (1:1000). Alexa Fluor 488 or Alexa Fluor 647 conjugated secondary antibodies (1:500) were applied for 1 h, and cell nuclei were stained with DAPI (1:1000).
For immunohistochemistry, preprocessed colonic sections were labeled with primary antibodies of anti-MMP9 (1:500), anti-IL-17A (1:900), anti-IFN-γ (1:200), or anti-α-SMA (1:900) overnight, followed by secondary antibodies and a color reaction using a DAB Kit (Maxim). Cell nuclei were counterstained with hematoxylin. All images were acquired using a Pannoramic digital slide scanner (3D HISTECH), and analyzed with ImageJ.
Flow cytometry analysis of colonic cell subgroups
Colonic lamina propria lymphocytes were isolated by digesting tissue with Liberase (31.25 μg/mL, Roche) and DNase I (50 μg/mL, Roche) at 37 °C for 1 h with rotation. Single-cell suspensions were obtained through a 70-μm cell strainer (Biosharp), and 1 × 106 cells were separated from each group for staining. After Fc receptor blocking, the cells were incubated with cell surface fluorescent antibodies containing BV421-CD45, FITC-CD3, and APC/FireTM 750-CD4 antibodies. For transcription factor staining, cells were fixed and permeabilized using a commercial FOXP3 staining kit (eBioscience) according to the manufacturer’s protocol, followed by incubation with PE-FOXP3 antibody. For intracellular cytokine staining, isolated lymphocytes were stimulated with a Cell Activation Cocktail (BioLegend) for 6 h at 37 °C before cell surface staining. Subsequently, PE-anti-IL-17 antibody and APC-anti-IFN-γ antibody were used. The proportions of Th1, Th17, and Treg cells in each sample were analyzed using a flow cytometer. Finally, all data were processed using FlowJo V10.8.1.
ELISA assay
Serum and colonic tissues were collected from IBD mice. Colon segments were homogenized in PBS using a homogenizer at 4 °C. Homogenates were centrifuged at 10,000×g for 10 min, and supernatants were analyzed for cytokines and MMP9 using ELISA kits following manufacturer protocols.
Reverse transcription quantitative real-time PCR (RT-qPCR)
Total RNA was extracted from frozen colon tissues using TRIzol, quantified on a NanoDrop 2000 spectrophotometer (Thermo Fisher, USA), and reverse-transcribed using HiScript III RT SuperMix (Vazyme). Then, qPCR was performed on the CFX Connect Real-Time PCR Detection System with SYBR Green Master Mix. Primer pairs for target mouse genes are shown in Supplementary Table 1. Relative gene expression was calculated using the 2−ΔΔCt method, normalized to GAPDH.
RNA sequencing analysis
On day 8, mice were euthanized, and their colon tissues were excised (n = 5). Total RNA from colon tissues was extracted using TRIzol. After total RNA was extracted, eukaryotic mRNA was enriched using Oligo (dT) beads. Following mRNA enrichment, RNA was processed for cDNA synthesis and library construction with the MGIEasy RNA Library Prep Kit. Libraries were sequenced on the MGISEQ-2000 sequencing platform (Beijing Genomic Institution, BGI). Raw reads were filtered with SOAPnuke 1.5.6, and clean reads were aligned to the mouse genome (GRCm38.p6) using HISAT2. Gene expression counts were obtained with feature counts, and differential expression analysis was performed using DESeq2 (v1.4.5) between two different groups. The genes with the parameter of |log2(fold change)|≥1 and adjusted P ≤ 0.05 were considered as differentially expressed genes (DEGs). Functional enrichment analyses, including heatmap clustering, GSEA, GO, and KEGG analysis, were performed on the BGI Dr. Tom platform (https://biosys.bgi.com). GSEA was conducted using the GSEA software v4.0.3 to determine whether predefined gene sets from the KEGG pathways were significantly enriched between two groups.
Western blot analysis
Protein expression was quantified using an automated Digital Capillary CE-Western system (JS4167, ProteinSimple, USA) according to the manufacturer’s protocol. Briefly, colon segments from mice were homogenized in RIPA buffer to extract protein, and protein concentration was determined using a BCA assay kit. Protein samples were added to each well, followed by the corresponding primary antibodies (p53 at 1:50, p21 at 1:50, Bax at 1:50, and GAPDH at 1:50) and HRP-conjugated secondary antibody. Relative protein levels were calculated by measuring the area under the peak of a chemiluminescent chromatogram and normalized to GAPDH. All analyses were carried out using Compass for Simple Western software (v 6.0.0, ProteinSimple). Uncropped scans are available in the Source Data file.
16S rRNA microbiota sequencing assay
On day 7, mouse feces were collected for gut microbiota analysis via 16S rRNA gene sequencing at Genesky Biotechnologies Inc. (Shanghai, China). Total DNA was extracted from fecal content using a fecal DNA extraction kit. The hypervariable V3-V4 regions of the bacterial 16S rRNA gene were amplified by PCR, and products were purified with the Agencourt AMPure XP Kit (Beckman Coulter, CA, USA). Libraries were constructed with Illumina adapters, quantified via Qubit 3.0 Fluorometer, and validated with an Agilent Bioanalyzer 2100 system. Sequencing was performed on an Illumina NovaSeq platform to generate 250 bp paired-end reads. Data analysis was conducted on the Genesky Cloud Platform (http://cloud.geneskybiotech.com), with microbial composition analyzed at the family level.
Statistical analysis
Data are presented as mean ± SD (standard deviations). For comparisons between two groups, a two-tailed Student’s t-test was applied. For multiple comparisons, one-way ANOVA followed by Tukey’s post hoc test was used. Survival curves were analyzed by the log-rank test. All statistical analyses were performed using GraphPad Prism (v9.0). P < 0.05 was considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The 16S rRNA gene sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under accession code PRJNA1400654. The RNA-seq data used in this study are available in the NCBI SRA database with accession number PRJNA1400962. All data underlying this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Hodson, R. Inflammatory bowel disease. Nature 540, S97 (2016).
Maloy, K. J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Neurath, M. F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 20, 970–979 (2019).
Plichta, D. R. et al. Therapeutic opportunities in inflammatory bowel disease: mechanistic dissection of host-microbiome relationships. Cell 178, 1041–1056 (2019).
Alsoud, D. et al. Breaking the therapeutic ceiling in drug development in ulcerative colitis. Lancet. Gastroenterol. Hepatol. 6, 589–595 (2021).
Kobayashi, T. & Hibi, T. Improving IBD outcomes in the era of many treatment options. Nat. Rev. Gastroenterol. Hepatol. 20, 79–80 (2023).
D’Haens, G. et al. Challenges in the pathophysiology, diagnosis, and management of intestinal fibrosis in inflammatory bowel disease. Gastroenterology 162, 26–31 (2022).
D’Alessio, S. et al. Revisiting fibrosis in inflammatory bowel disease: the gut thickens. Nat. Rev. Gastroenterol. Hepatol. 19, 169–184 (2022).
Yaeger, R. et al. Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology 151, 278–287 (2016).
Rajamäki, K. et al. Genetic and epigenetic characteristics of inflammatory bowel disease–associated colorectal cancer. Gastroenterology 161, 592–607 (2021).
Shah, S. C. & Itzkowitz, S. H. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology 162, 715–730.e713 (2022).
Verstockt, B. et al. IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 20, 433–446 (2023).
Lee, J. Y. et al. Serum amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell 180, 79–91 (2020).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
D’Haens, G. et al. Mirikizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 388, 2444–2455 (2023).
Rubin, D. T. et al. Guselkumab in patients with moderately to severely active ulcerative colitis (QUASAR): phase 3 double-blind, randomised, placebo-controlled induction and maintenance studies. Lancet. 405, 33–49 (2025).
Adedokun, O. J. et al. Pharmacokinetics and exposure response relationships of ustekinumab in patients with Crohn’s disease. Gastroenterology 154, 1660–1671 (2018).
Dikiy, S. & Rudensky, A. Y. Principles of regulatory T cell function. Immunity 56, 240–255 (2023).
Sakaguchi, S. et al. Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008).
Dominguez-Villar, M. & Hafler, D. A. Regulatory T cells in autoimmune disease. Nat. Immunol. 19, 665–673 (2018).
Ferreira, L. M. R. et al. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 18, 749–769 (2019).
Clough, J. N. et al. Regulatory T-cell therapy in Crohn’s disease: challenges and advances. Gut 69, 942–952 (2020).
Raffin, C., Vo, L. T. & Bluestone, J. A. Treg cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 20, 158–172 (2020).
Tian, Y. et al. Reduction of choroidal neovascularization via cleavable VEGF antibodies conjugated to exosomes derived from regulatory T cells. Nat. Biomed. Eng. 5, 968–982 (2021).
Okoye, I. sobelS. et al. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 41, 89–103 (2014).
Barnert, J. & Messmann, H. Diagnosis and management of lower gastrointestinal bleeding. Nat. Rev. Gastroenterol. Hepatol. 6, 637–646 (2009).
Chang, J. T. Pathophysiology of inflammatory bowel diseases. N. Engl. J. Med. 383, 2652–2664 (2020).
Zundler, S. et al. Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances. Gut 68, 1688–1700 (2019).
Fischer, A. et al. Differential effects of α4β7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut 65, 1642–1664 (2016).
Hu, C. M. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526, 118–121 (2015).
Scherlinger, M. et al. The role of platelets in immune-mediated inflammatory diseases. Nat. Rev. Immunol. 23, 495–510 (2023).
de Bruyn, M. & Ferrante, M. Failure of MMP-9 antagonists in IBD: demonstrating the importance of molecular biology and well-controlled early phase studies. J. Crohns Colitis 12, 1011–1013 (2018).
Tong, F. et al. MMP-2-triggered, mitochondria-targeted PROTAC-PDT therapy of breast cancer and brain metastases inhibition. Nat. Commun. 15, 10382 (2024).
Fucikova, J. et al. Induction of tolerance and immunity by dendritic cells: mechanisms and clinical applications. Front. Immunol. 10, 2393 (2019).
Cifuentes-Rius, A. et al. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat. Nanotechnol. 16, 37–46 (2021).
Morante-Palacios, O. et al. Tolerogenic dendritic cells in autoimmunity and inflammatory diseases. Trends Immunol. 42, 59–75 (2021).
Kenison, J. E., Stevens, N. A. & Quintana, F. J. Therapeutic induction of antigen-specific immune tolerance. Nat. Rev. Immunol. 24, 338–357 (2024).
Tung, S. L. et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 8, 6065 (2018).
Koo, S. et al. Ceria-vesicle nanohybrid therapeutic for modulation of innate and adaptive immunity in a collagen-induced arthritis model. Nat. Nanotechnol. 18, 1502–1514 (2023).
Izcue, A. et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28, 559–570 (2008).
Coenen, D. M., Mastenbroek, T. G. & Cosemans, J. M. E. M. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood 130, 2819–2828 (2017).
Neurath, M. F. Strategies for targeting cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 24, 559–576 (2024).
Neurath, M. F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14, 329–342 (2014).
Günther, C. et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011).
Schreiber, S. et al. Therapeutic interleukin-6 trans-signaling inhibition by olamkicept (sgp130Fc) in patients with active inflammatory bowel disease. Gastroenterology 160, 2354–2366.e2311 (2021).
Chaudhry, A. et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578 (2011).
Jia, L. et al. Porphyromonas gingivalis aggravates colitis via a gut microbiota-linoleic acid metabolism-Th17/Treg cell balance axis. Nat. Commun. 15, 1617 (2024).
Shi, X. et al. A novel role of the splenic volume in Crohn’s disease: evaluating the efficacy of infliximab. Front. Pharmacol. 14, 1246657 (2023).
Citi, S. Intestinal barriers protect against disease. Science 359, 1097–1098 (2018).
Caruso, R., Lo, B. C. & Núñez, G. Host–microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 20, 411–426 (2020).
Guo, H. et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science 370, eaay9097 (2020).
Luo, Y. et al. Rational consideration of Akkermansia muciniphila targeting intestinal health: advantages and challenges. npj Biofilms Microbiomes 8, 81 (2022).
Cornick, S. et al. VAMP8-mediated MUC2 mucin exocytosis from colonic goblet cells maintains innate intestinal homeostasis. Nat. Commun. 10, 4306 (2019).
Kaakoush, N. O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 5, 84 (2015).
Hartl, K. et al. p53 terminates the regenerative fetal-like state after colitis-associated injury. Sci. Adv. 10, eadp8783 (2024).
Serra, P. & Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 37, 238–251 (2019).
Au, K. M. et al. An injectable subcutaneous colon-specific immune niche for the treatment of ulcerative colitis. Nat. Biomed. Eng. 8, 1243–1265 (2024).
Guo, P. et al. Engineered probiotic ameliorates ulcerative colitis by restoring gut microbiota and redox homeostasis. Cell Host Microbe 32, 1502–1518 (2024).
Temido, M. J. et al. Overcoming the challenges of overtreating and undertreating inflammatory bowel disease. Lancet. Gastroenterol. Hepatol. 10, 462–474 (2025).
Lee, Y. et al. Hyaluronic acid–bilirubin nanomedicine for targeted modulation of dysregulated intestinal barrier, microbiome and immune responses in colitis. Nat. Mater. 19, 118–126 (2020).
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (No. 2023YFD1800105, L.M. and F.Z., No. 2022YFE0139800, L.M.), National Natural Science Foundation of China (82472163, F.Z., 32401260, M.O., 82272154, L.M., and 32371458, Y.L.), Tianjin Science Fund for Distinguished Young Scholars (22JCJQJC00120, L.M.), China Postdoctoral Science Foundation (2024M750242, M.O., 2025T180974, M.O.), Natural Science Foundation of Tianjin (23JCYBJC00470, F.Z.), the Fundamental Research Funds for the Central Universities, Peking Union Medical College (3332025196, M.O.), Postdoctoral Fellowship Program of CPSF (GZB20230085, M.O.), State Key Laboratory of Advanced Medical Materials and Devices Grant (24ZXZSSS00200, L.M.), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (No. 2024-I2M-TS-028 and 2025-I2M-XHJC-048, L.M.), and Guangdong Basic and Applied Basic Research Foundation (2025A1515010411 and 2024A1515220045, F.Z.).
Author information
Authors and Affiliations
Contributions
L.M., M.O., F.Z., and J.C. conceived and designed the project. J.C. and M.O. conducted all the experiments and analyzed the data. R.L., R.M., W.L., B.Z., L.Y., Y.F., X.W., W.Z., H.Z., and Z.M. provided technical input on this project. J.C. and M.O. wrote the original manuscript. L.M., M.O., Y.L., and F.Z. reviewed and revised the manuscript. J.Z. provided support for the manuscript revision. L.M., M.O., Y.L., and F.Z. supervised the overall research. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Houyu Wang and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, J., Luo, R., Miao, R. et al. Engineered exosome nanovesicles for delivery of antibodies to treat inflammatory bowel disease. Nat Commun 17, 2737 (2026). https://doi.org/10.1038/s41467-026-69382-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69382-4
