
Abstract
Wheat stem rust is a disease of global importance caused by the fungal pathogen Puccinia graminis f. sp. tritici (Pgt). Here we generate chromosome-level, nuclear-phased genome references for Pgt isolates ETH2013-1 and ITA2018-1, representing races TKTTF and TTRTF respectively, that have caused major epidemics in Africa and Europe. The nuclear haplotypes of ETH2013-1 and ITA2018-1 are unique and unrelated to those of Ug99 and Pgt21. Pgt nuclear haplotypes show extensive variation in sequence and copy number of six known Avr genes and AvrSr33, which we identify through an effector gene library screen. Recognition properties of 22 novel Avr gene variants explain the race virulence phenotypes and the outbreak of TTRTF on durum cultivars containing Sr13b, since ITA2018-1 carries a homozygous deletion of AvrSr13. This work establishes an Avr gene atlas for Pgt that can inform wheat breeding and enable development of sequence-based virulence diagnostic tools for pathogen surveillance.
Similar content being viewed by others


Introduction
The sudden emergence of novel strains of plant pathogens that overcome previously resistant cultivars is a major challenge in the management of crop diseases1. In the case of wheat stem rust disease caused by Puccinia graminis f. sp. tritici (Pgt), the emergence of novel races has led to multiple epidemics in recent years resulting in substantial yield losses in the most vulnerable wheat-growing regions2,3,4. For instance, Pgt race TTKSK (known as Ug99) was first detected in Uganda in 19985 and caused major epidemics in eastern Africa during the 2000’s. TTKSK and other races in the Ug99 lineage remain a significant threat for wheat production as they continue to spread into South Asia6,7. More recent epidemics in Africa and Europe have been caused by other races of Pgt with different virulence profiles, including TKTTF and TTRTF8,9, presenting further challenges for disease resistance breeding.
Genetic resistance to the rust fungi in small grain cereal hosts is typically conferred by race-specific resistance (R) genes encoding immune receptors that recognise pathogen effector proteins encoded by avirulence (Avr) genes in a gene-for-gene manner10,11. Thus, the evolution of new rust races relies on genetic changes such as mutation, somatic nuclear exchange and sexual recombination12,13. Over 20 stem rust resistance genes have been isolated from wheat and barley14 and the identification of Avr genes from Pgt is accelerating with the emergence of new tools and improved genome resources15,16. Currently six Avr genes have been identified: AvrSr13, AvrSr2215, AvrSr2717, AvrSr3518, AvrSr5019, and AvrSr6220. However, there is a need to systematically characterise the diversity and function of allelic and paralogous variants of these Avr genes in Pgt populations, particularly in major epidemic-causing lineages. For example, Ortiz et al.21 examined AvrSr50 allelic diversity by assembling sequence variants from Illumina sequence data for a range of isolates, identifying 14 different sequence variants. However, the dikaryotic nature of rust fungi, with each isolate containing two independent haploid nuclei, has been a challenge for broader Avr genotype assignment13. Recently, the combination of Hi-C chromatin contact analysis and highly accurate long-read DNA sequencing has enabled resolution of the two haploid genomes and assembly of nuclear-phased genome references for several rust species22,23,24,25,26. This approach was first adopted for Pgt, generating nuclear haplotype-phased assemblies for isolates Ug99 and Pgt21-022, which belong to clonal lineages designated as clades I and IX, respectively, based on SNPchip and sequence-based phylogenies of global isolates27. Genome comparison revealed that these two isolates share one near-identical nuclear haplotype genome (designated haplotype A), while the second nuclear haplotype in each isolate (C and B, respectively) are highly divergent22. These data suggest that the Ug99 (clade I) lineage (nuclear haplotypes A and C) arose via somatic hybridization and nuclear exchange between a clade IX isolate (nuclear haplotypes A and B) and an unknown donor containing haplotype C. Notably, an isolate collected in Iran in 2015 (IR-01) was shown to contain the C genome and could represent the other parental lineage of Ug9922. Guo et al.27 also identified a lineage of Pgt (designated as clade II) present in Africa prior to the emergence of Ug99 as a candidate for the C genome donor based on a genome admixture analysis. However, the relationship between clade II and IR-01 is not known.
Other important new races of Pgt have emerged more recently. In 2013, an epidemic broke out in Ethiopia caused by race TKTTF that possesses novel virulence for the resistance gene SrTmp present in the widely grown wheat cultivar Digalu8. Based on genotyping with a SNPchip ( ~ 1000 nucleotide polymorphisms), these isolates of race TKTTF were divided into two genetic groups, designated as clades IV-A and IV-B, raising questions about their relationship8. Interestingly, clade IV-A isolates of race TKTTF had also been detected in Turkey in 201228, Germany in 2013 and Georgia in 20142, suggesting an earlier origin and migration to Ethiopia of this lineage. Patpour et al.29 detected TKTTF isolates of clade IV-B in Europe from 2017 onwards. Lewis et al.30 also described an isolate of race TKTTF from the UK in 2013. Subsequent analysis of isolates collected during stem rust outbreaks in Germany and Georgia led to the designation of six additional clade IV subgroups2,31.
In 2016, a wheat stem rust outbreak occurred in Sicily caused by a novel Pgt race, TTRTF9, which overcame the Sr13b resistance in both durum and bread wheat2,32. Race TTRTF subsequently became the most prevalent Pgt virulence type in Europe between 2017 and 202129 and has also been detected in Iran33 and Ethiopia34. Olivera et al.2 also detected isolates of race TTRTF in Georgia in 2014 and 2015, again suggesting a broader dispersal prior to the epidemic in Sicily. The TTRTF isolates were assigned to clade III-B by SNPchip and simple-sequence repeat marker analysis and are genetically distinct from the TKTTF clade IV isolates2,29.
To investigate the genomes and virulence genotypes of Pgt races causing these recent epidemics, we generated haplotype-phased genome assemblies for isolates from Ethiopia (ETH2013-1 of race TKTTF clade IV-B subgroup) and Sicily (ITA2018-1 of race TTRTF). These genome resources enabled us to identify variants of known Avr genes in these isolates, assign their recognition properties through functional assays, and corelate their genotypes to virulence phenotypes. To facilitate sequence-based detection of virulence genotypes in Pgt lineages and enhance surveillance capabilities, we initiated an Avr gene atlas that includes Avr gene variants and associated recognition profiles.
Results
Haplotype-phased genome references for two Pgt isolates responsible for recent epidemics
Two Pgt isolates were obtained from regions where recent epidemics occurred: ETH2013-1 was collected during the 2013 epidemic growing on wheat cultivar ‘Digalu’ in Ethiopia and ITA2018-1 was collected in 2018 in Sicily, Italy. Inoculation of these isolates onto a standard wheat differential set confirmed their races as TKTTF and TTRTF, respectively (Supplementary Data 1). Both isolates were also inoculated onto transgenic wheat lines containing the stem rust resistance genes Sr13c, Sr22, Sr27, Sr33, Sr35, or Sr50 genes and the wheat introgression line Zahir 1644, which carries Sr62. ITA2018-1 was virulent on Sr13c and Sr35, but avirulent on the other Sr genes, while ETH2013-1 was avirulent on all lines (Supplementary Fig. 1). To determine genetic relationships of these isolates to those previously characterized, whole genome short read sequence data were generated and used in a phylogenetic analysis with publicly available sequences for 144 Pgt isolates, including 105 with whole genome data, 38 with RNAseq data and one with both whole genome and RNAseq data (Fig. 1, Supplementary Fig. 2). ETH2013-1 was placed in a clonal lineage containing multiple European and African isolates30,35, including 13ETH23-1, which was one of the originally defined isolates of race TKTTF identified in Ethiopia and assigned to clade IV-B based on a SNPchip assay8. This clade also includes the isolate UK-01 (race TKTTF) detected in the UK in 201330. Other isolates collected from Ethiopia, also with race TKTTF, fall into a separate clonal lineage that is genetically divergent from the clade IV-B and includes isolates previously assigned to clade IV-A by SNPchip analysis8. The IV-A clade also includes an isolate from Israel collected in 2012 (12ISR2083), further confirming the wider dispersal of this lineage prior to the Ethiopian epidemic. ITA2018-1 groups in a clonal lineage with four isolates (IT-01, IT-02, IT-03, IT-04) collected in Sicily during the 2016 epidemic but with unknown race types30.

Maximum likelihood phylogenetic tree of 146 Pgt isolates based on 393,568 SNPs compared to the diploid Pgt21-0 reference assembly. Nodes are colored by the geographic region where isolates were collected. Node symbol indicates sequence data type: circle = whole genome sequencing, star = RNA-seq. Bootstrap values over 80% are shown as circles at branch midpoints. Scale is mean substitutions per site. Nuclear-phased references exist for isolates with bold names. Clade identifiers are based on previous reports using SNPchip analysis2,8,31.
PacBio HiFi and Hi-C sequencing data were generated for ETH2013-1 and ITA2018-1 and used to assemble nuclear haplotype-phased reference genomes for each isolate using Hifiasm (Supplementary Data 2). This resulted in two distinct haplotypes for each isolate, with no haplotype phase switches detected by NuclearPhaser23 in contigs of either assembly. Contigs were scaffolded using Hi-C contact data resulting in 18 chromosomes in each haplotype, with between 4 and 14 gaps per haplotype (Supplementary Data 3). Individual chromosomes in the ETH2013-1 and ITA2018-1 assemblies showed an average of 97.8% of trans Hi-C contacts to other chromosomes in the same designated nuclear haplotype (Supplementary Data 4; Supplementary Fig. 3), confirming accurate phasing and nuclear assignment. This was also supported by whole-genome Hi-C contact maps (Supplementary Fig. 4). Unplaced contigs were mostly small (N50 of 35.6 kbp and 39.7 kbp for ETH2013-1 and ITA2018-1, respectively) and had extremely high repeat content (82.33% and 89.29%; Supplementary Data 3). Nuclear haplotype sizes ranged from 82.9 to 86.5 Mbp (average 84.8 Mbp), similar to the previously assembled haplotypes for Pgt21-0 (83.9 and 85.9 Mbp)22 (Supplementary Data 3).
Here, we switched to a haplotype numbering scheme rather than maintain the A, B and C haplotype nomenclature established by Li et al.22 for isolates Pgt21 and Ug99. This allows for the future expansion of the Pgt pangenome to more than 26 haplotypes, noting that for instance over 50 haplotypes have now been assembled for the related species Puccinia coronata f. sp. avenae (Pca)24,36,37. We designated the A, B and C haplotypes of Pgt21 and Ug99 as hap01, hap02 and hap03 respectively, while the two haplotypes of ETH2013-1 were designated as hap04 and hap05 and those of ITA2018-1 as hap06 and hap07 (Fig. 2A). We also obtained Hi-C data for the Pgt isolate Ug99 and used this to scaffold the published contigs22 into chromosome assemblies for its hap01 and hap03 nuclear haplotypes (Supplementary Data 5). Genome alignments showed that the chromosomes for each haplotype were similar in size and highly syntenous in structure with no major rearrangements (Fig. 2B, Supplementary Fig. 5), except for hap02 which contains two large translocations between chromosomes 3 and 5 and between 8 and 16 as previously reported22. The BUSCO completeness scores of 95.6–96.4% and 0.8–1.1% duplication for each haplotype (Supplementary Data 3) were comparable to the haplotype-phased genomes obtained previously for Pgt22, P. triticina23,38, P. striiformis f. sp. tritici39 and Pca24,36,37. Pairwise whole genome alignment of haplotypes showed divergence between all pairs, with haplotype identity lowest between hap02 and hap07 (88.46% bases aligned, 95.09% identity), while hap02 and hap03 were the most similar (92.12% bases aligned, 97.63% identity) (Supplementary Data 6). Recombination block analysis revealed no evidence for recent recombination events, except for a few small regions (n = 22, 8.5 Mbp total length) shared between hap05 and hap07 (Supplementary Fig. 6; Supplementary Data 7).

A Haplotype designations (01 to 07) for haploid nuclei of the four haplotype-phased Pgt isolates. B Synteny plot of seven haplotypes (hap01 to hap07), with chromosomes represented on the x-axis. Hap02 is shown first as it has two large chromosomal translocations relative to the other haplotypes. Vertical coloured lines connecting chromosomes reflect four relationships between sequences: syntenic (grey), inverted (yellow), translocated (red), and duplicated (blue).
Variation at mating type loci in the newly designated haplotypes of Puccinia graminis f. sp. tritici
Mating type alleles at both the a and b loci were identified in all newly designated haplotypes (Supplementary Fig. 7A, B; Supplementary Data 8) using homology searches of previously published Pgt sequences22,40. Haplotypes hap05 and hap07 encode the STE3.2.2 a locus allele with the associated mfa2 pheromone precursor located 845 and 878 bp downstream, respectively. Hap04 and hap06 encode the STE3.2.3 receptor allele with the mfa3 pheromone precursor located 31,037 and 29,864 bp upstream, respectively. The STE3.2.2 and mfa2 allele in each isolate is identical to the sequence published for Pgt isolate 75-36-700-3-7a40. However, the STE3.2.3 alleles from isolates Pgt21-0, ETH2013-1, and ITA2018-1 differ from 75-36-700-3-7a by ten nucleotides including four non-synonymous substitutions leading to amino acid differences (I332M, H365Q, D373N, and Q376P). The Ug99 STE3.2.3 allele encodes a protein truncated by 48 amino acids due to a single nucleotide deletion as reported by Li et al.22. Several clade I isolates share this mutation (04KEN15604, 06KEN19V3, 07KEN24-4, 14KEN58-1, Pgt-60, ET-01, and ET-02), although some others (Pgt-55, Pgt-59, Pgt-61, 72ETH11-4; Supplementary Data 9) do not, suggesting a recent mutation event in this lineage (Supplementary Fig. 7C). Interestingly, this polymorphism corresponds to the separation between the Sr31 avirulent and Sr31 virulent isolates in this lineage. The mfa3 alleles all share the same single amino acid difference (N27D) from the published 75-36-700-3-7a allele40. K-mer containment for both of the a mating locus alleles (STE3.2.2 and STE3.2.3) was high in genomic and RNAseq sequence data from almost all isolates from the global collection, indicating uniform heterozygosity for these two alleles (Supplementary Data 9). Two Pgt isolates showed low shared k-mers for both STE3.2 alleles (69.10–87.60%), but this was likely due to their low mapped read coverage (76WA1295A – 3.5X, DE-01 – 6.38X). The k-mer identity for STE3.2.3 was high in all samples, but percentage of shared k-mers was lower (73.20-89.90%) in 63 isolates. Comparisons of read mapping revealed conserved patterns of either nine or ten SNPs within STE3.2.3 exons in these isolates. The gene sequences with 10 SNPs matched the published STE3.2.3 allele from 75-36-700-3-7a, while those with nine differed by a single SNP. Both variants of STE3.2.3 are present in isolates from Asia, Africa, and Europe, while to date all isolates from North and South America have the 75-36-700-3-7a variant, and isolates from Australia possess only the hap01 variant.
The four haplotypes from ETH2013-1 and ITA2018-1 each have unique and novel b locus allele pairs (b5 to b8), bringing the total of known b locus alleles represented in Pgt genome references to eight (Supplementary Fig. 7A, B)22,27,40. Using a k-mer containment approach, one or two known b locus alleles were assigned to 113 of the 146 Pgt isolates, with all isolates showing heterozygosity at this locus (Supplementary Fig. 2; Supplementary Data 10). These data support the hypothesis of mating type being controlled by a bi-allelic a locus and multi-allelic b-locus as observed in other species of Puccinia36,38.
Identification of Puccinia graminis f. sp. tritici lineages with shared nuclear haplotypes
To identify potential somatic hybridization events between Pgt isolates/lineages, we performed k-mer containment analysis36,38 on all the available whole genome sequence data. Isolates in the Ug99 and Pgt21-0 lineages fully contain the k-mers from hap01, while two isolates from Pakistan and two from the Czech Republic fully contain k-mers from hap02 (Fig. 3; Supplementary Data 11), consistent with the haplotype specific phylogeny results reported previously22. All clade II isolates show full containment of hap03 k-mers. This includes isolate IR-01 (collected in Iran in 2015), which Li et al.22 identified as a carrier of hap03, as well as isolates from east Africa that predate Ug99 emergence and were postulated by Guo et al.27 as a possible donor lineage of hap03 to Ug99. These shared haplotype assignments were also supported by haplotype-specific phylogenetic trees (Supplementary Fig. 8A–C).

The containment of hap01 and hap02 (Pgt21-0), hap03 (Ug99), and hap04 and hap05 (ETH2013-1) in whole genome short-read sequences from 108 Pgt isolates. The k-mer identity (%) score was plotted against the shared k-mer (%) score for containment of the phased haplotypes for each isolate. Coloured data points represent isolates containing the respective haplotype (k-mer identity ≥ 99.89% and shared k-mers ≥ 97.67%), and data points labelled in grey represent isolates that do not contain that haplotype. Raw k-mer containment values are provided in Supplementary Data 11.
The k-mer containment analysis also showed that hap04 and hap05 (ETH2013-1) were shared by all the isolates placed in the clade IV-B clonal lineage (Fig. 1; Fig. 3; Supplementary Data 11). Hap05 was also shared by the Iranian isolate IR-06 (100% k-mer identity, 99.92% shared k-mers), which agrees with the phylogenetic tree based on the hap05 reference (Supplementary Fig. 9A, B; Supplementary Data 11). No isolates were identified that shared hap06 or hap07 of ITA2018-1 by k-mer containment analysis (Supplementary Data 11) or using haplotype-specific trees (Supplementary Fig. 9C, D). Results from the b mating locus allele assignments were consistent with the postulated presence of hap02 in Czech and Pakistani isolates and hap05 in IR-06, as these isolates contained the bW1bE1 (hap02) or bW6bE6 (hap05) alleles respectively (Supplementary Figs. 8B, 9B; Supplementary Data 10).
Variation in avirulence effector genes underlies virulence phenotypes
To understand the genotypic differences behind the virulence phenotypes of isolates ETH2013-1 and ITA2018-1, and build an Avr gene catalogue, we examined sequence variation in known Avr genes and adopted a consistent nomenclature to refer to these variants. Avr variants in the reference genomes were assigned a two-digit identifier based on their unique protein sequence, with previously characterized variants renamed to fit this scheme (e.g., AvrSr50-A1 became AvrSr50-01) (Supplementary Data 12). Variants encoding the same protein sequence but with synonymous nucleotide changes were distinguished by an additional suffix (e.g., AvrSr22-02.1 and AvrSr22-02.2). Figure 4 shows a summary of the nuclear-resolved genotypes of isolates Pgt21-0, Ug99, ETH2013-1 and ITA2018-1 for known Avr loci in Pgt. Detailed maps of Avr loci are shown in Supplementary Figs. 10–14.

Schematic showing Avr gene variants encoded by haplotypes of the AvrSr13, AvrSr22, AvrSr27, AvrSr33, AvrSr35/50, AvrSr62 loci in Pgt isolates Pgt21, Ug99, ETH2013-1 and ITA2018-1. Nuclear haplotypes of each strain are shown at left. Genes are shown as arrows and numbers indicate variant designation. Blue denotes Avr variants recognized by the corresponding Sr gene in functional assays, while pink denotes unrecognized variants. Blue and pink striped arrows denote Avr variants which gave an intermediate response in functional assays. Light blue fill with dashed outline indicates recognized alleles with zero or very low expression. Triangles represent insertional mutants previously described in ref. 22. Gene IDs for Avr variants are provided in Supplementary Data 12.
At the AvrSr13 locus (Supplementary Fig. 10), ETH2013-1 has two novel gene variants that encode proteins (AvrSr13-02, -03) that differ by a single amino acid in the signal peptide region and both differ by six amino acids from AvrSr13-0115. However, ITA2018-1 has null alleles for this locus in both haplotypes due to deletion events. This explains the virulence of this isolate/lineage on Sr132,32 (Supplementary Data 1). For AvrSr22, both ITA2018-1 and ETH2013-1 isolates carry one allele encoding an identical protein sequence to AvrSr22-0215 and one unique allele encoding a protein (AvrSr22-03) with a single amino acid difference from AvrSr22-02 (Supplementary Fig. 11). Substantial variation exists at the AvrSr27 locus, with differences in the encoded amino acid sequence (six novel alleles detected in ETH2013-1 and ITA2018-1), gene copy number (one or two per haplotype) and gene orientation (Fig. 4; Supplementary Fig. 12). The AvrSr50/AvrSr35 locus in all haplotypes from ETH2013-1 and ITA2018-1 is most structurally similar to hap02 (Supplementary Fig. 13). ETH2013-1 is homozygous for AvrSr50-03 (previously AvrSr50-A2) which is recognised by Sr5021 and explains its avirulent phenotype on this resistance gene. ITA2018-1 is heterozygous for a novel recognized allele AvrSr50-04 (see below) and the known recognized AvrSr50-05 allele (previously AvrSr50-B4). ETH2013-1 is homozygous for AvrSr35-01, which is identical to the originally isolated allele from 99KS7618, while ITA2018-1 is homozygous for the novel AvrSr35-02 allele, with unknown recognition. The AvrSr62 locus in Pgt21-0 is complex, with four paralogs in hap01 and three in hap02, of which two in each haplotype are recognized (01, 04, 05 and 07)20. Hap03, hap04, hap06 and hap07 contain similar AvrSr62 haplotypes to hap02, each containing three paralogs with some minor sequence differences (Supplementary Fig. 14). The AvrSr62 locus in hap05 is similar to that in hap01 but with one additional intact gene copy (AvrSr62-09) that corresponds to a pseudogene in hap01 (inactive due to a splice site mutation). Altogether, four novel variants of AvrSr62 (-08 to -11) were identified.
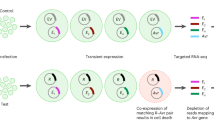
To assess the potential avirulence function of the effector proteins encoded by the novel Avr gene variants, we generated expression constructs for each variant and tested them by co-expression with the corresponding Sr genes in wheat protoplasts and in Nicotiana benthamiana. Proteins AvrSr13-02, AvrSr22-03, AvrSr27-05 to -10, and AvrSr50-04 all triggered cell death when co-expressed with the corresponding Sr protein but not when expressed alone, confirming their recognition (Fig. 5; Supplementary Figs. 15–19). Likewise, recombinant Barley Stripe Mosaic Virus (BSMV) expressing any of these Avr gene variants was unable to infect wheat lines containing the corresponding Sr genes (Fig. 5B; Supplementary Figs. 15, 17, 19). The AvrSr35-02 protein failed to trigger cell death when co-expressed with Sr35 in N. benthamiana and induced a weaker response in protoplasts than AvrSr35-01, consistent with the Sr35-virulent phenotype of ITA2018-1 (homozygous for avrsr35-02) (Supplementary Fig. 20A–C). There are 40 single amino acid differences and 2 indels between AvrSr35-01 and -02, but only one of these lies in the interaction interface with Sr3541,42, S349 (AvrSr35-01) to R352 (AvrSr35-02). Experimental data in N. benthamiana shows exchanging this single residue is sufficient to abolish recognition in AvrSr35-01 and restore recognition in AvrSr35-02 (Supplementary Fig. 21D, E). For the AvrSr62 locus, each haplotype contained two variants that were both recognised when co-expressed with Sr62 in N. benthamiana or wheat protoplasts (Supplementary Fig. 21), and one to three unrecognized variants, similar to Pgt21-0.

A Transient expression in protoplasts of the wheat cultivar Fielder. The x-axis shows genes used for transformation while the y-axis shows fluorescence values normalized to the YFP reference (RFU). Grey columns show mean values from three biological replicates. Error bars indicate the standard error. Black dots represent the result for each replicate of a given treatment. Two-tailed t-tests were performed for pairwise comparisons between specific treatments, ‘***’ indicates significance at 0.001 level (P-value = 2.165e-4, 1.99e-4), ‘ns’ non-significant. A t-test significance value directly above a column indicates a comparison with the YFP reference. Raw data, detailed analysis and t-test P-values are provided in Source data file. B Transient expression of AvrSr22-03 and negative control iLOV in wheat cultivars Schomburgk (Sr22) and Cadenza (sr22) using the BSMV-based protein over-expression system. Fractions under leaf pictures indicate the number of plants presenting Barley Stripe Mosaic Virus (BSMV) symptoms over the total number of inoculated plants. C Co-expression assay in Nicotiana benthamiana. Dotted lines mark out the area infiltrated with an Agrobacterium tumefaciens suspension. D Cell death score in all infiltrated N. benthamiana leaves represented as a box and whiskers plot with the number (n) of replicates indicated in the graph. The x-axis indicates the different treatments, the y-axis represents the cell death score out of 5 (0 = no cell death, 5 = cell death over the whole inoculated area). Letters indicate samples that do not differ significantly (Welch’s ANOVA followed by Dunnett’s T3 multiple comparisons test, pairwise P-values in Source Data File).
Given that virulence on Sr27 was associated with low expression of the AvrSr27-3 allele despite its recognition in functional assays17, we examined the expression levels for the corresponding variants in the two isolates during host infection. Both ETH2013-1 and ITA2018-1 possess at least one gene copy that was expressed, with most copies showing TPM > 20. There were two exceptions; AvrSr27-05 (hap04) was not expressed (TPM < 1), and AvrSr27-10 (hap07) had very low expression (average TPM = 5.21) during infection. Both gene variants occur in haplotypes with an adjacent expressed gene; thus, it is likely that both strains are homozygous for avirulence at this locus.
Thus, ETH2013-1 contains avirulence alleles corresponding to each of these Sr genes, consistent with its avirulent infection phenotype (Supplementary Data 1), while ITA2018-1 contains avirulence alleles for Sr22, Sr27, Sr50 and Sr62, but is homozygous for virulence alleles for Sr13 and Sr35, consistent with its infection profile.
Identification of AvrSr33 and characterization of genetic variants
To further expand the Avr gene catalogue, we also used the Sr33 resistance gene43 to screen a library of 1,373 effector gene candidates from Pgt21-0 by co-expression in wheat protoplasts15,20. A single effector gene construct (clone #0956) showed significantly reduced expression in the presence of Sr33 relative to an empty vector control (Fig. 6A; Supplementary Fig. 22), suggesting immune recognition-induced protoplast cell death. We further tested this candidate (AvrSr33-01) by individual co-expression with Sr33 in wheat protoplasts, in N. tabacum and in N. benthamiana, as well as by VOX in wheat (Fig. 6B–E), which confirmed its specific recognition. The AvrSr33 locus contains 0, 1, or 2 homologous copies of this gene in each haplotype, with a total of eight sequence variants identified (Fig. 4, Supplementary Fig. 23). Of these, AvrSr33-02, -03 and -04 were each recognized strongly by Sr33 in co-expression assays in N. benthamiana and wheat protoplasts, AvrSr33-05 and -08 led to weak responses both in protoplasts and N. benthamiana, while AvrSr33-06 and -07 were not recognized in either assay (Fig. 6; Supplementary Figs. 24, 25). Thus, isolates Pgt21-0, ETH2013-01 and ITA2018-1 are assigned as homozygous for avirulence to Sr33, while Ug99 is heterozygous with a null allele in hap03 due to a 3,109 bp deletion relative to hap06.

A Differential gene expression analysis of a library comprising 1373 predicted effector genes from P. graminis f. sp. tritici (Pgt) divided into two pools, part A and part B, after co-expression in wheat protoplasts of cultivar Fielder with Sr33 compared to an empty vector. The volcano plot shows differential expression (x-axis) versus adjusted P-value (y-axis) for each effector construct (dots). The effector gene construct that showed significantly reduced expression is represented in red. Shown here is the result for library part A (718 effector candidates). The result for part B (655 effector candidates) can be found in Supplementary Fig. 22. B Validation of the interaction between Sr33 and AvrSr33 in protoplasts of wheat cultivar Fielder. The x-axis represents the different treatments, the y-axis represents fluorescence normalized to the YFP reference (RFU). Grey columns show mean values from three biological replicates. Error bars indicate the standard error. Black dots represent the result for each replicate of a given treatment. Analysis of variance (ANOVA, two-tailed Tukey post hoc test) test detected significant differences between samples (P-value = 4.06E-08) and letters indicate samples that do not differ significantly (pairwise P-values in Source Data File). C Infection of wheat cultivars Lorikeet (Sr33) and Cadenza (sr33) with BSMV expressing AvrSr33-01 or the negative control iLOV. Fractions under leaf pictures indicate the number of plants presenting Barley Stripe Mosaic Virus (BSMV) symptoms over the total number of inoculated plants. D Validation of the interaction between Sr33 and AvrSr33 by co-expression in Nicotiana tabacum. Dotted lines mark out the area infiltrated with an Agrobacterium tumefaciens suspension. E Validation of the interaction between Sr33 and AvrSr33 by co-expression in N. benthamiana. Dotted lines mark out the area infiltrated with an A. tumefaciens suspension.
Discussion
Nuclear haplotype-phased reference genomes in cereal rust fungi are crucial for understanding genetic diversity in these dikaryotic organisms44. Previously, only two nuclear haplotype phased references were available for Pgt, derived from isolates Pgt21-0 and Ug99, with only the former scaffolded to chromosome level22. Here, we have expanded the genomic resources for Pgt by generating haplotype-resolved reference genomes for two isolates (ETH2013-1 and ITA2018-1) representing lineages that caused recent epidemics in Ethiopia and Italy8,9 and which have both spread to become predominant races in Europe and Africa29. We also assembled full chromosomes for the Ug99 haplotypes. These resources have enabled the establishment of an Avr gene atlas compiling genotypic variation in known Avr loci across these important Pgt strains.
The four nuclear haplotype sequences of ETH2013-1 and ITA2018-1 are unique and highly diverged from those of the previous reference genomes (Ug99 and Pgt21-0). Thus, there are now a total of seven fully phased nuclear haplotypes available for Pgt (designated hap01 to hap07), with hap01 shared between Pgt21-0 and Ug99 due to a nuclear exchange event22. Another chromosome-scale genome assembly was generated for the US isolate 99KS7627, but this still contains phase swaps, with each of the two pseudo-haplotypes (designated E and F) containing a mixture of sequences derived from either nucleus (Henningsen et al., unpublished data). Previous SNPchip analysis of isolates derived from the 2013 stem rust outbreak in Ethiopia revealed the existence of two distinct lineages amongst isolates sharing the TKTTF pathotype, clades IV-A and IV–B8. Phylogenetic analysis placed isolate ETH2013-1 in clade IV-B. This clade also includes an isolate detected in the UK in 201330, suggesting the widespread dispersal of the clade IV-B TKTTF lineage prior to the Ethiopian outbreak in 2013, as observed for the clade IV-A lineage2,28. Genome sequence data confirms that the IV-A clade is genetically distinct to IV-B with no evidence of shared nuclear haplotypes, despite sharing the same race type (TKTTF). Other isolates collected in Germany and Georgia displayed similar pathotypes to TKTTF but SNPchip genotyping placed them into other distinct genetic subgroups within clade IV (IV-C, IV-D, etc), and it was suggested that this set of lineages may derive from a sexually recombining population2,31. Genome assembly of additional isolates from these clade IV subgroups will help to resolve their genetic relationships and the evolutionary origins of this clade.
We previously showed that the Ug99 lineage arose via somatic hybridization and nuclear exchange, with hap01 donated by Pgt21-0 and hap03 by an unknown parent22. Although the IR-01 isolate contains the hap03 nuclear genome it was first isolated in 2015, well after the emergence of Ug99 lineage; thus, it was not clear whether it represented a parental or derivative lineage22. Guo et al.27 proposed the clade II lineage, which was present in Africa prior to the Ug99 epidemic, as a possible donor of hap03. Here we found that IR-01 is a member of clade II and confirmed that these isolates do fully contain hap03, suggesting nuclear exchange between these lineages. However, the earliest collected isolate confirmed as part of the Ug99 lineage was from 1972 (72ETH11-4), while the earliest collected isolate placed in clade II was from 1984 (84KEN8B). The question of which is the parental or derived lineage remains open. We also found that hap05 in ETH2013-1 is fully contained in IR-06, which may indicate another nuclear exchange event. However, an alternative explanation is that hap05 is a direct sexual product of IR-06 that is fully derived from the two unknown IR-06 haplotypes by meiotic recombination (Supplementary Fig. 26). Henningsen et al.36 found such a relationship between haplotypes in two lineages of the oat crown rust fungus (Pca) from Australia. These lineages showed a non-reciprocal haplotype-sharing arrangement in that one haplotype of lineage L11 (hap25) was fully contained in genomic sequences from lineage L18, but neither haplotype of L18 (hap03 and hap04) was fully contained in L11. Recombination block analysis of the haplotype-resolved genome assemblies revealed that hap25 was 50% derived from each of hap03 and hap04, consistent with it being the F1 product of meiotic recombination in the L18 parent. Given the postulated sexual origin of the clade IV lineages2, a similar relationship may exist between ETH2013-1 and IR-06. Preliminary data suggests that IR-06 belongs to clade IV-C and full genome assembly of IR-06 or another member of this lineage will be required to answer this question. Isolates CZ-02,03 and PK01,02 represent two different clonal lineages that each share hap02 from Pgt21-022 (Supplementary Fig. 8B, Supplementary Data 10). Given the age of the Pgt21-0 lineage ( > 100 years), it is most likely that these lineages acquired hap02 as a result of nuclear exchange. Again, full genome assembly of isolates from these lineages would clarify this relationship.
Like other Puccinia species22,36,38,40,45, Pgt seems to contain a bi-allelic a (or PR) mating locus and multi-allelic b (or HD) locus with eight alleles now defined at the sequence level. All 146 Pgt isolates with available genome sequence data show heterozygosity at the a mating locus (STE3.2.2/STE3.2.3). While the b locus alleles of the seven reference haplotypes are all distinct, this is not sufficient to distinguish them from other haplotypes. For example, isolates IT-05, IR-06, IR-02 and SC-01 all share the bW6bE6 allele with hap05 of ETH2013-1 but only IR-06 contains hap05.
The identification of Avr genes from Pgt has been accelerating with the emergence of new screening tools15,16. Here we identified AvrSr33 in a protoplast screen from a library of 1,373 Pgt effector candidates and confirmed its recognition using VOX assays in wheat and co-expression with Sr33 in N. benthamiana. This brings the total number of Avr loci identified in Pgt to seven. The fully haplotype-phased genome references generated here provide an ideal resource for assessing genetic variation at these loci. We identified the genotypes of the seven Avr loci in each of the Pgt reference haplotypes and functionally characterized 22 novel Avr gene variants for recognition by the corresponding Sr genes. This allowed assignment of Avr genotypes to these epidemiologically important lineages of the wheat stem rust pathogen which can explain their virulence phenotypes on wheat lines carrying the corresponding Sr genes. Isolates of the TTRTF race from Italy were previously reported to be virulent on Sr13b, but avirulent on a wheat differential line (ST464) containing Sr13c2,9,32. However, we found that in addition to being virulent on Sr13b, ITA2018-1 was also virulent on a transgenic Fielder line containing Sr13c, which is consistent with the homozygous deletion of AvrSr13 in this strain. This suggests that the ST464 line may contain an additional Sr gene (or genes) effective against this strain, possibly explaining some of the reported differences in recognition specificity between Sr13b and Sr13c, which encode NLR receptor proteins that differ by only two amino acids. This highlights the value of using near-isogenic wheat lines carrying single R genes for accurate assignment of pathogen virulence phenotypes. ITA2018-1 was also virulent on transgenic wheat line Fielder expressing a Sr35 and is homozygous for the novel avrsr35-02 allele, which was not recognized by Sr35 in co-expression assays.
Apart from AvrSr13 and AvrSr35 in ITA2018-1 each of the four reference genomes contains at least one recognized allele at each of the Avr loci, consistent with their avirulent phenotypes on wheat lines carrying the corresponding Sr genes. As noted by Chen et al.20, Avr62-05 and -07 gave strong recognition responses in functional assays, and we found similar strong responses for the closely related variants Avr62-08 and -11. However, AvrSr62-01 and -04 gave intermediate recognition responses20, so it is still not clear if hap01 and hap05 containing only these two recognized variants express an avirulent phenotype on Sr62 wheat. AvrSr22 was the only locus at which all the isolates were homozygous for strongly recognized alleles. This means that evolution of virulence to Sr22 in these lineages would require two independent mutations. Thus, Sr22 may provide more durable resistance against these Pgt strains.
Our findings indicate that it is critical to detect nuclear haplotype exchanges as these will impact the overall evolution of the pathogen and virulence profiles of rust populations. Given that avirulence is a dominant trait in rust fungi, nuclear exchanges may be unnoticed by rust surveillance practices that rely solely on virulence phenotyping across the relatively small wheat differential set. This is exemplified by the presence of Pgt21-0 in Australia since the 1950’s which carries a nuclear haplotype present in the Ug99 lineage22. Characterizing the genetic diversity of Avr genes among major Pgt races causing epidemics is important for monitoring rust evolution and prioritizing resistance breeding strategies. The catalogue of Avr genes and recognition properties provides insight into the virulence mechanisms behind major disease outbreaks, such as the deletion of AvrSr13. This change allowed the TTRTF race group to infect durum wheat lines containing Sr13b, which were prevalent in the field during the outbreak in Sicily in 20169. Continued development of a comprehensive Avr gene atlas with additional reference haplotypes and new Avr genes will provide a valuable resource to enable virulence prediction from sequence-based surveillance tools such as the MARPLE platform46,47.
Methods
Wheat stem rust isolate sampling and virulence phenotyping
The Pgt isolate ETH2013-1 was collected from the Bale region of Ethiopia in 2013 from the bread wheat (Triticum aestivum L.) cultivar (cv) Digalu. Isolate ITA2018-1 was collected from Ciminna, Sicily, Italy in 2018 from the bread wheat cv Antonello. Urediniospores of ETH2013-1 and leaf samples infected with ITA2018-1 were sent to the University of Minnesota and processed within the Biosafety Level 3 facility (St Paul, MN, USA). Single pustules of each isolate were selected from the susceptible wheat cv McNair 701 and increased several times to obtain pure cultures as described by Li et al.22. Isolates were typed for race based on the infection types they exhibited on the wheat stem rust standard differential set48 (Supplementary Data 1). Infection types ranging from 0 to 2 and combinations thereof were indicative of low compatibility (avirulent) and those from 3 to 4 were indicative of high compatibility (virulent) based on the 0 to 4 scale of Stakman et al.49. Isolates were also phenotyped for their infection types on transgenic wheat lines of cv Fielder containing the single genes Sr13c, Sr22, Sr27, Sr33, Sr35, and Sr50. Additionally, isolates ETH2013-1 and ITA2018-1 were assayed for their infection types on the wheat introgression line Zahir 1644, containing Sr62 derived from the wild wheat Aegilops sharonensis20 (Supplementary Data 1). To generate the Sr13c transgenic line, a 9,016 bp genomic sequence (NCBI accession number KY924305.1) comprising 2,913 bp upstream and 2,590 bp downstream of the Sr13c coding sequence was synthesized (Epoch, USA) and cloned into pD12 donor vector as previously described in ref. 50. The Sr13c gene was transferred into a binary vector with Bar selection using GatewayTM LR ClonaseTM Enzyme mix (Invitrogen, USA) and transformed into wheat cv Fielder as previously described in ref. 51.
Genome sequencing
DNA for short read sequencing was extracted from 30 mg of urediniospores using the OmniprepTM Genomic DNA isolation kit (#786-136, G-Biosciences), according to the manufacturer’s instructions. Samples were sequenced by the University of Minnesota Genomics Center (UMGC, St. Paul, MN) using 150 bp paired-end Nextera Flex library preparation and the Illumina NovaSeq 6000 platform. High molecular weight DNA for long-read sequencing was extracted from ~450 mg of urediniospores as described by Li et al.22 and sequenced on the PacBio Sequel II platform at UMGC. For Hi-C sequencing of ETH2013-1, ITA2018-1, and Ug99, 350 mg of urediniospores were cross-linked in 1% formaldehyde (20 min at ~22 °C) and then glycine was added to a 125 mM final concentration. After 15 min incubation, urediniospores were washed twice with distilled H2O, centrifuged at 3500 g for 5 min, and the supernatant removed. Washed urediniospores were ground in liquid N2 and shipped on dry ice to Phase Genomics Inc. (Seattle, WA) for Hi-C library preparation. Paired-end libraries were sequenced with Illumina NovaSeq at Azenta Life Sciences (South Plainfield, NJ).
RNA sequencing
Susceptible wheat plants (cv McNair 701) were inoculated with freshly collected urediniospores and maintained as above. At five- and seven-days post inoculation, infected leaves were sampled in triplicate and frozen in liquid N2. Germinated urediniospore samples were prepared by dusting 200 mg of fresh spores over a germination solution as previously described in ref. 52, collected after 10 h, and frozen in liquid N2. All tissues were ground to a fine powder using a mortar and pestle in liquid N2 and the RNA extracted using the Qiagen RNeasy Plant Mini kit (Cat No: 74904). Samples were sequenced at UMGC on the Aviti Cloudbreak platform after preparation of 150 bp paired-end stranded mRNA libraries (high output sequencing kit).
Genome assembly and scaffolding
PacBio HiFi reads were assembled using hifiasm53 v0.19.8 in Hi-C-integration mode using corresponding Hi-C reads with default parameters. The initial assembly was cleaned as described in Sperschneider et al.38 and Henningsen et al.36. Briefly, HiFi reads were aligned to the assembly using minimap254 v2.26 and coverage was assessed using the pileup.sh tool from bbmap v39.01 (https://sourceforge.net/projects/bbmap/). Low coverage contigs were removed from assemblies after determining cut-offs (7x for ETH2013-1, 10x for ITA2018-1) with a custom R script (https://github.com/JanaSperschneider/GenomeAssemblyTools/tree/master/CollapsedGenomicRegions). Mitochondrial and contaminant contigs were identified using BLASTn55 and removed from the assemblies. The NuclearPhaser v1.1 pipeline was used to assess phasing (https://github.com/JanaSperschneider/NuclearPhaser). Scaffolding was performed using information from SALSA56 v2.2, Hi-C-Pro57 v3.1.0, Hicexplorer58, and synteny to hap01 from isolate Pgt21-0 (GenBank accession GCA_008522505.1) assessed by alignment with D-GENIES59. BUSCO360 v3.0.2 was used to assess completeness of the assemblies (-l basidiomycota_odb9). Telomeres were detected using the findtelomeres.py script (https://github.com/JanaSperschneider/FindTelomeres). The published Ug99 contigs22 were also phased and scaffolded into chromosomes as described above.
Gene annotation
Genes and repeats were annotated in the ETH2013-1 and ITA2018-1 genomes as described by Sperschneider et al.38 and Henningsen et al.36. Briefly, RepeatModeler v2.0.2a (-LTRStruct) and RepeatMasker v4.1.2p1 were run to model and mask repeats in the genomes (http://www.repeatmasker.org). The full Repeatmodeler library was used to mask the genomes for statistics, whereas for gene prediction a library of only classified repeat families was used. RNAseq reads were aligned to their respective genomes with HISAT261 v2.2.1 and transcripts assembled with genome-guided Trinity62 v2.13.2 and Stringtie63 v2.2.1. CodingQuarry64 v2.0 was run in pathogen mode on the transcripts. We then ran funannotate65 v1.8.5 predict and supplied Trinity transcripts, CodingQuarry predictions, and Pucciniomycotina EST clusters from JGI MycoCosm (http://genome.jgi.doe.gov/pucciniomycotina/pucciniomycotina.info.html). To capture additional genes encoding secreted proteins, we ran TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder) on the StringTie infection transcripts and selected complete ORFs encoding proteins with a signal peptide predicted by SignalP66 v4.1 and no transmembrane domains predicted by TMHMM67 v2.0. Novel secreted protein encoding genes were added to the annotation unless they were >90% contained in a repetitive region.
Whole genome comparison and k-mer containment analysis
Whole genome alignment was performed with MUMmer68 v4.0.0rc1 (default nucmer parameters), and sequence divergence was assessed with dnadiff68. Synteny plots of Pgt chromosomes were generated with MUMmer68 v4.0.0rc1 (nucmer --maxmatch -c 100 -b 500 -l 50; delta-filter -m -i 60 -l 1000; show-coords -THrd), syri69 v1.6.3 and plotsr70 v1.1.0. The long arm of chromosome 3 in hap01 from isolate Pgt21-0 in NCBI was found to have an incorrect inversion from a scaffolding gap (position = 4,014,136 bp) to the end of the chromosome, which was corrected for the synteny visualization. Recombination analysis was performed using the pangenome-graph approach described by Henningsen et al.36 using cactus71 v2.9.0. Mash72 v2.0 was used to measure k-mer containment of sketched reference haplotype-phased sequences (-s 100000) in genomic Illumina reads from Pgt isolates. Isolates with k-mer identity of ≥ 99.85% and shared k-mers ≥ 97.50% were considered as likely containing the screened haplotype.
Phylogenetic analysis
In addition to the Illumina data generated for isolates ETH2013-1 and ITA2018-1, we also utilized publicly available RNAseq and genomic data from 151 Pgt isolates18,19,22,27,30,35,73,74,75 (Supplementary Data 13). Genomic data for seven isolates were excluded because SNP allele frequency and k-mer containment analysis showed signs of sample contamination (Supplementary Data 13). Illumina reads were processed using fastp76 v.0.23.2. Genomic reads were aligned to the haplotype-phased reference genomes of isolates Pgt21-0, Ug9922, ETH2013-1, and ITA2018-1 using bwa-mem277 v.2.2.1. RNAseq reads were aligned to the same references with STAR78 v.2.7.9a. Samtools79 was used to prepare alignment files for variant calling with Freebayes80 v1.3.6 (--use-best-n-alleles 6). Variants were filtered with vcflib81 v1.0.1 (-f “QUAL > 20 & QUAL / AO > 10 & SAF > 0 & SAR > 0 & RPR > 1 & RPL > 1 & AC > 0”) and vcftools82 v0.1.16 (--min-alleles 2 --max-alleles 2 --max-missing 0.9 --maf 0.05). The vcf file was converted to Phylip format using vcf2phylip (https://github.com/edgardomortiz/vcf2phylip/blob/master/vcf2phylip.py). Maximum likelihood tree search and bootstrapping were conducted with RAxML83 v8.2.12 (options -f -a -m GTRCAT -p 12345 -x 12345 -#500 –no-bfgs).
Assessment of mating type loci
To identify putative mating loci in isolates ETH2013-1 and ITA2018, protein sequences of STE3.2.2, STE3.2.3, bW-HD1, and bE-HD2 from Pgt21-022 were BLASTed (tblastn) against genome references with BLAST + 55 v2.13.0. Proteins overlapping the best hits were examined for conserved domains and intron/exon count22,40,84. Protein sequences for bW-HD1 and bE-HD2 alleles (Supplementary Data 8) were aligned with CLUSTALW85, and phylogenetic trees were generated with the ‘raxml-bootstrap’ option. Protein trees were rooted on alleles from Pca isolate Pca203 (Pca bW1 and Pca bE1)24 with R package ‘ape’86 and visualized with iTOL87. To detect STE3.2 and HD alleles in Pgt isolates, the coding sequences of individual exons were extracted, sketched together with mash72 v2.0 (-s 1000), and then screened with whole genome or RNAseq Illumina reads for 146 Pgt isolates (Supplementary Data 13). The b locus alleles were considered contained at or above 99% k-mer identity, and 90% shared k-mers.
High throughput effector screening in wheat protoplasts
A library of 1,373 effector candidates from Pgt15,20 was screened in two pools (part A and part B, containing 718 and 655 clones, respectively). Pooled libraries were propagated in ElectroMAX™ Stbl4™ competent cells, and DNA was isolated using the QIAGEN EndoFree Plasmid Giga kit. The reporter (pTA22-YFP), library proxy (pTA22-AvrSr50), empty vector (pTA22), controls pTA22-Sr50 and pTA22-Sr35, and pTA22-Sr33 plasmids were isolated from E. coli DH5α using the Macherey Nagel NucleoBond Xtra Midi Plus EF kit. A total of 300,000 protoplast cells of wheat cv Fielder were transformed with a pooled effector library at a multiplicity of transfection (MOT) of 0.14 million molecules per cell (mmpc) for each construct, corresponding to 162 µg and 163 µg for parts A and B of the library, respectively, along with either the empty vector pTA22 (MOT = 36mmpc, 54 µg), controls pTA22-Sr50 (MOT = 36mmpc, 86 µg) and pTA22-Sr35 (MOT = 7mmpc, 17 µg), or pTA22-Sr33 (MOT = 36mmpc, 100 µg) in triplicate as described in ref. 15. pTA22-Sr35 was used at a lower MOT to reduce cell death caused by autoactivity of the Sr35 protein in protoplasts. A control protoplast sample (300,000 cells) was transformed with pTA22-YFP (MOT = 36mmpc, 62 µg) and pTA22-AvrSr50 (MOT = 96mmpc, 157 µg) as a library proxy to assess transfection efficiency by fluorescence as above.
Total RNA was extracted from transformed protoplasts after 20 hours using the Maxwell® RSC Plant RNA Kit (Cat. #AS1500) with the Maxwell® RSC 48 Instrument (Cat. #AS8500) following the manufacturer’s protocol with minor modifications. Briefly, 500 µL of homogenization buffer was added to each sample along with 250 µL of lysis buffer and then 600 µL of the cleared lysate was transferred to a Maxwell® RSC Cartridge for RNA extraction. The RNA was eluted in 55 µL of nuclease-free water. Library-specific cDNA synthesis and PCR was carried out on 1 µg RNA using the Invitrogen SuperScript IV One-Step RT-PCR System with ezDNase kit (Cat. # 12595100) with the forward and reverse primers ZmUbi1_5UTR_F3b (5’-GCACACACACACAACCAG-3’) and FS_cDNA_R (5’-TGCTAGATCTCGACAGTACG-3’) as previously described in ref. 15. The cDNA was sent to the ACRF Biomolecular Resource Facility, The John Curtin School of Medical Research, Australian National University, for Illumina library preparation and sequencing on the NovaSeq X Series with a 1.5B flow cell with 100 bp single-end reads.
Sequencing reads were cleaned using fastp76 v0.23.2. The clean reads were aligned to the coding sequences of the effector candidates with STAR78 2.7.9 (--alignIntronMax 1, --outFilterMultimapNmax 10). Read counts were compiled with samtools79 idxstats and imported into DESeq288 for differential expression analysis using default parameters followed by lfcShrink (type = “apeglm”) to compare the Sr and empty vector treatments. Volcano plots were produced with EnhancedVolcano (https://github.com/kevinblighe/EnhancedVolcano). Syntenic regions in the Pgt genome assemblies were called with minimap254 (-X -N 50 -p 0.1 -c) and plots were produced with gggenomes (https://github.com/thackl/gggenomes).
Identification of Avr gene alleles
Avr gene alleles were identified by a tblastn search (NCBI toolkit version 25.2.0) with the phased genome sequences as query sequences against a local protein sequence database comprised of AvrSr13 (PGT21_021053), AvrSr22 (PGT21_017626), AvrSr27 (PGT21_006532), AvrSr35 (PGT21_019625) AvrSr50 (PGT21_020314), AvrSr33 and AvrSr62 (PGT21_030464, PGT21_030510, PGT21_030812, PGT21_030832, PGT21_036621, PGT21_036648, PGT21_036655). The genomic coordinates of the best matches were used to extract the corresponding Avr coding sequences using samtools79 faidx. Exon/intron boundaries were established from RNAseq data aligned to the assemblies using TopHat289. Protein sequences were aligned with ClustalW85 v2.1 and ML trees were generated with the ‘raxml-bootstrap’ option. Synteny plots were generated with R package gggenomes90 v1.0.1 using alignments generated with minimap254 v2.25 (options -X -N 1 -g 300 -m 20 -O7 -E7).
Transient expression in Nicotiana benthamiana and wheat protoplasts
The coding sequences (excluding signal peptides) of Avr gene alleles were codon optimized for wheat and synthesised either directly in pDONR207 (Epoch Life Science) or as G-blocks with Gateway overhangs (Integrated DNA Technologies) and cloned into pDONR207 and subsequently into expression vectors via a Gateway BP and LR reactions (Invitrogen) respectively. Amino acid mutations were introduced by Dpn1-mediated site-directed mutagenesis using Phusion High-Fidelity DNA polymerase (NEB, Cat. #M0530L). Plasmids used in this study are listed in Supplementary Data 14. Nicotiana benthamiana and N. tabacum plants were grown in a growth chamber at 23 °C with a 16 h light period and used for agroinfiltration at the 4-week-old stage. Agrobacterium tumefaciens cultures of strains GV3103-pMP90RK (pAM constructs) and GV3101-pMP90RK (pBIN19 constructs) were grown at 28 °C overnight with shaking at 200 RPM in Luria-Bertani liquid medium with appropriate antibiotic selections. Cells were pelleted by centrifugation and resuspended to a final concentration of OD600 = 0.2 for the Agrobacterium strains carrying R genes and OD600 = 0.5 for those carrying Avr genes in 10 mM MES pH 5.6, 10 mM MgCl2, 150 μM acetosyringone and incubated at 20–23.5 °C for 2 h before infiltration. Leaves were photographed or scanned 3 to 5 days after infiltration.
For protoplast assays, plasmids were extracted using the NucleoBond Xtra Midi EF kit (Macherey-Nagel). Wheat seedlings (cv Fielder) were grown in Martins Seed Raising and Cutting Mix at 24 °C under a 12 h light (∼130 µmol m−2 s−1) and 12 h dark cycle. At seven days, the first leaf was harvested for protoplast isolation and cells transformed as described in ref. 15. Following a 20 h incubation, protoplasts were transferred to 2 mL tubes and left to settle for 30 m. Half of the volume of supernatant was removed and the protoplasts resuspended in the remainder. 130 μL of each sample was transferred in duplicate to a Black 96-Well Immuno Plate (Thermofisher, Cat. #9502867). YFP fluorescence was measured using a CLARIOstar Plus plate reader (BMG Labtech; the top optic was used, optic settings: presetname = YFP, excitation = 497-15, dichroic filter = 517.2, emission = 540-20, focal height = 5.7 mm, scan settings: number of flashes per scan = 4, scan mode = matrix scan, scan matrix dimension = 30 x 30, scan width = 7 mm). The gain was set based on fluorescence scanning of YFP control samples (scan settings as above except: number of flashes per scan = 100, scan mode = spiral average). Fluorescence was normalized against the mean of the YFP control samples for each experiment.
Virus-mediated in planta effector expression
We employed the Barley Stripe Mosaic Virus (BSMV)-based protein overexpression system comprising three T-DNA binary plasmids: pCaBS-α, pCaBS-β, and pCa-γb2A-LIC19,91. Coding sequences of mature Avr protein variants (no SP) were synthesized (Twist Biosciences, South San Francisco, CA, USA), PCR-amplified using primers listed in Supplementary Data 15 and then inserted into the pCa-γb2A-LIC vector. BSMV:iLOV was used as a negative control92,93. Each of the three BSMV binary vectors was introduced into A. tumefaciens strain GV3101 (pMP90) and N. benthamiana plants (3-4 weeks old) were agro-infiltrated as previously described91. Infected tissue was used to prepare sap for mechanical inoculation of wheat plants carrying the appropriate Sr resistance genes (Supplementary Data 16) at the three-leaf stage. Inoculated wheat plants were maintained under controlled growth conditions with day/night temperatures of 23 °C/20 °C, 60-65% relative humidity, a 16 h photoperiod, and light intensity of approximately 220 μmol m−2 s−1. AvrSr13 assays were conducted at slightly elevated day/night temperatures of 25 °C/22 °C, as Sr13 resistance is more effective at higher temperature94. Systemic leaves were inspected at 10,16 and 24 days post-inoculation, and the presence or absence of viral symptoms was recorded. For each construct, at least ten plants were tested per experiment, with all experiments independently repeated and yielding consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequence data are available under NCBI BioProject PRJNA1267768. Assembly and annotation files are deposited in the CSIRO data access portal (https://data.csiro.au/collection/csiro:65828). Source data are provided with this paper.
Code availability
Scripts are available at https://github.com/henni164/epidemic_pgt (Zenodo https://doi.org/10.5281/zenodo.18322261).
References
Fones, H. N. et al. Threats to global food security from emerging fungal and oomycete crop pathogens. Nat. Food 1, 332–342 (2020).
Olivera, P. D. et al. Presence of a sexual population of Puccinia graminis f. sp. tritici in Georgia provides a hotspot for genotypic and phenotypic diversity. Phytopathology 109, 2152–2160 (2019).
Meyer, M. et al. Wheat rust epidemics damage Ethiopian wheat production: A decade of field disease surveillance reveals national-scale trends in past outbreaks. PLoS One 16, e0245697 (2021).
Bhavani, S., Singh, R. P., Hodson, D. P., Huerta-Espino, J. & Randhawa, M. S. Wheat Rusts: Current Status, Prospects of Genetic Control and Integrated Approaches to Enhance Resistance Durability. in Wheat Improvement 125–141 (Springer International Publishing, Cham, 2022). https://doi.org/10.1007/978-3-030-90673-3_8.
Pretorius, Z. A., Singh, R. P., Wagoire, W. W. & Payne, T. S. Detection of virulence to wheat stem rust resistance gene Sr31 in Puccinia graminis f. sp. tritici in Uganda. Plant Dis. 84, 203–203 (2000).
Dean, R. et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430 (2012).
Patpour, M. et al. First report of Ug99 wheat stem rust caused by Puccinia graminis f. sp. tritici in South Asia. Plant Dis. 108, 2253–2588 (2024).
Olivera, P. D. et al. Phenotypic and genotypic characterization of race TKTTF of Puccinia graminis f. sp. tritici that caused a wheat stem rust epidemic in southern Ethiopia in 2013–14. Phytopathology 105, 917–928 (2015).
Bhattacharya, S. Deadly new wheat disease threatens Europe’s crops. Nature 542, 145–146 (2017).
Flor, H. H. Current status of the gene-for-gene concept. Annu Rev. Phytopathol. 9, 275–296 (1971).
Dodds, P. N. From gene-for-gene to resistosomes: Flor’s enduring legacy. Mol. Plant Microbe 36, 461–467 (2023).
Figueroa, M. et al. Changing the game: using integrative genomics to probe virulence mechanisms of the stem rust pathogen Puccinia graminis f. sp. tritici. Front. Plant Sci. 7, 205 (2016).
Figueroa, M., Dodds, P. N. & Henningsen, E. C. Evolution of virulence in rust fungi — multiple solutions to one problem. Curr. Opin. Plant Biol. 56, 20–27 (2020).
Dracatos, P. M., Lu, J., Sánchez-Martín, J. & Wulff, B. B. H. Resistance that stacks up: engineering rust and mildew disease control in the cereal crops wheat and barley. Plant Biotechnol. J. 21, 1938–1951 (2023).
Arndell, T. et al. Pooled effector library screening in protoplasts rapidly identifies novel Avr genes. Nat. Plants 10, 572–580 (2024).
Lubega, J., Figueroa, M., Dodds, P. N. & Kanyuka, K. Comparative analysis of the avirulence effectors produced by the fungal stem rust pathogen of wheat. Mol. Plant-Microbe Interact. 37, 171–178 (2024).
Upadhyaya, N. M. et al. Genomics accelerated isolation of a new stem rust avirulence gene–wheat resistance gene pair. Nat. Plants 7, 1220–1228 (2021).
Salcedo, A. et al. Variation in the AvrSr35 gene determines Sr35 resistance against wheat stem rust race Ug99. Science 358, 1604–1606 (2017).
Chen, J. et al. Loss of AvrSr50 by somatic exchange in stem rust leads to virulence for Sr50 resistance in wheat. Science 358, 1607–1610 (2017).
Chen, R. et al. A wheat tandem kinase activates an NLR to trigger immunity. Science 387, 1402–1408 (2025).
Ortiz, D. et al. The stem rust effector protein AvrSr50 escapes Sr50 recognition by a substitution in a single surface-exposed residue. N. Phytol. 234, 592–606 (2022).
Li, F. et al. Emergence of the Ug99 lineage of the wheat stem rust pathogen through somatic hybridisation. Nat. Commun. 10, 5068 (2019).
Duan, H. et al. Physical separation of haplotypes in dikaryons allows benchmarking of phasing accuracy in Nanopore and HiFi assemblies with Hi-C data. Genome Biol. 23, 84 (2022).
Henningsen, E. C. et al. A chromosome-level, fully phased genome assembly of the oat crown rust fungus Puccinia coronata f. sp. avenae: a resource to enable comparative genomics in the cereal rusts. G3-Genes Genom. Genet 12, jkac149 (2022).
Sperschneider, J. et al. A Chromosome-Scale Genome Assembly of the Flax Rust Fungus Reveals the Two Unusually Large Effector Proteins, AvrM3 and AvrN. Mol. Plant-Microbe Interact. 38, 677–688 (2025).
Tam, R. et al. Long-read genomics reveal extensive nuclear-specific evolution and allele-specific expression in a dikaryotic fungus. Genome Res 35, 1364–1376 (2025).
Guo, Y. et al. Population genomics of Puccinia graminis f. sp. tritici highlights the role of admixture in the origin of virulent wheat rust races. Nat. Commun. 13, 6287 (2022).
Mert, Z., Karakaya, A., Duşunceli, F., Akan, K. & Çetin, L. Determination of Puccinia graminis f. sp. tritici races of wheat in Turkey. Turkish J. Agriculture Forestry 36, 107–120 (2012).
Patpour, M. et al. Wheat stem rust back in Europe: Diversity, prevalence and impact on host resistance. Front Plant Sci 13, 882440 (2022).
Lewis, C. M. et al. Potential for re-emergence of wheat stem rust in the United Kingdom. Commun. Biol. 1, 13 (2018).
Olivera Firpo, P. D. et al. Characterization of Puccinia graminis f. sp. tritici isolates derived from an unusual wheat stem rust outbreak in Germany in 2013. Plant Pathol. 66, 1258–1266 (2017).
Patpour, M. et al. First report of race TTRTF of wheat stem rust (Puccinia graminis f. sp. tritici) in Eritrea. Plant Dis. 104, 973 (2020).
Omrani, A. & Roohparvar, R. First report of TTRTF race of the wheat stem rust pathogen, Puccinia graminis f. sp. tritici from Iran (Northwest, Cold Zone). J. Appl. Res. Plant Prot. 9, 101–103 (2021).
Tesfaye, T., Chala, A., Shikur, E., Hodson, D. & Szabo, L. J. First report of TTRTF race of wheat stem rust, Puccinia graminis f. sp. tritici, in Ethiopia. Plant Dis. 104, 293–293 (2020).
Tsushima, A., Lewis, C. M., Flath, K., Kildea, S. & Saunders, D. G. O. Wheat stem rust recorded for the first time in decades in Ireland. Plant Pathol. 71, 890–900 (2022).
Henningsen, E. C. et al. A high-resolution haplotype collection uncovers somatic hybridization, recombination and intercontinental movement in oat crown rust. PLoS Genet 20, e1011493 (2024).
Henningsen, E. C. et al. Haplotype-resolved genomes of diverse oat crown rust isolates reveals both global dispersion of long-lived clonal haplotypes and limited recombination between haplotypes. bioRxiv (2025).
Sperschneider, J. et al. Nuclear exchange generates population diversity in the wheat leaf rust pathogen Puccinia triticina. Nat. Microbiol 8, 2130–2141 (2023).
Wang, J. et al. A fully haplotype-resolved and nearly gap-free genome assembly of wheat stripe rust fungus. Sci. Data 11, 508 (2024).
Cuomo, C. A. et al. Comparative analysis highlights variable genome content of wheat rusts and divergence of the mating loci. G3-Genes Genom. Genet 7, 361–376 (2017).
Förderer, A. et al. A wheat resistosome defines common principles of immune receptor channels. Nature 610, 532–539 (2022).
Zhao, Y.-B. et al. Pathogen effector AvrSr35 triggers Sr35 resistosome assembly via a direct recognition mechanism. Sci Adv 8, eabq5108 (2022).
Periyannan, S. et al. The Gene Sr33, an Ortholog of Barley Mla Genes, Encodes Resistance to Wheat Stem Rust Race Ug99. Science 341, 786–788 (2013).
Henningsen, E. C. et al. Genome Biology of Rust Fungi. Annu Rev. Phytopathol. 63, 529–552 (2025).
Holden, S. et al. Uncovering the history of recombination and population structure in western Canadian stripe rust populations through mating type alleles. BMC Biol. 21, 233 (2023).
Radhakrishnan, G. V. et al. MARPLE, a point-of-care, strain-level disease diagnostics and surveillance tool for complex fungal pathogens. BMC Biol. 17, 65 (2019).
Savva, L. et al. A portable, nanopore-based genotyping platform for near real-time detection of Puccinia graminis f. sp. tritici lineages and fungicide sensitivity. BMC Genomics 26, 327 (2025).
Jin, Y. et al. Detection of Virulence to Resistance Gene Sr24 Within Race TTKS of Puccinia graminis f. sp. tritici. Plant Dis. 92, 923–926 (2008).
Stakman, E. C. & Levine, M. N. The determination of biologic forms of Puccinia graminis on Triticum spp. in Bulletin No. 8 (University of Minnesota Agricultural Experiment Station, St. Paul, 1922).
Luo, M. et al. A five-transgene cassette confers broad-spectrum resistance to a fungal rust pathogen in wheat. Nat. Biotechnol. 39, 561–566 (2021).
Richardson, T., Thistleton, J., Higgins, T. J., Howitt, C. & Ayliffe, M. Efficient Agrobacterium transformation of elite wheat germplasm without selection. Plant Cell, Tissue Organ Cult. (PCTOC 119, 647–659 (2014).
Barnes, C. W. & Szabo, L. J. Detection and identification of four common rust pathogens of cereals and grasses using real-time polymerase chain reaction. Phytopathology 97, 717–727 (2007).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 421 (2009).
Ghurye, J., Pop, M., Koren, S., Bickhart, D. & Chin, C.-S. Scaffolding of long read assemblies using long range contact information. BMC Genomics 18, 527 (2017).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Ramírez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018).
Cabanettes, F. & Klopp, C. D.-G. E. N. I. E. S. dot plot large genomes in an interactive, efficient and simple way. PeerJ 6, e4958 (2018).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Testa, A. C., Hane, J. K., Ellwood, S. R. & Oliver, R. P. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics 16, 170 (2015).
Palmer, J. M. & Stajich, J. Funannotate v1.8.1: eukaryotic genome annotation. Preprint at https://doi.org/10.5281/zenodo.4054262 (2020).
Nielsen, H. Predicting Secretory Proteins with SignalP. in Protein Function Prediction (ed. Kihara, D.) vol. 1611 59–73 (Humana Press, New York, NY, 2017).
Möller, S., Croning, M. D. R. & Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17, 646–653 (2001).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol. 14, e1005944 (2018).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277 (2019).
Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 2922–2926 (2022).
Hickey, G. et al. Pangenome graph construction from genome alignments with Minigraph-Cactus. Nat. Biotechnol. 42, 663–673 (2024).
Ondov, B. D. et al. Mash Screen: high-throughput sequence containment estimation for genome discovery. Genome Biol. 20, 232 (2019).
Duplessis, S. et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. P Natl. Acad. Sci. USA 108, 9166–9171 (2011).
Upadhyaya, N. M. et al. Comparative genomics of Australian isolates of the wheat stem rust pathogen Puccinia graminis f. sp. tritici reveals extensive polymorphism in candidate effector genes. Front Plant Sci. 5, 759 (2015).
Kangara, N. et al. Mutagenesis of Puccinia graminis f. sp. tritici and selection of gain-of-virulence mutants. Front. Plant Sci. 11, 570180 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Vasimuddin, M., Misra, S., Li, H. & Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. in 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) 314–324 https://doi.org/10.1109/IPDPS.2019.00041 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 1207.3907v2 (2012).
Garrison, E., Kronenberg, Z. N., Dawson, E. T., Pedersen, B. S. & Prins, P. A spectrum of free software tools for processing the VCF variant call format: vcflib, bio-vcf, cyvcf2, hts-nim and slivar. PLoS Comput Biol. 18, e1009123 (2022).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Kües, U., James, T. Y. & Heitman, J. 6 Mating Type in Basidiomycetes: Unipolar, Bipolar, and Tetrapolar Patterns of Sexuality. in Evolution of Fungi and Fungal-Like Organisms 97–160 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2011). https://doi.org/10.1007/978-3-642-19974-5_6.
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49, W293–W296 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Hackl, T., Ankenbrand, M., van Adrichem, B., Wilkins, D. & Haslinger, K. gggenomes: effective and versatile visualizations for comparative genomics. ArXiv (2024) https://doi.org/10.48550/arXiv.2411.13556.
Kanyuka, K. Virus-mediated Protein Overexpression (VOX) in monocots to identify and functionally characterize fungal effectors. in Effector-Triggered Immunity (eds. Kufer, T. A. & Kaparakis-Liaskos, M.) vol. 2523 93–112 (Humana, New York, 2022).
Chapman, S. et al. The photoreversible fluorescent protein iLOV outperforms GFP as a reporter of plant virus infection. Proc. Natl. Acad. Sci. USA 105, 20038–20043 (2008).
Franco-Orozco, B. et al. A new proteinaceous pathogen-associated molecular pattern (PAMP) identified in Ascomycete fungi induces cell death in Solanaceae. N. Phytologist 214, 1657–1672 (2017).
Zhang, W. et al. Identification and characterization of Sr13, a tetraploid wheat gene that confers resistance to the Ug99 stem rust race group. Proc. Natl. Acad. Sci. USA 114, E9483–E9492 (2017).
Acknowledgements
This project was supported by USDA-NIFA award 2022-67013-36505 to BJS and BBSRC grant BB/W018403/1 to KK as part of the NSF/BBSRC Lead Agency Agreement, the CSIRO Research Office to JS, PND and MF (OD-227545, OD-235285), the CSIRO Synthetic Biology Future Science Platform to TV (OD-213047), the Grains Research and Development Corporation project CSP2403-014RTX to PND, the Lieberman-Okinow Endowment at the University of Minnesota to BJS, the 2Blades Foundation to MF and the Gatsby Foundation to PND, MA, MF. ECH was supported by an ANU University Research Scholarship and an ANU/CSIRO Digital Agriculture PhD Supplementary Scholarship. We acknowledge Dr David Hodson at CIMMYT for providing the rust isolate ETH2013-1, as well as Dr Matthew J Moscou and Kim-Phuong Nguyen for discussions and feedback as well as technical support. We truly appreciate their unwavering support. We thank Stephanie Dahl and Aubree Kees for their assistance at the University of Minnesota Biosafety Level-3 containment facility, the Minnesota Supercomputing Institute, and CSIRO High Performance Computing Services for computational resources, and Peter Tyson and Joel Hansen for providing computational support. We thank Biagio Randazzo (AS.A.R. - Società Semplice Agricola Randazzo, Baucina (PA), Italy) and Massimo Palumbo (CREA - Council for Agricultural Research and Economics, Research Center for Cereal and Industrial Crops, Acireale (CT), Italy) for their involvement in processing the rust isolate ITA2018-1.
Funding
Open access funding provided by CSIRO Library Services.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.K., P.N.D., B.J.S., M.F.; Data curation: R.S., E.C.H., C.L.P., J.S.; Formal analysis: R.S., E.C.H., C.L.P., J.C., J.L., O.M., J.S.; Investigation: R.S., E.C.H., C.L.P., J.C., J.L., O.M., D.L., L.C.C., Z.S., A.F., E.S.N., F.L., M.A.O.; Project Administration: R.S., C.L.P., P.N.D., B.J.S., M.F.; Software: R.S., E.C.H., J.S.; Methodology: E.C.H., C.L.P., J.L., M.A.O., T.A., T.V., K.K., J.S., P.N.D., B.J.S., M.F.; Visualization: R.S., E.C.H., C.L.P., J.C., J.L., O.M.; Validation: C.L.P., J.C.; Writing – original draft: R.S., E.C.H., C.L.P., J.S., P.N.D., M.F.; Writing – review & editing: R.S., E.C.H., C.L.P., J.C., J.L., O.M., D.L., L.C.C., Z.S., A.F., E.S.N., F.L., M.A.O., T.A., T.V., N.V., M.L., M.A., E.S., K.K., J.S., P.N.D., B.J.S., M.F.; Resources: T.A., T.V., N.V., M.L., M.A., B.J.S.; Supervision: C.L.P., J.C., M.A.O., T.V., E.S., K.K., J.S., P.N.D., B.J.S., M.F.; Funding Acquisition: E.S., K.K., P.N.D., B.J.S., M.F.
Corresponding authors
Ethics declarations
Competing interests
T.A, T.V., P.N.D., M.F., and J.S. are inventors on patent application WO2024103117 filed by CSIRO and relating to the identification of protein-protein interactions by protoplast screening. The remaining authors declare that they have no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Spanner, R.E., Henningsen, E.C., Langlands-Perry, C. et al. Allelic variation of Avr genes in highly virulent strains explains severe wheat stem rust epidemics. Nat Commun 17, 2718 (2026). https://doi.org/10.1038/s41467-026-69508-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69508-8
