
Abstract
Nanopore-based single-molecule sensing holds immense promise for revolutionizing proteomics, however, its practical application remains constrained by low throughput, suboptimal library preparation, and limited analytical power for complex, stochastic signals. To overcome these challenges, we develop a high-throughput nanopore sensing platform that couples a streamlined peptide library preparation strategy with an AI-driven analytical workflow, enabling accurate peptide differentiation and protein identification. Our analytical framework captures the distinct statistical signatures of peptides within massive single-molecule event streams, transforming them into reliable, information-rich fingerprints to achieve remarkable classification accuracy. We also apply this platform to establish a rapid and cost-effective workflow for antibody validation, facilitating precise epitope screening and semi-quantitative affinity determination. Critically, in a blinded study, this high-throughput sensing system demonstrates its robustness by unambiguously identifying multiple proteins from their complex enzymatic digests. By establishing an end-to-end pipeline from native proteins/peptides modification, parallel sensing to their identification, this work develops a scalable and powerful method for proteomics research.
Similar content being viewed by others

Introduction
Nanopore-based technologies, initially transformative for single-molecule nucleic acid sequencing by enabling direct, real-time analysis of single molecules, are now poised to revolutionize protein analysis1,2. Extending this paradigm to proteins promises a groundbreaking leap in proteomics, offering high-throughput, sensitive, and accurate detection within complex biological mixtures3,4,5,6,7. Initial breakthroughs have adapted strategies from DNA sequencing, such as employing motor proteins to guide oligo-conjugated peptides through pores, thereby improving translocation control and signal-to-noise ratios8,9,10,11. More recently, the successful fingerprinting of long peptide fragments on nanopore arrays using molecular motors like ClpX underscored the accelerating progress in this field, bringing routine nanopore-based protein analysis closer to reality12,13,14. Despite promising proof-of-concept demonstrations, translating nanopore-based peptide and protein analysis into a robust, widely applicable technology faces several fundamental limitations8,9,10,14,15,16. A primary limitation lies in the reliance on model systems, which frequently use chemically modified peptides or engineered proteins with specific sequences. While valuable for initial validation, these approaches have left a gap in effective strategies for analyzing unmodified native samples. Secondly, most methods are analyte-specific, often demanding bespoke functionalized pores or narrow biochemical conditions, which severely restricts inter-study comparability and cross-validation. Thirdly, the low data throughput from prevalent custom, single-channel platforms necessitates data curation, a potential source of inconsistency that can affect the robustness of subsequent statistical conclusions. Collectively, the absence of an integrated workflow for native sample preparation, high-throughput data acquisition, and automated analysis means that nanopore-based peptide/protein analysis still lacks practical applications.
Building on the principles and instrumentation of the nanopore DNA sequencing platform, our goal is to develop a parallel, high-throughput sensing platform for the analysis of natural proteins/peptides, providing a comprehensive end-to-end solution. This work introduces a versatile peptide library preparation protocol coupled with an optimized analysis workflow, which significantly enhances the accurate profiling of native peptides and their post-translational modifications (PTMs). Capitalizing on the high sensitivity of the nanopore sensing platform, we also demonstrate its application in the characterization of antibodies and native proteins. The reliability of immunoassays in proteomics is particularly challenged by the inconsistent quality of commercial antibodies, which rarely disclose detailed epitope information, provide vague affinity ranges, and lack third-party verification17,18,19. By integrating antibody-mediated enrichment, our platform enables rapid epitope screening and semi-quantitative affinity determination. Finally, using a peptide fingerprinting strategy, we demonstrate direct unambiguous differentiation of proteins.
Results
Library preparation and peptide sensing
Motor protein-driven nanopore sensing of peptides has previously employed peptidyl-oligonucleotide conjugates, achieving controlled translocation but often relying on libraries built from pre-modified peptides8,9,10,20. Here, we introduce a streamlined method to construct Oligo-Peptide-Oligo (OPO) conjugates directly from native proteins/peptides, ensuring compatibility with DNA motor-driven nanopore sensing systems. It is also applicable to synthetic peptides that still retain at least one primary amine group at their N- or C-termini. The library preparation workflow encompasses three key steps: (1) protein fragmentation and peptide terminal modification, (2) DNA-templated OPO conjugation, and (3) sequencing adaptor ligation and library purification (Fig. 1a). To circumvent harsh carboxyl modification chemistries that may compromise native peptide stability, we utilize LysC endopeptidase digestion, yielding peptides with C-terminal lysine residues. This allows for efficient dual-terminal functionalization using fluorosulfuryl azide (FSO2N3), a mild and effective diazo transfer reagent21. Treatment under ambient conditions (room temperature, 30 min) converts both the N-terminal α-amine and C-terminal ε-amine of lysine residues to azides (Fig. 1b; Supplementary Fig. 1), ready for click chemistry. This approach exhibited high efficiency across more than 50 peptides tested, although reduced yields were observed for peptides with multiple N-terminal tyrosine (likely steric hindrance) and no product for N-terminal proline (lacking a primary α-amine) (Supplementary Fig. 2 and Fig. 3). For sequential OPO assembly, we adapted the DNA-templated conjugation (DTC) strategy22,23. A rationally designed DNA template comprising two DBCO-modified oligonucleotides (DNA1, DNA2) was annealed to a complementary strand (DNA3) featuring a central abasic (AP) site (Supplementary Table 1). This flexible design accommodates peptides of various lengths (Fig. 1b). The template spatially pre-organizes the reactants, enhancing the probability that conjugation of one peptide terminus to DNA1 promotes subsequent conjugation of the other terminus to DNA2 via proximity effects (Supplementary Fig. 4). Efficient dual-terminal conjugation was demonstrated using a series of model peptides of varying lengths, with shorter peptides exhibiting higher conjugation efficiency (Supplementary Fig. 5 and Supplementary Table 2). Notably, additional AP sites incorporated within DNA1 and DNA2 are designed to generate distinct sensing signals, serving as markers flanking the peptide-specific signal during sequencing (Fig. 1b, c). After ligation with sequencing adaptors that have attached T4 Dda helicase motors, the final OPO library is enriched through beads purification to remove byproducts and excess adaptors. (see “Methods”, Supplementary Figs. 6 and 7). This dual-terminal conjugation for obtaining OPO reads facilitates library purification, improves capture efficiency, optimizes signal resolution, and enables objective peptide blockade extraction.

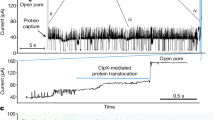
a Workflow for OPO library preparation: colored lines represent different peptide fragments featuring a lysine residue at the C-terminus; red dots represent the terminal azido modification; light green anchor integrated with a red dot is to describe the click chemistry linkage; sequencing adaptor is marked as double-stranded blue lines. LysC digestion and azidation modification typically require 0.5 h, though total reaction time may extend to 3 h for hard-to-digest proteins or peptides with limited solubility. b (top) Azido conditions: Peptide and FSO2N3 in 1:3 molar ratio; KHCO3 (100 mM), MTBE/DMF/H2O (10 μL, 95:2:3, v/v/v), room temperature (RT), 30 min; the incubation may be extended up to 3 h to enhance yield. (bottom) Library design and DNA-templated conjugation (DTC): DBCO modifications present at the 3’-end of DNA1 and the 5’-end of DNA2; Two AP sites, represented as short yellow markers, are placed in both DNA1 and DNA2, and another AP site is in DNA3; Click chemistry reaction was carried out in 1×PBS buffer at RT. c A typical sensing reads from peptide hp1_5 (VERIK): AP site-induced two current peaks (red star) of up to 110 pA enable the algorithmic extraction of OPO signal windows by establishing a threshold at 0.4698 × I0. d (left) Schematic of nanapore sensing of the OPO libraries. An insulating membrane containing an embedded CsgG nanopore separates cis and trans chambers equipped with electrodes. A sequencing adapter of the OPO library molecule, attached with a T4 Dda helicase motor, is captured by the nanopore. Under optimized ionic conditions (including ATP and MgCl2), the ATP-dependent helicase motor controls the translocation of the OPO structure through the nanopore; (middle) The linear correlation of the length of uncharged peptides vs. τoff with R2 = 0.9601; (right) The linear correlation of the molar volume of uncharged peptides vs. the value of (1 − I/I0) × τoff with R2 = 0.9476.
To enable high-throughput sequencing of the OPO libraries, we employed a sensing platform based on the CsgG nanopore, capitalizing on its proven utility of DNA sequencing in arrayed formats14,24,25 over previously explored pores like MspA (Mycobacterium smegmatis porin A), α-HL (α-Hemolysin), and aerolysin26,27,28,29. Our prototype device features a microwell array of 256 independent microwells ( ~ 200 µm pitch, ~28.9 pores/mm² density), where CsgG were inserted into lipid bilayers formed over integrated microelectrodes (Supplementary Fig. 8). A schematic of the nanopore sensing platform for OPO libraries is shown in Fig. 1d. Using a helicase motor under optimized ionic conditions (0.5 M KCl, 10 mM HEPES at pH 8.0, 0.5 mM ATP, 1 mM MgCl2, 180 mV bias, 5 kHz sampling), translocation events were acquired over 100,000 per each library in 2 h. To further validate this integrated workflow, we employed a defined set of synthetic peptides with specific sequences, along with peptides derived from LysC digests of three model human proteins: hNEDD8 (hp1), hCOMMD6 (hp2), and hGABARAPL2 (hp3)30,31,32, for subsequent model training and testing (Supplementary Table 2). A representative signal trace from a peptide (hp1_5: VERIK) (Fig. 1c) illustrates the characteristic current levels: the open pore baseline (Io), distinct signals corresponding to the adaptor and DNA portions, the central peptide blockade, and transient high-current peaks ( ~ 110 pA) associated with AP-sites within the DNA carrier. The known flanking DNA/adaptor sequences provide conserved signal anchors, enabling automated algorithmic extraction of the intervening peptide-specific electrical signature.
Following rigorous quality filtering of the raw data, a process that removed reads exhibiting anomalous durations, signal spikes, or incomplete DNA flanking sequences (Supplementary Fig. 9 and Supplementary Table 3), the central peptide blockade was extracted based on AP site-specific signals. Characterization of OPO reads focused on I/I0 (mean peptide blockade current to open pore current), STD (standard deviation of I/I0), and the dwell time τoff (redefined as the ratio of the peptide blockade duration to the total read duration to minimize variability). The I/Io and τoff distributions closely approximated log-normal forms, while STD followed a Gaussian profile (Supplementary Fig. 10). Intriguingly, we found that τoff exhibited a strong positive linear correlation with peptide length, but this trend was specific to uncharged peptides (Fig. 1d). In contrast, no significant correlation between τoff and length was observed for charged peptides. Notably, the current blockade effect induced by peptide is more pronounced than that of DNA (Fig. 1c). Furthermore, we observed a significant positive linear correlation between the average molar volume of uncharged peptide and the (1 − I/Io) × τoff (Fig. 1d). This suggests that, the observed sensing signal more likely corresponds to a current blockage feature generated as the entire peptide translocases through the pore in a ‘blob’ conformation, reflecting its overall volume and electrical properties33.
CNN-DM analysis
These collective variabilities mentioned above consequently manifest as substantial overlap when plotting conventional single-point metrics (I/Io, STD, and τoff), especially when coupled with the inherent randomness in peptide orientation during OPO library preparation, limiting the discriminatory power for closely related peptides. While initial quality control removes obvious artifacts like unstable baselines or misidentified AP sites, even stringent statistical filtering—selecting reads within one standard deviation of the mean for all three metrics (‘3D filtering’, Fig. 2a) to isolate the core signal population—does not fully resolve this ambiguity. Intriguingly, a detailed examination of these filtered, high-quality reads revealed a more subtle yet powerful discriminant: the temporal profile of the current blockade. Overlaid signal traces (I/Io vs. normalized time) for a given peptide, despite individual variations, collectively form distinctive, characteristic temporal morphologies (Fig. 2b). To quantitatively capture this distinct spatio-temporal signature, we developed a ‘Density Matrix’ (DM) representation. DM generation involved initial low-pass filtering and uniform downsampling (to 100 points) of each trace. A Soft-DTW barycenter was then computed from these processed traces to establish a representative temporal profile, robust for non-linear time warping. Subsequently, each downsampled trace was aligned to this barycenter using dynamic time warping (DTW), thereby compensating for variations in individual translocation rates. The DM itself was constructed by binning the I/Io values of these aligned traces at each of the 100 corresponding time points. This process yielded a 2D histogram (density map) that encapsulates the statistical distribution of signal amplitude across the normalized translocation event (Fig. 2b). Strikingly, DMs generated in this manner exhibit distinct features for different peptides, effectively transforming the seemingly noisy temporal signal into a stable, information-rich fingerprint.

a A 3D filtered scatter plot from the data points of Pep1: SDVTNQLVDFQWK (blue), Pep2: LGMAVSSDTCRSLK (orange), and Pep3: YPYVAVMLK (green) falling within mean ± one standard deviation of their τoff, I/I0, and STD, comprising 20,000 peptide reads for each peptide. b (top left) Examples of trend distribution map by overlaying all 3D filtered peptide reads of Pep1 (blue), Pep2 (orange), and Pep3 (green). (right) Algorithmic flow for transforming 3D-filtered superposition maps into density matrices (DM). (bottom left) The heatmap of the DMs corresponding to the peptides presented on top. c (left) Illustration of the CNN architecture and the training process. During the training phase, 70% and 10% of the data were used for training and validation, respectively. Then, the CNN performance was tested with the 20% test set during the inference phase. (right) The process of DM reconfirmation relies on a similarity assessment between the peptide-sensing trace and its designated DM. As illustrated for two reads initially classified as Pep1 by CNN: the red one, showing high similarity to the Pep1 DM reference, is retained; in contrast, the blue one, originally belonging to Pep2 but misclassified as Pep1, is filtered out due to low similarity. d For Pep1, Pep2 and Pep3, the classification accuracy of CNN alone was 97.5%, whereas the CNN-DM improved the accuracy to 99.2%.
While direct application of a Convolutional Neural Network (CNN) to the resulting high-quality dataset, partitioned into training (70%), validation (10%), and testing (20%) subsets, proved effective for initial peptide identification, we achieved enhanced performance by incorporating DMs as a secondary validation filter (Fig. 2c, “Methods”). In this two-step process, putative peptide identities assigned by the CNN were cross-validated against the corresponding reference DM. Reads were retained for downstream analysis only if their temporal signal patterns exhibited strong concordance with the probabilistic distribution of the reference DM, exceeding a predefined similarity score threshold. This integrated CNN-DM approach improved classification accuracy for Peptides 1, 2, and 3 from 97.5% (CNN alone) to 99.2% (Fig. 2d). This DM-augmented validation, therefore, not only improves identification accuracy but also provides a robust foundation for developing comprehensive reference libraries and advanced algorithms capable of filtering unknown analytes from complex biological signals.
Peptide profiling
Building upon this established workflow, our initial investigations focused on a set of model peptides featuring single amino acid variants (SAAVs) within a GSGSXSGSGK palindromic sequence (X being the variable residue) (Supplementary Table 2). OPO libraries incorporating all 20 canonical SAAVs at this position were sequenced, yielding substantial read depths. Although initial attempts to classify all 20 SAAVs directly showed limited success (Supplementary Fig. 11), the grouping by physicochemical properties revealed a striking sensitivity to amino acid charge. Classification accuracy exceeded 95% for distinguishing: positively charged (K and R), negatively charged (D and E), and neutral amino acids (A, C, F, G, H, I, L, M, N, P, Q, S, T, V, W and Y) (Fig. 3a). Furthermore, analysis of volumetric effects revealed a compelling trend for neutral amino acids. The system distinguished amino acids with large (I, L, M, F, Y and W) from those with small (A, C, G, N, P, S and T) side chains with over 92% accuracy34. Conversely, residues of intermediate volume (V, H and Q) proved challenging to resolve, often being miscategorized (Fig. 3a). This indicates that the sheer volume of data enables CNN-DM to discern subtle, yet consistent, physicochemical signatures related to peptide charge and volume, thereby achieving remarkable discriminatory power.

a Confusion matrix for the classification of SAAVs based on amino acid charge and volume. The “others” category represents the aggregation of all uncharged amino acids excluding K, R, D, and E. And S, M, and L represent small (A, C, G, N, P, S and T), medium (V, H and Q) and large (I, L, M, F, Y, and W) amino acid categories within SAAVs, respectively34. b Classification for SAAVs involving leucine (L) and isoleucine (I), and their corresponding DMs for “3I” and “3L”. c Classification of positional variants involving the charged amino acid D and the bulky amino acid Y, along with their corresponding DMs. d Confusion matrix for the classification of PTMs (phosphorylation, acetylation, methylation, and octanoylation) on the serine residue of 1S peptide (GSGSSSGSGK). e Illustration of classification and corresponding DMs induced by potential natural modifications and mutations at specific sites within a model protein, hp1 and hp3.
Subsequently, we extended our investigation to the more challenging task of discriminating peptide isomers: positional and isobaric. Focusing first on isomers, notably leucine (L) and isoleucine (I), remains a formidable challenge in proteomics. As anticipated, the classification accuracy for distinguishing single L from I residues hovered around 70%. Interestingly, when challenged with distinguishing between LLL vs. III (GSGXXXGSGK), the classification accuracy slightly improved to 81%, even though their density matrices remained largely undifferentiated (Fig. 3b and Supplementary Table 2). This suggests that minute, cumulative volumetric and/or conformational differences between the consecutive propyl (L) and sec-butyl (I) side chains become sufficiently amplified to generate subtly divergent, yet learnable, nanopore signatures. The differentiation of positional isomers, we relocated the variable residue X to the C-terminus (GSGSGSGSXK), thereby introducing asymmetry in the peptide’s charge or volume (Supplementary Table 2). Reflecting the stochastic N- or C-terminal conjugation inherent to our OPO library, and given that the “blob-like” conformation of peptides during translocation, one would presume they would exhibit minimal significant signal resolution. Remarkably, however, the CNN successfully achieved high classification accuracies for all investigated positional isomers. The accuracies for distinguishing 1D/1D’, 1Y/1Y’, 1 R/1 R’ and 1 W/1 W’ are 97.8%, 97.9%, 98.8% and 98.1%, respectively (Fig. 3c and Supplementary Fig. 12). These subtle differences are also reflected in variations in their DMs, this phenomenon that is particularly pronounced for charged or large amino acids. Notably, for peptides with asymmetric charge distribution, discernible differences in signal traces (reflected in their DMs) were observed among the N-terminal entry, C-terminal entry, and mixed-entry libraries (Supplementary Fig. 13). This observation underscores the potential of, and perhaps the necessity for, developing more efficient, site-directed conjugation strategies to further enhance peptide sensing resolution by ensuring uniform peptide entry.
Furthermore, the detection of PTMs represents a key frontier in nanopore sensing. While strategies employing modified nanopore or Phos-tag-mediated retardation effects for phosphorylation sensing have been reported20,35,36, the pronounced sensitivity of our system to charge and volume alterations suggested a direct avenue for PTMs discrimination. Indeed, investigations into phosphorylation and caprylation, inducing substantial negative charge or large side chain shifts (modification occurs at the underlined serine (S) residue in the peptide sequence GSGSSSGSGK), yielded high classification accuracies, further corroborated by distinct differences in their respective DMs. Conversely, methylation and acetylation, which impart more subtle changes, proved challenging to resolve; even when assaying peptides with three consecutive modifications of these types (GSGSSSGSGK), classification accuracy plateaued around 87% (Fig. 3d and Supplementary Table 2). Extending this to naturally occurring modifications in hp1 and hp3, we readily detected the deamidation of Q40 to E in hp1_7, a modification induced by bacterial effector proteins known to disrupt cellular function, owing to its significant charge alteration37. Similarly, both single and dual phosphorylation at S87 and S88 in hp3_11, modifications that critically destabilize its complex with the processing enzyme ATG438, were accurately detected and distinguished, with their DMs also reflecting these clear distinctions (Fig. 3e and Supplementary Table 2).
Our work shows that even with the CsgG nanopore, typically employed for DNA sequencing, strategic analysis of large datasets allows for the differentiation of critical peptide features. It is evident that the resulting sensing signals are significantly influenced by a combination of peptide properties, such as their entry orientation relative to the pore, and the intramolecular distribution of charge and volume. Furthermore, the implementation of a two-step analytical framework, integrating CNN-based classification with DM-based verification, proves highly effective. This approach not only captures nuanced distinctions from complex signal profiles but also systematically filters out low-confidence signals, thereby enhancing the robustness and reliability of the classification data for downstream biological insights.
Peptide identification
The peptide profiling results demonstrate that integrating efficient peptide library preparation, high-throughput sensing, and a CNN-DM analysis pipeline enables precise discrimination of peptides with minimal variations. This technological advancement enables precise peptide identification within diverse yet closely related peptide mixtures, thereby facilitating the development of customized solutions tailored for anti-peptide antibody validation, ensuring both specificity and selectivity.
Choosing C-peptide—a 31-amino acid (aa) segment released during proinsulin processing—as a model, we designed six overlapping peptides, tiled every four residues across the entire C-peptide sequence (Fig. 4b and Supplementary Fig. 14). Individual OPO conjugates were prepared for these six peptides, and subsequent CNN-DM analysis achieved high classification accuracy of 97.2% (Fig. 4b). These six OPO libraries were firstly pooled equally and incubated with magnetic beads coated with different commercially sourced antibodies (37 °C for 1 h) (Fig. 4a). Subsequently, the bound OPO conjugates were dissociated for nanopore sensing and analysis, revealing distinct epitope preferences for different antibodies. For instance, Fig. 4b illustrates the distribution of the relative fold-change of OPO reads percentage for typical antibodies, showing that antibodies Ab1 and Ab2 (Sino Biological, Inc.) are both significantly enriched for CP-6 peptide (LQPLALEGSLQ). While the supplier’s information indicated that these two antibodies recognize distinct epitopes and are suitable for use as an antibody pair in sandwich ELISA assays, our validation clearly suggested that this antibody pair is evidently not optimal for such use. In contrast, antibody Ab3 (HeavyBio, Inc.) preferentially bound to the CP-5 peptide (GAGSLQPLAL), while antibody Ab4 (HeavyBio, Inc.) showed a primary enrichment for the CP-3 peptide (QVELGGGPGA). This suggests that this antibody pair may be an ideal candidate for the sandwich immunoassay. Antibody Ab5 (Medix Biochemica, Inc.) enriched both CP-5 and CP-6, suggesting a primary binding site towards LQPLAL (residues 21–26). This finding demonstrates a more refined epitope information compared to that provided by the manufacturer (the binding site spans residues 12–30). An independent antibody enrichment assay and LC-MS analysis corroborate the consistency of epitope information with our nanopore detection.

a C-peptide antibody validation workflow: Synthesized C-peptide fragments were used to generate their respective OPO conjugates, and pooled in equimolar amounts. The mixed OPO conjugates were then captured via antibody-conjugated magnetic beads. After removing the supernatant, the enriched OPO conjugates were eluted. b (left) Average accuracy predicted by CNN-DM reached 97.2%. (right) Six colored lines correspond to the C-peptide sequence regions covered by the CP-1 to CP-6 peptides. The mean enrichment ratios of C-peptide fragments by five different antibodies were normalized against the control group. n = 6 technical replicates, values represent mean ± SD. c Schematic of anti-FLAG antibody affinity characterization: FLAG peptide and F1 to F4 fragments with point mutations were mixed in equimolar amounts and incubated with anti-FLAG M2 and FLAG-Tag Mouse mAb antibodies separately. After ultrafiltration, the retentate was collected and used to prepare an OPO library. d (left) Average accuracy predicted by CNN-DM reached 96.4%. (right) The sequence of FLAG mutants F1-F4 and the FLAG peptide, and the average fold change of read percentage for different FLAG mutants (normalized against the control group). n = 3 technical replicates, values represent mean ± SD.
An intriguing observation from the C-peptide experiments was the apparent correlation between the enrichment efficiency for different peptides by the same antibody and the corresponding read output (e.g., Ab5 antibody enriched CP-5 and CP-6, yielding 8178 and 4170 reads, respectively). Likely, the affinity of the antibody-peptide interaction might quantitatively correlate with the final OPO library read output. To quantitatively test this hypothesis, we employed the well-characterized FLAG-tag system. Equimolar mixtures of the FLAG peptide and its four mutants (F1–F4) were incubated with the anti-FLAG M2 (Sigma) and FLAG-Tag Mouse mAb (Yeasen Biotechnology (Shanghai) Co., Ltd.) antibodies separately. After enrichment via ultrafiltration, the retained peptides were used to prepare OPO libraries for parallel sensing (Fig. 4c). The analysis was underpinned by a CNN-DM model that identified these analogous peptides with an average accuracy of 96.4% (Fig. 4d). For the anti-FLAG M2, the relative nanopore reads abundance for each peptide showed a strong positive correlation with their reported binding affinities. This finding was also consistent with Surface Plasmon Resonance (SPR) validation, except for the F4 mutant, which exhibited an affinity comparable to that of the wild-type FLAG—rather than the superior affinity previously reported (Fig. 4d and Supplementary Fig. 15)39,40. And FLAG-Tag Mouse mAb displayed a selectivity profile with a preference for the FLAG, F4 and F1 peptides, a result that was also validated by LC-MS (Fig. 4d and Supplementary Fig. 15). To conclude, based on the integrated nanopore platform as well as the CNN-DM analysis workflow, our methodology enables the accurate identification of a series of closely related peptide mixtures. This approach establishes a rapid, cost-effective, and highly effective approach for comprehensive antibody characterization, offering precise epitope screening and semi-quantitative affinity assessment. This significantly streamlines the validation of antibody specificity, selectivity, and relative affinity, thereby accelerating immunoassay development and expanding the practical applications of nanopore sensing.
Protein identification
The established capacity of our nanopore sensing system for accurate peptide identification within complex mixtures suggested its potential application to protein identification. Adopting a peptide fingerprinting strategy, the workflow proceeds as follows: First, synthetic peptides derived from LysC-digested targets are prepared as OPO libraries and analyzed to train a CNN-DM model, establishing reference fingerprint profiles. Next, purified protein samples undergo LysC digestion and analogous library assembly to generate experimental OPO reads. Finally, the CNN-DM model classifies these reads against the reference profiles, compiling full peptide signatures to enable protein identification. To demonstrate the platform’s efficacy in distinguishing native proteins, twenty-four synthetic peptides derived from LysC digests of three human proteins (hp1, hp2, and hp3) were individually used to prepare OPO libraries, and their respective DMs were characterized (Supplementary Figs. 16–19 and Supplementary Table 2). Our analytical strategy, leveraging a CNN-DM framework, achieved a classification accuracy of 96.3%—a significant improvement over the 93.5% accuracy from CNN alone (Fig. 5a; Supplementary Figs. 20 and 21).

a The confusion matrix of the test set based on the CNN-DM filtered dataset. Label hp1_1 represents the first LysC-derived peptide fragment from the hp1 protein (etc.). Data with an error rate lower than 1% is not shown. b Schematic of the single-blind protein identification workflow. Anonymously labeled protein samples (Protein 1, 2, or 3) are individually subjected to LysC digestion. The resulting peptide mixtures are then azidated with FSO2N3, followed by OPO library preparation. Nanopore sensing and CNN-DM analysis then allow for the assessment of their distribution characteristics and the prediction of the protein’s identity as one of three candidates (hp1, hp2, or hp3). c Distribution of predicted OPO reads for three model proteins generated from CNN-DM-based classification of protein 1 (dark green), protein 2 (yellow), and protein 3 (light green), which were identified as hp1 (n = 9 peptides), hp2 (n = 6 peptides) and hp3 (n = 9 peptides), respectively. The Tukey box plot summarizes OPO reads’ distribution, where the box represents the interquartile range (IQR) from the 25th to the 75th percentile, the central line indicates the median, the whiskers extend to the highest and lowest values within 1.5 times the IQR, and any data points beyond the whiskers are identified as outliers.
To challenge our platform with a blind identification task, we prepared three anonymized samples (Protein 1, 2, and 3) from Lys-C digests of proteins hp1, hp2 (both expressed in Escherichia coli BL21(DE3)) and hp3 (Sino Biological, Inc.), with digestion confirmed by SDS-PAGE (Supplementary Note). After azide modification and OPO library construction, nanopore sequencing generated >300,000 reads for each sample. When analyzed with our pre-trained CNN-DM model, the reads from each sample correctly clustered into three distinct and predictable classifications (Fig. 5b, c). These assignments were independently validated by LC-ESI-MS (Supplementary Note), demonstrating the robustness of our approach for protein identification.
Despite the high overall accuracy, we observed a lower relative abundance for peptides with high net negative charges (e.g., -5 net charge at pH 8.0 for hp1_4; -3 net charge at pH 8.0 for hp3_3). This suggests that the efficiency of DTC can be attenuated for highly anionic peptides, likely due to electrostatic repulsion to the DNA backbone, particularly in complex mixtures where species compete. Furthermore, distinguishing between dipeptides differing by a single amino acid (e.g., hp1_2 and hp2_6), represents a current resolution limit (Fig. 5c). Although an unexpected observation was the consistent detection of reads classified as hp3_10 within our recombinantly expressed hp1 and hp2 samples, it is crucial to note that such isolated artifacts are statistically unlikely to mimic the full peptide signature of an entire protein and therefore do not impede robust protein-level identification (Fig. 5c). Collectively, this work validates our methodology’s capacity for the successful blind identification and classification of proteins from their complex enzymatic digests. These results demonstrate the practical viability of our end-to-end analytical pipeline for proteomic applications, confirming that multi-peptide signatures acquired via nanopore sensing are sufficient for accurate protein-level assignments in biologically relevant mixtures.
Discussion
In this study, we present a streamlined nanopore parallel sensing platform for peptide profiling and protein identification. Our method integrates mild modification chemistries with a DNA-templated conjugation strategy, enabling the rapid (≤6 h) generation of high-purity OPO libraries directly from native proteins or peptides. This is complemented by high-throughput data acquisition (≤ 2 h) and subsequent analysis via a CNN-DM pipeline, which achieves high-confidence classification of both synthetic and native biological samples. Crucially, the designed library incorporates features facilitating purification, further enhancing OPO reads yield, and distinct AP site-specific signals that enable automated peptide reads extraction. The “sandwiched” OPO library structure and helicase-based translocation system are potentially compatible with commercial sequencers (e.g., Oxford Nanopore Technology, CycloneSEQ), enabling automated, large-scale data acquisition41,42. This scalability generates datasets of sufficient magnitude for robust AI model training, addressing a critical limitation of previous single-channel nanopore approaches. The demonstrated timeliness, reliability, and broad applicability of this platform provide a robust foundation for future advancements in rapid biomarker detection and clinical diagnostics, paving the way for diverse proteomic applications.
Nevertheless, the non-directional nature of the DTC reaction in our protocol permits peptides to enter the nanopore via either their N- or C-termini. This mixed entry orientation may introduce signal heterogeneity, an issue particularly pronounced for peptides with significant charge or volumetric asymmetry. Optimizing directional peptide modification strategies, such as N-terminal specific approaches43 combined with FSO₂N₃ treatment, may assist in increasing sensing resolution. While even with enzymatic control, once a peptide rapidly slides from the helicase, it may transiently form unpredictable, compact structures within the pore. Additionally, the effective read length is fundamentally limited by the distance between the helicase and the nanopore constriction10. These factors pose a significant challenge to achieving stable peptides’ translocation. Consequently, individual peptides, despite typically having smaller molecular diameters than single-stranded DNA, can paradoxically generate often-deeper current blockades that correlate linearly with peptide molar volume. This interpretation resonates with recent explorations of peptide ‘blob’ formation during uncontrolled nanopore translocation33. While mitigating this effect might be possible by further slowing translocation or using a smaller pore, the intrinsic flexibility—influenced by amino acid composition, chain geometry, solution ionic strength, and nanoscale confinement and the interactions with intra-pore residues—is expected to introduce additional complex translocation dynamics and signal uncertainty9,44. Therefore, reliable classification and interpretation of peptide signals necessitate statistical analysis of sufficient sensing events to overcome this inherent variability. Such an approach, capable of extracting distinct signal patterns from raw data, surpasses the analysis of limited events. This underscores the critical need for, and thus validates our development of, a high-throughput sensing platform to fully realize this technology’s potential for practical peptide analysis.
In summary, by leveraging massively parallel sensing and advanced large data analytics, we have demonstrated that CsgG nanopores are capable of accurately profiling and classifying complex peptide samples, including SAAVs, isomers, PTMs, and mixtures of highly similar peptides. Notably, this work represents the application of a nanopore sensing platform for antipeptide antibody validation and epitope screening. This approach also facilitates the semi-quantitative assessment of antigen-antibody affinity. The nanopore-based platform provides a rapid, low-cost, scalable, and end-to-end solution for antibody analysis, serving as a valuable complementary tool to conventional methods such as MS or SPR. Additionally, we introduce a streamlined methodology for the differentiation of native proteins through classification against reference libraries. This approach successfully identifies three blind-labeled proteins (hp1, hp2, and hp3) as their respective targets. However, OPO construction can be compromised by lysine-deficient C-terminal peptides or specific PTMs (e.g., N-acetylation/lysine modifications), which limit achievable coverage. The pursuit of full-sequence coverage is not only computationally prohibitive but is also often unnecessary for answering focused biological questions. Integration with targeted enrichment thus renders this methodology practical for investigating specific peptide regions of interest. Prospectively, the integration of DNA barcoding with protein encoding strategies holds considerable promise for enabling larger-scale proteomic analyses. Future efforts, building on the demonstrated utility of this technology, will focus on developing a dedicated, automated, and integrated platform derived from our laboratory prototype or adapting this methodology for existing commercial sequencing platforms. Such advancements are poised to equip the scientific and clinical communities with a powerful tool to accelerate proteomics research.
Methods
General materials
All oligonucleotides and primers used for nanopore sensing and plasmid construction were synthesized by GCATbio Co., Ltd (Changzhou, Jiangsu, China) and Sangon Biotech (Shanghai, China). All peptides were synthesized by GCATbio Co., Ltd (Changzhou, Jiangsu, China), GenScript (Nanjing, Jiangsu, China) and Wuxi AppTec (Shanghai, China). All kits, reagents, solvents, buffers, and dyes were purchased from GCATbio Co., Ltd (Changzhou, Jiangsu, China), BBI Life Sciences (Shanghai, China), Beyotime (Shanghai, China) and Sangon Biotech (Shanghai, China). Other material sources have been indicated in the main text.
OPO library preparation
Azide modification reaction was carried out at room temperature for 0.5 h with a peptide (10 mM in 1× PBS) and FSO2N3 in a 1:3 molar ratio. The final product could be stored at −20 °C for weeks for subsequent processing. Annealed DNA template (25 μM) and azide-modified peptide were mixed in a 1:3 molar ratio in 1×PBS and incubated at room temperature for 3 h. Sequencing adapters that have attached T4 Dda helicase motors (M1G/E94C/C109A/C136A/K194L/A360C, Supplementary Table 1) were ligated to the OPO conjugates using T4 DNA ligase (GCATbio Co., Cat: LS-EZ-E-00008P) at 25 °C for 0.5 h. Subsequently, the OPO library was enriched through bead purification to remove reaction byproducts and excess adapters. Capture beads were obtained from an incubation of Streptavidin-coated (SA) magnetic beads (10 μL, Nanjing Vazyme Biotech Co., Ltd, Cat: N512-02) and capture DNA (20 pmol) in a binding buffer (50 mM Tris, 150 mM NaCl, 0.1 mM EDTA) at room temperature for 0.5 h and washed with wash buffer (50 mM Tris, 150 mM NaCl, 0.05% Tween-20) twice. Subsequently, the ligation products were incubated with 10 μL capture beads in a binding buffer at 30 °C for 0.5 h and washed twice with 1× PBS. The bead pellet was incubated in a 10 μL 1× RNaseH buffer containing 0.2 μL RNaseH (Yeasen Biotechnology (Shanghai) Co., Ltd., Cat: 12906ES76) at 37 °C for 20 min and washed with 1× PBS. The OPO library was finally liberated from the beads by incubation with 0.5 μL RNaseA (Yeasen Biotechnology (Shanghai) Co., Ltd., Cat: 10405ES03) in 10 μL 1× PBS at 30 °C for 40 min. The enriched OPO library could be stored at 4 °C for weeks.
Peptide sensing data acquisition
The prototype device incorporates 256 independent channels compatible with a microwell chip array containing 256 discrete microwells (~ 200 µm pitch, ~28.9 pores/mm² density). Before each experiment, the CsgG transmembrane protein mutant (Y51A/F56Q/R97W/R192D, Supplementary Table 1) was inserted into lipid bilayers. Sequencing buffer (0.5 M KCl, 10 mM HEPES at pH 8.0, 0.5 mM ATP, 1 mM MgCl2) was perfused through the system to remove excess transmembrane proteins. To collect peptide sensing data, 300 μL sequencing buffer containing 5 μL OPO library and 3 μL anchor (1 μM) was slowly pumped into the chip from the sample loading port. After incubation at 25 °C for 10 min, a voltage of 180 mV was applied, and sequencing data were recorded at a sampling rate of 5 kHz. The sequencing data of each 2 h were saved as separate files for subsequent data analysis. A reversed membrane potential was used to help remove blockages from the pores (Supplementary Fig. 8).
Signal normalization
Given a time series \(R=\left[{r}_{1},{r}_{2},\ldots,{r}_{L}\right]\) with length L, we first apply uniform downsampling to obtain a reduced signal \(S=\left[{s}_{1},{s}_{2},\ldots,{s}_{N}\right]\) with length N, where
Here, we define \(R[j]={r}_{j}\). Subsequently, the downsampled signal S is normalized by dividing each element by its open-pore current I0, yielding the normalized current signal \(N=\left[{n}_{1},{n}_{2},\ldots,{n}_{N}\right]\), where
We denote this normalization process as \({N{orm}}_{N}()\) hat scales and normalizes a time series R into N with a length of N. Thus, the normalized signals have the same length of N and fall within the range of 0 to 1, as signals exhibiting any negative currents or values exceeding I0 have been filtered out during preprocessing. For computational efficiency, we set N = 100 to prepare input for DM construction and N = 1000 for CNN classification.
CNN model training
A CNN containing convolutional layers, batch normalization, activations, squeeze-and-excitation blocks, spatial attention blocks and fully-connected layers was developed to perform peptide reads classification. Its architecture follows a hierarchical pattern across six primary blocks. Each block consists of a convolutional layer (typically with a kernel size of 3, a stride of 1, and padding of 1), batch normalization, ReLU activation, a Squeeze-and-Excitation (SE) module, a spatial attention module and a max-pooling layer with a kernel size of 2 and a stride of 2. Following the convolutional backbone, the feature maps are flattened into a vector. A dropout layer with a rate of 0.5 is applied for regularization to mitigate overfitting. The classification head consists of two fully connected layers: the first projects the high-dimensional features onto a 256-unit representation, followed by a ReLU activation, and the final layer maps this to the output logits corresponding to the number of target classes. Initially, all current signals from the windows of clean peptide reads (Supplementary Fig. 9) were normalized to 1000 time points and normalized against I0, generating a dataset in which each read had 1000 features ranging from 0 to 1 and a known label indicating what peptide it came from. The dataset was randomly split into training (70%), validation (10%) and test (20%) sets (Fig. 2c). Adam was used as the optimizer with an initial learning rate of 5 × 10−3 and cross-entropy loss was applied as our optimized target for training. The performance of CNN and traditional machine learning classifiers was compared using nine peptides from the hp1 dataset, with CNN demonstrating superior performance over other classifiers (Supplementary Fig. 22).
DM reconstruction
Let the set of original d traces from a specific peptide be denoted as \(\{{R}^{1},{R}^{2},\ldots,{R}^{d}\}\), where \({R}^{i}=[{r}_{1}^{i},{r}_{2}^{i},\ldots,{r}_{{L}_{i}}^{i}]\) represents the i-th trace (typically current signals) with length \({L}_{i}\). At this time, traces are not the same length. Each trace undergoes initial low-pass filtering through median filter followed by normalization to a length of 100:
where \(L{PF}()\) denotes the median filter operation from the scipy Python package with a parameter size of 100 for high-frequency noise removal. Then a representative temporal profile is established by computing the Soft-DTW barycenter from the normalized trace set \(\{{N}^{1},{N}^{2},\ldots,{N}^{d}\}\):
where \({S{oftDTW}}_{\gamma }()\) denotes the soft dynamic time warping distance with regularization parameter γ and \(B\in {[{\mathrm{0,1}}]}^{100}\) represents the barycenter sequence with a length of 100, providing robustness to nonlinear time warping. We set the γ as 0.001 here. After obtaining barycenter sequence, all normalized signals are aligned to barycenter and generate new aligned sequences as \(\{{A}^{1},{A}^{2},\ldots,{A}^{d}\}\), where
Here, \({\pi }_{j}^{i}\) denotes the set of time points in B that are aligned to the time point j of \({N}^{i}\) using DTW. Lastly, we bin the values of the aligned traces \(\{{A}^{1},{A}^{2},\ldots,{A}^{d}\}\) into 100 equal-width bins at each of the 100 time points and calculate occurrence frequencies of all bins as DM matrix D:
where \({b}_{0},{b}_{1},\ldots,{b}_{100}\) are the boundaries of 100 bins with \({b}_{0}=1\) and \({b}_{100}=0\), and is the indicator function. This process yields a two-dimensional density map that encapsulates the statistical distribution of signal amplitude across normalized translocation events.
DM reconfirming
As mentioned previously, we filtered out CNN results by the distance to DMs (Fig. 2c). Specifically, we discarded a read if CNN classified it as a peptide and its distance to the DM of the predicted peptide is smaller than the cutoff of this peptide (Supplementary Fig. 18). To calculate the distance between a read and a DM, we first uniformly downsampled the read to 100 time points, then normalized it based on its I0 to scale its values between 0 and 1. Second, the normalized current was superimposed onto the DM (Fig. 2b). Lastly, we took the logarithm of each DM element value traversed by the read, accumulated these logarithmic values, and finally took the negative of this sum as the distance. We kept reads with distances smaller than the cutoff of this peptide, which was defined as the distance value corresponding to the peak of the distribution of distances between all filtered reads from this peptide and its DM. Thus, the final results are supported by the CNN model and further reconfirmed by similarity to DMs.
Peptide identification
C-peptide Antibody Validation: Synthetic C-peptide fragments (CP-1 to CP-6) were used to generate their respective OPO conjugates, according to the protocol described above (Supplementary Table 2). The data from the parallel sensing of the six OPO libraries were then used for CNN-DM modeling. Five commercial antibodies (Ab1: Sino Biological, Inc., Cat: 110121-MM12; Ab2: Sino Biological, Inc., Cat: 110121-MM15; Ab3: HeavyBio, Inc., Cat: HA127-1M; Ab4: HeavyBio, Inc., Cat: HA127-2M; Ab5: Medix Biochemica, Inc., Cat: 100113) were biotinylated using NHS-PEG4-Biotin. The labeling ratios (biotin:protein) ranged from 1.5 to 2.5, as verified by a Biotin Quantitation Assay Kit (Beyotime, Cat: P0371M). 50 μg biotinylated antibodies were incubated with SA magnetic beads (1 mg, Nanjing Vazyme Biotech Co., Ltd, Cat: N512-02) at RT for 1 h in 1× binding buffer. The antibody-coated beads were then washed twice with wash buffer and stored in binding buffer for subsequent use. The six OPO conjugates were quantified and quality-controlled using a dsDNA HS Assay Kit (Yeasen Biotechnology (Shanghai) Co., Ltd., Cat: 12640ES76) and PAGE gel analysis. The six OPO conjugate samples were then pooled in equimolar amounts to a final concentration of 2.0 μM. The OPO conjugate pool was divided into equal aliquots. And they were individually incubated with the individual antibody-coated SA magnetic beads in 50 μL of 1× binding buffer at 4 °C with 1000 rpm shaking for 3 h, while keeping one aliquot as a control. Subsequently, the beads were washed twice with 1× wash buffer. The bound OPO conjugates were dissociated in 10 μL of elution buffer (2 M NaCl) at 37 °C for 1 h. Finally, the control sample and the dissociated products were diluted to 60 μL and ligated with sequencing adaptors, ready for high-throughput sensing and data acquisition. Each sample underwent six replicate tests.
Antibody Affinity Profiling: OPO libraries were generated from a synthetic FLAG peptide and its four variants (F1-F4) using the procedure described above (Supplementary Table 2). The resulting high-throughput sensing data from each library were subsequently used for CNN-DM modeling. For the affinity selection, the five synthetic peptides (FLAG and F1-F4) were premixed in equimolar amounts to a final concentration of 500 μM. The peptide pool was then divided into three equal aliquots. One aliquot was kept as a control, while the other two were incubated with anti-FLAG M2 antibody (0.5 mg/mL, Sigma, Cat: F1804) and FLAG-Tag Mouse mAb (0.5 mg/mL, Yeasen Biotechnology (Shanghai) Co., Ltd., Cat: 30505ES90) in 1× binding buffer at 4 °C with 1000 rpm shaking for 3 h. After incubation, the mixture was diluted with 1× PBS by centrifuging in a 30 kDa MWCO ultrafiltration column three times (14,000⨯g for 10 min at 4 °C) to remove unbound peptides. Following, target peptides were dissociated from the antibodies in 25 μL 1× PBS via competitive elution with an excess of native 2× FLAG peptide. The control sample and the two dissociated products were then used for OPO library preparation, nanopore sensing and data analysis. Each sample underwent three replicate tests.
Model protein preparation and LysC digestion
GABARAPL2, referred to as hp3, was commercially obtained from Sino Biological, Inc. (Cat: 14563-H07E). For hp1 and hp2, the codon-optimized gene fragments for NEDD8 and COMMD6 (BGI Write: Beijing, China) were synthesized and cloned into pET vectors using the In-Fusion Cloning technique. Both expression vectors (pET-NEDD8-6×His and pET-6×His-SUMO-COMMD6) were chemically transformed into E. coli BL21(DE3) competent cells to afford expression seeding stocks. Expression starters were prepared with a small amount of each seeding stock in 50 mL LB medium containing 50 μg/mL kanamycin. Overnight starter of each protein was inoculated into LB medium containing 50 μg/mL kanamycin using 1:100 (v:v) ratio and allowed to grow at 37 °C until OD600 reached 0.6 – 0.9. Afterwards, 0.5 mM IPTG was added to induce the expression at 37 °C for 4-5 h. Upon saturation, cells were collected by centrifugation (8000 × g, 30 min, 4 °C) and stored at −80 °C if not lysed directly. NEDD8-6×His and 6×His-SUMO-COMMD6 were further purified by AKTA Protein Purifier. COMMD6 was obtained by further 6×His-SUMO removal using ULP protease. LysC digestion reaction was carried out in 10 mM ammonium bicarbonate (pH 8) solution by protein (500 ng/μL): LysC = 200:1 (m:m) ratio and incubated at 37 °C for 0.5 ~ 3 h. The digested products were then purified via a 5000 MWCO spin filter column to remove LysC. The flow-through was dispensed into several aliquots for either subsequent reactions or lyophilization, and stored at −80 °C for future use (Supplementary Note).
Statistical analysis
All analyses and visualizations were conducted using GraphPad Prism Version 8.0.2 and Python (v3.8), with the four most relevant packages: numpy (v1.26.4), pandas (v2.2.3), matplotlib (v3.9.2) and pytorch (v2.7.0). For nanopore sensing, all raw reads underwent rigorous quality filtering, which removed reads with anomalous dwell times, signal spikes, or incomplete flanking DNA sequences. No statistical method was employed to predetermine sample size. Randomization was not applicable to this study. The protein identification assay was performed in a single-blind manner; blinding was not used for other experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study have been deposited into the CNGB Sequence Archive (CNSA)45 with accession number CNP0006016. The source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
The source code is released on GitHub under the BSD-2-Clause License in this link [https://github.com/BGINPS/npspy].
References
Ying, Y.-L. et al. Nanopore-based technologies beyond DNA sequencing. Nat. Nanotechnol. 17, 1136–1146 (2022).
Dorey, A. & Howorka, S. Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics. Nat. Chem. 16, 314–334 (2024).
Asandei, A. et al. Nanopore-based protein sequencing using biopores: current achievements and open challenges. Small Methods 4, 1900595 (2020).
Cressiot, B., Bacri, L. & Pelta, J. The promise of nanopore technology: advances in the discrimination of protein sequences and chemical modifications. Small Methods 4, 2000090 (2020).
Hu, Z.-L., Huo, M.-Z., Ying, Y.-L. & Long, Y.-T. Biological nanopore approach for single-molecule protein sequencing. Angew. Chem. 133, 14862–14873 (2021).
Alfaro, J. A. et al. The emerging landscape of single-molecule protein sequencing technologies. Nat. Methods 18, 604–617 (2021).
Wei, X. et al. Engineering biological nanopore approaches toward protein sequencing. ACS Nano 17, 16369–16395 (2023).
Yan, S. et al. Single molecule ratcheting motion of peptides in a Mycobacterium smegmatis porin A (MspA) nanopore. Nano Lett. 21, 6703–6710 (2021).
Brinkerhoff, H., Kang, A. S. W., Liu, J., Aksimentiev, A. & Dekker, C. Multiple rereads of single proteins at single–amino acid resolution using nanopores. Science 374, 1509–1513 (2021).
Chen, Z. et al. Controlled movement of ssDNA conjugated peptide through Mycobacterium smegmatis porin A (MspA) nanopore by a helicase motor for peptide sequencing application. Chem. Sci. 12, 15750–15756 (2021).
Wanunu, M. Back and forth with nanopore peptide sequencing. Nat. Biotechnol. 40, 172–173 (2022).
Nivala, J., Marks, D. B. & Akeson, M. Unfoldase-mediated protein translocation through an α-hemolysin nanopore. Nat. Biotechnol. 31, 247–250 (2013).
Nivala, J., Mulroney, L., Li, G., Schreiber, J. & Akeson, M. Discrimination among protein variants using an unfoldase-coupled nanopore. ACS Nano 8, 12365–12375 (2014).
Motone, K. et al. Multi-pass, single-molecule nanopore reading of long protein strands. Nature 633, 662–669 (2024).
Dutt, S. et al. High accuracy protein identification: fusion of solid-state nanopore sensing and machine learning. Small Methods 7, 2300676 (2023).
Soni, N. et al. Full-length protein classification via cysteine fingerprinting in solid-state nanopores. Nat. Nanotechnol. 20, 1482–1490 (2025).
Edfors, F. et al. Enhanced validation of antibodies for research applications. Nat Commun 9, 4130 (2018).
Kahn, R. A., Virk, H. S. & McPherson, P. S. Heed a decade of calls for antibody validation. Nature 620, 492–492 (2023).
Lund-Johansen, F. A strong case for third-party testing. eLife 12, e93329 (2023).
Nova, I. C. et al. Detection of phosphorylation post-translational modifications along single peptides with nanopores. Nat. Biotechnol. 42, 710–714 (2024).
Meng, G. et al. Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 574, 86–89 (2019).
Venkatesan, N. & Kim, B. H. Peptide conjugates of oligonucleotides: synthesis and applications. Chem. Rev. 106, 3712–3761 (2006).
Rosen, C. B., Rodriguez-Larrea, D. & Bayley, H. Single-molecule site-specific detection of protein phosphorylation with a nanopore. Nat. Biotechnol. 32, 179–181 (2014).
Motone, K., Cardozo, N. & Nivala, J. Herding cats: Label-based approaches in protein translocation through nanopore sensors for single-molecule protein sequence analysis. iScience 24, 103032 (2021).
Cardozo, N. et al. Multiplexed direct detection of barcoded protein reporters on a nanopore array. Nat. Biotechnol. 40, 42–46 (2022).
Bakshloo, M. A. et al. Polypeptide analysis for nanopore-based protein identification. Nano Res. 15, 9831–9842 (2022).
Zhang, Y. et al. Peptide sequencing based on host–guest interaction-assisted nanopore sensing. Nat. Methods 21, 102–109 (2024).
Zhang, M. et al. Real-time detection of 20 amino acids and discrimination of pathologically relevant peptides with functionalized nanopore. Nat. Methods 21, 609–618 (2024).
Wang, K. et al. Unambiguous discrimination of all 20 proteinogenic amino acids and their modifications by nanopore. Nat. Methods 21, 92–101 (2024).
Papakyriacou, I. et al. Loss of NEDD8 in cancer cells causes vulnerability to immune checkpoint blockade in triple-negative breast cancer. Nat. Commun. 15, 3581 (2024).
Yang, M. et al. Prognosis and modulation mechanisms of COMMD6 in human tumours based on expression profiling and comprehensive bioinformatics analysis. Br. J. Cancer 121, 699–709 (2019).
Zhang, Z. et al. GABARAPL2 is critical for growth restriction of Toxoplasma gondii in HeLa cells treated with gamma interferon. Infect. Immun. 88, e00054–20 (2020).
Sauciuc, A. et al. Blobs form during the single-file transport of proteins across nanopores. Proc. Natl. Acad. Sci. USA 121, e2405018121 (2024).
Pommié, C., Levadoux, S., Sabatier, R., Lefranc, G. & Lefranc, M.-P. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recognit. 17, 17–32 (2004).
Lan, W.-H., He, H., Bayley, H. & Qing, Y. Location of phosphorylation sites within long polypeptide chains by binder-assisted nanopore detection. J. Am. Chem. Soc. 146, 24265–24270 (2024).
Restrepo-Pérez, L., Wong, C. H., Maglia, G., Dekker, C. & Joo, C. Label-free detection of post-translational modifications with a nanopore. Nano Lett. 19, 7957–7964 (2019).
Yu, C. et al. Gln40 deamidation blocks structural reconfiguration and activation of SCF ubiquitin ligase complex by Nedd8. Nat. Commun. 6, 10053 (2015).
Herhaus, L. et al. TBK1-mediated phosphorylation of LC3C and GABARAP-L2 controls autophagosome shedding by ATG4 protease. EMBO Rep. 21, e48317 (2020).
Layton, C. J., McMahon, P. L. & Greenleaf, W. J. Large-scale, quantitative protein assays on a high-throughput DNA sequencing chip. Mol. Cell 73, 1075–1082.e4 (2019).
Wegner, G. J., Lee, H. J. & Corn, R. M. Characterization and optimization of peptide arrays for the study of epitope−antibody interactions using surface plasmon resonance imaging. Anal. Chem. 74, 5161–5168 (2002).
Zhang, J.-Y. et al. A single-molecule nanopore sequencing platform. https://doi.org/10.1101/2024.08.19.608720 (2024).
Ip, C. L. C. et al. MinION analysis and reference consortium: phase 1 data release and analysis. F1000Res 4, 1075 (2015).
Rosen, C. B. & Francis, M. B. Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol. 13, 697–705 (2017).
Vaitheeswaran, S. & Thirumalai, D. Interactions between amino acid side chains in cylindrical hydrophobic nanopores with applications to peptide stability. Proc. Natl Acad. Sci. USA 105, 17636–17641 (2008).
Wang, W. et al. The China National GeneBank Sequence Archive (CNSA) 2024 update. Hortic. Res. 12, uhaf036 (2025).
Acknowledgements
This work is supported by the National Key R&D Program of China (2024YFC3406300), Shenzhen Science and Technology Program (KQTD20221101093603011) and (JCYJ20230807153500001), and “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2024C03004). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the paper. We also thank GCATbio Co., Ltd, for support of DNA oligo and peptide synthesis and CNSA for providing data storage and access services.
Author information
Authors and Affiliations
Contributions
Ji Wang and Junyi Chen designed the experiment, managed the project, and wrote the paper; Wenbing Qin, Yuqing Deng, and Liang Shen examined the azide modification conditions; Huixian zeng, Yishuo Zhang, Yunfeng Zhang optimized the DTC experiment; Xiaojing Sheng, Qingqing Xie, and Dapeng Wang, performed the pore protein preparation and instrument setup; Fengqin Luo, Yuchen Qiao, and Xilong Yuan managed the sensing experiment and data acquisition; Hailin Pan, Zhiwei Zhai, and Qianhua Zhu conducted the machine learning algorithms; Xu Yan provided AI arithmetic support, Tao Zeng, Yuning Zhang, Yinqi Bai, Mengzhe Shen, Bo Teng, Ou Wang, Yuxiang Li, and Chuanyu Liu provided valuable input during the conceptual development of the project, Yuliang Dong, Siqi Liu and Xun Xu co-supervised the project.
Corresponding authors
Ethics declarations
Competing interests
J. Wang, F. Luo, Y. Qiao, Y. Deng, T. Zeng, O. Wang, and Y. Dong are listed as inventors on three related patent applications (WO/2024/182947, WO/2025/129587, and WO/2025/260292), which disclose methods for OPO library construction and purification. J. Wang, F. Luo, Y. Qiao, X. Yan, T. Zeng, Y. Li, and Y. Dong are listed as inventors on patent applications (WO/2025/123211 and WO/2025/123212), which disclose methods for peptide signal extraction and classification. J. Wang, F. Luo, W. Qin, Y. Deng, O. Wang, and Y. Dong are listed as inventors on the patent application (WO/2025/138302), which discloses a method for protein detection. All applications were filed by BGI Research, Shenzhen. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Chen, J., Pan, H. et al. Nanopore-based massively parallel sensing for peptide profiling and protein identification. Nat Commun 17, 3058 (2026). https://doi.org/10.1038/s41467-026-69628-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69628-1
