
Abstract
Each amino acid except two is encoded by multiple synonymous codons, but at unequal frequencies. Such codon usage bias (CUB) is observable in almost all species, and commonly assumed as the result of natural selection towards an optimal CUB that matches the cellular tRNA supply. Here we hypothesize instead that the optimal CUB of a gene should slightly mismatch the tRNA supply to avoid excessive translational costs, while ensuring adequate functional payoff. By modifying the CUB of a resistance gene expressed in bacteria under antibiotic selection, we demonstrate that a small mismatch with the tRNA supply confers faster bacterial growth than those with minimized or large CUB-tRNA mismatches. Intriguingly, the optimal degree of CUB-tRNA mismatch increases as the resistance gene becomes less important in media with lower antibiotic concentrations, which is explainable by our model as a shift in the balance between the gene’s functional payoff and translational cost. Furthermore, genomic analyses in model organisms suggest that the optimal degree of CUB-tRNA mismatch is larger for endogenous genes with lower functional importance and higher mRNA abundance, respectively supporting the impact of functional payoff and translational cost. Finally, we find that mutations increasing or decreasing the CUB-tRNA mismatch of native genes are both predominantly deleterious, such that the CUB-tRNA mismatch is likely selectively maintained rather than minimized to that achievable in the presence of genetic drift and mutational bias. These results challenge the commonly assumed unidirectional selection on CUB and highlight the CUB-modulated balance between functional payoff and translational cost.
Similar content being viewed by others


Introduction
Eighteen out of twenty amino acids are encoded by two or more synonymous codons. The mutation of one codon to its synonymous codon, or synonymous mutation, is traditionally regarded as functionally neutral, since it does not alter the sequence of a protein. However, there is currently a debate regarding whether and why synonymous mutations may be non-neutral1,2,3. In this context, some “preferred” codons are used more frequently in the genome than their synonymous counterparts, a phenomenon commonly known as codon usage bias (CUB)4. Other than some special cases affecting only limited neighboring regions5,6,7,8, the prevailing hypothesis for the widely observed CUB is the selection-mutation-drift model9,10, which proposes that translational selection favors an optimal CUB, whereas deviations from the optimal CUB persist as a result of genetic drift (or in other words, the drift-barrier11) and mutational bias. For translational selection, it is commonly assumed that the optimal CUB maximally matches the cellular tRNA supply so that the translation is more efficient and/or more accurate10,12,13,14,15. In this context, we can quantify the CUB-tRNA mismatch using a previously16 proposed metric, D, which is based on the Euclidean distance between synonymous codons’ demand and supply of tRNA (see Methods). Accordingly, the aforementioned assumption can be expressed as unidirectional translational selection towards the optimal D = 0 (Fig. 1a). Although this assumption has not been directly tested, it is often considered supported by the widely observed patterns that highly expressed genes, for which translational selection is stronger than lowly expressed genes within the same genome, tend to exhibit a better CUB match for the cellular tRNA supply10,13.

a The conventional model assumes unidirectional selection towards the minimal mismatch between a gene’s codon usage and the cellular tRNA supply. b We propose a novel model in which the optimal CUB mismatches the cellular tRNA slightly, thereby preventing excessive translational costs. In both panels, the x axis represents D, a measure of the disparity between a gene’s codon usage and the cellular tRNA supply (see Methods), and the y axis represents cellular fitness. The black curves represent the x-y relationship assumed in the two models, while the red dot, the gray arrow, and the horizontal dashed line represent the optimal CUB, the direction of translational selection, and the genetic drift/drift barrier, respectively.
Nevertheless, as we16,17 and others18,19,20 have recently showed, the heterologous expression of exogenous genes with CUB overly matching the tRNA supply, and therefore excessively small D, may be detrimental to the fitness of the host cell. One plausible molecular mechanism behind such phenomenon is that the expression of exogenous genes trans-regulates the expression of other genes in the cell via tRNA depletion. An exogenous gene with excessively small D will have a very high protein expression level due to an increased abundance of mRNA21,22 and/or more efficient translation20,23,24,25, but at the same time, will impose a heavy translational cost on the cell, thus hindering the translation of other genes within the same cell16,20. In theory, the same logic applies to endogenous genes native to the cell, since they also trans-regulates the expression of other genes26. However, endogenous genes are critically different from exogenous genes in that they contribute to the native function or fitness of the cell. Consequently, a D-decreasing mutation in an endogenous gene would both exacerbate its translational cost and enhance its functional payoff for the cell. Whether translational selection on CUB is dominated by the functional payoff and thus unidirectional towards maximally matching the tRNA supply (the “conventional model” in Fig. 1a), or a balance between the translational cost and functional payoff so that a slight mismatch with the tRNA supply would be optimal (the “novel model” in Fig. 1b), remains to be determined.
To answer this question, we evaluated the functional payoff and translational load of 53 synonymous variants of gentamicin resistance gene GmR exhibited on Escherichia coli MG1655 strain grown in Luria-Bertani (LB) media containing 120 μg/ml gentamicin, where GmR mimics an endogenous gene in terms of contributing to cellular fitness. As compared to variants with moderately small D, those with minimal D have greater functional payoffs, higher translational cost, and a lower overall fitness, suggesting that the optimal CUB does not correspond to a perfect match with tRNA supply. Similar experiments conducted under various gentamicin concentrations (ranging from 0 to 180 μg/ml) revealed that the optimal D increases as the GmR gene becomes functionally less important in media with lower gentamicin concentration, indicating that optimal CUB indeed shifts with the balance between GmR’s functional payoff and translational cost. Additional experiments using Ribo-Seq, tRNA supply modification, or the ampicillin resistance gene AmpR provided consistent support. To further test whether our new model applies to evolution in nature, we analyzed endogenous genes of several model organisms under the assumption that the wildtype codon usage has been evolutionarily optimized to the maximum extent possible in the presence of genetic drift. We found that the optimal D decreases for genes with greater functional importance and increases for genes with higher translational costs, respectively supporting their roles in determining the optimal D of actual endogenous genes. Lastly, we found that a substantial proportion of the point mutations occurred during neutral evolution of the mutation accumulation (MA) lines reduces D, and that D-decreasing and D-increasing mutations in representative genes are equally likely to be deleterious. Both phenomena are compatible with the existence of substantial D-increasing selection in the wildtype genome. Collectively, these results challenged the conventional view that translational selection for codon usage is unidirectional towards a maximum match with cellular tRNA supply, and suggested that a slight mismatch to tRNA supply is beneficial, a previously unrecognized complexity in the evolution of codon usage bias.
Results
A codon usage that maximally matches the cellular tRNA supply is not optimal
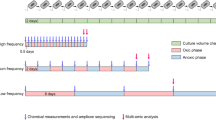
We used the gentamicin resistance gene GmR as a model to examine how the codon usage of a gene alters its translational cost and functional payoff, as well as the growth rate of the cell. To this end, a collection of synonymous variants of GmR consisting of three distinct sequences for each of 19 unique D values were designed. Fifty-two of them, together with the wildtype GmR, were successfully synthesized and individually constructed into an expression cassette (see below), covering a wide range of CUB from a near-perfect match with (D = 0.078) to a large mismatch with tRNA supply (D = 0.967) (see Fig. 2a and Supplementary Data 1). Variants with maximal mismatch (the theoretical maximal D is 1.414) were not considered since their expression is most likely too low for an observable functional payoff. All GmR variants were connected to a strong promoter J23119, fused with mCherry for the direct readout of its protein expression level, and then inserted to an expression vector containing a YFP driven by a weaker promoter J23114 (Fig. 2b, see Methods and Supplementary Table 1). Here the YFP is used as a measure of the overall translational efficiency of the cell and is therefore used to inversely estimate the translational cost imposed by GmR. We individually transformed the 53 vectors into Escherichia coli MG1655 strain, and then cultured these strains in Luria-Bertani (LB) media supplemented with 120 μg/ml gentamicin. This concentration is below the minimum inhibitory concentrations (MICs) (see Methods and Supplementary Table 2) of 48 out of the 53 strains, thereby ensuring the measurability for the majority of the strains.

a Distribution of D values of the designed GmR synonymous variants and the one wildtype GmR sequence. They were divided into three groups containing similar numbers of distinct D values, as indicated by the color tiles at the bottom. b The structure of the expression vector. c The expression level of the GmR protein as measured by the fluorescence of mCherry, which represents the functional payoff of the gene (y axis). d The translational load imposed by GmR expression, as measured by the reduction in YFP fluorescence relative to its maximum value observed in the same environment. e The relative fitness of cells expressing GmR synonymous variants of various CUBs. In (c)–(e), results were obtained in LB media supplemented with 120 μg/ml gentamicin. Statistical significance of two-tailed Wilcoxon Rank Sum Tests are indicated as *P < 0.1, **P < 0.05, or ***P < 0.01. We conducted three biological as well as three technical replicates on each strain, whose mean + standard error is presented. Source data are provided as a Source Data file.
The 53 synonymous variants of GmR were divided into three groups of minimum, small and large mismatch between the codon usage and cellular tRNA supply (Fig. 2a). It was found that the variants with minimum D showed the highest level of GmR expression as indicated by mCherry fluorescence (Fig. 2c), which is consistent with the cis-regulatory effect of CUB on the gene’s expression and functional payoff for the cell. As for CUB’s trans-regulatory effect for other genes, the YFP fluorescence suggests a greater translational cost for GmR variants with smaller D (Fig. 2d). These observations collectively suggest that a D-decreasing mutation would elevate both the functional payoff and the translational cost of a gene. Most importantly, the net functional payoff combining the two effects above, which is approximated by a direct measurement of cellular growth rate, appears to be the highest for the variants that have a small, but not minimum D (Fig. 2e). These results suggest that, due to the balance between functional payoff and translational cost, a gene’s optimal CUB should exhibit a slight mismatch with the cellular tRNA supply.
The optimal degree of mismatch between codon usage and tRNA supply depends on the gene’s functional importance
Our model also predicts that genes of different functional importance will have different optimal CUBs (Fig. 3a). More specifically, for a gene with a given transcription level, its translational cost is determined by its codon usage (Fig. 3a middle row), but is unaffected by its functional importance. However, an important gene would have a greater functional payoff (Fig. 3a top row), which allows the gene to afford a more severe translational cost and, therefore, a codon usage more closely matches the cellular tRNA supply. Consequently, the optimal CUB of a more important gene would likely correspond to a smaller D, whereas the optimal CUB of a less important gene would likely correspond to a larger D (Fig. 3a bottom row). We emphasize that this prediction applies to synonymous variants of any particular gene as long as the gene exhibits a diminishing (relative to the increase of translational cost) increase of functional payoff as D decreases.

a Schematic diagram showing the theoretical expectation for the optimal CUB when the gene’s functional payoff shifts (see Results for details). b The functional payoff by GmR as estimated by its protein expression level, measured in LB media supplemented with 0–180 μg/ml gentamicin. c The functional payoff of GmR variants with minimum D relative to those with large D (y axis) increases with the functional importance of the GmR protein (x axis). The Spearman’s rank correlation coefficient (ρ) and its two-tailed P value were presented. d The translational cost by GmR as estimated by the reduction of YFP fluorescence relative to its maximal value observed in the same environment, measured in LB media supplemented with 0-180 μg/ml gentamicin. e The translational cost of GmR variants with minimum D relative to those with large D (y axis) increases with the functional importance of the GmR protein (x axis), but to a lesser extent compared to the functional payoff shown in c. The Spearman’s rank correlation coefficient (ρ) and its two-tailed P value were presented. f The relative fitness of cells expressing various GmR synonymous variants in LB media supplemented with 0-180 μg/ml gentamicin. In (b)/(d)/(f), statistical significance of two-tailed Wilcoxon Rank Sum Tests are indicated as *P < 0.1, **P < 0.05, or ***P < 0.01. We conducted three biological as well as three technical replicates on each strain, whose mean ± standard error is presented. Source data are provided as a Source Data file.
To test this prediction, we cultured the 53 strains expressing synonymous GmR variants in LB media supplemented with gentamicin at 12 different concentrations ranging from 0 to 180 μg/ml, so that the GmR gene would have different functional importance. The functional payoff of the GmR gene is estimated by the GmR-mCherry expression, which is rescaled so that the average functional payoff of the group with large D is 1 in each gentamicin concentration to facilitate comparison across conditions. The translational cost exerted by each GmR variant under a given gentamicin concentration is estimated by the fold reduction of YFP expression relative to the maximum YFP expression observed for the focal concentration of gentamicin, which is also further rescaled so that the average translational cost of the group with large D is always 1 in each gentamicin concentration. We found that the GmR variants with large D always exert the smallest functional payoff among the three groups (Figs. 3b and S1), which is compatible with CUB’s cis-regulatory effect. Consistent with GmR’s greater functional importance in media with higher gentamicin concentrations, variants with minimum D displayed a greater functional payoff therein, with 68.9% of the rank variations explained by the gentamicin concentration (Fig. 3c). The average translational cost of the GmR variants with large D is also always the lowest among the three groups regardless the concentration of gentamicin (Figs. 3d and S1). However, the dependency of translational cost on functional importance is much weaker than that of functional payoff, so that only 27% of the rank variation of translational cost imposed by the GmR variants with minimum D is explained by the gentamicin concentration (Fig. 3e). Most importantly, direct measurement of the fitness indicates that, as gentamicin concentration increases from 0 to 180 μg/ml and GmR becomes more important, the optimal CUB gradually shifts from a large D at concentrations of 0 to 60 μg/ml, to a small D at concentrations of 80 to 120 μg/ml, and finally to a minimum D at concentration of 180 μg/ml (Figs. 3f and S1). This observation demonstrates that the optimal CUB of a gene depends on the gene’s functional importance, and when the gene is less important, the optimal CUB should be one that mismatches the cellular tRNA supply to a greater extent. We note that even in media with high gentamicin concentrations, GmR synthesis cannot exceed a physical limit (e.g., as determined by cell resource uptake rate). Functional payoffs and fitness of the group with the minimum D may have been affected by this limit, thus making the difference with the group of minimum/small D less statistically significant. Nevertheless, the translational cost should have been less affected, resulting in a stronger pattern (right-most group in Fig. 3d).
We conducted four additional analyses to further strengthen our conclusion. First, under the assumption that natural selection has optimized the supply-demand balance of tRNAs for the whole transcriptome (but not necessarily for individual genes), we estimated the effective tRNA supply for each codon based on the combined transcriptomic codon demand of the top 10% highly expressed genes. However, it may be argued that direct measurement of tRNA transcript abundance would provide a better estimate of tRNA supply. Although it cannot capture factors such as tRNA charging, binding of EF-Tu and GTP, and codon-anticodon affinity, we still attempted to estimate tRNA supply based on high-throughput tRNA sequencing data27. As a result, we found minor changes in the value of D, which did not affect the grouping (Fig. S2) and therefore our results. Second, to demonstrate that the translation of GmR incurs a translational cost that is dependent on its codon usage and also affects endogenous genes, we used Ribo-Seq to measure the codon decoding time of wildtype and four E. coli strains expressing GmRs of different D (see Methods). For the two strains expressing GmRs of minimum D, decoding time of codons frequently used by GmR tend to increase, especially in endogenous genes with high translational activity (Fig. S3). This pattern is absent in the two strains that express GmRs of large D. Combined with the YFP-based data, they indicate that the translation of GmRs of minimum D imposes a genome-wide translational cost. Third, the GmR experiment only manipulated codon demand, while our model also predicts a similar effect for manipulation of tRNA supply. The supply of all tRNAs can be difficult to manipulate, but we attempted to increase the supply of a specific tRNA using a tRNA gene expression cassette (Fig. S4a), and examined two strains whose D changed from the other two groups to the small mismatch group (see Methods). As is predicted by the conventional model, a decrease in D caused by altered tRNA supply is always beneficial (or at least not detrimental in low or intermediate gentamicin concentrations). On the contrary, our model predicts that at intermediate gentamicin concentration, both strains should be fitter after the tRNA supply change, whereas fitness changes would be opposite at low or high gentamicin concentration (Fig. S4b). The experimental results support our model (Fig. S4c/d), although the fitness changes observed for the strain with increased D are not statistically significant, possibly due to the relatively small change in D. Fourth, to further exclude the possibility that the above observation is caused by gentamicin’s interference with the translation process9, we tested again the fitness effect of synonymous codon usage change using another antibiotic resistance genes, namely AmpR, the ampicillin resistance gene (Fig. S5a). We constructed 47 synonymous variants of AmpR (see Supplementary Data 2 and Methods), which were divided into three equal-sized groups of minimum, small, and large D. tested whether altered functional importance can result in a shift in optimal D as predicted by our model. In the presence of ampicillin at 100, 2000, and 4000 μg/ml concentration (see Supplementary Table 3), the fittest groups were respectively found to be large, small and minimum D (Fig. S5b), which again directly supports our model. Also, we note that these patterns observed for GmR and AmpR are also consistent with our previous observations that expression of viral genes with CUB excessively similar to host CUB is highly deleterious16, because viral genes in theory should have zero functional payoff to host cells. Combined, the manipulative experiments above demonstrated that the optimal CUB for a gene contributing to cellular fitness is not one that perfectly matches tRNA supply, but rather one that exhibits some degree of mismatch with tRNA supply.
The optimal CUBs of native endogenous genes are affected by both functional payoff and translational cost
We then ask whether the conclusion drawn from the manipulative experiments on resistance genes is applicable to native endogenous genes as well. Based on the same logic as above (Fig. 3a), our model predicts that a more important gene should have an optimal CUB that is more closely aligned with cellular tRNA supply, and therefore correspond to a smaller D. To gauge the optimal CUB for the native genes, we assumed that the native codon usages of endogenous genes have been optimized by translational selection to the maximum extent possible in the presence of genetic drift. Here, the relative strength of translational selection and genetic drift varies for different genes within the genome. By following previous practice28, we presumed that genetic drift (determined by effective population size) was constant across genes, and that translational selection was determined by mRNA expression levels. Therefore, our hypothesis predicts a negative correlation between a gene’s functional importance and the D value of its native codon usage, especially for highly expressed genes because they are under stronger translational selection that keeps their native codon usage close to their optimal CUB.
Using the genomic data of E. coli strain MG1655, we found that the functional importance of a gene, as estimated by the fitness reduction upon its replacement by KanR (kanamycin-resistance)29, is indeed anticorrelated with its native D value (Pearson’s R = −0.09, P < 10-6; Spearman’s ρ = −0.05, P < 0.01). The anticorrelation is stronger among genes whose mRNA expression levels are higher, which cannot be explained by the reduced number of genes since random downsampling to the same number of genes will not generate the same result (Fig. 4a. See Fig. S6a for tRNA expression-based D). The same results were observed using genomic data30,31 of Saccharomyces cerevisiae strain BY4742, for which functional importance of 4679 genes are also available from replacement by KanR (Figs. 4b and S6b). These results collectively supported the notion that genes with greater functional payoff can withstand a more severe translational cost and, therefore, have optimal CUB more closely matches the cellular tRNA supply.

The Spearman’s Rank Correlation between a gene’s functional importance and D of its native CUB (y axis) is estimated for E. coli (a) or S. cerevisiae (b) endogenous genes whose expression levels exceed a specific threshold (x axis). Error bars indicate standard deviations of the correlation assessed from 1,000 bootstraps (purple) or random samples (gray). An inset scatter plot represents one correlation from each panel. Statistical significance of two-tailed Wilcoxon signed-rank test between real and random gene sets is indicated as *P < 0.1, **P < 0.05, or ***P < 0.01. In E. coli, gene counts at decreasing cutoffs (100%, 50%, 30%, 10%, 5%, 4%, 3%) were N = 3318, 1659, 995, 331, 165, 132, and 99, while in S. cerevisiae, they were N = 4249, 2124, 1274, 424, 212, 169, and 127. The data are presented as mean values ± standard deviations. Source data are provided as a Source Data file.
While the above results supported the role of functional payoff in determining the optimal CUB, demonstrating the role of translational cost is more important. This is because, under the conventional model, where the optimal CUB matches tRNA supply perfectly, the anticorrelation between gene importance and native D would also be predicted because they are both confounded by gene expression level32. That is, highly expressed genes will likely have high functional importance and also be subjected to greater translational selection towards the optimal D = 0 (as posited by the conventional model), therefore exhibiting smaller D. Accordingly, the key feature of our model that differentiates it from the conventional model is the translational cost shown by CUB corresponding to excessively small D. More specifically, a D-decreasing mutation in a gene with high transcript abundance should result in a greater increase of translational cost than that in a gene with low transcript abundance, since a greater number of RNA molecules are mutated in the former case. Consequently, highly expressed genes should be more sensitive to reductions in D, resulting in a larger optimal D in highly expressed genes than lowly expressed genes of similar importance (Fig. 5a). Once again, this phenomenon should be more pronounced among genes with stronger translational selection, as their native CUB should more closely resemble their optimal CUB.

a A diagram illustrating why the optimal D would be higher for genes with greater translational costs. b–e For E. coli (b), S. cerevisiae (c), D. melanogaster (d) and H. sapiens (e), the Spearman’s Rank Correlation between a gene’s mRNA expression level and D of its native CUB (y axis) is estimated among endogenous genes whose expression levels exceeds a given threshold (x axis). Purple bars indicate results from the real set of genes above the expression threshold, while gray bars represent results from a randomly sampled set of genes consisting of the same number of genes as the real set. Error bars indicate standard deviations of the correlation assessed from 1000 bootstraps (purple) or random samples (gray). An inset scatter plot represents one correlation from each panel, with histogram to the right indicating the distribution of gene expression. Statistical significance of two-tailed Wilcoxon signed-rank test between real and random gene sets is indicated as *P < 0.1, **P < 0.05, or ***P < 0.01. The number of genes corresponding to successive cutoffs (100%, 50%, 30%, 10%, 5%, 4%, 3%) was N = (3832, 1916, 1149, 383, 191, 153, 114) for E. coli; N = (5520, 2760, 1656, 552, 276, 220, 165) for S. cerevisiae; N = (13555, 6777, 4066, 1355, 677, 542, 406) for D. melanogaster; and N = (17212, 8606, 5163, 1721, 860, 688, 516) for H. sapiens. The data are presented as mean values ± standard deviations. Source data are provided as a Source Data file.
Based on this logic, we tested the correlation between mRNA expression levels and the D value of native codon usage among E. coli endogenous genes whose expression exceeds a certain threshold. We found that D and expression level are positively correlated among the most highly expressed genes (Fig. 5b, purple bars. See Fig. S7a for tRNA expression-based D), supporting the effect of translational cost due to excessively small D. The positive correlation is not an artifact of the limited number of genes above the expression threshold, since randomly downsampling all genes to the same number as those retained at each expression threshold will not result in the same correlation (Fig. 5b, gray bars. See also Fig. S7a). Moreover, endogenous genes in yeast, fly, and human show similar patterns (Figs. 5c-e and S7b-d), and these patterns cannot be attributed to potential base composition biases (Fig. S8) or the dominance of an outlier amino acid (Fig. S9). Interestingly, the maximum correlation achieved is the weakest in human compared to the other three species, which is consistent with the weak translational selection observed for metazoan species33. It is evident from these observations that translational cost strongly influences the optimal D when a native gene’s CUB excessively matches the cellular tRNA supply. Together with the results that demonstrate the effect of functional payoff, we have found evidence for both facets of our model regarding an optimal CUB as dictated by a balance between translational cost and functional payoff.
The mismatch between endogenous gene’s codon usage and cellular tRNA supply is selectively maintained
To further verify our model, we set out to test the direction of selection on CUB of native endogenous genes, which is predicted in conventional model as a unidirectional selection towards a perfect match with the cellular tRNA supply, or optimal D = 0 (Fig. 6a). Our novel model, however, predicts converging selections from both directions, which maintained an optimal CUB with a slight mismatch with the cellular tRNA supply (Fig. 6b). In this context, it is possible to determine the type of selection acting on a trait from its change upon the removal of the selection34. Specifically, evolution in the absence of selection should tend to cause a rise in D if D has been selectively minimized to the drift barrier, as posited by the conventional model (Fig. 6a). On the contrary, if natural selection has maintained D at a moderately small level, the probabilities that D will increase or decrease upon the removal of selection are both substantial (Fig. 6b).

a The conventional model posits that CUB is under unidirectional selection towards a minimal D, therefore most mutations occurring during evolution in the absence of selection should be D-increasing. b Our new model proposes that CUB is under stabilizing selection towards an optimal D, so that the probabilities of D-increasing or D-decreasing mutations during evolution in the absence of selection are both substantial. c The fraction of D-increasing mutations found in the E. coli mutation accumulation lines34,36,37 (red arrow) is not significantly different from the random expectation (gray histogram. See Methods). Only the top 15% most highly expressed genes were used, since their native CUB closely resembled the optimal CUB due to strong translational selection, and this fraction (15%) allows sufficient statistical power (see Fig. S10a). d Similar to c except that the average effect size (changes in D) of D-increasing mutations is compared with its random expectation. e Similar to c except that the average effect size of D-decreasing mutations is compared with its random expectation. f–h Similar to (c)–(e), except that synonymous mutations from S. cerevisiae mutation accumulation lines were analyzed. In each of (c)–(h), the P-value was calculated using a permutation test in which the observed value (red arrow) was compared against a null distribution derived from 10,000 simulations (gray histogram). i The fitness effect of synonymous mutations in 21 representative yeast genes1 were examined. For each gene (x axis, in ascending order of expression), the fraction of deleterious synonymous mutations (y axis) is presented. Statistical significance against a binomial null of 50% chance deleterious is shown by the color of the bar, as indicated by the color legend at the top left corner. j The same set of synonymous mutants as in panel i is separated into D-decreasing and D-increasing. The fitness of each mutant was presented by a gray dot, and the average fitness effect of all mutations on a gene is shown by a black dot, with error bars indicating standard deviation of the mutants. The red dashed line indicates y = 1. At the bottom left corner of each panel, two-tailed Spearman’s correlation coefficient and P value between gene expression and fitness of the mutants were indicated. The blue lines represent linear models fitted towards either D-decreasing (N = 1063) mutations or D-increasing (N = 803) mutations. Source data are provided as a Source Data file.
Following this logic, we extracted the single nucleotide synonymous mutations observed in E. coli and S. cerevisiae mutation accumulation (MA) lines. These mutations were observed in wildtype or mismatch repair deficient strains (∆mutL for E. coli and ∆MSH2 for S. cerevisiae) evolving with minimized natural selection34,35,36,37. Take E. coli an example, among the top 15% highly expressed genes, whose sequence supposedly has been nearly optimized by translational selection before MA, we found that 49.4% of single nucleotide mutations are D-increasing (Fig. 6c, red arrow). This fraction does not differ from its expected value of 49.2 ± 3%, which is determined by sampling the same number and types of mutations on the same genes at random (Fig. 6c, gray histogram. See Methods). This observation is not caused by a lack of statistical power, since statistical significance can be achieved when a “positive control” set of mutations constructed by random mutations in genes with minimum D values was subjected to the same test (Fig. S10. see Methods). D-increasing and D-decreasing mutations found in E. coli MA lines have average effect sizes of 0.00257 ± 0.000196 and 0.00298 ± 0.000273 on D, respectively, which again show no difference from their respective random expectations (Fig. 6d, e). These observations made with E. coli are all consistently recapitulated in S. cerevisiae (Figs. 6f–h and S10). Combined, these results strongly suggest that D is not selectively minimized in the ancestor of MA lines.
Additionally, a recent study on the fitness effects of point mutations based on genome editing has enabled us to examine synonymous mutations directly in 21 representative yeast genes1 and distinguish between our model and the conventional model. In particular, while both our model and the conventional model predict that D-increasing synonymous mutations are mostly deleterious, only our model predicts that D-decreasing synonymous mutations are also mostly deleterious. As a result of analyzing all synonymous mutations in the dataset, we found that an overwhelmingly large percentage of both D-decreasing and D-increasing mutations are deleterious (Fig. 6i). A more quantitative analysis of the mutations’ fitness effects shows that both D-decreasing and D-increasing mutations are deleterious on average, with greater deleterious effects in genes with higher expression levels (Figs. 6j and S11), which is consistent with stronger translational selection38. All these results are recapitulated by tRNA expression-based D (Fig. S12). Collectively, these observations strongly suggest that the native codon usage of endogenous genes is selectively maintained at a slight mismatch with cellular tRNA supply, thus supporting our hypothesis and rejecting the conventional model.
Discussion
In this study, we propose that the degree of mismatch (D) between an endogenous gene’s CUB and the cellular tRNA supply affects both the gene’s functional payoff and translational cost. Consequently, the payoff-cost trade-off results in an optimal CUB corresponding to a moderately small D that allows the cell to achieve the optimal growth rate. Using 53 synonymous variants of the gentamicin resistance gene GmR as a model, we showed that E. coli strains expressing variants with moderately small D indeed grow faster than other variants with minimum or larger D in a culture with intermediate gentamicin concentration. Moreover, the optimal D increases when the GmR becomes functionally less important in media supplemented with less gentamicin, indicating that a less important gene is more sensitive to translational costs resulting from an excessive match between CUB and tRNA supply. Further supporting our model, we found that the optimal D of native endogenous genes from multiple species decreases for genes with higher functional importance and increases for genes with higher translational costs. Lastly, evidence from both mutation accumulation lines and genome-editing-based mutational effects suggest that the CUB of wildtype genes correspond to D values that are selectively maintained at an intermediate level instead of being minimized. Our results contradicted the conventional model in which codon usage of endogenous genes are being selectively optimized towards a perfect match with the cellular tRNA supply, and revealed a previously unrecognized complexity in translational selection and evolution of CUB.
Our results have a couple of limitations that merit discussion. First, the GmR genes were inserted into a high copy number vector, a configuration that mimics common natural conditions39,40 but could compromise the comparability among strains because of differences in plasmid copy number. However, the observed CUB-related patterns appeared unexplainable by plasmid copy number changes in response to gentamicin concentrations, since the DNA copy number of GmR gene relative to dxs by qPCR using genomic DNA as template (see Methods) is not correlated with D (Figure S13a), and the correlation between D and functional payoff/translational cost/fitness remains qualitatively unchanged in partial correlations that controls for the plasmid copy number (Figure S13b). Second, we used D to measure the degree of mismatch between a gene’s CUB and the cellular tRNA supply. Designed with the notion that an optimal CUB proportional to cellular tRNA supply would maximize expression of the focal gene and minimize the translational cost impacting other genes due to differential depletion of tRNAs for synonymous codons, this metric D has been successful in revealing CUB’s cis-regulatory effects on viral gene expression and trans-regulatory effects on host translation and fitness16,17. Other commonly used CUB measures, such as the Codon Adaptation Index (CAI), have different premises in that the optimal CUB corresponds to the maximal use of the preferred codons13,41, but are not appropriate for our analyses for two reasons. On the one hand, some previous studies have indicated that CAI is not a major determinant of protein expression level42,43. On the other hand, CAI does not capture the supply-demand relationship between tRNAs and codons, and therefore is insufficient for describing the trans-regulatory effects on other genes due to tRNA depletion. For example, the optimal CAI ( = 1), in which preferred codons are maximally used, cannot in theory be considered optimal from a supply-demand perspective, since the supply of tRNA for unpreferred codons is never used. Third, in terms of the molecular mechanism behind our observation, our working model is tRNA depletion triggered by excessive translation of genes with minimum D. Although the results from our experiments with GmR and AmpR, as well as those obtained from tRNA overexpression and Ribo-Seq, can parsimoniously be explained by this model, it has not been proven conclusively. Moreover, we did not rule out other molecular mechnisms that could contribute to GmR’s translational cost, such as increased plasmid copy number, transcription level, or translational initiation rate. Future research will be necessary to fully elucidate the molecular mechanisms involved.
By maintaining the optimal degree of mismatch between a gene’s CUB and the cellular tRNA supply, translational selection will leave evolutionary signatures that are clearly distinguishable from those expected by the conventional model. For example, in the conventional model, which suggests that further decreases in D are prevented by genetic drift, most D-decreasing synonymous mutations on wildtype genes would be effectively neutral. According to our novel model, however, a significant fraction of D-decreasing synonymous mutations should be deleterious as they will increase translational costs, which outweigh the spontaneous increase in functional payoff. Consistent with this notion, we showed that both D-decreasing and D-increasing synonymous mutations are mostly deleterious using a recent dataset1 of mutational effects in representative yeast genes. Together with the observations we made with the MA lines, it is strongly suggested that the synonymous codon usage bias is subject to stabilizing/converging selection towards an optimal D rather than unidirectional selection towards a minimal D. Additionally, as translational cost is jointly determined by transcription and translation, and transcription level also exhibits a payoff-cost trade-off44, further exploration is required to determine how the evolution of a gene’s promoter sequences and its CUB will interact45,46. Regardless, natural selection most likely affected all levels of gene expression, be it translation or transcription. Therefore, the regulatory effect of CUB as demonstrated in this current study must have played a role in regulatory evolution. Last but not least, our novel model can explain the more pronounced selection on CUB in housekeeping genes compared to other human genes47. This is because housekeeping genes are more likely to have constant functional importance and translational costs, and thus constant optimal CUB across tissues, thereby allowing more efficient selection.
We have previously shown that when an exogenous gene, such as those on an infecting virus, is expressed in a host cell, an excessive match between its CUB and the cellular tRNA supply may be detrimental to the host cell due to their elevated expression and tRNA-depletion effects, thus causing a repulsion between the CUB of virus and host during their coevolution16,17. In this work, we further demonstrated that this concept can be extended to endogenous genes native to the genome. In other words, to ensure optimal cellular growth, any gene expressed in a cell, including those that are functionally very important, should have a CUB that slightly deviates from the cellular tRNA supply, though the specific direction and level of deviation may differ for each gene. We believe that this principle also applies to other scenarios, such as horizontally transferred genes entering or adapting to new hosts, where the trade-off between their functional payoff and expression cost determines the optimal CUB.
Besides the conceptual novelty for the evolutionary genetics of synonymous codon usage, our results are also of practical relevance. For example, codon usage optimization is known to play a crucial role in antigen design of mRNA therapeutics and vaccines48,49. A common practice based on the conventional model emphasizes the necessity of matching the mRNA’s codon usage with the tRNA supply in the host cell in order to ensure sufficient expression of the translational product. In contrast, our novel model illustrates cellular toxicity due to translational costs resulting from excessive CUB matches with the host tRNA supply. Therefore, our model may explain some of the cellular toxicity shown by mRNA drugs and vaccines48,49, as well as offer conceptually novel directions for minimizing such toxicity. Another example appears in genetic engineering, where codon optimization is commonly used to enhance the expression of heterologous genes. Typical strategies for this task, such as codon harmonization50, focus on increasing the translational and folding efficiency of heterologous proteins by maximally matching the codon usage of the heterologous gene with the cellular tRNA supply, but ignore the collateral increase of translational cost as demonstrated in this study. We proposed instead a slight mismatch between the codon usage of the heterologous gene and the tRNA supply of the host cell should allow a better balance between host cell proliferation and heterologous protein production, especially with heterologous genes with high levels of mRNA expression. A third example is the evolution of antibiotic resistance genes, whose expression and therefore codon usage of small D values are favored under antibiotic selection but are costly in the absence of antibiotics51.
Methods
Measuring codon usage preference
Codon usage of a gene was measured by the disparity between the codon demand of the focal gene and the tRNA supply of the cell as follows:
Here i represents one of the 18 amino acids that has two or more synonymous codons; ni is the number of synonymous codons for amino acid i; Yij represents the relative demand of synonymous codons, as approximated by the fraction of codon j among all synonymous codons of amino acid i found in the open reading frame (ORF) of the focal gene; Xij represents the tRNA supply of the cell, approximated by the fraction of codon j among the synonymous codons of amino acid i for the highly expressed genes in the transcriptome of the cell15. It is important to note that we are using the transcriptome, not the genome, which means the codon usage has been weighted by the expression of those genes before being summed up. Using all genes would not alter our result as the highly expressed genes dominated the transcriptomic codon usage. Essentially, Di is the Euclidean distance between the codon demand and tRNA supply among synonymous codons encoding the amino acid i. Finally, D equals the geometric average of all 18 \({D}_{i}\). It should be noted that cellular tRNA supply in this context encompasses factors such as tRNA expression levels, the rate of tRNA aminoacylation, and the formation of the EFTu•GTP•aminoacylated-tRNA tertiary complex, as well as codon-anticodon binding affinity considering wobble base pairs, etc. In other words, D measures how well the translational demand of a codon matches the cell’s effective supply of tRNAs, which might not be accurately reflected by tRNA expression profile, and is a better fit for our codon-centered analysis than a tRNA-centered approach. Nevertheless, we extracted tRNA expression levels of E. coli27, S. cerevisiae20, H. Sapiens and D. melanogaster52 from previous publications and re-calculated D using tRNA expression-based Xij, and obtained qualitatively unchanged results (Figs. S2, S6, S7 and S12).
Construction of E. coli strains
A set of 53 synonymous mutants were designed based on the reference sequence of GmR (encoding gentamicin-3-acetyltransferase), with similar guanine and cytosine content (GC%, 46% ~ 55%) but 19 different levels of codon usage bias (D ranges from 0.078 to 0.967. Figure 2a and Supplementary Data 1). All synonymous mutations among these 53 mutants are located in the nucleotides 60 to 531 of the open reading frame (ORF). This design facilitates easier construction of expression cassettes for the mutants (see below), and avoids modifying the first 57 nucleotides of the ORF, which may have an impact on protein expression in a tRNA-independent manner42,53.
A template reporter cassette (Fig. 2b and Supplementary Table 1) consisting of a GmR fused with mCherry in the forward strand and a YFP in the reverse strand was synthesized and ligated to the pUC57 vector (Supplementary Table 1). The other synonymous GmR mutants were directly synthesized from the 37th nucleotide in the ORF to the 22nd nucleotide downstream of its stop codon (the stop codon itself is excluded, giving rise to a total length of 517 nucleotides), and amplified using primers GmRm-F and GmRn-R listed in Supplementary Table 4. Using the ClonExpress II One Step Cloning Kit (Vazyme, no. C112) according to the manufacturer’s instructions, these segments were ligated to other elements on the template reporter cassette (Fig. 2b and Supplementary Table 1), which were amplified from the template using primers PUC-F and PUC-R in Supplementary Table 4. These elements include (i) a strong constitutive promoter (J23119) that drives the expression of GmR-mCherry fusion protein, ensuring that GmR has a sufficient impact on cellular translation; (ii) a YFP gene serving as a readout for the overall translational efficiency of the cell, which is driven by a relatively weak promoter (J23114)54 so that its impact on host tRNA supply is negligible. The expression cassette also include two efficient and strong ribosome binding sites (RBS)55,56, as well as the bidirectional E. coli tonB-P14 transcription terminator57. The 53 constructed vectors were individually transformed into E. coli MG1655 cells (ZOMANBIO, ZC1040-2) by the typical calcium chloride method58, and selected on LB with 50 μg/mL ampicillin. The MICs of gentamicin for the 53 constructed strains were determined using the agar dilution method and interpreted using breakpoints defined by the Clinical and Laboratory Standards Institute59.
We aimed to overexpress one tRNA by inserting it into the high copy number PUC57 vector, and we assumed this would increase the supply of this specific tRNA at least twice as much as the supply of the most abundance synonymous tRNAs in the wildtype strain (I.e., set the \({X}_{{ij}}\) of the affected codon \(j\) to twice the maximum \({X}_{{ij}}\) of synonymous codons for amino acid \(i\), then rescale all \({X}_{{ij}}\) of amino acid \(i\)). We selected the argWCCT tRNA gene and two specific GmR variants (D = 0.328, in the minimum D group, and D = 0.778, in the large D group. See Supplementary Data 1), as these variants will be moved into the small D group as a result of the tRNA overexpression. Via homologous recombination (ClonExpress Ultra One Step Cloning Kit, Vazyme, C115), we inserted the argWCCT tRNA gene, driven by its native promoter, into the respective GmR plasmids, resulting in two tRNA-overexpressed strains. Primers used are listed in Supplementary Table 4.
We repeated the assessment for optimal D for different functional importance of the gene using AmpR, the ampicillin resistance gene. Specifically, we replaced the AmpR region native to the pUC57-GmR vector by the Mobilized Colistin Resistance-1 gene (MCR-1, see Supplementary Table 1), which confers polymyxin resistance, via homologous recombination. Next, the 47 synonymous variants of AmpR (Supplementary Data 2) were synthesized and similarly used to replace the GmR region in the vector (Supplementary Table 1) through homologous recombination. Using polymyxin as a selection marker, we successfully obtained 47 strains expression synonymous variants of the AmpR. Primers used are listed in Supplementary Table 4.
Fluorescence reporter gene assays
To evaluate the cis-regulatory effect of GmR’s codon usage on GmR expression, as well as the trans-regulatory effects on other endogenous genes within the cell, the 53 strains were first cultured in LB media supplemented with 50 μg/ml ampicillin (Sangon Biotech, A100304-0005) and 0, 10, 20, 30, 40, 60, 70, 80, 100, 120, 140, 160, or 180 μg/ml gentamicin (BioFroxx, 1146GR001). Then the expression levels of GmR-mCherry and YFP proteins were measured in >300,000 cells in log-phase growth for each of the 53 strains using flow cytometry (Attune N×T, Life Technologies). The fluorescence signals of mCherry and YFP were evaluated with a 15 nm bandpass centered on 620 nm and a 30 nm bandpass centered on 530 nm, respectively. The experiments were conducted with three biological and three technological replicates. E. coli cells with mCherry and YFP fluorescence signals ten times greater than those of the MG1655 negative control cells were retained for further analyses. The forward scatter (FSC), as well as the fluorescence signals of mCherry and YFP were measured for all cells. The expression levels of the fluorescent proteins were determined by dividing their fluorescence signals by FSC. The fluorescence levels were not measured for the AmpR strains since ampicillin compromises membrane integrity and causes cell content leakage, making fluorescence levels unreliable for determining the real cost and payoff.
Growth rate measurements
A spectrophotometer was used to measure the growth rates of the 53 strains cultured in LB media supplemented with 50 μg/ml ampicillin and 0, 10, 20, 30, 40, 60, 80, 100, 120, 140, 160, or 180 μg/ml gentamicin. Cells were cultured in LB medium at 37°C and 250 rpm overnight, then diluted to OD600 = 0.15 ~ 0.2 and transferred to 96-well plates. Growth was monitored on Epoch2 microplate reader (BioTek) at 37°C for 12 h by measuring absorbance (OD600) every 10 min. The doubling time (DT) was calculated by contrasting the starting and ending OD of a given time period via the following equation60
Here, \({{{{\rm{OD}}}}}_{{t}_{1}}\) and \({{{{\rm{OD}}}}}_{{t}_{2}}\) are, respectively, the starting and ending OD600 of the focal time period from t1 to t2. Considering the fact that different strains and gentamicin selections might exhibit different exponential growth phases in terms of OD600 ranges, it is imperative to identify the exponential growth phase of each strain using a unified, subjective method. To this end, for each sample, all combinations of OD600 measurements in three or more consecutive time points within the range of 0.2 < OD600 < 0.6 were fitted with linear models using the “lm” function in R. A combination with the most significant P value and a R2 value among the top 40% of all combinations is considered to be best described by exponential growth. The minimum DT in this exponential phase was used as the DT of the sample under consideration. The DT of each strain was measured in three biological and three technological replicates. Finally, the relative Wrightian fitness of each strain is calculated by dividing the mean DT of all strains cultured in gentamicin-free LB by the mean DT of the focal strain. This same procedure was used to measure the growth rate of AmpR strains cultured in LB media supplemented with 3 μg/ml polymyxin and 100, 2000, or 4000 μg/ml ampicillin.
The relative copy numbers of GmR plasmid and the abundance of YFP mRNA
The 53 GmR strains were lysed as previously described61,62. Specifically, the strains were cultured overnight at 37 °C and 250 rpm in 4 ml of LB medium containing 50 μg/ml of ampicillin and 0 or 60 μg/ml gentamicin until saturation. They were diluted to an OD600 of 0.15 ~ 0.2 and then grown to the logarithmic phase. After centrifuging the bacterial cultures, the supernatants were discarded. A sterile PBS solution (pH 7.4) was used to resuspend the cell pellets to an OD600 of 1. Subsequently, cells were lysed, via 95 °C incubation for 10 minutes followed by immediate freezing in −20 °C. The β-lactamase gene (bla) on the plasmid and d-1-deoxyxylulose 5-phosphate synthase gene (dxs) on the E. coli chromosome were amplified by qPCR, using 3 μl suspension of the lysed cells, iTaq Universal SYBR Green Supermix (Bio-Rad, 1725122), and previously described63 primers (Bla-F and Bla-R, Dxs-F and Dxs-R, listed in Supplementary Table 4). The relative copy number of the plasmid was determined by the qPCR-based abundances of bla relative to that of dxs.
To determine the YFP mRNA abundance, total RNA samples were isolated from the log phase of all strains in LB media using the Bacteria RNA Extraction Kit (Vazyme, no. R403) according to the manufacturer’s instructions. Complementary DNAs were reversely transcribed from RNAs using PrimeScript RT reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, no. RR047A) according to the manufacturer’s instructions. Finally, RT–qPCR was carried out with cDNAs and primers (Supplementary Table 4) using the iTaq Universal SYBR Green Supermix 1000 Rxn (Bio-Rad, no. 1725122) according to the manufacturer’s instructions, measured as abundance relative to that of dxs.
Ribosome profiling (Ribo-Seq)
Single colonies of E. coli strains were inoculated and cultured overnight at 37 °C with shaking at 220 rpm. They were then subcultured into fresh medium and incubated under the same conditions (37 °C, 220 rpm) for 4–5 hours until the OD600 reached 0.6–0.7. A portion of the culture was tested for mycoplasma using the Mycolor One-Step Mycoplasma Detector (Vazyme, no. D201). Mycoplasma -free samples were treated with chloramphenicol (Selleck, no. S1677) at a final concentration of 100 μg/ml. The cultures were then incubated again for 10 minutes under the same condition. After cooling on ice, the cells were centrifuged at 4 °C and 3200 × g for 10 minutes to collect cell pellets. After being flash frozen in liquid nitrogen for 10 minutes, the pellets were sent to Novogene for Ribo-Seq library preparation and sequencing.
Raw Ribo-Seq data were processed by removing adapters using Cutadapt64, aligning to the reference genome (GCF_000005845.2) and corresponding GmRs using STAR65, and quality-controlled with RiboParser66 to determine the read (ribosome-protected fragment, or RPF) counts per codon for each gene. Translational activity (TA) of a gene was defined as its average number of RPFs per codon after excluding the 20% highest and 20% lowest RPF values. This trimming removes extreme pauses and provides a length-normalized indicator for the translational activity of the gene, which combines both mRNA abundance and translational initiation rate per mRNA molecule. The codon consumption of a gene was calculated by multiplying the number of codon occurrences in the gene by the corresponding gene’s TA. The relative decoding time for each codon was calculated in two steps. First, the normalized ribosome density of a codon in a gene is calculated by dividing its average RPF counts within the gene by the gene’s TA. Second, the relative decoding time was calculated by averaging the normalized ribosome density of the same codon from different genes. Finally, the relative decoding times estimated from the GmR-expressing strains were divided by those estimated from the wildtype strain to reflect the changes in decoding time caused by the expression of GmR (Fig. S3).
Genomic analysis for model organisms
Four model organisms were analyzed, namely E. coli, S. cerevisiae, Drosophila melanogaster, and Homo sapiens. Annotations and sequences of protein-coding genes are downloaded from EnsEMBL67 and NCBI68. The gene expression levels were derived from previous studies69,70,71,72. Using only the longest transcript with expression levels available for each gene, we obtained 3832, 5520, 13555, and 17212 genes for E. coli, S. cerevisiae, D. melanogaster, and H. sapiens, respectively. For the calculation of D values of genes in each species, only the top 10% highly expressed genes are considered for the Xij of the host (see above). Using all genes or tRNA expression levels would not change the conclusion.
The growth rate of E. coli gene deletion strains was defined as the average values of strains cultured in 30 environments obtained from a previous study29. The growth rate of S. cerevisiae gene deletion strains was defined as the average values of strains cultured in 11 environments (excluding UV-treatment, which is lethal to most deletion strains) from www.yeastphenome.org31. In both species, the relative growth rate was estimated by dividing the growth rate of a gene deletion strain by the growth rate of the wildtype strain. We then calculated the functional importance of each gene in E. coli and S. cerevisiae by subtracting 1 (the relative growth rate of the wildtype strain) from the corresponding gene deletion strain.
Analysis of mutation accumulation lines
The propensity of D-increasing mutations in the mutation accumulation (MA) lines is compared with its random expectation. Single nucleotide synonymous mutations occurring in MA lines are extracted from previous reports34,36,37. By comparing the D value of the mutant sequence to that of the wildtype, each mutation is classified as either D-increasing or D-decreasing. Considering that selection intensifies as gene expression level increases32,38, we restricted the analysis to m highly expressed genes above a certain threshold, and calculated the observed fraction of D-increasing mutations in MA lines. To approximate the random expectation of the observed fraction, for each highly expressed gene with n single nucleotide mutations, n other single nucleotide mutations were randomly selected from the focal gene. We required that the randomly selected mutations matched the observed mutations in terms of mutation types (one of A/T → C/G, A/T → G/C, A/T → T/A. C/G → A/T, C/G → G/C, C/G → T/A). We combined these random mutations from all m highly expressed genes into an “expectation” set of mutations. Using 10,000 such expectation sets, we estimated the random expectation of the fraction of D-increasing mutations. To demonstrate that this test has sufficient power to detect potential enrichment of D-increasing mutations in MA lines, we extracted the m genes with the smallest D values. Such constructed “positive control set” were subjected to the aforementioned observed (the positive control set) versus expectation (expectation sets reconstructed according to the positive control set) test for the enrichment of D-increasing mutations (Fig. S10a/b). Another “negative control set” is constructed (Fig. S10c/d) by a group of weakly expressed genes (defined as 20% least expressed genes) that matched the m highly expressed genes in terms of the number of genes, number of mutations of each type in a gene and GC content (±15%) of a gene. Because they have not undergone sufficiently strong translational selection, they are considered “negative controls” in the sense that they should show no enrichment of D-increasing mutations compared to random expectation.
Analysis of the fitness effects of synonymous mutations
A previous study1 measured growth rates in YPD for yeast strains containing one of 1866 synonymous mutations in 21 representative genes. The growth rates were obtained from this previous study. Mutations that appeared to be deleterious in all four experimental replicates were considered deleterious mutations in our study.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon request. Raw sequencing reads from Ribo-Seq are available in NCBI BioProjects under the accession number PRJNA1335396. Source data for the figures and Supplementary Figs. are provided as a Source Data file. Source data are provided with this paper.
Code availability
Custom R scripts were used in data analysis and are available on GitHub (https://github.com/chenfengokha/GmRevol) or Zenodo (https://zenodo.org/records/18425169).
References
Shen, X., Song, S., Li, C. & Zhang, J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 606, 725–731 (2022).
Kruglyak, L. et al. Insufficient evidence for non-neutrality of synonymous mutations. Nature 616, E8–E9 (2023).
Nyerges, A. et al. Synthetic genomes unveil the effects of synonymous recoding. bioRxiv (2024).
Plotkin, J. B. & Kudla, G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32 (2010).
Pechmann, S., Chartron, J. W. & Frydman, J. Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat. Struct. Mol. Biol. 21, 1100–1105 (2014).
Tuller, T. et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141, 344–354 (2010).
Xu, Y. et al. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature 495, 116–120 (2013).
Zhou, M. et al. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115 (2013).
Hahn, F. E. & Sarre, S. G. Mechanism of action of gentamicin. J. Infect. Dis. 119, 364–369 (1969).
Hershberg, R. & Petrov, D. A. Selection on codon bias. Annu Rev. Genet 42, 287–299 (2008).
Lynch, M. et al. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet 17, 704–714 (2016).
Gingold, H. & Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 7, 481 (2011).
Qian, W., Yang, J. R., Pearson, N. M., Maclean, C. & Zhang, J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet 8, e1002603 (2012).
Xia, X. Maximizing transcription efficiency causes codon usage bias. Genetics 144, 1309–1320 (1996).
Xia, X. A major controversy in codon-anticodon adaptation resolved by a new codon usage index. Genetics 199, 573–579 (2015).
Chen, F. et al. Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 4, 589–600 (2020).
Chen, F. & Yang, J.-R. Distinct codon usage bias evolutionary patterns between weakly and strongly virulent respiratory viruses. iScience 25, 103682 (2022).
Love, A. M. & Nair, N. U. Specific codons control cellular resources and fitness. Sci. Adv. 10, eadk3485 (2024).
Guo, X. et al. Non-adaptive evolution in codon usage of human-origin monkeypox virus. Comp. Immunol. Microbiol Infect. Dis. 100, 102024 (2023).
Chu, D., Barnes, D. J. & von der Haar, T. The role of tRNA and ribosome competition in coupling the expression of different mRNAs in Saccharomyces cerevisiae. Nucleic Acids Res 39, 6705–6714 (2011).
Chen, S. et al. Codon-Resolution Analysis Reveals a Direct and Context-Dependent Impact of Individual Synonymous Mutations on mRNA Level. Mol. Biol. Evol. 34, 2944–2958 (2017).
Zhou, Z. et al. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 113, E6117–E6125 (2016).
Weinberg, D. E. et al. Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep. 14, 1787–1799 (2016).
Yu, C. H. et al. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell 59, 744–754 (2015).
Hersch, S. J., Elgamal, S., Katz, A., Ibba, M. & Navarre, W. W. Translation initiation rate determines the impact of ribosome stalling on bacterial protein synthesis. J. Biol. Chem. 289, 28160–28171 (2014).
Frumkin, I. et al. Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. USA 115, E4940–E4949 (2018).
Mordret, E. et al. Systematic Detection of Amino Acid Substitutions in Proteomes Reveals Mechanistic Basis of Ribosome Errors and Selection for Translation Fidelity. Mol. Cell 75, 427–441.e5 (2019).
Cope, A. L. & Gilchrist, M. A. Quantifying shifts in natural selection on codon usage between protein regions: a population genetics approach. BMC Genomics 23, 408 (2022).
Tong, M. et al. Gene Dispensability in Escherichia coli Grown in Thirty Different Carbon Environments. mBio 11 (2020).
Yoshikawa, K. et al. Comprehensive phenotypic analysis for identification of genes affecting growth under ethanol stress in Saccharomyces cerevisiae. FEMS Yeast Res 9, 32–44 (2009).
Turco, G. et al. Global analysis of the yeast knockout phenome. Sci. Adv. 9, eadg5702 (2023).
Zhang, J. & Yang, J. R. Determinants of the rate of protein sequence evolution. Nat. Rev. Genet 16, 409–420 (2015).
Benitiere, F., Lefebure, T. & Duret, L. Variation in the fitness impact of translationally optimal codons among animals. Genome Res 35, 446–458 (2025).
Liu, H. & Zhang, J. The rate and molecular spectrum of mutation are selectively maintained in yeast. Nat. Commun. 12, 4044 (2021).
Wei, W. et al. Rapid evolution of mutation rate and spectrum in response to environmental and population-genetic challenges. Nat. Commun. 13, 4752 (2022).
Liu, H. & Zhang, J. Yeast Spontaneous Mutation Rate and Spectrum Vary with Environment. Curr. Biol. 29, 1584–1591.e3 (2019).
Zhu, Y. O., Siegal, M. L., Hall, D. W. & Petrov, D. A. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl. Acad. Sci. USA 111, E2310–E2318 (2014).
Wu, Z. et al. Expression level is a major modifier of the fitness landscape of a protein coding gene. Nat. Ecol. Evol. 6, 103–115 (2022).
Blair, J. M. A., Webber, M. A., Baylay, A. J., Ogbolu, D. O. & Piddock, L. J. V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015).
Wein, T. & Dagan, T. Plasmid evolution. Curr. Biol. 30, R1158–R1163 (2020).
Sharp, P. M. & Li, W. H. The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15, 1281–1295 (1987).
Kudla, G., Murray, A. W., Tollervey, D. & Plotkin, J. B. Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258 (2009).
Nieuwkoop, T. et al. Revealing determinants of translation efficiency via whole-gene codon randomization and machine learning. Nucleic Acids Res 51, 2363–2376 (2023).
Cherry, J. L. Expression level, evolutionary rate, and the cost of expression. Genome Biol. Evol. 2, 757–769 (2010).
Frumkin, I. et al. Gene Architectures that Minimize Cost of Gene Expression. Mol. Cell 65, 142–153 (2017).
Mittal, P., Brindle, J., Stephen, J., Plotkin, J. B. & Kudla, G. Codon usage influences fitness through RNA toxicity. Proc. Natl. Acad. Sci. USA 115, 8639–8644 (2018).
Ma, L., Cui, P., Zhu, J., Zhang, Z. & Zhang, Z. Translational selection in human: more pronounced in housekeeping genes. Biol. Direct 9, 17 (2014).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Bitounis, D., Jacquinet, E., Rogers, M. A. & Amiji, M. M. Strategies to reduce the risks of mRNA drug and vaccine toxicity. Nat. Rev. Drug Discov. 23, 281–300 (2024).
Angov, E., Legler, P. M. & Mease, R. M. Adjustment of codon usage frequencies by codon harmonization improves protein expression and folding. Methods Mol. Biol. 705, 1–13 (2011).
Guo, Z. et al. Assessment of the reversibility of resistance in the absence of antibiotics and its relationship with the resistance gene’s fitness cost: a genetic study with mcr-1. Lancet Microbe, (2024).
Behrens, A., Rodschinka, G. & Nedialkova, D. D. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol. Cell 81, 1802–1815.e7 (2021).
Tuller, T., Waldman, Y. Y., Kupiec, M. & Ruppin, E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. 107, 3645–3650 (2010).
Vo, P. L. H. et al. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol. 39, 480–489 (2021).
Elowitz, M. B. & Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).
Olins, P. O. & Rangwala, S. H. A novel sequence element derived from bacteriophage T7 mRNA acts as an enhancer of translation of the lacZ gene in Escherichia coli. J. Biol. Chem. 264, 16973–16976 (1989).
Postle, K. & Good, R. F. A bidirectional rho-independent transcription terminator between the E. coli tonB gene and an opposing gene. Cell 41, 577–585 (1985).
Green, M. R. & Sambrook, J. Molecular cloning: a laboratory manual. Anal. Biochem. 186, 182–183 (2001).
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, (2023).
Murakami, C. & Kaeberlein, M. Quantifying yeast chronological life span by outgrowth of aged cells. J Vis Exp (2009).
Anindyajati, Artarini, A. A., Riani, C. & Retnoningrum, D. S. Plasmid Copy Number Determination by Quantitative Polymerase Chain Reaction. Sci. pharmaceutica 84, 89–101 (2016).
Skulj, M. et al. Improved determination of plasmid copy number using quantitative real-time PCR for monitoring fermentation processes. Micro. Cell Fact. 7, 6 (2008).
Lee, C., Kim, J., Shin, S. G. & Hwang, S. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 123, 273–280 (2006).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Ren, S., Li, Y. & Zhou, Z. RiboParser/RiboShiny: an integrated platform for comprehensive analysis and visualization of Ribo-seq data. J Genet Genomics (2025).
Harrison, P. W. et al. Ensembl 2024. Nucleic Acids Res 52, D891–D899 (2024).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 50, D20–D26 (2022).
Greenbaum, D., Colangelo, C., Williams, K. & Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 4, 117 (2003).
Papatheodorou, I. et al. Expression Atlas: gene and protein expression across multiple studies and organisms. Nucleic Acids Res 46, D246–D251 (2018).
Sastry, A. V. et al. The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat. Commun. 10, 5536 (2019).
Viswanatha, R., Li, Z., Hu, Y. & Perrimon, N. Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells. Elife 7 (2018).
Acknowledgements
This work was supported by the National Key R&D Program of China (grant number 2022YFA1106700 to F.C., 2021YFA1302500 to J.-R. Y.), the National Natural Science Foundation of China (32122022, 32361133555 to J.-R. Y., 32000401 and 32270681 to F.C.), Guangdong Basic and Applied Basic Research Foundation (2023A1515011926 to F.C.), the Science and Technology Planning Project of Guangdong Province, China (2024B1212070013 to J.-R. Y.), and the Science and Technology Projects in Guangzhou (2025A04J5439 to F.C.).
Author information
Authors and Affiliations
Contributions
J.-R.Y. conceived the idea and supervised the study; F.C., J.L., X. F., J. C. and J.-R.Y. designed the study and conducted formal data analyses; Y.L., Z.Z., J.L., X. F., Y.H. and Y. C. conducted experiments and acquired data; F.C. and J.-R.Y. wrote the paper with inputs from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Yitzhak Pilpel, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, F., Liu, Y., Zhou, Z. et al. A slight mismatch between a gene’s codon usage and the cellular tRNA supply is beneficial. Nat Commun 17, 3371 (2026). https://doi.org/10.1038/s41467-026-69643-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69643-2
