
Abstract
Aqueous photocatalytic CH4 oxidation offers a promising route for converting natural gas into oxygenates, a process governed by multi-electron and proton transfer at the catalyst-water interface. Here, we demonstrate that spatially confining water within Au/TiO2@pSiO2 core-shell catalysts—by reducing silica pore size to 1.7 nm—increases CH4 conversion three-fold and H2O2 production 22-fold compared to Au/TiO2. This strategy is generalizable to other semiconductors and cocatalysts, with Pt/TiO2@pSiO2-1.7 exhibiting oxygenate yields of 32.7 mmol g-1 h-1 and a 14.1% apparent quantum yield at 365 nm. Spectroscopic studies and molecular dynamics simulations reveal that water confined within pores, with a weakened hydrogen-bonding network, alters proton-coupled electron transfer pathways. Water oxidation transits to a concerted pathway, favoring •OH production for CH4 conversion, while oxygen reduction shifts to a two-electron process, directly producing H2O2. This work highlights the potential of water confinement for designing efficient photocatalysts for CH4 conversion.
Similar content being viewed by others


Introduction
Aqueous photochemical processes are vital for energy conversion and environmental applications, such as water splitting1, O2/CO2 reduction2, and pollutants removal3. These processes typically involve multi-electron and multi-proton transfer steps that generate reactive oxygen species (ROS) as key intermediates3,4,5. Photocatalytic methane oxidation has emerged as a green route to directly transform abundant natural gas into valuable fuels and chemicals, such as methanol6,7,8. This approach utilizes photon energy to overcome the high activation barriers of CH4, enabling room-temperature conversion via hydroxyl radicals (•OH) and bypassing the energy-intensive syngas route6,9. Moreover, the aqueous environment offers unique advantages in improving product selectivity by facilitating the desorption of partially oxygenated products9,10. Although extensive research has focused on developing photocatalysts with enhanced charge separation and surface reactions regulation through band structure engineering11,12,13 and cocatalyst modification9,14,15,16,17,18, the potential of tuning the aqueous microenvironment surrounding the semiconductor to boost CH4 conversion remains largely unexploited.
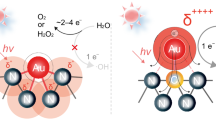
Aqueous photocatalytic CH4 oxidation involves a complex network of electron transfer (ET) and proton transfer (PT) reactions at the semiconductor-water interface (Fig. 1, Supplementary Fig. 1), where water serves as a reactant, proton source, and solvent19,20,21. For hydrophilic oxide semiconductors like TiO2, water is the primary hole acceptor, generating •OH via proton and electron transfer (Fig. 1a)9,20. These •OH radicals activate CH4 through hydrogen atom transfer (HAT), forming methyl radicals (•CH3). Simultaneously, O2 is reduced to superoxide radicals (•OOH), which further contribute to •CH3 conversion into oxygenates15. These ET and PT events can occur stepwise or concertedly via proton-coupled electron transfer (PCET) mechanisms, which are highly sensitive to the solvent environment due to the significant energy required for solvent reorganization—especially in water22,23. While studies over model reactions have shown examples of enhanced catalytic performance by tuning water structure24,25,26,27,28,29,30,31,32,33, its application to complex photocatalytic reactions such as methane oxidation remains largely unexplored. Therefore, designing a photocatalyst that modulates water structure near the semiconductor interface presents a promising avenue for tuning proton/electron transfer pathways and optimizing methane oxidation kinetics.

a Schematic representation of PCET and CH4 activation pathways in photocatalytic CH4 oxidation over Au/TiO2 in bulk water. b Schematic representation of PCET and CH4 activation pathways in photocatalytic CH4 oxidation over Au/TiO2@pSiO2 with confined water. The black dashed rectangle and blue dashed lines show a schematic of the disrupted water structure inside the nanopore. In (a, b), colored spheres represent specific atoms (light gray: H; dark gray: C; ice blue: O). PT: proton transfer; ET: electron transfer; PCET: proton-coupled electron transfer; CPET: concerted proton-electron transfer; HAT: hydrogen atom transfer; h+: holes; e−: electrons.
Spatial confinement via tunable nanopores offers an effective strategy to alter water structure22,34, and consequently, the dynamics of these ET and PT processes35. Confined water is known to exhibit distinct hydrogen-bonding (HB) networks, physical properties and reactivities compared to bulk water35,36. In this work, we demonstrate that water confinement by silica nanopores in Au/TiO2@pSiO2 catalysts modulates PCET pathways and achieves efficient CH4 conversion and H2O2 production simultaneously (Fig. 1b). We select TiO2 as the semiconductor material for its ability to oxidize water to •OH radicals12,20 and employ a noble metal cocatalyst to enhance CH4 conversion9,14,37. Water confinement is achieved by installing nanoporous and UV-transparent SiO2 enclosures on the Au/TiO2 core. Reducing the silica pore size from 3.1 nm to 1.7 nm leads to an approximately three-fold enhancement in CH4 conversion and a 22-fold increase in H2O2 production compared to Au/TiO2. This confinement strategy is applicable to other metal cocatalysts and semiconductors, as demonstrated by Pt/TiO2@pSiO2-1.7, achieving maximum oxygenate yields of 32.7 mmol g−1 h−1 with 95.5% selectivity and an apparent quantum yield (AQY) of 14.1% at 365 nm. Mechanistic investigations and molecular dynamics (MD) simulations with machine learning potentials (MLPs) reveal that the weakened HB network of confined water modulates PCET pathways for both water oxidation reaction (WOR) and oxygen reduction reaction (ORR). Specifically, WOR shifts from stepwise to concerted PCET, enhancing •OH radical production for CH4 conversion, while ORR shifts from a one-electron (1e−) to a 2e− process, promoting H2O2 production. These findings underscore the potential of engineering water structures via spatial confinement to promote photocatalytic activity and selectivity.
Results
Synthesis and characterization of Au/TiO2@pSiO2 catalysts
TiO2 is a well-established semiconductor for photocatalytic methane oxidation, with a valence band position sufficient for oxidizing water to •OH radicals12,20. Noble metal cocatalyst modification is known to promote CH4 conversion9,14,37. Therefore, we selected Au/TiO2 as the model catalyst to study the effect of water confinement. We chose the SiO2 enclosure due to its facile growth, controllable pore size, optical transparency in the UV region, and good stability. Furthermore, our previous studies have shown that a thin, non-porous silica shell can prevent oxygenates overoxidation while still allowing •OH radicals transport and convert CH420, indicating good compatibility of silica with the TiO2-based photocatalysts.
Gold nanoparticles (NPs) were loaded onto TiO2 (P25) via NaBH4 reduction9,38, and a nanoporous silica shell was constructed on Au/TiO2 using the Stöber method with alkylammonium surfactants as templates (Fig. 2a)39. By varying surfactant carbon chain length or adding auxiliary organics40,41, we obtained Au/TiO2@pSiO2-X with different pore sizes (X = pore size). The surface area and porosity of Au/TiO2@pSiO2-X were characterized by nitrogen physisorption (Fig. 2e, Supplementary Table 1). Using cetyltrimethylammonium bromide (C16TAB) resulted in mesopores ~2.6 nm, while a shorter surfactant, dodecyltrimethylammonium bromide (C12TAB), yielded smaller pores around 1.7 nm (Fig. 2f). Adding hexane as a pore-expanding agent yielded larger pores around 3.1 nm. For comparison, Au/TiO2@SiO2 without nanopores were synthesized similarly but without alkylammonium surfactants (Supplementary Fig. 2).

a Schematic illustration of the preparation of Au/TiO2@pSiO2-X catalysts. The black dashed box highlights the template agents used in material synthesis: C12TAB, dodecyltrimethylammonium bromide; C16TAB, cetyltrimethylammonium bromide. b HR-TEM image of Au/TiO2@pSiO2-1.7 catalysts. The white dashed line outlines the porous SiO2 shell, and the white arrow points to the SiO2 shell. c A High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Au/TiO2@pSiO2-1.7 catalysts. White arrows point to the highly dispersed Au NP and TiO2 core, respectively. d EDS mapping images of Au/TiO2@pSiO2-1.7. Different colors correspond to different elements (red: Ti; green: Si; yellow: Au; cyan: O). e N2 adsorption-desorption isotherms of Au/TiO2 and Au/TiO2@pSiO2-X. f Pore size distribution of Au/TiO2 and Au/TiO2@pSiO2-X. g UV-Vis DRS of Au/TiO2 and Au/TiO2@pSiO2-X.
Transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDS), and X-ray diffraction (XRD) confirmed full encapsulation of Au/TiO2 by a uniform, amorphous SiO2 shell across all Au/TiO2@pSiO2-X catalysts (Fig. 2b–d, Supplementary Figs. 3–5). The silica layer had an average thickness of 8 nm (Fig. 2b–d), which is tunable by varying the amount of tetraethyl orthosilicate (TEOS) (Supplementary Fig. 6). Au NPs ranging 2–6 nm were randomly dispersed on TiO2 (Fig. 2b–d, Supplementary Fig. 3), with a loading of ~0.1 wt% (Supplementary Table 2). Contact angle measurements demonstrate that catalysts with different pore sizes exhibit similar hydrophilic properties (Supplementary Fig. 7). UV-visible diffusive reflective spectra (UV-Vis DRS) of Au/TiO2 and Au/TiO2@pSiO2-X were nearly identical (Fig. 2g, Supplementary Fig. 8), suggesting minimal effect of the SiO2 coating on light absorption. X-ray photoelectron spectra (XPS) showed similar Ti oxidation states among all samples (Supplementary Fig. 9). These results indicate that the nanoporous coating does not significantly modify the intrinsic properties of the semiconductors.
Pore-size dependent CH4 photocatalytic oxidation by Au/TiO2@pSiO2-X
Photocatalytic reactions were conducted in a batch reactor with 100 mL of distilled water under 2.5 MPa CH4 and 1 MPa O2 at 25 °C (Supplementary Fig. 10). Liquid products (CH3OOH, CH3OH, and HCOOH) were analyzed by 1H-NMR, while HCHO was quantified by colorimetry methods (Supplementary Fig. 11). Control experiments, including dark and catalyst-free controls (Supplementary Table 3), and a 13CH4 isotope-labeling study (Supplementary Fig. 12), validated that oxygenates were generated from photocatalytic methane oxidation over Au/TiO2@pSiO2. Compared to bare Au/TiO2, Au/TiO2@pSiO2 significantly enhanced oxygenate yields, which increased as pore size decreased (Fig. 3a, b). Au/TiO2@pSiO2-3.1 exhibited a total oxygenates yield of 18.0 ± 0.4 mmol g−1 h−1, with 92.5% selectivity. Further reducing the pore size to 1.7 nm led to higher yields, achieving 27.4 ± 0.6 mmol g−1 h−1—approximately triple that of Au/TiO2. The AQY for Au/TiO2@pSiO2-1.7 was 10.9 ± 0.3% at 365 nm (Supplementary Fig. 13, Supplementary Table 4), substantially exceeding Au/TiO2 (3.3 ± 0.2%). Furthermore, this performance trend remained consistent under a broad-spectrum Xenon lamp, demonstrating that the catalytic improvement is independent of the specific light source (Supplementary Fig. 14). Notably, Au/TiO2@pSiO2 also produced a significant amount of H2O2 as a valuable byproduct (Fig. 3a, c, Supplementary Figs. 15–17 and Supplementary Note 1). The H2O2 production also increased with decreasing pore size, reaching 44.6 ± 1.3 mmol g−1 h−1 for Au/TiO2@pSiO2-1.7 (Fig. 3c), representing a 22-fold increase over Au/TiO2 and competitive with state-of-the-art catalysts (Supplementary Table 5).

a Schematics of products formation from CH4 and O2 over Au/TiO2@pSiO2-X. b Yields of oxygenates from CH4 oxidation over different photocatalysts. c H2O2 production over different photocatalysts. Reaction conditions for (b, c): 10 mg catalysts, 100 mL H2O, 2.5 MPa CH4, 1 MPa O2, 1 h reaction time, reaction temperature of 25 ± 3 °C, light source of 365 nm UV LED, 80 mW/cm2. d O2 partial pressure dependence on Au/TiO2@pSiO2-2.6. Reaction conditions: 10 mg catalysts, 100 mL H2O, 2.5 MPa CH4, 1 h reaction time, reaction temperature of 25 ± 3 °C, light source of 365 nm UV LED, 80 mW/cm2. e Productivity assessment of Au/TiO2@pSiO2-1.7 obtained at different light intensities. Reaction conditions: 10 mg catalysts, 100 mL H2O, 2.5 MPa CH4, 1 MPa O2, 1 h reaction time, reaction temperature of 25 ± 3 °C, light source of 365 nm UV LED. In (b–e), bars represent product yields (left y-axis), while the pink diamonds and corresponding pink arrow indicate oxygenate selectivity (right y-axis). The error bars in (b–d) represent the standard deviations derived from three independent experiments.
Further optimization of parameters, including light intensity, water volume, and Au loading, was performed (Fig. 3e, Supplementary Figs. 18, 19). By increasing the light flux to 500 mW/cm2, we achieved a yield of 62.9 mmol g−1 h−1 with 80% selectivity for oxygenates (Fig. 3e). Au/TiO2@pSiO2-1.7 remained stable over ten cycles, with consistent production and selectivity (Supplementary Fig. 20). XRD and TEM showed no obvious structure changes in the spent catalyst (Supplementary Fig. 21), and N2 adsorption isotherms showed pore size distribution and surface area similar to those of fresh catalysts (Supplementary Fig. 22). ICP-OES analysis verified that the gold loading remained unchanged after cycles, with negligible gold leaching observed (Supplementary Table 6).
To elucidate the role of the nanoporous silica shell, a series of control experiments was performed. A physical mixture of Au/TiO2 and porous SiO2 (pSiO2-1.7) showed similar catalytic performance to Au/TiO2 alone, highlighting the importance of the core-shell architecture (Supplementary Figs. 23, 24). Notably, Au/TiO2@SiO2 without pores yielded more than Au/TiO2 but less than the nanoporous catalysts (Fig. 3b), probably due to the greater diffusion barrier in non-porous silica. This trend signifies the importance of controlling silica pore size for efficient methane conversion.
Silica thickness also significantly impacts performance, showing a non-monotonic effect. For Au/TiO2@pSiO2 catalysts with an identical pore size and surface hydrophilicity, an 8 nm shell was optimal (Supplementary Figs. 6, 25–27). A 30 nm shell reduced yields (Supplementary Fig. 27), likely due to an increased diffusion barrier. However, thinner shells (3.6 nm and 6 nm) also resulted in lower yields; the catalyst with a 3.6 nm shell, in particular, was substantially less active. This is likely attributed to an ultra-thin shell failing to provide complete and uniform encapsulation over TiO2, thus being unable to establish an effective water confinement environment. These findings reveal a critical trade-off: the shell must be thick enough to create an effective confined microenvironment but thin enough to minimize mass transport resistance. Thus, a shell architecture with both optimal porosity and thickness is essential.
Given the critical role of the nanoporous shell, we investigated its influence on gaseous reactant transport. Temperature-programmed desorption of CH4 (CH4-TPD) showed weak adsorption on both Au/TiO2 and Au/TiO2@pSiO2-1.7 (Supplementary Fig. 28), with the latter showing an even lower adsorption temperature. These results rule out CH4 enrichment as the source of improved catalytic performance. We then examined the effect of O2, whose direct participation in oxygenates formation was confirmed by 18O isotopic labeling (Supplementary Fig. 29). Both Au/TiO2 and Au/TiO2@pSiO2-X exhibited low yield under anaerobic conditions, with yields increasing with O2 pressure (Fig. 3d, Supplementary Fig. 30). Au/TiO2 achieved maximum yield at 0.042 MPa O2, whereas Au/TiO2@pSiO2-2.6 and Au/TiO2@pSiO2-1.7 required higher pressures (up to 1 MPa), suggesting hindered O2 transport by the silica shell (Fig. 3d, Supplementary Fig. 30a, b and Supplementary Note 2). Furthermore, TiO2 and TiO2@pSiO2 required higher O2 pressures to attain optimal yields compared to their Au-incorporated counterparts (Supplementary Fig. 30c, d), and relocating Au NPs to the SiO2 surface decreased product yields (Supplementary Fig. 31). These results indicate that the enhanced performance of Au/TiO2@pSiO2 is not due to O2 enrichment from its high surface area. Instead, O2 transport within silica shell is hindered in aqueous solution; however, the adverse effect of limited O2 transport on overall product yield is mitigated by a combination of increasing O2 pressure and effective O2 utilization enabled by the supported Au cocatalysts. The function of Au as an electron acceptor for O2 reduction was confirmed using in situ XPS, where the Au 4 f signal shifted to lower binding energies upon illumination (Supplementary Fig. 32).
The counter-intuitive observation that the SiO2 shell promotes catalytic performance despite impeding O2 transport implies the enhancement originates not from gaseous reactant enrichment. We therefore hypothesized that the origin is the confinement of water layers within the hydrophilic nanopores. To further decouple these factors, we systematically modified the optimal Au/TiO2@pSiO2-1.7 catalyst with methyltrimethoxysilane (MTMS) to increase its surface hydrophobicity while preserving the pore structure (Supplementary Figs. 33–36). Fourier transform infrared (FTIR) spectra confirmed the successful surface grafting (Supplementary Fig. 37). At low modification levels (e.g., 0.1–0.5 wt%), the shell thickness, pore size, and surface area of the catalyst remained unchanged (Supplementary Figs. 33, 34), while a higher 5% MTMS loading caused a decrease in surface area, likely attributed to pore blockage. In principle, a more hydrophobic surface is expected to enrich nonpolar reactants like CH4 and O2 and potentially accelerate the reaction42,43,44. However, the catalytic results show the opposite trend: oxygenate yields decreased drastically compared to the unmodified, hydrophilic catalyst (Supplementary Fig. 36). Even a tiny 0.1 wt% MTMS modification, corresponding to an estimated Si–CH3 surface coverage of <0.5% (Supplementary Table 7), was sufficient to significantly suppress the reaction. This extreme sensitivity to a miniscule fraction of hydrophobic groups provides strong evidence that the interfacial water environment, rather than the mass transport of gaseous reactants, plays the dominant role in the catalytic enhancement.
Generalization of the nanoporous confinement strategy
We explored this confinement strategy with different metal cocatalysts and semiconductors. Substituting Au with Pd, Pt, or Rh exhibited a similar trend: confined pores around metal-TiO2 surface significantly promoted oxygenates and H2O2 yields compared to bare metal-TiO2 catalysts, with smaller pores yielding higher productions (Fig. 4a, Supplementary Figs. 38, 39 and Supplementary Note 3). Among these, Pt/TiO2@pSiO2-1.7 achieved oxygenate yields of 32.7 ± 0.8 mmol g−1 h−1 and an AQY of 14.1 ± 0.3% for CH4 oxidation at 365 nm, representing competitive performance relative to previously reported catalysts (Fig. 4, Supplementary Tables 4, 8). We further extended our study to other semiconductors, including BiVO4, ZnO and anatase TiO2 (A-TiO2) (Fig. 4a, Supplementary Figs. 40–42 and Supplementary Note 3). Applying the porous silica coating improved oxygenate yields and H2O2 production for these catalysts. For example, Au/A-TiO2@pSiO2-1.7 achieved oxygenate and H2O2 formation rates approximately 5 and 21 times higher than those of Au/A-TiO2, respectively (Fig. 4a). These results underscore the versatility of the nanoporous confinement design, demonstrating its broad applicability across different photocatalyst systems.

Yields of oxygenated product and H2O2 from photocatalytic methane oxidation over core-shell photocatalysts with different metal cocatalysts and semiconductors. Reaction conditions: 10 mg catalysts, 100 mL H2O, 2.5 MPa CH4, 1 MPa O2, 1 h reaction time, reaction temperature of 25 ± 3 °C, light source: 365 nm UV LED, 80 mW/cm2. The error bars represent the standard deviations derived from three independent experiments. The vertical dashed lines categorize the different cocatalysts and semiconductor supports.
Key ROS for photocatalytic CH4 oxidation
We also investigated the role of SiO2 shell in photocarrier separation using photoluminescence (PL) and photocurrent measurements, and electrochemical impedance spectroscopy (EIS). Steady-state and time-resolved PL (TRPL) spectroscopy revealed that the silica shell has a negligible impact on photocarrier separation and lifetimes (Supplementary Figs. 43, 44). Furthermore, Au/TiO2 showed a higher photocurrent and lower impedance compared to Au/TiO2@pSiO2 (Supplementary Figs. 45, 46). These results, together with findings on gas transport and surface modifications (Supplementary Note 2), demonstrate that improved performance in Au/TiO2@pSiO2 is not due to improved photocarrier separation or gas transport, but rather to effects associated with the structure of the confined water layers in contact with the semiconductors.
To unravel the underlying mechanism, we next sought to identify the key ROS responsible for methane activation and H2O2 formation within the confined environment. This can be achieved by introducing sacrificial agents that selectively quench different ROS and monitor the change in methane conversion15,45. Both holes and •OH radicals are capable of activating methane9,14,15. We used salicylic acid and (NH4)2C2O4 to scavenge •OH radicals and holes (h+), respectively15. The addition of salicylic acid nearly completely inhibited CH4 conversion, showing a stronger suppression than (NH4)2C2O4 (Supplementary Fig. 47). This suggests that •OH radicals, rather than h⁺, are the main reactive species for CH4 activation.
The Au/TiO2@pSiO2 catalyst with smaller pores produced more •OH radicals, as monitored by in situ electron paramagnetic resonance (EPR), which correlates with their increased performance in CH4 conversion (Fig. 5a, b). Coumarin was also employed as a fluorescent probe to assess •OH production15, which further supports the findings from EPR (Supplementary Figs. 48, 49). Typically, there are two distinct pathways by which •OH radicals are formed: one involves H2O oxidation by holes (1e− WOR), while the other involves O2 reduction by electrons (ORR) (Supplementary Fig. 1). To identify its origin, we performed 18O isotopic labeling with benzoic acid as a probe to capture •OH (Supplementary Fig. 50). Under H216O/18O2 conditions, over 95% of products corresponded to H16OC6H4CO2H (m/z = 137), while with H218O/16O2, approximately 80% of products corresponded to H18OC6H4CO2H (m/z = 139). This revealed that •OH radicals primarily originate from 1e− WOR rather than from O2 reduction over Au/TiO2@pSiO2-1.7 (Fig. 5c, Supplementary Fig. 51). In contrast, for Au/TiO2 and TiO2, approximately half of the •OH radicals derive from H2O oxidation and the other half from O2 reduction (Supplementary Figs. 52, 53). Thus, the enhanced 1e− WOR to •OH radicals within confined SiO2 pores are responsible for the improved CH4 conversion over Au/TiO2@pSiO2.

a In situ EPR spectra of DMPO-OH for monitoring the generation of •OH radicals over Au/TiO2 and Au/TiO2@pSiO2-X. b Correlation between CH4 conversion rate with •OH radical production rate over different photocatalysts. Black and red arrows denote the trends in methane conversion and •OH generation versus pore size, respectively. c 18O isotopic studies on the origins of •OH radicals over Au/TiO2@pSiO2-1.7, with benzoic acid used as a probe to capture •OH in situ. d In situ EPR spectra of DMPO-OOH for monitoring the generation of •OOH active species over Au/TiO2 and Au/TiO2@pSiO2-X. e Correlation between H2O2 generation and •OOH radical production rate over different photocatalysts. Blue and green arrows denote the trends in •OOH and H2O2 generation versus pore size. f 18O isotopic studies on the origins of H2O2 over Au/TiO2@pSiO2-1.7.
EPR spectra of DMPO-OOH show that Au/TiO2@pSiO2 with smaller pores produced less •OOH radicals (Fig. 5d), consistent with their enhanced H2O2 production as the main product in ORR (Fig. 5e). Electron scavenger experiments using AgNO3 or NaIO3 and 18O2/H218O isotope labeling confirmed that H2O2 is generated exclusively through ORR (Fig. 5f, Supplementary Figs. 54, 55). The ORR selectivity to H2O2 is 75 % over Au/TiO2@pSiO2-1.7, compared to 11% over Au/TiO2 (Supplementary Fig. 13, Supplementary Note 4). The high H2O2 production and negligible •OOH production over Au/TiO2@pSiO2 suggest that the silica nanopores confinement alters the ORR pathway from a 1e− process to a 2e− process.
Thus, facilitated 1e− WOR producing •OH and an altered ORR pathway account for the improved catalytic performance of Au/TiO2@pSiO2. Similar enhancements in •OH production from 1e− WOR and altered ORR pathways were also observed with Pt/TiO2@pSiO2 (Supplementary Figs. 56–58, Supplementary Note 3). These results indicate that this alteration is a general phenomenon when water is spatially confined within nanopores. We hypothesize that both the accelerated 1e− WOR and the modified ORR pathway result from the distinct structure of water confined within these nanopores compared to bulk water.
KIE investigations for CH4 oxidation process
H/D kinetic isotope effect (KIE) studies were used to analyze whether PT is involved in the rate-determining step (RDS)46,47,48, which would further shed light on the mechanism of altered reactivity and selectivity. For the 1e− WOR that produces •OH radicals, Au/TiO2@pSiO2–1.7 shows a pronounced KIE value of 2.0–2.3, indicating a strong involvement of PT in the RDS. In contrast, Au/TiO2 exhibits a low KIE value of 1.1–1.2 (Fig. 6a, Supplementary Figs. 59, 60), suggesting a distinctly different 1e− WOR pathway. The absence of KIE for Au/TiO2 is consistent with the widely accepted stepwise proton-electron transfer (SPET) mechanism32,49,50, where water readily deprotonates on TiO2 before being oxidized by the photogenerated hole, making the electron transfer step (OH− + h+ → •OH) the RDS. Conversely, the large KIE for Au/TiO2@pSiO2–1.7 suggests that molecular water confined in the nanopores acts directly as the hole acceptor, generating •OH radicals through a concerted proton-electron transfer (CPET) pathway (H2O + h+ → •OH + H+). As a result, the slower dissociation and deuteron transfer in D2O compared to H2O greatly reduce the •OH radical production.

a Comparison in •OH radical production over Au/TiO2 and Au/TiO2@pSiO2-1.7 measured in H2O and D2O. b Comparison in CH4 conversion rate over Au/TiO2 and Au/TiO2@pSiO2-1.7 measured in H2O and D2O. c Comparison in H2O2 generation rate over Au/TiO2 and Au/TiO2@pSiO2-1.7 measured in H2O and D2O. d DRIFTS analysis of the OH stretching band of the water in different photocatalysts. e DRIFTS analysis of the HOH bending band of the water in different photocatalysts. In (d, e), colored curves represent the deconvoluted components of specific HB environments (blue: DA; orange: DDAA; green: DDA; purple: DAA); The black lines denote the raw data, while the hollow circles (yellow in (d), gray in (e)) represent the cumulative fitted curves. f Spectroscopically observable hydrogen-bonding configurations of H2O. g Mean HB state of water in Au/TiO2 and Au/TiO2@pSiO2-X samples derived from DRIFTS. Dashed lines represent hydrogen bonds between O (red) and H (gray). h Free energy profiles for water dissociation and •OH generation/deprotonation on the anatase TiO2(101) surface in bulk water. i Corresponding free energy profiles in confined water. Both energy profiles (h, i) were calculated assuming a hole concentration of Ch+ = 10−9 ML.
At a molecular level, this mechanistic divergence stems from the preferential reactivity of photogenerated holes with either molecular water or hydroxide anions (OH−). The SPET mechanism on Au/TiO2 suggests extensive water dissociation, populating the surface with OH− that are highly reactive toward holes. In contrast, the shift to a CPET mechanism in Au/TiO2@pSiO2–1.7 indicates that confined water resists dissociation, reacting instead in its molecular form. This change can be attributed to the weakened HB network and reduced solvation effect within the nanopores26,32. The elevated •OH production over Au/TiO2@pSiO2–1.7 suggests that the CPET mechanism provides a more effective route for generating the •OH radicals necessary for CH4 activation than the two-step SPET pathway. Thus, confinement-driven changes in water reactivity and PCET pathways are key to enhanced CH4 conversion for Au/TiO2@pSiO2–1.7.
As the WOR-producing •OH is key to CH4 activation, its KIE influences the KIE of overall CH4 oxidation. Consequently, Au/TiO2 does not show KIE effect for CH4 conversion, and Au/TiO2@pSiO2-1.7 shows KIE value of 1.3–1.6 (Fig. 6b, Supplementary Figs. 61–64). The KIE decreases over time for Au/TiO2@pSiO2–1.7, likely due to H/D exchange between CH4 and D2O during reactions (Supplementary Fig. 64, Supplementary Note 5).
On the ORR side, a large KIE of 2.97 is also observed for Au/TiO2@pSiO2–1.7 (Fig. 6c), which is expected for the 2e− process that produces H2O2 from oxygen and proton. In stark contrast, a reverse KIE for H2O2 production was observed over Au/TiO2, indicating that the confined water significantly modulates the PT and ET dynamics. Notably, the observed shift in ORR pathway from the 1e− process for Au/TiO2 to the 2e− process for Au/TiO2@pSiO2 can also be attributed to facilitated proton transfer due to water confinement. Compared to the 1e− process, the 2e− process is thermodynamically more viable but requires simultaneous transfer of two protons. Therefore, efficient proton transfer, along with increased protons generated from the enhanced WOR process, is responsible for the shift to the 2e− process and enables efficient production of H2O2.
Confined water characterization
The weakened HB network of the confined water was analyzed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurement at various relative humidity (RH) levels (Fig. 6d–g, Supplementary Figs. 65, 66). The broad OH stretching band (2800−3600 cm−1) is sensitive to the HB network configuration and can be deconvoluted into distinct Gaussian sub-bands (Fig. 6f)31,34. From Au/TiO2 to Au/TiO2@pSiO2-1.7, water in the smaller pore shows a significantly larger portion of DA to DDAA ratio along with a lower average number of HBs (<NHB>) (Fig. 6g, Supplementary Table 9), indicating a weakened HB network. A similar trend was observed in the HOH bending (1500–1800 cm−1) region (Fig. 6e), where deconvolution of the spectra can give the ratio between two peaks at 1620 cm−1 and 1680 cm−1, attributed to DA and DDAA water clusters31. These results suggest that H2O molecules within smaller pores adopt a more linear arrangement with a weaker HB network, leading to more isolated water species that are prone to oxidation and a reduced solvation that better stabilizes the semi-hydrophobic •OH radicals.
To rationalize the role of water structure, we performed MD simulations using MLPs on the anatase TiO2(101) surface44,51. Two different water environments were modeled: one containing 128 water molecules to represent the bulk system and the other with an interfacial layer of 39 molecules designed to mimic the restricted content of the confined system within the 1.7 nm pores (Supplementary Fig. 67a–c, Supplementary Data 1). Simulated water structures yielded DA/DDAA ratios in reasonable agreement with the experimental DRIFTS data (Fig. 6g, Supplementary Fig. 67d), supporting the qualitative consistency of our computational model. Using this framework, we computed the energetics of the SPET and CPET pathways, corresponding to the oxidation of surface hydroxide (*OHt−, reactions (1)–(3)) and adsorbed molecular water (*H2Oad, reaction (4)), respectively (Supplementary Fig. 68, Supplementary Data 1)49.
Although the reaction barriers for hole-driven oxidation are low under standard conditions (barrierless for *OH⁻; ~12.5 kJ/mol for *H2O), the reaction rate is governed by the extremely low concentration of surface-reaching holes (Ch+ = 10−9 monolayer (ML)) under realistic photocatalytic conditions49,50,52,53. To account for this, we converted this concentration constraint into an equivalent energetic penalty, which adds an additional barrier of ~51.3 kJ/mol to the hole-involving steps (Supplementary Note 6). In bulk water, exergonic dissociation (−5.2 kJ/mol) populates the surface with *OHt−, resulting in a lower effective barrier for SPET (46.1 kJ/mol) compared to CPET (63.8 kJ/mol) (Fig. 6h, Supplementary Figs. 69, 70). Under confinement, however, water dissociation becomes unfavorable (20.3 kJ/mol), which stabilizes molecular *H2O (Fig. 6i, Supplementary Figs. 69, 70). This raises the effective barrier of SPET to 71.6 kJ/mol, thereby shifting the reaction to the CPET pathway. This mechanistic distinction is robust across a wide range of hole concentrations, even at a concentration of 10−4 ML (Supplementary Fig. 71). These computational findings align well with our experimental KIE results.
Furthermore, the nanoconfined environment enhances the stability of the generated •OH radicals. In bulk water, •OH radical is prone to deprotonation to form surface oxygen radicals (•Ot−) through reaction (5) (Supplementary Fig. 68)53, a process with a barrier (45.2 kJ/mol) slightly lower than that of radical production, facilitating rapid consumption (Fig. 6h). However, the disrupted hydrogen-bond network in the confined system hinders this process. Quantitative analysis shows a substantial increase in both the free energy and activation barrier for this step, rising from ~45 kJ/mol in bulk to >60 kJ/mol under confinement (Fig. 6i). This suggests that the deprotonation barrier exceeds the •OH production barrier in nanopores, potentially extending the lifetime of semi-hydrophobic •OH species and facilitating their participation in CH4 activation.
Therefore, both enhanced methane conversion and H2O2 production of the Au/TiO2@pSiO2 can be explained by the weakened HB network of the confined water in porous silica (Fig. 7): (a) for h+ driven WOR, confined water with disrupted HB networks suppresses dissociation and preferentially reacts with h+ in its molecular form, causing a shift from SPET to CPET mechanism that generates more •OH radical for CH4 activation; (b) for e− driven ORR, confined water with weakened HB transfers proton more efficiently, enabling a shift from 1e− to 2e− process that directly produces H2O2.

The schematic illustrates the distinct PCET pathways over Au/TiO2 in bulk water (left) versus Au/TiO2@pSiO2-1.7 with confined water (right). Over Au/TiO2, water oxidation to •OH radical proceeds via SPET, and O2 reduction follows a 1e− pathway; In contrast, the confined water with disrupted HB networks promotes the CPET pathway for water oxidation to •OH radicals and 2e− pathway for O2 reduction. H atom: light gray; O atom: ice blue.
Discussion
In summary, we have demonstrated a core-shell photocatalyst design that leverages spatial confinement to modulate water structure, leading to a significant enhancement in both CH4 conversion and H2O2 production. The confined water with weaker HB networks significantly modulates the PT and ET dynamics in the reaction involving photogenerated electrons and holes. Specifically, the confined water suppresses dissociation and preferentially reacts with h+ in WOR, causing a shift from SPET to CPET mechanism and generating more •OH radicals to activate CH4. Concurrently, the weakened HB network facilitates more efficient proton transfer in ORR, enabling a shift from the 1e− to the 2e− process and directly producing H2O2. This study provides a fundamental understanding of how interfacial water structure can be engineered to optimize photocatalytic performance, opening new avenues for photocatalyst design.
Methods
Materials
All reagents were commercially obtained without purification. Titanium dioxide (P25, ≥99.5%), gold (III) chloride trihydrate (HAuCl3•3H2O, 99.99%), chloroplatinic acid (H2PtCl6, ≥99.5%), palladium chloride (PdCl2, 99.9%), and Sodium sulfate (Na2SO4, ≥99%) were purchased from Sigma Aldrich. Rhodium(III) chloride trihydrate (RhCl3•3H2O, 98%), methyltrimethoxysilane (98%), dodecyltrimethylammonium bromide (C12TAB, ≥99%), cetyltrimethylammonium bromide (C16TAB, ≥99%), n-hexane (95%), tetraethyl silicate (TEOS, ≥99%), ammonium acetate (≥99%), coumarin (≥98%), 7-hydroxycoumarin (≥99%), acetylacetone (≥99.5 %), benzoic acid (≥99%), deuterium water (≥99.9atom%), H218O (≥97%), hydroxybenzoic acid (≥99%), 5,5-dimethyl-1-pyrrolidinium N-oxide (DMPO, ≥97%), Tetra-methylpiperidine N-oxide (TEMPO, ≥99%) were purchased from Aladdin. Ammonium hydroxide (NH3•H2O, 25–28%), dimethyl sulfoxide (≥99.9%), ethanol (99%), sodium borohydride (NaBH4, 98%), sodium iodate (NaIO3, 99.8%), ammonium oxalate (NH4)2C2O4, 99.5%) and salicylic acid (HOC6H4COOH, 99%) were purchased from Sinopharm Chemical Reagent Co., Ltd., China. The Ag/AgCl electrode was purchased from Shanghai Chenhua.
Preparation of M/TiO2 photocatalyst
Metal cocatalyst (Au, Pt, Pd, Rh) was loaded onto TiO2 (P25) using a simple NaBH4 reduction method9,38. Typically, 50 mg of TiO2 was ultrasonically dispersed in 30 mL of deionized water, after which a specific amount of metal precursor solution (HAuCl3•3H2O, H2PtCl6, PdCl2, RhCl3•3H2O) was added. Following 10 min of stirring, 2 mL of freshly prepared 0.1 M NaBH4 was added. After an additional 30 min of stirring, the product was collected by centrifugation, washed with deionized water, and dried in an oven at 60 °C. The metal loadings for the catalyst are 0.1 wt%, unless specifically noted.
Preparation of M/TiO2@pSiO2-X
To synthesize M/TiO2@pSiO2-1.7 (M denoted to Au, Pd, Pt, Rh), 100 mg of C12TAB was used as the templating agent, which was dissolved in 10 mL of deionized water and 40 mL of anhydrous ethanol. Subsequently, 50 mg of M/TiO2 was added, sonicated for 30 mins, followed by the addition of TEOS. After 15 min of stirring, 0.5 mL ammonium hydroxide solution (25–28%) was added, and the mixture was stirred overnight. The product was collected via centrifugation, washed twice with DI H2O and ethanol, dried at 60 °C, and calcined at 550 °C for 2 h in static air to remove the C12TAB templates. TEOS volume controlled the SiO2 thickness; 300 μL produced an 8 nm SiO2 shell. For the M/TiO2@pSiO2-2.6 sample, the synthesis process was identical, except C16TAB was used as the template. The preparation of M/TiO2@pSiO2-3.1 followed the same procedure as M/TiO2@pSiO2-2.6, with the exception that 10 mL of n-hexane was added to the mixture prior to the addition of TEOS. The metal loadings for the catalysts are 0.1 wt%, unless specifically noted.
Preparation of TiO2@pSiO2 and TiO2@pSiO2-Au
The synthesis procedure for TiO2@pSiO2 was the same as the synthesis of M/TiO2@pSiO2 except using TiO2 as the starting material. The synthesis procedure for TiO2@pSiO2-Au was the same as the synthesis of Au/TiO2, except using TiO2@pSiO2 as the supporting material.
Hydrophobic modification of Au/TiO2@pSiO2-1.7
The Au/TiO2@pSiO2-1.7 catalyst (50 mg) was ultrasonically dispersed in 30 mL of n-hexane, followed by the addition of methyltrimethoxysilane (MTMS) at varying mass ratios (y wt% relative to the catalyst’s mass). The resulting suspension was ultrasonicated for 2 h and subsequently stirred at room temperature for 24 h. The solid product was then collected by centrifuge, washed sequentially with n-hexane (three times) and anhydrous ethanol (three times), and dried at 80 °C. The obtained catalyst was denoted as Au/TiO2@pSiO2-y wt% MTMS. The y wt% is the added MTMS amount; the actual MTMS loading should be lower.
Characterization
The crystal structure of the catalysts was characterized using a powder X-ray diffractometer D2 (Bruker, Switzerland) with Cu Kα radiation (λ = 1.542 Å). Morphologies of the photocatalysts were studied using TEM (JEM-1400plus) and scanning electron microscopy (SEM, JEOL JEM-7800F). High-resolution TEM (HRTEM) images, HAADF-STEM images, and corresponding EDS maps were collected using a JEOL JEM-F200 field-emission microscope operated at 200 kV. Nitrogen sorption isotherms were collected on a BELSORP-Max 2 apparatus. The metal loading in the photocatalysts was determined by an inductively coupled plasma optical emission spectrometer (ICP-OES, Icap7400, Thermo). UV-Vis DRS were recorded on a Cary 5000 spectrometer (Agilent) equipped with an integrating sphere. The absorption spectra were obtained using the Kubelka–Munk transformation. XPS were collected using a Thermo Fisher ESCALAB 250Xi spectrometer with monochromatic Al-Kα X-rays as the excitation source. All spectra were calibrated to the C 1 s peak at 284.8 eV. In-situ XPS spectra were measured at BL02B01 in the Shanghai Synchrotron Radiation Facility (SSRF), where the data were collected under dark or during irradiation. All spectra were calibrated to an Au foil. The hydrophobicity of the as-prepared catalyst was measured using a contact angle goniometer (SL200KS). Steady-state PL spectra were recorded on the Horiba Fluorolog-3 Spectrometer with an excitation wavelength of 350 nm. Time-resolved PL spectra were measured on the same instrument with an excitation wavelength of 367 nm. All electrochemical measurements (photocurrent, EIS, and Mott-Schottky tests) were performed on a Gamry Instrument Interface 1010E workstation using a three-electrode system. A 1 M Na2SO4 solution (pH = 6.8 ± 0.1) served as electrolyte, with Ag/AgCl (3 M KCl) as the reference electrode, platinum plate as the counter electrode and catalysts loaded on indium-tin oxide (ITO) as the working electrode. Automatic iR compensation was applied for Mott-Schottky measurements.
The solution resistance (Rs) was determined via EIS at open-circuit potential (100 kHz –3 Hz, 10 mV amplitude). The high-frequency intercept of the Nyquist plot yielded an average Rs of 19.2 ± 0.2 Ω. All potentials in this work are converted to the RHE scale by E (vs RHE) = E (vs Ag/AgCl) + 0.210 V + 0.0591 × pH.
Photocatalytic activity measurements
Evaluation of photocatalytic activity for CH4 oxidation was performed using a high-pressure autoclave (250 mL) with an optical sapphire window. After ultrasonically dispersing the catalyst (10 mg) in deionized water (100 mL) for 30 min, the reactor was purged with argon (99.999%). The gaseous reactants, CH4 (99.999%) and O2 (99.999%), were subsequently introduced into the vessel at partial pressures of 2.5 MPa and 1.0 MPa, respectively. The reactor was irradiated from the top using an LED lamp (365 nm, PLS-LED100C, Beijing Perfectlight Technology Co., Ltd.). The photocatalytic reaction was conducted for 1 h at 25 °C. A thermocouple was inserted into the solution to directly detect the temperature of the solution. The photoreactor was directly connected to a gas chromatograph (Agilent GC 8890) equipped with a flame ionization detector (FID) and a methanizer for the gas product analysis of CO2 and ethane. The liquid products (CH3OOH, CH3OH, and HCOOH) were analyzed by 1H-NMR (Bruker 400 MHz), with DMSO added as an internal standard.
HCHO quantification
The amount of HCHO was quantified using the acetylacetone color-development method37,54. Typically, 0.5 mL of the sample liquor, 2 mL of deionized water and 0.5 mL of reagent solution (consisting of 15 g of ammonium acetate, 0.3 mL of acetic acid, and 0.2 mL of acetylacetone dissolved in 100 mL of H2O) were mixed and placed in a water bath at 35 °C for 40 min. The absorption value of the mixture at 412 nm was then measured using UV-visible absorption spectroscopy (PerkinElmer, Lambda 265).
H2O2 quantification
The concentration of produced H2O2 was determined using the titanium oxalate spectrophotometric method55. To prepare the test solution, 0.636 g of potassium titanium oxalate and 0.02 mL of concentrated sulfuric acid were dissolved in 99.98 mL of DI H2O. Then, 1 mL of the above test solution was mixed with 1 mL of the reaction sample. The absorption of the mixture was measured at 390 nm using UV-visible absorption spectroscopy (PerkinElmer, Lambda 265).
CH4 conversion rates and oxygenates selectivity calculation
The methane conversion and oxygenates selectivity in this process are calculated according to the following equations:
The initial methane amount in the system is calculated according to the following equation (V is the volume of the headspace of the reactor):
AQY of CH4 conversion was calculated according to the following equation:
where N(electrons) and N(photons) represent the number of reacted electrons and the number of incident photons, respectively. N(photons) = IAt/E, where I, A, t and E represent incident light intensity (W/cm2), irradiation area (cm2), light incident time (s) and photo energy (J), respectively. N(electrons) was calculated based on the following reported work9,20: N(electrons) = n(CH3OOH) + 3n(CH3OH) + 5n(HCHO) + 7n(HCOOH) + 9n(CO2), where n(CH3OOH), n(CH3OH), n(HCHO), n(HCOOH) and n(CO2) represent the number of moles of produced CH3OOH, CH3OH, HCHO, HCOOH and CO2, respectively. The AQY for H2O2 production is calculated similarly, with N(electrons) = 2n(H2O2), where n(H2O2) represents the number of moles of H2O2 produced. The light intensity was calibrated using a PL-MW2000 optical power meter (Beijing Perfectlight Technology Co., Ltd.). Error bars represent the standard deviation (SD) from three independent experiments. Data points without error bars represent single measurements.
Catalyst cycling test
To study the reusability of the catalyst, the solid catalyst was separated by centrifugation after each reaction run. The catalysts were re-used in the next run after drying at 90 °C overnight under vacuum and annealing at 300 °C in air to remove any adsorbed organic species. Aq
CH4-TPD experiments
CH4-TPD were performed using a Chemisorption analyzer (ChemStar). Catalysts were pretreated at 150 °C for 30 min under a He flow of 30 sccm. After cooling to 50 °C, adsorption was conducted under a pure CH4 atmosphere for 120 min. The sample was then purged with He for 30 min, and the TPD profile was recorded at a heating rate of 5 °C/min under a He flow.
Analysis of photogenerated •OH radicals
The •OH production was measured using PL with coumarin, which reacts with •OH to form fluorescent 7-hydroxycoumarin (7-HC) (Supplementary Fig. 48). 2 mg catalyst was dispersed in 100 mL of 1.0 mM coumarin solution in the dark, irradiated for 10 min, then centrifuged and analyzed with a Horiba FluoroLog-3 spectrofluorometer at 332 nm excitation.
Isotope experiments for identifying the origin of products
For carbon source investigation with isotopically labeled 13CH4: 10 mg Au/TiO2@pSiO2 was dispersed in 5 mL H2O, and the reactor was purged with Ar to remove air. The photoreactor was then filled with 0.55 MPa 13CH4 (99%), 0.45 MPa 12CH4 (99.999%) and 0.2 MPa O2 (99.999%). After 2 h of light irradiation, gas products were analyzed by gas chromatograph-mass spectroscopy (GC-MS, Shimadzu), and liquid products by 1H-NMR.
For oxygen source investigation with 18O isotope labeling with 18O2/H216O and 16O2/H218O: 10 mg Au/TiO2@pSiO2 was dispersed in 10 mL H216O or (H218O), and the reactor was purged with Ar. The photoreactor was charged with 2.5 MPa CH4 (99.999%) and 0.5 MPa 18O2 (98%) (or 16O2). Products were analyzed by GC-MS after 2 h of light irradiation.
EPR measurements
In situ electron paramagnetic resonance (EPR) spectra were acquired using a Bruker ESR5000 spectrometer equipped with a 300 W xenon lamp (PLS-SXE300 + /UV, Perfectlight) for illumination. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) served as the radical trap. For •OH radical detection, 5 mg of catalyst was dispersed in 5 mL of distilled H2O in the dark and purged with ultrapure argon (99.999 vol.%) for 2 min. For •OOH radical detection, 5 mg of catalyst was dispersed into a 5 mL of DMSO with O2 bubbling for 2 min.
Investigation of the photochemical pathway of H2O2 production
To determine if H2O2 production originates from WOR or ORR, liquid samples from 18O2 or H218O isotope CH4 oxidation experiments were analyzed. As shown in Supplementary Fig. 55, 2 mL of the liquid samples was injected into a sealed vial (Vial A) and purged with He for 30 min. Another vial (Vial B), containing 200 mg of MnO2 and 5 mL H2O, was also purged with He for 30 min. Then, 2 mL from Vial A was injected into Vial B. After complete decomposition of H2O2 over MnO2, 1 mL of gas from the headspace of Vial B was extracted for GC-MS analysis.
Investigation of the photochemical pathway for •OH generation
To analyze photochemical pathways of •OH production,18O2 isotope experiments and H218O isotope experiments were conducted. In the 18O2 experiments, 2 mg of catalyst was dispersed in 1 mL of 1 mM benzoic acid solution (H216O as solvent). The reactor was purged with Ar to remove air and filled with 0.1 MPa 18O2 (98%). The reaction was carried out under UV irradiation for 600 s. The products were analyzed by liquid chromatography–mass spectrometry (LC–MS, Thermofisher). Similarly, for the H218O isotope experiments, the setup was identical except using benzoic acid dissolved in H218O and 0.1 MPa 16O2.
KIE measurements
For •OH radical generation, 5 mg of catalyst were dispersed in 2 mL of either D₂O or H₂O, and in situ EPR measurements were performed to track the formation of •OH radicals. For CH4 conversion, 10 mg of catalysts were dispersed in 10 mL of D2O or H2O. The reactor vessel was then pressured to 2.5 MPa CH4 and 1 MPa of O2, followed by light irradiation, and liquid products were collected at specific time intervals for analysis. For H2O2 generation, 10 mg of catalysts were dispersed in 10 mL of D2O or H2O. The reactor vessel was then pressured to 1 MPa of O2, followed by light irradiation. The resulting liquid product was collected for analysis.
Active species scavenging experiments
The experiments were conducted by adding different types of radical scavengers was used to understand the role of radical species involved in the photocatalytic CH4 oxidation. Tetra-methylpiperidine N-oxide (TEMPO) as a scavenger for all radicals, and ammonium oxalate ((NH4)2C2O4), sodium iodate (NaIO3), salicylic acid (HOC6H4COOH) as scavengers for photogenerated holes, photogenerated electrons, hydroxyl radicals, respectively15. In a typical experiment, 10 mg photocatalyst was dispersed in 100 mL distilled water containing 1 mmol radical scavengers. After being finely sealed, the suspension was purged with ultrapure Ar (99.999%) for 20 min to completely remove air. Then O2 (99.999%) and CH4 (99.999%) were injected to acquire the desired pressure. The reaction was conducted for 10 mins. Products including CH3OH, CH3OOH, HCHO, HCOOH and CO2 were measured according to the method shown above.
In situ DRIFTS measurements
In situ DRIFTS measurements were performed using a Thermo Scientific iS50 FTIR spectrometer equipped with a liquid-nitrogen-cooled MCT/A detector. An infrared cell (Harrick) with ZnSe windows was used for measurements. Each spectrum was recorded by averaging 64 scans at a resolution of 4 cm–1. Samples were first pretreated at 673 K (5 K min−1) for 30 min in flowing O2 (20 mL min−1) to remove impurities adsorbed on the surface. Background spectra were then taken at 303 K in dry Ar. Wet Ar (Ar/H2O with controlled humidity) was subsequently introduced (20 mL min−1), and spectra were acquired at the 303 K. By using the following equation proposed by previous work31,34, we determined the average number of hydrogen bonds of each water molecule:
MLP-MD simulations
All MD simulations were conducted using LAMMPS56 with an implementation of an MLP trained on the optB88-vdW density functional, which accurately captures the energetics and dynamics of water dissociation on TiO2 surfaces. This approach, developed by Zeng et al51, enables large-scale simulations of water-TiO2 interfaces with near DFT accuracy. As MLPs are trained on DFT data, they suffer from the same limitations as DFT57. A timestep of 1 fs was used for all simulations. Free-energy landscapes at 300 K were evaluated using metadynamics, as implemented in LAMMPS patched with PLUMED49. Metadynamics simulations were performed to explore the free-energy surfaces associated with •OH generation and subsequent transformations. The PLUMED input file specifying the collective variables (CVs) is provided in Supplementary Table 10. All simulations were carried out in the NVT ensemble using a Nosé-Hoover thermostat. Each independent metadynamics trajectory was run for 20 ns. Following G. Bussi et al58, we obtained the average 1D free energy profile after tfill that all the free energy minima are filled with Gaussian hills, and then computed the values of the free energy barrier and reaction free energy. According to the free energy barrier and reaction free energy curves shown in Fig. 6h, i, Supplementary Fig. 71, we determined the tfill as 10 ns for these simulations.
Structure of Water/Anatase TiO2 (101) interface in MD simulations
The anatase TiO2(101) surface exhibits a terraced morphology characterized by step-edge twofold-coordinated oxygen atoms (O2c), which is critical for proton transfer, and fivefold-coordinated titanium sites (Ti5c) for water adsorption. The slab size of p(1 × 3) TiO2(101) surface is 10.5 Å × 11.5 Å × 48.6 Å (Supplementary Fig. 67). This surface was represented using a ten-trilayer O-Ti-O slab comprising 180 atoms, a thickness validated to eliminate slab-size artifacts in surface energetics and water adsorption behavior. To mimic the bulk water environment surrounding Au/TiO2, a cell containing 128 water molecules was constructed, corresponding to a density of 1 g cm−3. To model the confined water for Au/TiO2@pSiO2-1.7, a system with 39 water molecules, corresponding to a three-layer hydration film with reduced water content, was constrained at the interface between the TiO2(101) surface and a virtual silica pore wall. The HB network of water molecules was analyzed using a workflow developed on top of the MD Analysis toolkit59. For each trajectory snapshot, the HB topology was determined by first assigning to each oxygen atom its two nearest hydrogen atoms. The remaining hydrogen atom in each water molecule was subsequently associated with the nearest oxygen atom based on the shortest O–H distance. HBs were identified using standard geometric criteria, namely an O1–O2 distance of <3.0 Å and an O1–H2 distance of <2.5 Å, where subscripts 1 and 2 refer to atoms belonging to the hydrogen-bond acceptor and donor, respectively. Interfacial hydrogen bonds were quantified using a donor-acceptor distance cutoff of 3.0 Å together with an angular cutoff of 30°, following established protocols60.
Data availability
All the data that support the findings of this study are included within the article and its Supplementary Information files. Source data are provided with this paper.
References
Maeda, K. & Domen, K. Photocatalytic water splitting: recent progress and future challenges. J. Phys. Chem. Lett. 1, 2655–2661 (2010).
Li, K., Peng, B. & Peng, T. Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 6, 7485–7527 (2016).
Chong, M. N., Jin, B., Chow, C. W. K. & Saint, C. Recent developments in photocatalytic water treatment technology: A review. Water Res. 44, 2997–3027 (2010).
Chen, C., Shi, T., Chang, W. & Zhao, J. Essential roles of proton transfer in photocatalytic redox reactions. ChemCatChem 7, 724–731 (2015).
Teng, Z. et al. Atomically dispersed low-valent Au boosts photocatalytic hydroxyl radical production. Nat. Chem. 16, 1250–1260 (2024).
Li, X., Wang, C. & Tang, J. Methane transformation by photocatalysis. Nat. Rev. Mater. 7, 617–632 (2022).
Li, Q., Ouyang, Y., Li, H., Wang, L. & Zeng, J. Photocatalytic conversion of methane: recent advancements and prospects. Angew. Chem. Int. Ed. 61, e202108069 (2022).
Wang, P., Shi, R., Zhao, J. & Zhang, T. Photodriven methane conversion on transition metal oxide catalyst: recent progress and prospects. Adv. Sci. 11, 2305471 (2024).
Song, H. et al. Direct and selective photocatalytic oxidation of CH4 to oxygenates with O2 on cocatalysts/ZnO at room temperature in water. J. Am. Chem. Soc. 141, 20507–20515 (2019).
Latimer, A. A., Kakekhani, A., Kulkarni, A. R. & Nørskov, J. K. Direct methane to methanol: the selectivity–conversion limit and design strategies. ACS Catal. 8, 6894–6907 (2018).
Jiang, Y. et al. Elevating Photooxidation Of Methane To Formaldehyde via TiO2 crystal phase engineering. J. Am. Chem. Soc. 144, 15977–15987 (2022).
Feng, N. et al. Efficient and selective photocatalytic CH4 conversion to CH3OH with O2 by controlling overoxidation on TiO2. Nat. Commun. 12, 4652 (2021).
Han, C. et al. Selective cleavage of chemical bonds in targeted intermediates for highly selective photooxidation of methane to methanol. J. Am. Chem. Soc. 145, 8609–8620 (2023).
Jiang, Y. et al. Enabling specific photocatalytic methane oxidation by controlling free radical type. J. Am. Chem. Soc. 145, 2698–2707 (2023).
Luo, L. et al. Binary Au–Cu reaction sites decorated ZnO for selective methane oxidation to C1 oxygenates with nearly 100% selectivity at room temperature. J. Am. Chem. Soc. 144, 740–750 (2022).
Zheng, K. et al. Room-temperature photooxidation of CH4 to CH3OH with nearly 100% selectivity over hetero-ZnO/Fe2O3 porous nanosheets. J. Am. Chem. Soc. 144, 12357–12366 (2022).
Zhou, Q. et al. Selective photocatalytic oxidation of methane to methanol by constructing a rapid O2 conversion pathway over Au–Pd/ZnO. ACS Catal. 14, 955–964 (2024).
Gong, H. et al. Selective photocatalytic aerobic oxidation of methane to methyl hydroperoxide by ZnO-loaded single-atomic ruthenium oxide catalyst. J. Am. Chem. Soc. 147, 9134–9146 (2025).
Zhai, G. et al. Direct photocatalytic oxidation of methane to formic acid with high selectivity via a concerted proton–electron transfer process. J. Am. Chem. Soc. 147, 2444–2454 (2025).
Xie, C. et al. Transport mediating core–shell photocatalyst architecture for selective alkane oxidation. Nano Lett. 23, 2039–2045 (2023).
Fan, Y. et al. Selective photocatalytic oxidation of methane by quantum-sized bismuth vanadate. Nat. Sustain. 4, 509–515 (2021).
Resasco, D. E., P, C. S., Bin, W. & White, J. L. Interaction of water with zeolites: a review. Catal. Rev. 63, 302–362 (2021).
Marcus, R. A. On the theory of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys. 24, 966–978 (1956).
Yao, J. et al. Interfacial hydrogen-bond network regulation tuned water dissociation enables selective chlorination of alkenes. J. Am. Chem. Soc. 147, 8024–8031 (2025).
Li, P. et al. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat. Catal. 5, 900–911 (2022).
Yang, W. et al. Effect of the hydrogen bond in photoinduced water dissociation: a double-edged sword. J. Phys. Chem. Lett. 7, 603–608 (2016).
Chen, X. et al. Revealing the role of interfacial water and key intermediates at ruthenium surfaces in the alkaline hydrogen evolution reaction. Nat. Commun. 14, 5289 (2023).
Zhang, H., Gao, J., Raciti, D. & Hall, A. S. Promoting Cu-catalysed CO2 electroreduction to multicarbon products by tuning the activity of H2O. Nat. Catal. 6, 807–817 (2023).
Wang, T. et al. Enhancing oxygen reduction electrocatalysis by tuning interfacial hydrogen bonds. Nat. Catal. 4, 753–762 (2021).
Dubouis, N. et al. Tuning water reduction through controlled nanoconfinement within an organic liquid matrix. Nat. Catal. 3, 656–663 (2020).
Verduci, R. et al. Water structure in the first layers on TiO2: a key factor for boosting solar-driven water-splitting performances. J. Am. Chem. Soc. 146, 18061–18073 (2024).
Ren, G., Zhou, M. & Wang, H. Weakened interfacial hydrogen bond connectivity drives selective photocatalytic water oxidation toward H2O2 at water/brookite-TiO2 interface. J. Am. Chem. Soc. 146, 6084–6093 (2024).
Ma, X. et al. Hydrogen-bond network promotes water splitting on the TiO2 surface. J. Am. Chem. Soc. 144, 13565–13573 (2022).
Bregante, D. T. et al. The shape of water in zeolites and its impact on epoxidation catalysis. Nat. Catal. 4, 797–808 (2021).
Wang, T. et al. Confined water for catalysis: thermodynamic properties and reaction kinetics. Chem. Rev. 125, 1420–1467 (2025).
Pascal, T. A., Goddard, W. A. & Jung, Y. Entropy and the driving force for the filling of carbon nanotubes with water. Proc. Natl. Acad. Sci. 108, 11794–11798 (2011).
Song, H. et al. Selective photo-oxidation of methane to methanol with oxygen over dual-cocatalyst-modified titanium dioxide. ACS Catal. 10, 14318–14326 (2020).
Zhang, X. et al. Selective photocatalytic oxidative ethane dehydrogenation on AuPd nanoparticle-decorated TiO2. ACS Appl. Mater. Interfaces 17, 14119–14128 (2025).
Xie, C. et al. Tandem catalysis for CO2 hydrogenation to C2–C4 hydrocarbons. Nano Lett. 17, 3798–3802 (2017).
Tennakoon, A. et al. Catalytic upcycling of high-density polyethylene via a processive mechanism. Nat. Catal. 3, 893–901 (2020).
Raman, N. K., Anderson, M. T. & Brinker, C. J. Template-based approaches to the preparation of amorphous, nanoporous silicas. Chem. Mater. 8, 1682–1701 (1996).
Tang, K. et al. Hydrophobization engineering of the Air–cathode catalyst for improved oxygen diffusion towards efficient Zinc–air batteries. Angew. Chem. Int. Ed. 61, e202202671 (2022).
Jin, Z. et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 367, 193–197 (2020).
Cao, B. et al. Zeolite encapsulation to enhance interfacial gas availability for photocatalytic hydrogen peroxide production. Angew. Chem. Int. Ed. 64, e202422495 (2025).
Cao, X. et al. A photochromic composite with enhanced carrier separation for the photocatalytic activation of benzylic C–H bonds in toluene. Nat. Catal. 1, 704–710 (2018).
Zhang, Y. et al. Pivotal role and regulation of proton transfer in water oxidation on hematite photoanodes. J. Am. Chem. Soc. 138, 2705–2711 (2016).
Liu, S. et al. Transition from sequential to concerted proton-coupled electron transfer of water oxidation on semiconductor photoanodes. J. Am. Chem. Soc. 145, 23849–23858 (2023).
Yu, S. et al. CO2-to-methanol electroconversion on a molecular cobalt catalyst facilitated by acidic cations. Nat. Catal. 7, 1000–1009 (2024).
Zhao, W.-N. & Liu, Z.-P. Mechanism and active site of photocatalytic water splitting on titania in aqueous surroundings. Chem. Sci. 5, 2256–2264 (2014).
Wang, D., Sheng, T., Chen, J., Wang, H.-F. & Hu, P. Identifying the key obstacle in photocatalytic oxygen evolution on rutile TiO2. Nat. Catal. 1, 291–299 (2018).
Zeng, Z. et al. Mechanistic insight into water dissociation on pristine low-index TiO2 surfaces from machine learning molecular dynamics simulations. Nat. Commun. 14, 6131 (2023).
Tamaki, Y. et al. Dynamics of efficient electron–hole separation in TiO2 nanoparticles revealed by femtosecond transient absorption spectroscopy under the weak-excitation condition. Phys. Chem. Chem. Phys. 9, 1453–1460 (2007).
Li, F., Chen, J.-F., Gong, X.-Q., Hu, P. & Wang, D. Subtle structure matters: the vicinity of surface Ti5c cations alters the photooxidation behaviors of anatase and rutile TiO2 under aqueous environments. ACS Catal. 12, 8242–8251 (2022).
Ding, J. et al. Asymmetrically coordinated cobalt single atom on carbon nitride for highly selective photocatalytic oxidation of CH4 to CH3OH. Chem 9, 1017–1035 (2023).
Agarwal, N. et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 358, 223–227 (2017).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995).
Cohen, A. J., Mori-Sánchez, P. & Yang, W. Insights into current limitations of density functional theory. Science 321, 792–794 (2008).
Bussi, G. & Laio, A. Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2, 200–212 (2020).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Hack, J. H. et al. Structural characterization of protonated water clusters confined in HZSM-5 zeolites. J. Am. Chem. Soc. 143, 10203–10213 (2021).
Acknowledgments
We thank Prof. P. Liu, Prof. Y. Huang, and Prof. P. Hu at ShanghaiTech University for the insightful discussion. We thank Prof. F. Yang and Prof. Y. Yang at ShanghaiTech University for help with FTIR experiments; Dr. S. Chen for help with the EPR measurements; and Prof. G. Ma, Dr. J. Zhang, and Ms. M. Liu for help with photoelectrochemical measurements. This work was financially supported by the Science and Technology Commission of Shanghai Municipality Sailing Program (23YF1426600) and start-up funding from ShanghaiTech University. We thank the Shanghai Synchrotron Radiation Facility of BL02B01 (31124.02.SSRF.BL02B01) for the assistance with in situ XPS measurements. The TEM experiments are supported by the Center for High-resolution Electron Microscopy (CℏEM) at ShanghaiTech University. We also thank the HPC Platform of ShanghaiTech University for computing time.
Author information
Authors and Affiliations
Contributions
C.X. conceived the project. F.L. performed the catalyst preparation, characterization, and catalytic tests. S.W. and B.Y. performed the MD simulations. X.Y.W. performed the HR-TEM, HAADF and EDS characterization. Y.Y. and X.Y.W. helped on TEM image analysis. C.Q. performed the contact angle measurements. X.W. performed XPS experiments. X.L. helped on the in situ XPS experiments. C.X. and F.L. wrote the manuscript. All authors contributed to the data discussion and revision of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yuchen Hao and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.”
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lv, F., Wei, S., Wu, X. et al. Simultaneous promotion of photocatalytic CH4 conversion and H2O2 production via nanopore water confinement. Nat Commun 17, 2119 (2026). https://doi.org/10.1038/s41467-026-69719-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69719-z
