
Abstract
Lithium halide solid electrolytes have garnered significant attention owing to their high ionic conductivity and positive electrode compatibility. However, achieving target ionic conductivity typically requires high lithium concentration (>4.3 wt%) within optimal structure, which raises costs and exacerbates air sensitivity. Here, we leverage anion clusters to synthesize a series of amorphous halide electrolytes xLi2SO4-ZrCl4, with optimal ionic conductivities of 1.5 mS cm-1 at 30 °C and a significantly reduced lithium content of 2.4 wt%, alongside good air stability. Through neutron/synchrotron X-ray experiments, first-principles calculations and machine learning-accelerated molecular dynamics simulations, we reveal a disordered backbone of [ZraCl4a(SO4)]2- (1 ≤ a ≤ 4) that enables fast Li-ion diffusion via under-coordinated oxygen sites. All-solid-state lithium batteries employing these electrolytes and LiNi0.8Co0.1Mn0.1O2 positive electrode exhibit 81.1% capacity retention after 1400 cycles at 1 C (60 min) and 30 °C. Our findings reveal anion-cluster chemistry as an approach that transforms solid electrolyte design for advanced batteries, bridging materials science with practical energy storage innovation.
Similar content being viewed by others

Introduction
Next-generation electronics and electric vehicles demand advanced all-solid-state lithium batteries (ASSLBs) for their safety, high energy density, and long cycle life1,2. The cornerstone of ASSLB technology is the development of Li-ion solid electrolytes (SEs)3. Oxides, sulfides and halides, have been explored over the past decades4,5. Oxides (e.g., Li7La3Zr2O12) exhibit excellent thermal and oxidative stability, but require high-temperature sintering due to their rigidity structure6,7,8. Sulfides (e.g., Li10GeP2S12, Li6-xPS5-xCl1+x), while offering high room-temperature ionic conductivity via cold pressing, have limited electrochemical stability and poor environmental resistance9,10,11. Halide SEs, commonly represented by ternary Li-M-X systems (M = Y, In, Sc, Zr, Sm, La, Al, Ta, or Nb; X = F, Cl, Br, or I), have garnered significant research interest due to their high ionic conductivity, wide electrochemical window, and reasonable deformability12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27.
Lithium concentration is a key factor influencing ionic conduction in Li-M-X SEs. In general, a high lithium content (>4.3 wt%) is required in these high-conducting halide SEs, as shown in Fig. 1c. For instance, Li2ZrCl628 is a promising low-cost SE but has a limited ionic conductivity of ~0.4 mS cm−1. Substitution of Zr4+ by trivalent cations (e.g., Fe3+, In3+, Sc3+, or Al3+)28,29,30 increases lithium content, enhancing ionic conductivity from 0.4 mS cm−1 to typically >1.0 mS cm−1. Theoretical study suggests that an optimal 40-60% Li octahedral occupancy (corresponding to a lithium weight percentage of ~4.8–6.7% in representative halides, Supplementary Table 1) is crucial for achieving high ionic conduction in close-packed halide SEs31. Very recently, this finding has been verified experimentally32. However, achieving high conductivity while reducing lithium content remains a significant challenge.

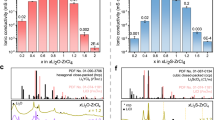
a The ionic conductivities at 30 °C and corresponding activation energies of xLi2SO4-ZrCl4 SEs. b The ionic conductivities at 30 °C and corresponding activation energies of 2xLiCl-ZrCl4 SEs. c Lithium content and raw material costs of 0.5Li2SO4-ZrCl4 compared to the state-of-the-art halide and sulfide SEs. Literature data sources are provided in Supplementary Table 6. d Ionic conductivities at 30 °C of 0.5Li2SO4-ZrCl4 and 2LiCl-ZrCl4 electrolytes measured before and after exposure in dry room or Ar atmosphere with different humidity. Comparison of the digital photos of the macroscopic morphology changes of 0.5Li2SO4-ZrCl4 and 2LiCl-ZrCl4 powders after exposure to air for different durations (e), and the corresponding mass growth ratio (f). g XRD patterns of the xLi2SO4-ZrCl4. h The TEM images and inserted SAED of 0.5Li2SO4-ZrCl4.
The high lithium content will increase the SE cost, especially when considering the limited global lithium reserves (Supplementary Fig. 1) and their upward fluctuations33. For example, the price of LiCl (anhydrous, 98%) in 2024 (Supplementary Tables 2 and 3) is about 20 times higher than that in 202134. Therefore, it is crucial to explore new strategies for decreasing a SE’s ultimate cost by decreasing lithium content, owing to the multiple variables like supply-demand fluctuations, synthesis and manufacturing complexities, etc5. Additionally, reducing the Li-ion content is crucial for improving the air stability of SE, as the high charge density of small Li+ cations leads to strong electrostatic interactions with polar H2O molecules35. As a result, spontaneous hydrolysis reactions occur when halide SEs are exposed to air, leading to a drastic decay in performance, as seen in all reported halide SEs36,37,38. This issue presents a significant challenge for large-scale production and processing.
In this work, we report a family of low-lithium-content amorphous SEs by combining clusters of [SO4]2- and [ZrCl4] using a simple ball milling method. By regulating the anion proportions, these materials achieve a significantly reduced Li content of 2.4 wt% while maintaining high Li+ conductivity of 1.5 mS cm−1 at 30 °C. To uncover the structural origin of the high conductivity in this low-lithium-content amorphous system, we performed pair distribution function (PDF) analysis based on the neutron total scattering and synchrotron X-ray data, integrated with multi-scale modeling under a self-constructed accurate and efficient workflow, including first-principles calculations and molecular dynamics simulations (MD) based on machine-learning force field (MLFF). We revealed an interesting, disordered backbone consisting of four complex anions clusters of [ZraCl4a(SO4)]2- (1 ≤ a ≤ 4) interconnected by sulfate ions, forming a percolation network for Li-ion with frustrated energy landscapes. The ionic conduction in the amorphous electrolyte is primarily attributed to Li-ion hopping between sites with low oxygen coordination. ASSLBs utilizing the SEs and LiNi0.8Co0.1Mn0.1O2 positive electrode demonstrate good cycling stability for over 1400 cycles at 1.0 C and 30 °C. In addition, the SEs deliver decent air stability under 30% relative humidity. Our work redefines the design paradigm for halide SEs, demonstrating that strategic anion-cluster manipulation in amorphous systems can circumvent traditional lithium-content limitations, thereby advancing cost-effective, high-energy-density all-solid-state batteries.
Results and discussion
Synthesis and conduction properties
The xLi2SO4-ZrCl4 SEs (0 <x ≤ 1.0) with different lithium content were mechanochemical synthesized from a stoichiometric mixture of Li2SO4 and ZrCl4. Arrhenius plots of xLi2SO4-ZrCl4 SEs, derived from the electrochemical impedance spectroscopy (EIS) at different temperatures, were compared for varying compositions (x) in Supplementary Figs. 2–3. The resultant ionic conductivities and activation energies (Ea) are calculated as shown in Fig. 1a. The ionic conductivity increased with the gradually increasing Li2SO4-to-ZrCl4 ratio, reaching to the highest value of 1.5 mS cm−1 when x = 0.5, and then decreased with further increasing Li2SO4 content. Correspondingly, 0.5Li2SO4-ZrCl4 show the lowest activation energy (0.33 eV), which is comparable with typical halide SEs like Li3ScCl6 (0.33 eV)19 and Li3InCl6 (0.347 eV)39.
A series of 2xLiCl-ZrCl4 SEs (0 <x ≤ 1.0) with the same lithium molar ratio were also prepared for comparison. As shown in Fig. 1b, the ionic conductivities of 2xLiCl-ZrCl4 SEs are generally lower than that of xLi2SO4-ZrCl4, and increased with the increase of x. The highest ionic conductivity of 0.58 mS cm−1 is achieved at x = 1.0 (2LiCl-ZrCl4), which is identical with the reported results34,40. When 2xLiCl-ZrCl4 system adopts the same low Li/Zr ratio as that in 0.5Li2SO4-ZrCl4, that is LiCl-ZrCl4 (x = 0.5), the ionic conductivity is only 0.24 mS cm-1, one order of magnitude lower than that of 0.5Li2SO4-ZrCl4. These results suggest that reduced lithium content in Li-Zr-Cl system is impractical for achieving high ionic conductivity, which however is achievable in the dual-anion clustered structure proposed in this work.
It’s worth noting that the lithium content in our 0.5Li2SO4-ZrCl4 is only 2.4 wt%, substantially lower than that in representative halide (5 ~ 6 wt%) and sulfide (typically >7 wt%) SEs with the desirable high ionic conductivity, as shown in Fig. 1c and Supplementary Tables 2–8. Notably, Zr-based halide SEs inherently possess a cost advantage over halide SEs based on other metals15, and our design further amplifies this benefit through two key features: first, the low-lithium-content configuration reduces reliance on uncertain lithium resources; Second, unlike the precursors of other selected polyanion-based SEs, the use of cost-effective Li2SO4 as a precursor enables our electrolyte to maintain stable ionic conductivity even when lower-purity raw materials are employed (Supplementary Figs. 4–5). This distinctive combination not only drives a significant reduction in material costs but also enhances the practicality of 0.5Li2SO4-ZrCl4 for future large-scale applications, strengthening its competitiveness in the SE landscape.
In addition, the electronic conductivities of xLi2SO4-ZrCl4 SEs were measured by direct current (DC) method and displayed in Supplementary Fig. 6. The electronic conductivities of xLi2SO4-ZrCl4 SEs are at the level of 10−9 S cm−1, which are significantly lower than their ionic conductivities and supports the application of these materials as a separator for ASSLBs.
Air stability
Beyond conduction property, practical application of these materials requires environmental stability. Conventional halide-based SEs often suffer from moisture sensitivity, which induces by-product formation and reduces ionic conductivity, thereby hindering scalable production and storage. Encouragingly, the 0.5Li2SO4-ZrCl4 SE demonstrates good air stability. As shown in Fig. 1d and Supplementary Figs. 7–8, the fresh 0.5Li2SO4-ZrCl4 SE didn’t display noticeable change in both ionic conductivity and phase after exposure in dry room or Ar with a relative humidity (RH) of ~1% at room temperature (dew point: −40 °C), suggesting the structural robust under dry air. Moreover, 2LiCl-ZrCl4 reference sample was also tested under the same condition for comparison. It has been recognized for the decent air stability among all the halide SEs34, and displayed similarly high air stability in dry room. However, under a harsher air condition (e.g., 30% RH), 2LiCl-ZrCl4 shows a more pronounced conductivity decrease (Fig. 1d), accompanied with a faster moisture absorption (Fig. 1e–f), reflecting the higher air stability of 0.5Li2SO4-ZrCl4.
Moreover, in-situ XRD patterns reveal striking differences in moisture stability between the two electrolytes. 0.5Li2SO4-ZrCl4 exhibits minimal changes in XRD patterns even after ~60 min of exposure, with only trace amounts of Li2SO4 and ZrOCl2·6H2O (PDF#00-018-1498) detected (Supplementary Fig. 9a–b). In contrast, 2LiCl-ZrCl4 undergoes complete phase transition within ~15 min (Supplementary Fig. 9c–d). Analysis of decomposition products after air exposure (Supplementary Fig. 9 and Supplementary Note 2) showed a significant amount of lithium salts decomposed from 2LiCl-ZrCl4, which correlates with its lower ionic conductivity. Given its better air stability with relative low lithium content (Supplementary Fig. 10-11, and Supplementary Note 2), 0.5Li2SO4-ZrCl4 alleviates the stringent requirements for low dew points typically necessary for the fabrication and storage of halide SEs. Coupled with its highly competitive cost, 0.5Li2SO4-ZrCl4 emerges as a promising SE candidate for large-scale, industrially viable production.
Amorphous structure analysis
Given that xLi2SO4-ZrCl4 possess high ionic conductivity, the relationship between this property and their structure is of interest. X-ray diffraction (XRD) patterns of the as-prepared xLi2SO4-ZrCl4 are shown in Fig. 1g. The XRD pattern of the 0.5Li2SO4-ZrCl4 sample displays no distinct Bragg diffraction peaks except for a weak Li2SO4 peak (Supplementary Fig. 12), indicating the dominating amorphous structure. An impurity phase of trigonal Li2ZrCl6 appeared with x > 0.5, while ZrCl4 reactant remained with x < 0.5. The evolution of phase in products and the corresponding ionic conductivities described above indicate that the higher ionic conductivity is most likely arise from the amorphous structure, which is further verified by the decreased ionic conductivity of calcinated xLi2SO4-ZrCl4 with higher degree of crystallinity (Supplementary Fig. 13). For comparison, the single-anion system 2xLiCl-ZrCl4 maintain crystalline phase under varying x (Supplementary Fig. 14), with significantly lower ionic conductivities compared to 0.5Li2SO4-ZrCl4.
Furthermore, the microstructure of 0.5Li2SO4-ZrCl4 was analyzed by transmission electron microscopy (TEM) measurement. No obvious lattice fringe can be found in the TEM images and the corresponding selective area electron diffraction (SAED) pattern shows merged rings with halo feature (Fig. 1h), confirming the amorphous nature of the sample. TEM energy dispersive spectra (EDS) elemental mapping for 0.5Li2SO4-ZrCl4 in Supplementary Fig. 15 demonstrates that the elements Zr, S, Cl and O are homogeneously distributed. Synchrotron-based XRD (sXRD) further confirm the amorphous nature (Supplementary Fig. 16). These results further demonstrate that the raw materials fully react with each other, and formed a highly amorphous structure of 0.5Li2SO4-ZrCl4.
The amorphous structure of 0.5Li2SO4-ZrCl4 was then investigated by using Raman spectroscopy. As shown in Fig. 2a, Raman spectra of 0.5Li2SO4-ZrCl4 represents a weak polarized band at 404 cm-1 and a depolarized band of medium intensity in the range of 50-150 cm-1, similar to the Ag modes of zirconium chloride octahedron in ZrCl441. It is reasonable to speculate that the octahedron units from the ZrCl4 precursors are likely to be presented in the amorphous 0.5Li2SO4-ZrCl4. Notably, ZrCl4 adopts monoclinic structure with a P2/c (C2h4) space group, composing of zigzag polymer chains of [(ZrCl4/2)Cl2]n with bridged octahedra. Three pairs of distinct Zr–Cl bonds (two bridging bonds and one terminal bond) form as a result of the highly distorted octahedra with edge sharing42. The bands at 281 and 217 cm−1 are attributed to the two bridging Zr-Clb bonds43, while the band at ~409 cm−1 for terminal Zr–Clt is similar to that (~412 cm−1) of the vapor complex ZrAl2Cl8 in which Zr is 6-fold coordinated44. Figure 2c exhibits the symmetric stretching vibrations (Ag), the antisymmetric stretching vibrations of Ag and Bg, and the bending motion (Ag, Bg) of six-coordinated zirconium chloride octahedron45. The vibration bands of ZrCl4 after ball-milling (BM-ZrCl4) were maintained but became broader, with a slight shift to lower energies in the 0.5Li2SO4-ZrCl4 product. These observations suggest the existence of octahedral fragments of ZrCl4 in 0.5Li2SO4-ZrCl4 that however has a more disordered structure27. Meanwhile, the bands at ~161 cm−1 and ~325 cm−1, characteristic of 2LiCl-ZrCl4 with face-sharing zirconium chloride octahedron, are likely still present. In addition, Raman vibration band in a range of 400–1250 cm−1 of 0.5Li2SO4-ZrCl4 displays weak signals compared to Li2SO4 (Fig. 2b). The vibrational modes of Li2SO4 in Fig. 2d shows a tetragonal Td symmetry for S-O tetrahedron46, including symmetric S–O stretching mode (ν1) at about 1010 cm−1, symmetric O–S–O bending mode (ν2) around 400–500 cm−1, asymmetric S–O stretching mode (ν3) at 1050 ~ 1250 cm−1, and asymmetric O–S–O bending mode (ν4) at 500 ~ 700 cm−1. Among them, the ν2 and ν4 modes represent the bending vibration modes related to the rotation of polyanion SO42-. It can be seen that these two vibration bands for BM-Li2SO4 and 0.5Li2SO4-ZrCl4 are highly broadened compared to those of Li2SO4 starting material, indicating that SO42- undergoes order-to-disorder transition during ball-milling in order to form the interconnected backbone47.

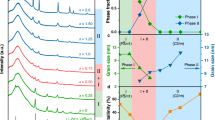
Raman spectra of the corresponding 0.5Li2SO4-ZrCl4 SE (a) and the raw materials before and after ball-milling (b). Vibrational modes of Zr–Cl octahedron (c) and S–O tetrahedron (d). e sPDF patterns for 0.5Li2SO4-ZrCl4, 2LiCl-ZrCl4, BM-ZrCl4 and ZrCl4 in a range of 20 Å, and the insert is [ZrCl4]2 dimer. Region A corresponds to the intrinsic local structure of the primary building blocks, indicative of a similar short-range order (SRO) that is consistent with those of ZrCl4 and 2LiCl-ZrCl4. Region B represents the interplay (i.e., coherence) between these aforementioned building blocks. f Experimental sPDF and nPDF patterns comparison for 0.5Li2SO4-ZrCl4 and 2LiCl-ZrCl4 in a range of 5.5 Å, respectively, and the bottom is simulated time-sampling (the blue background lines) and average (the front yellow and orange lines) sPDF and nPDF of the 0.5Li2SO4-ZrCl4 amorphous structure (detailed in Methods). g Simulation workflow illustrating the chemical transformation of Li2SO4 and ZrCl4 precursor clusters into 0.5Li2SO4-ZrCl4 monomer configurations, followed by MLFF-accelerated MD simulations to achieve the thermodynamically stable amorphous structure (Methods section for technical details).
The local structure of amorphous 0.5Li2SO4-ZrCl4 was further explored by pair distribution function (PDF) analysis based on the synchrotron X-ray and neutron total scattering data. As shown in Fig. 2e, the synchrotron PDF (sPDF) pattern of 0.5Li2SO4-ZrCl4 exhibits strong oscillation in the atomic pair distance range of r < 8.0 Å, and become significantly damped beyond 8.0 Å, indicating the completely random structure with loss of long-range order (LRO) in the amorphous product. In contrast, ZrCl4 and 2LiCl-ZrCl4 display sharp peaks in the sPDF pattern across a wide range of r (1 to 50 Å, Fig. 2e and Supplementary Fig. 17), indicating the presence of LRO. These observations align well with the previously discussed XRD and Raman results. Moreover, the PDF of the amorphous samples can be divided into two main regions of A and B. Region A (r < 5.31 Å) represents the internal local structure of the primary constituents or building blocks, suggesting a similar short-range order (SRO) structure consistent with that of ZrCl4 and 2LiCl-ZrCl4. Region B (5.31 Å <r < 8 Å) provides insight into the interplay (coherence) between these building blocks. A slight oscillation at 7.79 Å is still observable, consistent with the Cl–Cl distance (7.89 Å) in the edge-sharing [ZrCl4]2 dimer. This finding suggests that despite the less well-ordered state, the fundamental structure of amorphous 0.5Li2SO4-ZrCl4 remains primarily composed of these dimer units.
The comparison of nPDF and sPDF G(r) experimental results of 0.5Li2SO4-ZrCl4 and 2LiCl-ZrCl4, respectively, are displayed in Fig. 2f, and the simulated nPDF G(r) values of Li2ZrCl6, ZrCl4 and Li2SO4 (see Methods) are shown in Supplementary Fig. 18. Compared to the sPDF, the nPDF shows less resolved features owing to the strong incoherent scattering cross-sections of Cl (5.3 barn) and its high relative content48. Therefore, the more obvious Zr-Cl bond distance was observed in both sPDF tests, while the more obvious Cl–Cl bond distance was observed in nPDF, which indicates that the SRO structure of 0.5Li2SO4-ZrCl4 is similar to that of 2LiCl-ZrCl4 with face-sharing Zr-center octahedra. However, the Zr–Cl of 2.41 Å and Cl–Cl of 3.52 Å bond distances in 0.5Li2SO4-ZrCl4 become shorter relative to 2LiCl-ZrCl4, which implies a more contracted polyhedral structure. S–O of around 1.5 Å bond distance for 0.5Li2SO4-ZrCl4 is observed, indicating the strong bond in SO42- cannot be easily destroyed. In both nPDF and sPDF tests, the peaks corresponding to the Li–O and Li–Cl coordination cannot be observed, which is related to the contraction of the adjacent Zr–Cl bond. The contraction of Zr–Cl bond enlarges the size of the Li+ transport channel and ultimately promotes rapid ion diffusion.
To further elucidate the local coordination environment around Zr, we performed Zr K-edge X-ray absorption spectroscopy (XAS), including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements. Supplementary Fig. 19a compares the spectral features of 0.5Li2SO4-ZrCl4 along with reference spectra of ZrCl4 and ZrO2. The whiteline of 0.5Li2SO4-ZrCl4 showed three obvious splits, which were almost identical with the whiteline shape for ZrCl449. Thus, the amorphous structure in 0.5Li2SO4-ZrCl4 could be regarded as the fragmented ZrCl4 domains with three kinds of distinct bond lengths. Moreover, the more integrated whiteline shape of 0.5Li2SO4-ZrCl4 indicates that bond lengths heterogeneity within Zr–Cl/O polyhedrons becomes less pronounced after O substitution (from SO42-). To quantify the coordination environment around central Zr atoms, we employed Zr K-edge Fourier-transformed EXAFS (FT-EXAFS) spectroscopy (Supplementary Fig. 19b). Using single scattering information from ZrO2 (Zr–O bonds) and ZrCl4 (Zr–Cl bonds), we identified peaks corresponding to O and Cl scatterers around Zr. The Zr-O peak intensity increases, while the Zr–Cl peak intensity decreases, demonstrating that Cl in the immediate vicinity of Zr are replaced by O (from SO42-). In-depth analysis of phase-uncorrected wavelet transform (WT)-EXAFS (Supplementary Fig. 19c) further confirms the sequential coordination of Zr by O (~1.5 Å), Cl (~2 Å), and Zr (~3 Å). EXAFS fitting (Supplementary Fig. 19d) yields precise average coordination parameters: each central Zr atom is surrounded by 2.6 O atoms (2.23 Å) and 3.0 Cl atoms (2.45 Å) (Supplementary Table 9). This result is consistent with the contracted Zr–Cl polyhedral structure observed in PDF analysis, reinforcing the reliability of our structural interpretations.
In order to determine the atomic-level amorphous structure, we further developed a parameter-free, multi-scale modeling workflow (Supplementary Fig. 20), which is broadly applicable for constructing various amorphous materials. The workflow began by acquiring the initial chemical reaction between Li2SO4 to ZrCl4, referred to as cluster units, through ab-initio molecular dynamics (AIMD) simulations (Fig. 2g). Then, spatially arranged cluster units were relaxed using MD based on accurate machine-learning force field (MLFF, Supplementary Fig. 21), ultimately leading to the formation of final amorphous structure. The resulting amorphous structures are dense and free of voids (Fig. 2g) and the calculated density of 2.04 g/cm3 closely aligns with the experimental value of 2.05 g/cm3.
The simulated amorphous structures of 0.5Li2SO4-ZrCl4 demonstrate good agreement with the experimental PDF analysis (Fig. 2f). The theoretical sPDF and nPDF for the amorphous structure were calculated by averaging over 100 configurations equidistantly sampled from the MLFF-MD trajectory spanning 300 picoseconds (refer to Methods for details). All the peak positions are in alignment with the experimental values in Fig. 2f. As illustrated in Fig. 3a–d and Supplementary Fig. 22, the corresponding atomic pair contributions in the PDF are clearly resolved. The peak at 1.48 Å corresponds to S–O bonding in SO42-. Peaks at 2.15 Å represent Zr–O bonding between SO42- and ZrCl4. Peaks around 2.4 Å signify Zr–Cl bonding and the peak at 3.5 Å represents the Cl–Cl pair in the monomer of ZrCl4. The peak at 4 Å indicates Zr–Zr distribution in oligomers of [ZrCl4]n (n > 1), as clarified in Fig. 3c. The relatively weak peak observed between 4.5 Å and 5 Å is predominantly attributed to Zr–Cl and Cl–Cl atomic pairs. In summary, the PDF analysis indicates that SO42- are embedded within the ZrCl4 through the Zr–O bonds.

Four principal units selected from one MD snapshot labeled as the monomeric SO-Zr1 (a), the dimeric SO-Zr2 (b), the trimeric SO-Zr3 (c), and the tetrameric SO-Zr4 (d) with their relative proportions (e), respectively. The distances between labeled atomic pairs correlate with the signature peaks of the corresponding structural motifs, as evidenced by the PDF pattern in Fig. 2f. f The primary coordination environments of Li⁺ in MLFF-MD trajectory based on the number of coordinating oxygen atoms. g Arrhenius plots of Li+ conductivity derived from MLFF-MD simulations over a 300-ps trajectory. The room-temperature conductivity with a 3-ns trajectory is also contained. (See Supplementary Figs. 24-25 for MSD.) h Li⁺ probability distribution derived from MLFF-MD at 300 K over a 3-ns trajectory with highlighting partial Li+ diffusion pathways. The simulation in (h) was performed using a periodic box of ~37.6 × 26.6 × 30.1 Å3.
As depicted in Fig. 3a–e, four principal anion clusters consist of the amorphous structure frameworks: (1) Monomeric SO-Zr1: the SO42- are edge-sharing with ZrCl4 via two oxygen atoms, (2) Dimeric SO-Zr2: the SO42- bridges either an edge-sharing [ZrCl4]2 dimer or two isolated [ZrCl4] clusters through oxygen coordination; (3) Trimeric SO-Zr3: SO42- bridges an edge-sharing [ZrCl4]2 dimer while simultaneously binding to a third [ZrCl4] unit by chlorine, (4) Tetrameric SO-Zr4: SO42- bridges a corner-sharing [ZrCl4]3 trimer (pre-interconnected via chloride bridges) to a fourth [ZrCl4] cluster through oxygen coordination. Notably, the monomers SO-Zr, dimers SO-Zr2 and trimers SO-Zr3 constitute nearly 99% of this amorphous structure, whereas the tetramers SO-Zr4 are present in much lesser amounts (Fig. 3e). Apart from the involvement of SO42-, these units share a common feature: the coordination number of all zirconium atoms remains stable at 6 (Supplementary Fig. 23).
Li+ transport mechanism
The Arrhenius relation for the amorphous structure was calculated based on the MLFF-MD (see Methods). As shown in Fig. 3g, by analyzing the region where the mean squared displacement (MSD) curves exhibit a linear time dependence (Supplementary Fig. 24–25), the room-temperature ionic conductivity based on a 3-ns MLFF-MD simulation is about 1.7 mS cm−1, close to the experimental value of 1.5 mS cm−1. The activation energy derived from the simulation is 0.32 eV, which is in good agreement with experimental results (0.33 eV), further confirming the reliability of our computational results.
To elucidate the conduction mechanism of Li+ in the disordered framework, we initially examined the coordination environment of Li+. As shown in Fig. 3f, the coordination environment is complex and can be classified into four main types based on the number of coordinating oxygen atoms: (i) oxygen-free sites labeled as Li-Clx (x = 1–5), (ii) single-oxygen-coordinated Li-ClxO (x = 1–4), (iii) double-oxygen-coordinated Li-ClxO2 (x = 1–4), and (iv) triple-oxygen-coordinated Li-ClxO3 (x = 0-2). These coordination environments evolve over time due to the mobility of Li+ that is dictated by the kinetic energy of Li+ and the potential energy surface, potentially giving rise to more than a dozen distinct coordination environments, including Li-Cl5, Li-Cl4, Li-Cl3O, and Li-Cl2O, among others. All these local Li+ environments lack the periodically arranged LiCl6 sublattice observed in Li2ZrCl6, instead adopting a fully disordered configuration. Notably, the presence of diverse Li⁺ coordination environments creates a frustrated energy landscape50, which promotes fast ionic conduction by facilitating percolation networks for Li⁺ transport. The spatial distribution of Li+ probability at 300 K over the 3-ns trajectory in Fig. 3h reveals a percolating isotropic diffusion pattern, indicating efficient Li⁺ transport through interconnected pathways. Furthermore, in this amorphous framework, as shown in Supplementary Fig. 26a–b, approximately 50% of Li+ exhibit diffusion distances exceeding 2 Å, alongside 5% achieving distances greater than 8 Å in this frustrated energy landscape over the 3-ns trajectory. These diffusion processes are correlated with oxygen coordination. The shorter Li–O bond length (~2.0 Å compared to ~2.5 Å of Li–Cl from radial distribution functions in Supplementary Fig. 26e) arises from stronger Li–O interactions, which suggests that lower oxygen coordination environments favor Li+ diffusion, a trend corroborated by statistical analysis in Supplementary Fig. 26c. In summary, the amorphous framework creates various Li+ coordination environments that homogenizes the energy landscape, thereby enabling room-temperature superionic conductivity.
Cell performance
In addition to the high ionic conductivity, the 0.5Li2SO4-ZrCl4 SE also possess favorable deformability and good oxidation stability, which are two critical attributes for achieving high-energy density ASSLBs. The powder and pellet morphologies of 0.5Li2SO4-ZrCl4 were examined by scanning electron microscopy (SEM). As displayed in Supplementary Fig. 27, the powder samples, hundreds of nanometers to a few micrometers in size, can be easily cold-pressed into dense pellets, indicating good deformability. Atomic force microscopy (AFM) was further employed to assess the Young’s modulus of 0.5Li2SO4-ZrCl4, revealing a low average Young’s modulus of ~2.0 GPa (Supplementary Fig. 28), a value comparable to the representative soft electrolytes of AlOCl-LiCl51 and Li-Ta-Cl52, and lower than that of sulfide SEs (20 GPa)53. This low Young’s modulus ensures tight contact between SE particles, which is essential for minimizing interfacial resistance in ASSLBs. Moreover, the electrochemical stability of 0.5Li2SO4-ZrCl4 SE was evaluated through linear sweep voltammetry (LSV), as shown in Supplementary Fig. 29. The oxidation stability of 0.5Li2SO4-ZrCl4 is ~4.4 V vs. Li/Li+, which is identical to the calculated value (Supplementary Figs. 30–31), and higher than that of sulfide SEs (~2.4 V vs. Li/Li+) and competitive compared to the reported halides (~4.3 V vs. Li/Li+)14,54.
As a proof of concept, bulk-type ASSLBs utilizing 0.5Li2SO4-ZrCl4 SE were constructed to demonstrate the electrochemical performance of the amorphous halide-based SE. As shown in Fig. 4a, full cell with LiNi0.8Mn0.1Co0.1O2 (NCM811) positive electrode was measured between 2.8 and 4.3 V vs. Li/Li+ at 0.1 C (1 C = 190 mA g-1), delivering a high initial discharge capacity of 209.9 mAh g-1 and a high Coulombic efficiency of 96.7%, indicating a reliable stability of 0.5Li2SO4-ZrCl4 SE toward NCM811 positive electrode. As the specific current increased stepwise from 0.1 to 1.0 C, high discharge capacities can be maintained. Figure 4b, c exhibits the specific discharge capacities of 209.9, 188.2, 172.28, 157.5 and 121.9 mAh g−1 at the rates of 0.1, 0.2, 0.33, 0.5 and 1.0 C, respectively. Additionally, the specific discharge capacity of the full cell can recover to 194.7 mAh g−1 when the current density is returned to 0.1 C. Moreover, the full cell also shows good long-term cycling stability at 1.0 C, with a capacity retention of 81.1% w.r.t first cycle after 1400 cycles and maintaining a discharge capacity of 101.7 mAh g-1 after 2500 cycles with an average Coulombic efficiency over 99.99% (Fig. 4d).

a Initial charge and discharge voltage profiles at 0.1 C, with the Coulombic efficiency ηCoulomb denoted. Voltage curves (b) and Rate capability (c) at 0.1 C, 0.2 C, 0.33 C, 0.5 C and 1.0 C. d Long-term cycling performance at 1.0 C. The mass loading in (a–d) is 8.66 mg cm−2. e high-loading of 38.95 mg cm−2 loading at 0.1 C. f Cycling performance at 0.2 C within a voltage window of 2.8-4.6 V vs. Li/Li+. The specific currents of 1 C for NCM811 positive electrode materials are corresponding to 190 mA g−1. All the cycling tests were conducted at 30 °C.
To evaluate the practical relevance, we tested full cells with a high NCM811 loading of 38.95 mg cm−2. These achieved a high initial areal capacity of 6.4 mAh cm−2, and a robust capacity retention of 82.4% after 300 cycles (5.3 mAh cm−2). Moreover, full cell with the 0.5Li2SO4-ZrCl4 SE also can even endure a higher charge cut-off voltage of 4.6 V (Fig. 4f), delivering an initial capacity as high as 218.2 mAh g−1 under 0.2 C and maintaining a capacity retention of 70.0% over 100 cycles. The SE also enabled stable cycle performance with LiCoO2 positive electrode (Supplementary Figs. 32–35 and Supplementary Table 11). Overall, the performance of 0.5Li2SO4-ZrCl4-based cells is competitive with other representative halide SEs (Supplementary Table 10), underscoring its potential for high-energy-density ASSLBs.
In conclusion, we present a dual-anion clustered amorphous SE, xLi2SO4-ZrCl4 (x = 0.5) that combines a low lithium content of 2.4 wt% with good performance. The 0.5Li2SO4-ZrCl4 SE exhibits a high ionic conductivity of 1.5 mS cm−1, at 30 °C. This conductivity can be ascribed to the non-periodic structure of the anion clusters introduced via ball milling. An ab initio workflow integrating first-principles calculations and machine-learning molecular dynamics determined the amorphous atomic structure without empirical parameters. The calculated pair distribution function and temperature-dependent ionic conductivity show good agreement with experimental data. Within this structure, the migration of lithium ions is facilitated by the dual-anion clustered [ZraCl4a(SO4)]2- framework, where a reduced oxygen coordination is associated with enhanced ionic mobility. ASSLBs employing this SE with the NCM811 positive electrode demonstrate stable long-term cycling performance, with 81.1% capacity retention after 1400 cycles at 1.0 C, and achieve a high-loading performance with a stable capacity of 5.3 mAh cm−2 after 300 cycles at 0.1 C. Furthermore, 0.5Li2SO4-ZrCl4 also showcases improved air stability compared to that of 2LiCl-ZrCl4 at 30% RH. This anion-clustering strategy addresses key challenges of cost and stability for halide SEs, offering a viable design approach for practical high-energy-density batteries.
Methods
Synthesis of electrolytes
SEs with the general formula xLi2SO4-ZrCl4 (0 <x ≤ 1.0) were synthesized via a mechanochemical ball-milling. Stoichiometric mixture of Li2SO4 (Aladdin, 98.5%, titration) and ZrCl4 (Macklin, 99.5%) were used as the raw materials. For comparative studies, LiCl (Aladdin, 99.5%) replaced Li2SO4 to prepare 2xLiCl-ZrCl4. The starting materials were mixed in an argon-filled glovebox (H2O < 0.01ppm, O2 < 0.01ppm). Then, each mixture (1 g) was processed in a zirconia jar with 30 g grinding media using a FRITSCH Pulverisette 7 planetary mill. The milling protocol consisted of 500 rpm operation for 15 min, followed by 5 min pauses, with this cycle repeated for a total of 50 times. After ball-milling, the products were transferred into the glovebox for further use.
Cost analysis
The raw material costs for large-scale production (1000 kg) were estimated using the quantity-price scaling model55. This model relates the unit price (P) and purchase (Q) quantity through:
where a and b are constants for a given chemical. Laboratory-scale price data (P, Q) were obtained from Aladdin (https://www.aladdin-e.com/) and extrapolated to industrial quantities to calculated the scaled cost.
Structure characterization
Lab-based XRD was performed on a Rigaku Ultima IV diffractometer (Cu Kα1 radiation, λ = 1.5406 Å). Samples were sealed in Kapton films to prevent air/moisture exposure during measurement. The neutron total scattering data were acquired on the multi-physics instrument using a high-intensity time-of-flight diffractometer at the China Neutron Spallation Source (CSNS). The high-energy X-ray total scattering test was conducted at BL08, SPring-8 in Japan, utilizing a 16-inch two-dimensional detector (XRD1621) with a wavelength of 0.1087 Å. The simulated nPDF and sPDF G(r) values from CIF files are calculated by PDF gui software56. Scanning electron microscopy (SEM) images were acquired using a Hitachi S-4800 field emission SEM equipped with energy dispersive spectroscopy (EDS). Microstructures were further observed via transmission electron microscopy (TEM) (Talos F200X G2). X-ray photoelectron spectroscopy (XPS) data were recorded using a Thermo Scientific K-Alpha Spectrometer with a monochromatic Al Kα source. Raman spectra were obtained using a HORIBA Scientific LabRAM HR Raman spectrometer system (532.4 nm laser). X-ray absorption spectroscopy (XAS) measurements were performed at BL11B of the Shanghai Synchrotron Radiation Facility (SSRF). An atomic force microscopy (AFM) system (Bruker Dimension Icon) housed in an argon-filled glovebox was used to characterize the topography and modulus images of cold-pressed solid electrolyte (SE) pellets, which were formed under a pressure of 2 tons. For force spectroscopy measurements, an AFM tip (Bruker Corp.) with a spring constant (k) of 200 N·m−1 and a resonance frequency (f0) of 525 kHz was utilized.
Conductivity measurements
Prior to conductivity measurements, ~100 mg of the SE powders was placed in a polyetheretherketone (PEEK) mold (diameter 10 mm), and cold-pressed to form pellets under a pressure of 3 tons for 1 min using a hydraulic press (Xinnuo Instruments, SYP-12B). The thickness of the resulting pellets was controlled within the range of 0.06-0.07 cm. All pellet preparation steps were conducted in an argon-filled glovebox with water and oxygen contents <0.01 ppm. Ionic conductivity of the SE pellets was evaluated via EIS using a symmetric cell configuration with two stainless steel rods as blocking electrodes. EIS measurements were performed on a Bio-Logic MTZ-35 analyzer over a frequency range of 7 MHz to 1 Hz with alternating current (AC) amplitude of 10 mV. A total of 10 data points were collected per frequency decade to ensure sufficient data resolution. Electronic conductivity was assessed through a direct current (DC) polarization measurement. In this measurement, a voltage of 1.0 V was applied to the same symmetric cells. All these measurements temperature control was realized in an environment test chamber.
Computational details
Reactions between anion clusters
The AIMD unit reactions of ZrCl4 and Li2SO4 were calculated in ORCA57,58,59 using the r2SCAN-3c60 composite electronic-structure method (r2SCAN functional61 with triple-ζ Gaussian atomic orbital basis set plus refitted Grimme’s D4 dispersion62 and geometrical counter-poise corrections (gCP)63 for London-dispersion and basis set superposition error). Def2/J acceleration64 is enabled for the formation of the Coulomb matrix in the calculation. To investigate the dynamic behavior of these reactions, AIMD simulations were conducted in the NVT (constant particle number N, constant volume V and a temperature fluctuating around an equilibrium value T) ensemble at 350 K with a time step of 1.5 fs for 3000 steps, resulting in the generation of two structural cluster units.
Construction of training dataset
The training dataset was constructed via fourth-iterative (detailed in Supplementary Fig. 20) sampling AIMD trajectories from supercells containing 8 cluster units ([Li2Zr2Cl8SO4]8) using the CP2K software65. Simulations employed the r2SCAN functional61 with optimized norm-conserving Goedecker-Teter-Hutter pseudopotentials66 and the matched double-zeta polarized basis set67 within the framework of Gaussian Plane Waves (GPW). A plane-wave cutoff of 500 Ry was applied and each Gaussian function was mapped onto a grid with a cutoff of 60 Ry. Orbital transformation (OT) method68 was used to facilitate convergence. The self-consistent field energy was converged to a criterion of 1 × 10−5 Hartree. NVT AIMD simulations were performed from 300 K to 1200 K using the Bussi-Donadio-Parrinello (BDP) thermostat with a time step of 3 fs. All AIMD trajectory snapshots were recorded every two steps.
Machine learning force-filed finetuning
MLFF was constructed in the MACE framework69. We employed MACE-matpes-r2scan-omat-ft as base model and subsequently fine-tuned it for our specific system (detailed in Supplementary Fig. 20). The training dataset were randomly partitioned into 75% training, 5% validating and 20% testing sets. During fine-tuning, all model parameters remained frozen, with loss weights set to 1 for energy, 10 for forces, and 100 for stress. As shown in Supplementary Fig. 21, the root mean square error (RMSE) of the MLFF model for energy, forces and stresses is 0.4 meV/atom, 20.8 meV/Å and 254.5 bar, respectively, with R-squared values of 0.9838, 0.9990, and 0.9929.
Construction of amorphous structure
The multiscale modeling from the first-principles calculations to MLFF-MD were adopted for construction of amorphous structure. The workflow was shown in Supplementary Fig. 15. The initial amorphous structure (unrelaxed, [Li2Zr2Cl8SO4]64) was formed by stacking cluster units generated through AIMD. Subsequently, MLFF-MD was responsible for performing structural relaxation and amorphous reconstruction. MLFF-MD operates at the interface between the MACE and the Atomic Simulation Environment (ASE) software70.
The primary objective of the MLFF-MD simulations is to equilibrate the density of the system, a critical step in obtaining an accurate amorphous structure. This was accomplished using the NPT ensemble at room temperature, with gradual adjustments to the external pressure until the density difference between the simulation and experimental values was less than 0.1 g/cm3. A secondary goal was to equilibrate the structural configuration. To achieve this efficiently, we employed a method that involves initial heating followed by annealing, which could significantly reduce computational costs compared to traditional prolonged relaxation at room temperature. These processes resulted in a fully ab-initio-derived amorphous structure. Subsequent analyses, including structure, PDF and ionic conductivity, were conducted based on this amorphous structure. The optimized computational structures and the atomic configurations in AIMD and MLFF-MD simulations used in this study are available as Supplementary Data 1–3.
Ionic conductivity calculation
The ionic conductivity of the material was evaluated using MLFF-MD through the MACE-ASE interface, employing a time step of 3 femtoseconds for a total of 100,000 steps across a range of temperatures. Specifically, for room-temperature ionic conductivity, we performed MD simulations for a duration of 3 nanoseconds under the same timestep. Following the simulations, trajectory analysis (e.g., MSD) was conducted with the ASE, Kinisi71 and Pymatgen72,73 packages. The ionic conductivity (σ) at a specific temperature (T) is calculated from the self-diffusion coefficient (D) using the Einstein-Nernst relation σ = Dnq2/kBT, where kB is the Boltzmann constant, q is the charge of the diffusing ion, and n is the concentration of diffusing ion. The three-dimensional self-diffusion coefficient D is defined as \(D=\frac{1}{6}{{{\mathrm{lim}}}}_{{{{\rm{t}}}}\to \infty }\frac{{dMSD}(t)}{{dt}}\), here \({MSD}\left(t\right)=\frac{1}{N}\left\langle {\sum }_{i=1}^{N}\left[{r}_{i}({t}_{0}+t)\right]-{{r}_{i}({t}_{0})}^{2}\right\rangle\), N is the number of diffusing ion. \({r}_{i}({t}_{0}+t)\) and \({r}_{i}({t}_{0})\) denotes the atom position at time t + t0 and t0, respectively. 〈〉 denotes an average over time origins.
PDF calculations of amorphous structures
The neutron and X-ray PDFs were computed using the DebyeCalculator package74, with an isotropic atomic displacement factor (Debye-Waller factor) of 0.02. The final PDF was obtained by averaging the configurations from 100 structural snapshots sampled from the MLFF-MD simulations performed at a temperature of 300 K.
Electrochemical window and interfacial stability calculations
The chemical stability between the SE and electrode materials was assessed using a thermodynamic approximation method based on first-principles calculations14,75. The phase equilibria along the energy minimum path were determined by constructing and comparing the energies of all relevant phases within the corresponding compositional space. For a given phase with composition \(C\), its equilibrium phase composition under the chemical potential \({\mu }_{M}\) of element M is given by the grand potential phase diagram, denoted as \({C}_{{eq}}(C,{\mu }_{M})\). The decomposition reaction energy \(\Delta {E}_{D}\) of the SE in an open system at the lithium chemical potential \({\mu }_{{Li}}\) (referenced to Li metal) is calculated as:
where \(E\left({SE}\right)\) is the ground-state energy of the SE, and \(\Delta {n}_{{Li}}\) is the stoichiometric change in the number of Li atoms during the reaction.
The SE-electrode interface was treated as a pseudo-binary system76. The composition of the interfacial region, \({C}_{{interface}}\left({c}_{A},{c}_{B}\right)\), is expressed as:
Where \(x\) is the molar fraction of the SE, and \({c}_{A}\) and \({c}_{B}\) represent the specific compositions of the SE and the electrode, respectively. The total energy of this pseudo-binary interface system is approximated as a linear combination of the energies of its components:
Here, \(E\left({c}_{A}\right)\) and \(E\left({c}_{B}\right)\) are the ground state energy of the two components.
The mutual reaction energy \({\Delta E}_{D,{mutual}}\left({c}_{A},{c}_{B},x\right)\) between the SE and the electrode was evaluated by constructing a pseudo-binary phase diagram. This involves identifying the composition ratio (\(x\)) that yields the most negative reaction energy:
In this equation, \({E}_{{eq},{interface}}\) is the reaction energy of the equilibrium phase in the pseudo-binary system, \({E}_{D}\left({c}_{A}\right)\) and \({E}_{D}\left({c}_{B}\right)\) are the decomposition energies of the SE and the electrode, respectively, and N is the total number of atoms involved in the phase equilibrium, used for normalization.
Electrochemical measurements
Linear sweep voltammetry (LSV) test
The electrochemical stability of 0.5Li2SO4-ZrCl4 SE was assessed by the cell configuration: Li|Li6PS5Cl | SE | SE + CNT. Carbon nanotube (CNT) with a purity of 99.9% and a diameter of 20 nm was used as the conductive agent. Cell assembly was performed in an argon-filled glovebox (H2O, O2 < 0.01 ppm) as follow. Initially, 70 mg of SE powder was placed in a PEEK mold. A pressure of 2.0 tons was then applied to press the powder. Subsequently, the working electrode was prepared. For this, SE was manually mixed with CNT at a weight ratio of 7:3. A total of 10 mg of the resulting mixture was uniformly spread on one side of the SE pellet, and then pressed under a pressure of 3.0 tons. On the opposite side of the SE pellet, 30 mg of Li6PS5Cl powder was pressed at 2.0 tons. After that, a piece of Li foil (Tianjin Energy Lithium Co. LTD, 99.95%, 6 mm diameter, 20 μm thickness). was attached to serve as the counter electrode. LSV measurements were performed on a MTZ-35 analyzer (Bio-Logic) at a scan rate of 0.1 mV s−1 and 25 °C, scanning from open circuit to 6 V and 0 V, respectively. The Li6PS5Cl and CNT were provided by Heifei Kejing Star Technology Company Co., Ltd.
Assembly of ASSLBs
The composite LiCoO2 positive electrode material was prepared by hand mixing LiCoO2 with SE at a weight ratio of 70:30 for 10 min, without further additional processing. For the preparation of the composite NCM811 positive electrode material, the following steps were taken. Single-crystal NCM811, Vapor-Grown Carbon Fiber (VGCF) and 0.5Li2SO4-ZrCl4 were mixed at a weight ratio of 70:30:3. Subsequently, the composite NCM811 positive electrode material underwent ball-milling at a rotational speed of 300 rpm for 5 h. LiCoO2, NCM811 and VGCF were purchased from Guangdong Canrd New Energy Technology Co., Ltd.
The assembly process of ASSLBs initiated with the compression of 70 mg of SE powder in a PEEK mold. A pressure of 2.0 tons was applied for 1 min. After that, approximately 30 mg of Li6PS5Cl powder was uniformly dispersed on the other side of the SE layer, and then it was pressed at 2.0 tons for 1 min. Next, the composite positive electrode was evenly spread on one side of the cold-pressed SE pellet and then compressed under a pressure of 3.0 tons for 3 min. Finally, a piece of indium foil (diameter 10 mm, thickness 0.1 mm) was placed on top of the Li6PS5Cl layer. This was followed by the attachment of a piece of Li foil (diameter 6 mm, thickness 20 μm). Li-In alloy served as the negative electrode, verified to maintain a stable potential of +0.62 V versus Li/Li+18. The assembled cells were cycled using LAND CT2001A and Neware-CT-4008T battery testing systems. The LiCoO2-based cells were conducted at 25 °C and the NCM811-based cells were measured at 30 °C. The temperature control was achieved by placing the cells in an incubator (Tianjin Hongnuo Instrument, SPX-250B, with a temperature accuracy of ± 1 °C) during the cycling process. All the assembly operations were carried out in an Ar-filled glovebox (H2O, O2 < 0.01 ppm). To ensure the reproducibility of experimental results, at least 5 replicate cells were tested for each electrochemical experiment. Specific current and specific capacity values reported in this work are normalized to the mass of the active material in the positive electrode.
Air stability tests
Air stability evolution were systematically performed by exposing 500 mg powder samples of both 0.5Li2SO4-ZrCl4 and 2LiCl-ZrCl4 powder samples in open quartz crucibles to two controlled environments: (1) a dry room at −40 °C dew point (~1% RH) for 6 h; (2) Ar atmosphere of 1% RH for 24 h and humid air of 30% RH at 25°C for 6 h, respectively. EIS measurements were performed on the samples both before and after each exposure to characterize changes in ionic conductivity at 30 °C. The moisture absorption characteristics were evaluated by exposing 500 mg powder samples in open Petri dishes (100 mm diameter) under 30% RH, with precise mass measurements taken before and after exposure using an analytical balance (±0.1 mg resolution). In-situ X-ray diffraction (XRD) analysis was carried out to monitor structural evolution: 100 mg unexposed electrolyte pellets were subjected to 30% RH after ~2 min, and then XRD measurements were performed at a scan rate of 20°/min to track changes in crystal structure during exposure.
The measurement of gas evolution was conducted by Differential Electrochemical Mass Spectrometry (DEMS) using a QAS 100 mass spectrometer (Linglu Instruments, Shanghai). Three hundred milligrams of the powder samples were weighed, and compressed into round tablets with a diameter of 13 mm using a tablet press. The disc was then sealed in the battery mold. After that, the carrier gas system was connected, and a leak check was performed. The gas line was purged with a 2 mL/min Ar gas flow to expel all the air. The background argon gas flow rate was set to 2 mL/min, and the molecular masses of the gases to be measured were set as H2S, HCl, SO2, and Cl2. After the signal stabilizes, the formal collection of gas signals was initiated, with a collection time of 30 min.
Data availability
The authors declare that all the relevant data are available within the paper and its Supplementary Information file or from the corresponding author upon request. Source data are provided within this paper. The training datasets and the pretrained MACE model are available in https://doi.org/10.5281/zenodo.1834487277. Source data are provided with this paper.
References
Janek, J. & Zeier, W. G. A solid future for battery development. Nat. Energy 1, 1–4 (2016).
Wang, C. H., Liang, J. W., Kim, J. T. & Sun, X. L. Prospects of halide-based all-solid-state batteries: From material design to practical application. Sci. Adv. 8, eadc9516 (2022).
Famprikis, T., Canepa, P., Dawson, J. A., Islam, M. S. & Masquelier, C. Fundamentals of inorganic solid-state electrolytes for batteries. Nat. Mater. 18, 1278–1291 (2019).
Tuo, K., Sun, C. & Liu, S. Recent Progress in and Perspectives on Emerging Halide Superionic Conductors for All-Solid-State Batteries. Electrochem. Energy Rev. 6, 17 (2023).
Janek, J. & Zeier, W. G. Challenges in speeding up solid-state battery development. Nat. Energy 8, 230–240 (2023).
Sun, F. et al. Local Li+ framework regulation of a garnet-type solid-state electrolyte. ACS Energy Lett. 7, 2835–2844 (2022).
Jung, S.-K. et al. Unlocking the hidden chemical space in cubic-phase garnet solid electrolyte for efficient quasi-all-solid-state lithium batteries. Nat. Commun. 13, 7638 (2022).
Thangadurai, V., Narayanan, S. & Pinzaru, D. Garnet-type solid-state fast Li ion conductors for Li batteries: critical review. Chem. Soc. Rev. 43, 4714–4727 (2014).
Adeli, P. et al. Boosting solid-state diffusivity and conductivity in lithium superionic argyrodites by halide substitution. Angew. Chem. Int. Ed. 58, 8681–8686 (2019).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Li, G. et al. Sn-O Dual-Substituted Chlorine-Rich Argyrodite Electrolyte with Enhanced Moisture and Electrochemical Stability. Adv. Funct. Mater. 33, 2211805 (2022).
Nie, X., Hu, J. & Li, C. Halide-based solid electrolytes: The history, progress, and challenges. Interdiscip. Mater. 2, 365–389 (2023).
He, B. J. et al. Halogen chemistry of solid electrolytes in all-solid-state batteries. Nat. Rev. Chem. 7, 826–842 (2023).
Wang, S. et al. Lithium chlorides and bromides as promising solid-state chemistries for fast ion conductors with good electrochemical stability. Angew. Chem. Int. Ed. 58, 8039–8043 (2019).
Li, X. et al. Progress and perspectives on halide lithium conductors for all-solid-state lithium batteries. Energ. Environ. Sci. 13, 1429–1461 (2020).
Ishiguro, Y., Ueno, K., Nishimura, S., Iida, G. & Igarashib, Y. TaCl5-glassified ultrafast lithium ion-conductive halide electrolytes for high-performance all-solid-state lithium batteries. Chem. Lett. 52, 237–241 (2023).
Kim, K. et al. Material design strategy for halide solid electrolytes Li3MX6 (X = Cl, Br, and I) for all-solid-state high-voltage Li-Ion batteries. Chem. Mater. 33, 3669–3677 (2021).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Liang, J. et al. Site-occupation-tuned superionic LixScCl3+x halide solid electrolytes for all-solid-state batteries. J. Am. Chem. Soc. 142, 7012–7022 (2020).
Li, X. et al. Air-stable Li3InCl6 electrolyte with high voltage compatibility for all-solid-state batteries. Energ. Environ. Sci. 12, 2665–2671 (2019).
Zhang, S. et al. Advanced high-voltage all-solid-state Li-Ion batteries enabled by a dual-halogen solid electrolyte. Adv. Energy Mater. 11, 2100836 (2021).
Zhou, L. et al. High areal capacity, long cycle life 4 V ceramic all-solid-state Li-ion batteries enabled by chloride solid electrolytes. Nat. Energy 7, 83–93 (2022).
Zhou, L. et al. A new halospinel superionic conductor for high-voltage all solid state lithium batteries. Energ. Environ. Sci. 13, 2056–2063 (2020).
Yu, Y. et al. A high-voltage solid state electrolyte based on spinel-like chloride made of low-cost and abundant resources. Adv. Funct. Mater. 34, 2315512 (2024).
Fu, J. et al. Superionic conducting halide frameworks enabled by interface-bonded halides. J. Am. Chem. Soc. 145, 2183–2194 (2022).
Yin, Y.-C. et al. A LaCl3-based lithium superionic conductor compatible with lithium metal. Nature 616, 77–83 (2023).
Li, X. et al. The universal super cation-conductivity in multiple-cation mixed chloride solid-state electrolytes. Angewandte Chem. Int. Edn. 62, e202306433 (2023).
Kwak, H. et al. New cost-effective halide solid electrolytes for all-solid-state batteries: mechanochemically prepared Fe3+-substituted Li2ZrCl6. Adv. Energy Mater. 11, 2003190 (2021).
Kwak, H. et al. Li+ conduction in aliovalent-substituted monoclinic Li2ZrCl6 for all-solid-state batteries: Li2+xZr1-xMxCl6 (M = In, Sc). Chem. Eng. J. 437 135413 (2022).
Gao, K.-N., Bai, F., Sun, Z. & Zhang, T. Aliovalent substitution of Al3+ in Li2ZrCl6 solid electrolyte towards large-scale application. Energy Storage Mater. 70, 103444 (2024).
Liu, Y., Wang, S., Nolan, A. M., Ling, C. & Mo, Y. Tailoring the Cation Lattice for Chloride Lithium-Ion Conductors. Adv. Energy Mater. 10, 2002356 (2020).
Wang, C. et al. New insights into aliovalent substituted halide solid electrolytes for cobalt-free all-solid state batteries. Energ. Environ. Sci. 16, 5136–5143 (2023).
Choe, G. et al. Re-evaluation of battery-grade lithium purity toward sustainable batteries. Nat. Commun. 15, 1185 (2024).
Wang, K. et al. A cost-effective and humidity-tolerant chloride solid electrolyte for lithium batteries. Nat. Commun. 12, 4410 (2021).
Atkins, P., Overton, T. & Rourke, J. Shriver and Atkins’ Inorganic Chemistry 5th edn (Oxford Univ. Press, 2010).
Zhu, Y. & Mo, Y. Materials design principles for air-stable lithium/sodium solid electrolytes. Angew. Chem. Int. Ed. 59, 17472–17476 (2020).
Wang, S. et al. Air sensitivity and degradation evolution of halide solid state electrolytes upon exposure. Adv Funct Mater 32, 2108805 (2021).
Wang, K. et al. High-humidity-tolerant chloride solid-state electrolyte for all-solid-state lithium batteries. Adv. Sci. 11, 2305394 (2024).
Li, X. et al. Water-mediated synthesis of a superionic halide solid electrolyte. Angew. Chem. 131, 16579–16584 (2019).
Shi, J. et al. High-conductivity Li2ZrCl6 electrolytes via an optimized two-step ball-milling method for all-solid-state lithium-metal batteries. ACS Sustain Chem. Eng. 12, 2009–2017 (2024).
Photiadis, G. M. & Papatheodorou, G. N. Vibrational modes and structure of liquid and gaseous zirconium tetrachloride and of molten ZrCl4–CsCl mixtures ‡. J. Chem. Soc. Dalton Trans. 6, 981–990 (1998).
Photiadis, G. M. & Papatheodorou, G. N. Vibrational modes and structure of liquid and gaseous zirconium tetrachloride and of molten ZrCl-CsCl mixtures. J. Chem. Soc. Dalton 6, 981–989 (1998).
Taylor, D. R. & Larsen, E. M. Solution and vapor phase complex formation in the zirconium tetrachloride-aluminum chloride system. J. Inorg. Nucl. Chem. 41, 481–484 (1979).
Boghosian, S., Papatheodorou, G. N., Berg, R. W. & Bjerrum, N. J. Raman spectroscopic studies of vapor complexation in the MCl4-POCl3 and MCl4-AlCl3 (M = Zr or Hf) binary systems. Polyhedron 5, 1393–1403 (1986).
Rudra, M., Halder, S., Saha, S., Dutta, A. & Sinha, T. P. Temperature dependent conductivity mechanisms observed in Pr2NiTiO6. Mater. Chem. Phys. 230, 277–286 (2019).
Varghese, S. & Hariharan, K. Influence of quenching on the structural and conduction characteristics of lithium sulfate. Ionics 24, 2591–2599 (2017).
Li, Y. et al. Unraveling the dominance of structural vacancies in sodium ion conductivity in Na3SO4F. J. Phys. Chem. Lett. 14, 6832–6839 (2023).
Singh, B. et al. Critical role of framework flexibility and disorder in driving high ionic conductivity in LiNbOCl4. J. Am. Chem. Soc. 146, 17158–17169 (2024).
Zhang, S. et al. Amorphous oxyhalide matters for achieving lithium superionic conduction. J. Am. Chem. Soc. 146, 2977–2985 (2024).
Wang, S., Liu, Y. & Mo, Y. Frustration in super-ionic conductors unraveled by the density of atomistic states. Angewandte Chem. Int. Edn. 62, e202215544 (2023).
Duan, H. et al. Amorphous AlOCl compounds enabling nanocrystalline LiCl with abnormally high ionic conductivity. J. Am. Chem. Soc. 146, 29335–29343 (2024).
Li, F. et al. Amorphous chloride solid electrolytes with high Li-Ion conductivity for stable cycling of all-solid-state high-Nickel Cathodes. J. Am. Chem. Soc. 145, 27774–27787 (2023).
Sakuda, A., Hayashi, A. & Tatsumisago, M. Sulfide Solid Electrolyte with Favorable Mechanical Property for All-Solid-State Lithium Battery. Sci. Rep. 3, 2261 (2013).
Richards, W. D., Miara, L. J., Wang, Y., Kim, J. C. & Ceder, G. Interface stability in solid-state batteries. Chem. Mater. 28, 266–273 (2015).
Dr. Peter W. Hart & Dr. Jude T. Sommerfeld, P. Cost Estimation of Specialty Chemicals From Laboratory-Scale Prices. Cost Eng. 39, 31–35 (1997).
Farrow, C. L. et al. PDFfit2 and PDFgui: computer programs for studying nanostructure in crystals. J. Phys. Condensed Matter 19, 335219 (2007).
Neese, F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 8, e1327 (2018).
Neese, F. Software update: the ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Grimme, S., Hansen, A., Ehlert, S. & Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method.J. Chem. Phys. 154, 064103–06410 (2021).
Furness, J. W., Kaplan, A. D., Ning, J., Perdew, J. P. & Sun, J. Accurate and numerically efficient r2SCAN meta-generalized gradient approximation. J. Phys. Chem. Lett. 11, 8208–8215 (2020).
Caldeweyher, E., Bannwarth, C. & Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 147, 034112 (2017).
Kruse, H. & Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 136, 154101 (2012).
Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 24, 1740–1747 (2003).
Kühne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 114, 145–152 (2005).
VandeVondele, J. & Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (2007).
VandeVondele, J. & Hutter, J. An efficient orbital transformation method for electronic structure calculations. J. Chem. Phys. 118, 4365–4369 (2003).
Batatia, I. et al. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. ArXiv abs/2206.07697 (2022).
Hjorth Larsen, A. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
McCluskey, A. R., Coles, S. W. & Morgan, B. J. Accurate estimation of diffusion coefficients and their uncertainties from computer simulation. J. Chem. Theory Comput. 21, 79–87 (2023).
Ong, S. P. et al. Python materials genomics (pymatgen): a robust, open-source Python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Deng, Z., Zhu, Z., Chu, I.-H. & Ong, S. Data-driven first-principles methods for the study and design of alkali superionic conductors. Chem Mater 29, 281–288 (2016).
Johansen, F. et al. A GPU-accelerated open-source Python package for calculating powder diffraction, small-angle-, and total scattering with the Debye scattering equation. J. Open Source Softw. 9, 6024 (2024).
Zhu, Y., He, X. & Mo, Y. First principles study on electrochemical and chemical stability of solid electrolyte-electrode interfaces in all-solid-state Li-ion batteries. J. Mater. Chem. A 4, 3253–3266 (2016).
Wang, C.-W. et al. Engineering the interface between LiCoO2 and Li10GeP2S12 solid electrolytes with an ultrathin Li2CoTi3O8 interlayer to boost the performance of all-solid-state batteries. Energ. Environ. Sci. 14, 437–450 (2021).
Tang, W., Wang, F., Liang, S. et al. Polyanion-stabilized amorphous halide electrolytes with low lithium content for all-solid-state lithium batteries, flwang1998/NatComm-2026-Polyanion-stabilized-amorphous-halide-electrolytes, https://doi.org/10.5281/zenodo.18344872 (2026).
Acknowledgements
W.X. acknowledges the financial supports from the National Natural Science Foundation of China (22472079, W2441017), the Zhejiang Provincial Natural Science Foundation of China (LY23B030003), the Natural Science Foundation of Ningbo (2023J200). S.W. acknowledges the National Natural Science Foundation of China (52573249). F.W. acknowledges the China Postdoctoral Science Foundation (2025M780087). The calculations were supported by the High-performance Computing Platform of Eastern Institute of Technology, Ningbo. Synchrotron total scattering measurements were carried out on beamline BL08W at SPring-8 under proposal Nos. 2021B2006 and 2023A2341. The authors thank the staff members of the Multi-Physics Instrument (http://english.ihep.cas.cn/csns/fa/in/202109/t20210915_283259.html) at the China Spallation Neutron Source (CSNS), for providing technical support and assistance in data collection and analysis. The authors thank Wenhan Guo and Xue Tian from the Dongguan Key Laboratory of Interdisciplinary Science for Advanced Materials and Large-Scale Scientific Facilities, School of Physical Sciences, Great Bay University, for the help with the differential electrochemical mass spectrometry measurements.
Author information
Authors and Affiliations
Contributions
W.T. and W.X. conceived and designed the experiments. F.W., S.W. and W.X. developed the workflow for analyzing the amorphous structure and carried out all the calculations. W.T., S.L., and J.T. performed synchrotron data acquisition, analysis and discussion. J.L., W.Y. and W.X. conducted nPDF characterizations and analysis. Z.S. and Y.L. prepared air stability experiments. H.J., C.Z. and H.Z. provided guidance for data and image processing. F.H., P.Y. and F.R. participated in the discussions. W.T. and F.W. wrote the original manuscript. Z.M. and X.S. edited the manuscript. All authors commented the manuscript. X.S. and W.X. supervised the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dong-Hwa Seo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, W., Wang, F., Liang, S. et al. Polyanion-stabilized amorphous halide electrolytes with low lithium content for all-solid-state lithium batteries. Nat Commun 17, 3326 (2026). https://doi.org/10.1038/s41467-026-69737-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69737-x
