
Abstract
Third-generation long-read sequencing technologies, significantly improve metagenome assemblies. Highly accurate PacBio HiFi reads can yield hundreds of near-complete metagenome-assembled genomes (MAGs) from a single sample. Recently, the accuracy of the more cost-effective Oxford Nanopore Technologies (ONT) platform has increased to a per-base error rate of 1-2%. However, current metagenome assemblers are optimized for HiFi and do not scale to the large data sets that ONT enables. We present nanoMDBG, an evolution of metaMDBG, which supports the latest ONT reads through an error correction pre-processing step in minimizer-space. Across a range of ONT datasets, including a large 400 Gbp soil sample, nanoMDBG reconstructs up to twice as many high-quality MAGs as the next best ONT assembler, metaFlye, while requiring a third of the CPU time and memory. Critically, the latest ONT technology can now produce comparable MAG construction results as those obtained using PacBio HiFi at the same sequencing depth.
Similar content being viewed by others
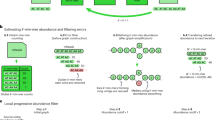

Data availability
The sequence data generated in this study have been deposited in the European Nucleotide Archive as the BioProject PRJEB88618. The individual accession numbers of all sequences used are: ERR15316007: Zymo ONT; ERR15285694: Human gut ONT; ERR15289757: Soil ONT; ERR15289675: Human gut HiFi; ERR15289804: Soil HiFi. Zymo mock reference genomes are available at https://s3.amazonaws.com/zymo-files/BioPool/D6331.refseq.zip. The ONT Zymo Fecal Reference data set is available at https://epi2me.nanoporetech.com/lc2024-datasets/. The HiFi Zymo Fecal Reference data set is available at https://www.pacb.com/connect/datasets/#metagenomics-datasets. Source data are provided with this paper.
Code availability
We implemented the nanoMDBG method in the metaMDBG software (https://github.com/GaetanBenoitDev/metaMDBG). The nanopore mode is activated using the input parameter (–in-ont), and the original PacBio HiFi mode using the parameter (–in-hifi). The analysis scripts used in this study to compare assemblers are available at https://github.com/GaetanBenoitDev/NanoMDBG_Manuscript.
References
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. & Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844 (2017).
Pinto, Y. & Bhatt, A. S. Sequencing-based analysis of microbiomes. Nat. Rev. Genet. 25, 829–845 (2024).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Pan, S., Zhao, X.-M. & Coelho, L. P. SemiBin2: self-supervised contrastive learning leads to better mags for short-and long-read sequencing. Bioinformatics 39, i21–i29 (2023).
Wang, Z. et al. Effective binning of metagenomic contigs using contrastive multi-view representation learning. Nat. Commun. 15, 585 (2024).
Chen, L.-X., Anantharaman, K., Shaiber, A., Eren, A. M. & Banfield, J. F. Accurate and complete genomes from metagenomes. Genome Res. 30, 315–333 (2020).
Bickhart, D. M. et al. Generating lineage-resolved, complete metagenome-assembled genomes from complex microbial communities. Nat. Biotechnol. 40, 711–719 (2022).
Feng, X., Cheng, H., Portik, D. & Li, H. Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nat. Methods 19, 671–674 (2022).
Benoit, G. et al. High-quality metagenome assembly from long accurate reads with metaMDBG. Nat. Biotechnol. 42, 1378–1383 (2024).
Moss, E. L., Maghini, D. G. & Bhatt, A. S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 38, 701–707 (2020).
Sereika, M. et al. Oxford Nanopore r10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 19, 823–826 (2022).
Sanderson, N. D. et al. Comparison of R9.4.1/Kit10 and R10/Kit12 Oxford Nanopore flowcells and chemistries in bacterial genome reconstruction. Microb. Genom. 9, mgen000910 (2023).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Ekim, B., Berger, B. & Chikhi, R. Minimizer-space de Bruijn graphs: whole-genome assembly of long reads in minutes on a personal computer. Cell Syst. 12, 958–968 (2021).
Quince, C. et al. STRONG: metagenomics strain resolution on assembly graphs. Genome Biol. 22, 214 (2021).
Portik, D. M. et al. Highly accurate metagenome-assembled genomes from human gut microbiota using long-read assembly, binning, and consolidation methods. Preprint at bioRxiv https://doi.org/10.1101/2024.05.10.593587 (2024).
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212 (2023).
Camargo, A. P. et al. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 42, 1303–1312 (2023).
Nayfach, S. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585 (2021).
Stanojević, D., Lin, D., Nurk, S., Florez de Sessions, P. & Šikić, M. Telomere-to-telomere phased genome assembly using HERRO-corrected simplex nanopore reads. Preprint at bioRxiv https://doi.org/10.1101/2024.05.18.594796 (2024).
Li, Y. et al. Repeat and haplotype aware error correction in nanopore sequencing reads with DeChat. Commun. Biol. 7, 1678 (2024).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Trigodet, F., Sachdeva, R., Banfield, J. F. & Eren, A. M. Troubleshooting common errors in assemblies of long-read metagenomes. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02971-8 (2026). Epub ahead of print.
Stewart, R. D. et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961 (2019).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Vicedomini, R., Quince, C., Darling, A. E. & Chikhi, R. Strainberry: automated strain separation in low-complexity metagenomes using long reads. Nat. Commun. 12, 4485 (2021).
Shaw, J., Gounot, J.-S., Chen, H., Nagarajan, N. & Yu, Y. W. Floria: fast and accurate strain haplotyping in metagenomes. Bioinformatics 40, i30–i38 (2024).
Kazantseva, E., Donmez, A., Frolova, M., Pop, M. & Kolmogorov, M. Strainy: phasing and assembly of strain haplotypes from long-read metagenome sequencing. Nat. Methods 11, 2034–2043 (2024).
Shaw, J., Marin, M. G. & Li, H. High-resolution metagenome assembly for modern long reads with myloasm. Preprint at bioRxiv https://doi.org/10.1101/2025.09.05.674543 (2025).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. methods 18, 170–175 (2021).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 41, 1474–1482 (2023).
Shaw, J. & Yu, Y. W. Fast and robust metagenomic sequence comparison through sparse chaining with skani. Nat. Methods 20, 1661–1665 (2023).
Sahlin, K., Baudeau, T., Cazaux, B. & Marchet, C. A survey of mapping algorithms in the long-reads era. Genome Biol. 24, 133 (2023).
Blanca, A., Harris, R. S., Koslicki, D. & Medvedev, P. The statistics of k-mers from a sequence undergoing a simple mutation process without spurious matches. J. Comput. Biol. 29, 155–168 (2022).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one 5, e9490 (2010).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Louca, S. & Doebeli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 34, 1053–1055 (2018).
Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinforma. 69, e96 (2020).
Wang, L.-G. et al. Treeio: an r package for phylogenetic tree input and output with richly annotated and associated data. Mol. Biol. Evol. 37, 599–603 (2020).
Xu, S. et al. ggtreeExtra: compact visualization of richly annotated phylogenetic data. Mol. Biol. Evol. 38, 4039–4042 (2021).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 1–11 (2010).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Lu, S. et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268 (2020).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis. (Springer, 2011).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11, e0163962 (2016).
Acknowledgements
C.Q. and S.R. acknowledge the support of the Biotechnology and Biological Sciences Research Council (BBSRC), part of UK Research and Innovation; Earlham Institute Strategic Program (ISP) Grant (Decoding Biodiversity) BBX011089/1 and its constituent work package BBS/E/ER/230002C; the Core Strategic Program Grant (Genomes to Food Security) BB/CSP1720/1 and its constituent work packages BBS/E/T/000PR9818 and BBS/E/T/000PR9817; and the Core Capability Grant BB/CCG2220/1. C.Q. and R.J. acknowledge the QIB Food Microbiome and Health ISP BB/X011054/1 and its constituent project BBS/E/F/000PR13631. The authors gratefully acknowledge the support of the QIB Colon Model Facility, which was funded by the BBSRC Core Capability Grant BB/CCG2260/1. R.C. was supported by ANR grants ANR-22-CE45-0007, ANR-19-CE45-0008, PIA/ANR16-CONV-0005, ANR-19-P3IA-0001, ANR-21-CE46-0012-03, and Horizon Europe grants No. 872539, 956229, 101047160 and 101088572 (ERC IndexThePlanet, also supporting G.B.). We acknowledge the assistance of Dr. Susheel Bhanu Busi (CEH, Wallingford) in organizing the soil sampling.
Author information
Authors and Affiliations
Contributions
G.B. devised and implemented the approach and performed analysis with assistance from S.R., R.J., and G.A. prepared DNA extracts for sequencing and constructed libraries. T.G. collected soil samples. G.B., R.C., and C.Q. conceived the study and supervised and coordinated the work. All authors wrote, reviewed, edited and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ben Woodcroft and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun (2026). https://doi.org/10.1038/s41467-026-69760-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-026-69760-y
