
Abstract
Genome annotation currently requires performing dozens of molecular assays in hundreds of cell and tissue samples, an expensive endeavor which is impractical to replicate across all species and conditions of interest. Here, we introduce BioSeq2Seq, a deep learning framework that infers cell-line-specific molecular assays widely used for genome annotation by leveraging a tri-modal input: evolutionarily conserved DNA sequence features, together with cell-line-specific transcriptional activity and directionality captured by a single run-on sequencing assay. BioSeq2Seq enables flexible genome annotation tasks through parameterized configurations of input features and output targets, combined with gradient-guided architectural refinement for specific biological objectives. Our model demonstrates high accuracy across four downstream tasks, showing improvements of 14.27% in histone modification prediction, 2.50% in functional element identification, and 2.90% in gene expression prediction compared to state-of-the-art methods. In transcription factor binding site (TFBS) prediction, it maintains performance comparable to that of leading existing approaches. By achieving competitive performance across tasks with single-cell-line input data, BioSeq2Seq provides an efficient and low-cost alternative for genome annotation.
Similar content being viewed by others
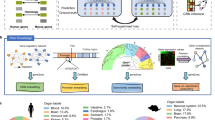


Data availability
All datasets used in this study are publicly available from the sources described in the Methods section, with detailed information on the RO-seq, histone modification ChIP-seq, RNA-seq, and TF ChIP-seq datasets used in this study provided in Supplementary Tables S2, S3, S7 and S8, respectively. Quality control reports for the data used in this study are available for download at https://zenodo.org/records/18234973/files/BioSeq2Seq_QC.zip?download=1. The Supplementary Information includes additional figures, tables, methods, and results, all of which are provided in Supplementary_Information.pdf. In addition, the model-predicted results have been deposited in Zenodo as follows: histone modification: [https://doi.org/10.5281/zenodo.18242398], functional element [https://doi.org/10.5281/zenodo.18242609], gene expression [https://doi.org/10.5281/zenodo.18243067], and TFBS [https://doi.org/10.5281/zenodo.18241447]. These data consist solely of model predictions generated from publicly available datasets and are provided to support reproducibility and reuse. All supplementary materials will be available upon publication. The source data underlying all figures in the manuscript are provided in Source_data.zip. Source data are provided with this paper.
Code availability
The code used to develop the model, perform the analyses, and generate the results in this study is publicly available and has been deposited in the GitHub repository BioSeq2Seq at https://github.com/zhichunlizzx/BioSeq2Seq81, under the Apache License 2.0, an OSI-approved open source license. The specific version of the code associated with this publication is archived on Zenodo and is accessible via [https://doi.org/10.5281/zenodo.18228811]. Pre-trained models for the four downstream tasks are available at https://zenodo.org/records/18234973/files/BioSeq2Seq_model.zip?download=1.
References
Dong, X. & Weng, Z. The correlation between histone modifications and gene expression. Epigenomics 5, 113–116 (2013).
Yin, Q., Wu, M., Liu, Q., Lv, H. & Jiang, R. Deephistone: a deep learning approach to predicting histone modifications. BMC Genom. 20, 193 (2019).
ALLFREY, V. G., FAULKNER, R. & MIRSKY, A. E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA. 51, 786–794 (1964).
Brehove, M. et al. Histone core phosphorylation regulates DNA accessibility. J. Biol. Chem. 290, 22612–22621 (2015).
Allen, D. M. Mean square error of prediction as a criterion for selecting variables. Technometrics 13, 469–475 (1971).
Binder, H. et al. Transcriptional regulation by histone modifications: towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 10, 026006 (2013).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
VerMilyea, M. D., O’Neill, L. P. & Turner, B. M. Transcription-independent heritability of induced histone modifications in the mouse preimplantation embryo. PLoS ONE 4, e6086 (2009).
Zhang, Y. & Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 (2001).
Lawrence, M., Daujat, S. & Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 32, 42–56 (2016).
Umarov, R. et al. ReFeaFi: genome-wide prediction of regulatory elements driving transcription initiation. PLOS Comput. Biol. 17, e1009376 (2021).
Nolis, I. K. et al. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl. Acad. Sci. USA. 106, 20222–20227 (2009).
West, A. G., Gaszner, M. & Felsenfeld, G. Insulators: many functions, many mechanisms. Genes Dev. 16, 271–288 (2002).
Ogbourne, S. & Antalis, T. M. Transcriptional control and the role of silencers in transcriptional regulation in eukaryotes. Biochem. J. 331, 1–14 (1998).
Cazares, T. A. et al. maxatac: Genome-scale transcription-factor binding prediction from atac-seq with deep neural networks. PLOS Comput. Biol. 19, e1010863 (2023).
Davidson, E. H. Emerging properties of animal gene regulatory networks. Nature 468, 911–920 (2010).
Nakato, R. & Sakata, T. Methods for chip-seq analysis: a practical workflow and advanced applications. Methods 187, 44–53 (2021).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
O’Connell, K. A. et al. Accelerating genomic workflows using NVIDIA Parabricks. BMC Bioinform. 24, 221 (2023).
Zhang, Z., Feng, F., Qiu, Y. & Liu, J. A generalizable framework to comprehensively predict epigenome, chromatin organization, and transcriptome. Nucleic Acids Res. 51, 5931–5947 (2023).
Benveniste, D., Sonntag, H.-J., Sanguinetti, G. & Sproul, D. Transcription factor binding predicts histone modifications in human cell lines. Proc. Natl. Acad. Sci. USA. 111, 13367–13372 (2014).
Quang, D. & Xie, X. FactorNet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods 166, 40–47 (2019).
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
Kelley, D. R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 (2018).
Quang, D. & Xie, X. Danq: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 44, e107 (2016).
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934 (2015).
Zhou, J. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 1250, 1171–1179 (2018).
Singh, R., Lanchantin, J., Sekhon, A. & Qi, Y. Attend and predict: understanding gene regulation by selective attention on chromatin. In Proc. Advances in Neural Information Processing Systems (NeurIPS) Vol. 30, 6785–6795 (Curran Associates, Inc., 2017).
Karollus, A., Mauermeier, T. & Gagneur, J. Current sequence-based models capture gene expression determinants in promoters but mostly ignore distal enhancers. Genome Biol. 24, 56 (2023).
Karbalayghareh, A., Sahin, M. & Leslie, C. S. Chromatin interaction-aware gene regulatory modeling with graph attention networks. Genome Res. 32, 930–944 (2022).
Feng, F., Yuan Yao and, X. Q., Wang, D., Zhang, X. & Liu, J. Connecting high-resolution 3d chromatin organization with epigenomics. Nat. Commun. 13, 2054 (2022).
Zhou, J. Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale. Nat. Genet. 54, 725–734 (2022).
Danko, C. G. et al. Identification of active transcriptional regulatory elements from gro-seq data. Nat. Methods 12, 433–438 (2015).
Wang, Z., Chu, T., Choate, L. A. & Danko, C. G. Identification of regulatory elements from nascent transcription using dreg. Genome Res. 29, 293–303 (2019).
Chu, T. et al. Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nat. Genet. 50, 1553–1564 (2018).
Wang, H. et al. H3k4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 615, 339–348 (2023).
Chu, T., Wang, Z., Chou, S.-P. & Danko, C. G. Discovering transcriptional regulatory elements from run-on and sequencing data using the web-based dreg gateway. Curr. Protoc. Bioinform. 66, e70 (2019).
Wang, Z. et al. Prediction of histone post-translational modification patterns based on nascent transcription data. Nat. Genet. 54, 295–305 (2022).
Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020).
Levin, M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 20, R754–763 (2010).
Long, H. K., Prescott, S. L. & Wysocka, J. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187 (2016).
Vaswani, A. et al. Attention is all you need. In Proc. Neural Information Processing Systems (NeurIPS) 5998–6008. https://arxiv.org/abs/1706.03762 (Curran Associates, Inc., 2017).
Zheng, S. et al. Rethinking semantic segmentation from a sequence-to-sequence perspective with transformers. In Proc. Computer Vision and Pattern Recognition (CVPR) 6877–6886 (IEEE, 2021).
Baisya, D. R. & Lonardi, S. Prediction of histone post-translational modifications using deep learning. Bioinformatics 26, btaa1075 (2020).
Freese, N. H., Norris, D. C. & Loraine, A. E. Integrated genome browser: visual analytics platform for genomics. Bioinformatics 32, 2089–2095 (2016).
Ozsolak, F. & Milos, P. M. Rna sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98 (2011).
Bergman, D. T. et al. Compatibility rules of human enhancer and promoter sequences. Nature 607, 176–184 (2022).
Consortium, T. E. P. The Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Maston, G. A., Evans, S. K. & Green, M. R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet. 7, 29–59 (2006).
Sandelin, A., Sandelin, A. & Danko, C. G. A unified architecture of transcriptional regulatory elements. Trends Genet. 31, 426–433 (2015).
Tome, J. M., Tippens, N. D. & Lis, J. T. Single-molecule nascent RNA sequencing identifies regulatory domain architecture at promoters and enhancers. Nat. Genet. 50, 1533–1541 (2018).
Scruggs, B. S. et al. Bidirectional transcription arises from two distinct hubs of transcription factor binding and active chromatin. Cell 58, 1101–1112 (2015).
Pundhir, S., Bagger, F. O., Lauridsen, F. B., Rapin, N. & Porse, B. T. Peak-valley-peak pattern of histone modifications delineates active regulatory elements and their directionality. Nucleic Acids Res. 44, 4037–4051 (2016).
Yashar, W. M. et al. Gopeaks: histone modification peak calling for cut & tag. Genome Biol. 23, 144 (2022).
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Core, L. J. et al. Analysis of nascent rna identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320 (2014).
Deng, X. et al. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat. Genet. 43, 1179–1185 (2011).
Tilgner, H. et al. Deep sequencing of subcellular rna fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 22, 1616–1625 (2012).
Singh, R., Lanchantin, J., Robins, G. & Qi, Y. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics 32, i639–i648 (2016).
Muhammad, T., Shehroz, K., James, D., Soichiro, Y. & Ahmed, A. TransformerChrome: transformer-based model for prediction ofgene expression from histone modifications. In Proc. Canadian Conference on Artificial Intelligence https://caiac.pubpub.org/pub/x9w6ew2i (2024).
Li, H. & Guan, Y. Fast decoding cell type-specific transcription factor binding landscape at single-nucleotide resolution. Genome Res. 31, 721–731 (2021).
Chen, C. et al. DeepGRN: prediction of transcription factor binding site across cell-types using attention-based deep neural networks. BMC Bioinform. 22, 38 (2021).
Korhonen, J., Martinmäki, P., Pizzi, C., Rastas, P. & Ukkonen, E. Moods: fast search for position weight matrix matches in DNA sequences. Bioinformatics 25, 3181–3182 (2009).
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. of the Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (NAACL-HLT) Vol. 1, 4171–4186 (Association for Computational Linguistics, 2019).
Beltagy, I., Peters, M. E. & Cohan, A. Longformer: the long-document transformer. Preprint at https://doi.org/10.48550/arXiv.2004.05150 (2020).
Manzil, Z. et al. Big Bird: transformers for longer sequences. In Proc. Advances in Neural Information Processing Systems (NeurIPS) Vol. 33, 17283-17297 (Curran Associates, Inc., 2020).
Yuan, J. et al. Native sparse attention: hardware-aligned and natively trainable sparse attention. In Proc. of the 63rd Annual Meeting of the Association for Computational Linguistics (ACL) Vol. 1, 23078–23097 (Association for Computational Linguistics, 2025).
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. Dnabert: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Gu, A. & Dao, T. Mamba: linear-time sequence modeling with selective state spaces. Preprint at https://arxiv.org/abs/2312.00752 (2023).
Schiff, Y. et al. Caduceus: bi-directional equivariant long-range DNA sequence modeling. In Proc. 41st International Conference on Machine Learning (ICML) 43632–43648 (Proceedings of Machine Learning Research, 2024).
Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 13, 705–719 (2012).
Lee, H.-G., Kahn, T. G., Simcox, A., Schwartz, Y. B. & Pirrotta, V. Genome-wide activities of polycomb complexes control pervasive transcription. Nat. Rev. Genet. 25, 1170–1181 (2015).
Pan, B. et al. Similarities and differences between variants called with human reference genome HG19 or HG38. BMC Bioinform. 20, 17–29 (2019).
Ernst, J. & Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 (2010).
Ernst, J. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011).
Federhen, S. The ncbi taxonomy database. Nucleic Acids Res. 40, 136–143 (2012).
Zhang, Y. et al. Model-based analysis for chip-seq (macs). Genome Biol. 9, R137 (2008).
Hu, W., Laber, E. B., Barker, C. & Stefanski, L. A. Assessing tuning parameter selection variability in penalized regression. Technometrics 61, 136–143 (2019).
Zhang, Z. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Zenodo, https://doi.org/10.5281/zenodo.18228811 (2026).
Acknowledgements
This work was supported by the LiaoNing Revitalization Talents Program (No. XLYC2002010) (Z.W.) and the AIS Project of the School of Future Technology, Dalian University of Technology (Z.W.). The authors would also like to express their sincere gratitude to the anonymous reviewers for their constructive comments and valuable suggestions, which have greatly improved the quality of this manuscript.
Author information
Authors and Affiliations
Contributions
Methodology: Z.Z., X.F., Z.W.; data curation and analysis: Z.Z., J.Z., L.J., C.Y.; results interpretation and visualization: Z.Z., J.Z., L.J., X.L.; software design and implementation: Z.Z., Y.H., Z.H., Z.W.; manuscript writing: Z.Z., X.F., Z.H., Z.W.; manuscript review and editing: S.-T.Y., R.W., C.G.D., Z.W.; supervision and conceptualization: R.W., C.G.D., Z.W.; all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dong Fang, who co-reviewed with Yu Liu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun (2026). https://doi.org/10.1038/s41467-026-70070-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-026-70070-6
