
Abstract
The compositional heterogeneity of post-consumer plastic waste, exemplified by prevalent polyethylene (PE)/polypropylene (PP) mixtures (>50% of the plastic market), severely complicates recycling. Kinetic disparities between PE and PP during chemical recycling create significant conversion gradients, limiting valued product yield and process viability. Here, leveraging strong interfacial coupling between ruthenium oxides and rutile TiO2, we construct highly active, epitaxial RuOx sites enabling efficient one-pot co-conversion of PE/PP mixtures with a high liquid yield of 95.02%, while maintaining a low 0.62% gas yield. Compared to conventional Ru nanoparticles, the epitaxial RuOx structure provides additional dehydrogenation sites for PP activation, which promotes carbon–metal back-donation to weaken C–C bonds, thus exhibiting comparable activation capabilities toward both 3C–2C bond in PP and 2C–2C bond in PE. This unique epitaxial catalyst enables highly efficient co-hydrogenolysis of mixed polyolefins, establishing a practical approach for their upcycling.
Similar content being viewed by others

Introduction
The improper disposal and large-scale emission of plastic waste pose pressing global challenges of resource depletion and environmental pollution1,2,3,4. Polyolefins, particularly polyethylene (PE, ~36% global production) and polypropylene (PP, ~21%), dominate plastic streams5,6,7. However, their inert C–H and C–C bonds limit traditional recycling efficiency (<20% recovery)8,9,10,11. Over the past decade, chemical upcycling has emerged as a promising strategy for polyolefin valorization12,13,14,15,16. While current catalytic systems demonstrate broad applicability to various types of polyolefin wastes, the substantial differences in intrinsic reactivity among substrates with diverse molecular structures in real waste streams often lead to reduced product yields and value17,18,19,20,21. Such variability remains a major obstacle to the development of efficient and robust industrial-scale upcycling processes22,23,24,25.
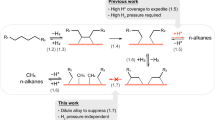
In polyolefin hydrogenolysis, PE and PP convert to liquid alkanes via sequential C–H activation, C–C cleavage, and hydrogenation on metal sites26,27,28,29,30. However, methyl substituents in PP create steric constraints (Fig. 1), requiring higher temperatures (ΔT ≈ 20 K) and longer reaction times than PE31,32,33,34,35,36. Consequently, during mixed polyolefin depolymerization, PE preferentially converts, generating excessive low-value gases18,37,38,39,40. Therefore, in co-hydrogenolysis systems involving PE and PP, the yield of liquid fuels is typically below 50%38. Mechanistically, cleaving 3C–xC bonds in PP—unlike 2C–2C bonds in PE—demands cooperative carbon adsorption and involves more thermodynamically challenging C–H activation steps, necessitating adjacent dehydrogenation sites41,42,43. Such contiguous sites, however, risk excessive C–C cleavage in PE, shifting products toward methane44,45. Thus, designing catalysts that enable integrated PE/PP co-hydrogenolysis while suppressing over-cleavage remains critical for efficient one-pot conversion of mixed wastes to liquid fuels.

Since PE and PP exhibit divergent hydrogenolysis kinetics on metal nanoparticle catalysts, the conventional co-hydrogenolysis leads to preferential over-cracking of PE into low-value gases before PP achieves full conversion. By engineering a catalyst structure with tailored active sites, our system enables simultaneous hydrogenolysis of both polymers into high-value liquid alkanes. The blue spheres represent carbon in the polyolefin backbone, and the red spheres represent the side methyl in PP.
Herein, we develop a selective catalyst through tailored interfacial coupling of ruthenium with rutile-phase TiO2, forming epitaxial RuOx nanoclusters. This design enables one-pot conversion of mixed PE/PP wastes to liquid fuels with high selectivity. Compared to conventional Ru nanoparticles, the extended RuOx structure provides additional dehydrogenation sites for PP activation, promoting carbon–metal back-donation to weaken C–C bonds. Concurrently, the nanoclusters’ confined size suppresses methane formation from PE. In co-hydrogenolysis of HDPE/PP mixtures, the catalyst achieves 84.6% liquid alkane yield with ~10.5% CH4 selectivity, delivering gasoline (C5–C12, 40.7%) and diesel-range (C13–C22, 43.8%) fractions. This work provides both fundamental insights into selective bond cleavage and a practical material design strategy for mixed polyolefin upcycling.
Results
Synthesis and geometric structural of Ru/TiO2-R(A) catalysts
We first calculated the lattice mismatch between RuO2 and the rutile TiO2 (TiO2-R) and anatase TiO2 (TiO2-A) to evaluate the potential for epitaxial overlayer formation. As illustrated by the pair of isostructural oxides RuO2 and TiO2-R (Supplementary Fig. 1), the lattice mismatch (δ) between them is only 2.65%, indicating superior interfacial compatibility (Supplementary Fig. 2a). In contrast, the significantly larger mismatch between RuOx and TiO2-A (δ = 17.11%) will lead to the reduction of RuO2 and encapsulation of Ru nanoparticles by the support during high-temperature reduction, which is characteristic of a strong metal–support interaction (SMSI)46. Furthermore, we calculated the formation energy of RuO2 on the TiO2-R surface. As shown in Supplementary Fig. 3, the formation energy of RuO2 decreases progressively with growth and eventually drops below 0 eV when a complete RuO2 layer is formed, indicating that the growth of RuO2 on TiO2-R is thermodynamically favorable. Therefore, high interfacial compatibility with a lattice deviation below 5% facilitates the formation of a RuOx interfacial layer46,47,48.
Based on the above theoretical simulations, we engineered Ru active sites with distinct structural characteristics by exploiting the metal–support interaction between Ru nanoparticles and different TiO2 crystal phases. Specifically, RuCl3 precursors were deposited onto rutile and anatase TiO2 supports via a freeze-drying method, achieving a Ru loading of ~2 wt% (Fig. 2a). The resulting catalysts underwent identical reduction pretreatments and exhibited comparable metal loadings (Supplementary Table 1), which are denoted as Ru/TiO2-R and Ru/TiO2-A, respectively.

a Schematic diagram of catalyst synthesis. b, c AC-HAADF-STEM images of b Ru/TiO2-R and c Ru/TiO2-A catalysts. d Elemental distributions of Ru/TiO2-R catalyst. e H2-TPR curves of Ru/TiO2-R(A) catalysts. f, g CO-DRIFTS spectra of f Ru/TiO2-R and g Ru/TiO2-A catalysts.
X-ray diffraction (XRD, Supplementary Fig. 4) analysis revealed no distinct diffraction peaks corresponding to Ru species in the reduced Ru/TiO2 catalysts; the diffraction patterns remained nearly identical to those of the pristine TiO2 supports (rutile and anatase phases), indicating that the Ru species are highly dispersed in the form of sub-nanometer particles. Transmission electron microscopy (TEM, Supplementary Fig. 5) further confirmed their high dispersions, showing that the reduced Ru species are uniformly anchored on the TiO2 surfaces as sub-nanometer nanoparticles. The average particle sizes on TiO2-R and TiO2-A were approximately 1.4 nm and 1.1 nm (Supplementary Fig. 6), respectively, consistent with the absence of Ru signals in the XRD patterns.
To directly visualize the geometric structure of metal particles on different supports, atomic-resolution characterization was conducted using aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM). As shown in Fig. 2b, high-contrast sub-nanometer particles are clearly observed on the surface of TiO2-R, exhibiting a layered structure integrated with the support. In contrast, the Ru species on TiO2-A appear as higher-contrast spherical structures (Fig. 2c), indicative of the formation of metal nanoparticles. Additionally, for the Ru/TiO2-R catalyst, elemental mapping (Fig. 2d) of the highlighted region (white dashed box) revealed the exclusive presence of Ru without detectable Ti. Furthermore, only weak Ru signal was detected on the surface RuO2 with no Ti signal on EELS spectra, confirming that the surface layer consists of Ru-based nanoparticles (Supplementary Fig. 7). Notably, these particles exhibit an ellipsoidal geometry with an average length/diameter ratio of ~1.47 (Supplementary Fig. 8), suggesting strong interfacial coupling between the metal particles and the TiO2-R surface, which promotes oriented growth along specific crystallographic planes and leads to the formation of quasi-two-dimensional layered structures49. In contrast, the Ru nanoparticles supported on TiO2-A display a typical spherical morphology (length/diameter ≈ 1.07) with uniform dispersion, indicating a pronounced influence of the TiO2 crystal phase on the geometric structure of the metal particles.
H2 temperature-programmed reduction (H2-TPR) analysis was conducted to probe the differences in metal–support interactions within the Ru/TiO2 catalysts (Fig. 2e). For Ru/TiO2-A, two distinct reduction peaks were observed at approximately 110 °C and 267 °C, corresponding to the reduction of RuO2 and TiO2-A, respectively28,50,51. In contrast, Ru/TiO2-R exhibited markedly different reduction behavior: the RuO2 reduction peak shifted to a higher temperature (~150 °C), indicating the stronger interfacial coupling between RuOx and TiO2-R. The second reduction peak at ~278 °C was attributed to the reduction of the TiO2 surface due to the hydrogen spillover52, and the reduction of the bulk TiO2-R support occurred at an elevated temperature of ~437 °C53. Furthermore, the H2 consumption was quantified by integrating the peak area using CuO as a reference. The H2/Ru molar ratio of Ru/TiO2-R was 0.88, which is lower than the theoretical value, indicating the presence of a considerable amount of oxidized Ru species (Supplementary Table 2). In contrast, the higher H2/Ru molar ratio of Ru/TiO2-A (1.73) suggests that Ru species were almost reduced to the metallic state. These results indicate that the strong interfacial coupling between RuOx and TiO2-R increases the reduction resistance of Ru and confirming the significant differences in metal-support interactions arising from the distinct crystallographic phases of TiO246.
After determining the geometric configurations of Ru species in the Ru/TiO2-R(A) catalysts, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) combined with CO probing was used to explore the chemical states of Ru species on different TiO2 supports. The samples were pretreated at 250 °C for 30 min in 5% H2/N2, and background spectra were collected at room temperature under N2 flow. CO was then introduced for adsorption, followed by about 40 min to ensure saturation, and N2 purging to remove physically adsorbed CO (Supplementary Fig. 9). As shown in Fig. 2f, the DRIFTS spectrum of Ru/TiO2-R displays three CO adsorption bands at ~2013, 2075, and 2137 cm−1, assigned to linearly adsorbed CO on Ru0, mono-adsorbed CO on partially oxidized Ruδ+, and multi-bound CO species on Ruδ+, respectively54,55. The relatively weak Ru0–CO band at 2013 cm−1 indicates that the surface Ru species on TiO2-R remain mainly oxidized even after high-temperature reduction56. In contrast, Ru/TiO2-A shows a single intense Ru0–CO band at ~2025 cm−1, suggesting that metallic Ru constitutes the dominant active site on anatase TiO2 (Fig. 2g).
Electronic structure of Ru/TiO2-R(A) catalysts
To gain deeper insight into the structural differences, X-ray absorption fine structure (XAFS) spectroscopy was employed to systematically analyze the electronic and coordination environments of the two catalysts. X-ray absorption near-edge structure (XANES) spectra (Fig. 3a) reveal that the absorption edge energies of both Ru/TiO2-R and Ru/TiO2-A lie between those of the Ru foil (Ru0) and RuO2 (Ru4+) reference samples, indicating that the Ru species exist in a mixed-valence state. Notably, Ru/TiO2-R exhibits a significant shift of the absorption edge toward higher energy, with spectral features more closely resembling those of RuO2. In contrast, Ru/TiO2-A displays spectra similar to Ru foil, directly confirming that TiO2-R effectively stabilizes oxidized Ru species, while TiO2-A favors the reduction of RuO2 to generate metallic Ru.

a XANES, b EXAFS and c WT contour plots of Ru/TiO2-R(A) and reference samples including RuO2 and Ru foil at Ru K edge. d, e NAP-XPS results of d Ru/TiO2-R and e Ru/TiO2-A at Ru 3 d orbital in the H2 atmosphere in the temperature range of 150–400 °C.
Extended X-ray absorption fine structure (EXAFS) analysis further elucidates differences in the local coordination environments (Fig. 3b). Both catalysts show coordination peaks at approximately 1.4 Å and 2.4 Å, corresponding to Ru–O and Ru–Ru coordination, respectively. As shown in Supplementary Fig. 10, the EXAFS data were fitted to obtain more accurate structural information on the Ru coordination environment. Fitting results (Supplementary Table 3) show that Ru/TiO2-R exhibits a Ru–O bond distance comparable to that of the RuO2 reference, with a coordination number of 5.68, indicating that the Ru species mainly exist in an oxidized RuOx. In contrast, the coordination environment of Ru/TiO2-A is dominated by Ru–Ru bonds with a coordination number of 6.03, while only a few O atoms are bonded to Ru, resulting in a larger fitting error bar, causing slight deviations in Ru–O band distance (1.93). Particularly, wavelet transform analysis (Fig. 3c) demonstrates a strong feature at k = 5 Å−1 (R = 1.4 Å) for Ru/TiO2-R, matching the Ru–O scattering path in RuO2. On the other hand, Ru/TiO2-A exhibits a dominant feature at k = 8 Å-1 (R = 2.4 Å), corresponding to metallic Ru–Ru coordination, which further demonstrates that TiO2-R can conduct an oxidized, Ru–O-rich surface structure through strong interfacial coupling, whereas TiO2-A cannot suppress the reduction of RuO2.
Near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) was further employed to investigate the in-situ electronic structure of the catalysts in H2 atmosphere. By monitoring the evolution of the Ru 3 d chemical states under different reduction temperatures, the reduction resistance of RuOx species on different TiO2 crystal phases was systematically studied. The Ru 3 d was analyzed instead of the Ru 3p because the latter exhibits lower signal intensity and overlaps with the Ti 2p signals57, which would reduce the accuracy of electronic structure analysis. As shown in Fig. 3d, e, upon in situ heating under a H2 atmosphere, the Ru 3d5/2 peak of Ru/TiO2-R shifted by approximately 0.8 eV, reaching ~280.5 eV at 400 °C, which remains higher than the characteristic binding energy of metallic Ru0 (~280.1 eV). This observation indicates the coexistence of metallic and oxidized Ru species, suggesting a partially reduced electronic structure. In contrast, Ru/TiO2-A exhibited a more pronounced shift of ~1.4 eV, with the peak position decreasing to 280.1 eV at 400 °C, corresponding to a predominantly metallic state. These findings reveal that the RuOx species on the Ru/TiO2-R surface exhibit greater resistance to H2-induced reduction, reflecting stronger interactions between RuOx and the TiO2-R support. Notably, the evolution of the Ti 2p spectra (Supplementary Fig. 11) further supports this difference. For Ru/TiO2-R, the Ti binding energy remained constant across the entire temperature range, suggesting a negligible reduction of the support. However, for Ru/TiO2-A, the Ti 2p peak gradually shifted from 459.0 eV to 458.5 eV, suggesting a Ti4+ → Ti3+ reduction process, which aligns with the electron transfer behavior identified by CO-DRIFTS, collectively confirming the presence of classical SMSI characteristics between anatase TiO2 and Ru nanoparticles.
Co-hydrogenolysis of PE and PP
We first evaluated the catalytic performance of different Ru structures in the hydrogenolysis of independent low-density polyethylene (LDPE) and PP over Ru/TiO2-R, Ru/TiO2-A, and commercial Ru/C catalysts. As depicted in Fig. 4a, under reaction conditions of 240 °C and 2 MPa H2 for 2 h, the Ru/TiO2-R catalyst featuring epitaxially grown RuOx active sites exhibits superior hydrogenolysis activity toward both polymers. The solid conversions of LDPE and PP reach 89.7% and 98.7%, while the selectivity toward gaseous products (C1–C4) is limited to 3.47% and 0.17%, respectively, indicating its remarkable selectivity for liquid fuel products. Notably, although PP shows a higher solid conversion, its product distribution differs significantly from that of LDPE. Gel permeation chromatography (GPC) analysis (Fig. 4b) reveals that the weight-average molecular weight (Mw) of PP hydrogenolysis products decreases from the initial 31,926 Da to 11,974 Da, with the polydispersity index (PDI) reduced from 6.65 to 1.86. In combination with the 1H NMR results (Fig. 4c), which show a relatively low proportion of -CH2- characteristic peaks (24.9%), all confirming that PP was primarily converted into heavy oil products. In contrast, the liquid products catalyzed from LDPE hydrogenolysis are predominantly light alkanes, with a carbon number distribution centered around C19 exhibiting a normal profile (Supplementary Fig. 12). Quantitative 1H NMR analysis reveals an A(-CH2-)/A(-CH3) ratio of 5.56, clearly distinguishing the product structure from that of PP. Therefore, the Ru/TiO2-R catalyst offers great potential for the co-conversion of mixed polyolefins into light and heavy oil products.

aSolid conversions of Ru/TiO2-R, Ru/TiO2-A and commercial Ru/C catalysts in hydrogenolysis of LDPE and PP. Reaction conditions: mRu = 1 mg, mLDPE/PP = 1 g, T = 240 °C, t = 2 h, PH2 = 2 MPa, stirring rate = 400 rpm. b GPC results of pristine PP powder and liquid products obtained from PP hydrogenolysis, and c 1H NMR results of the liquid products in LDPE and PP hydrogenolysis in Fig. 4a on Ru/TiO2-R catalyst. d Schematic illustration of the hydrogenolysis mechanisms of PE and PP over the three catalysts. The gray and light blue spheres represent branched and linear structures in PP and PE, as well as their corresponding reaction products, respectively. e Performance of Ru/TiO2-R in the co-hydrogenolysis of LDPE-PP mixtures with varying mass ratios. Reaction conditions: T = 240 °C, PH2 = 2 MPa, t = 3 h, mLDPE+PP = 1 g, mRu = 1 mg, stirring rate = 400 rpm. f Schematic illustration of the selective hydrogenolysis of squalane. g 2C–2C/3C–2C ratios and h liquid product distributions of different catalysts in squalane hydrogenolysis. Reaction conditions: mRu = 1 mg, msqualane = 1 g, T = 240 °C, t = 3 h, PH2 = 2 MPa, stirring rate = 400 rpm.
We investigated the time-dependent performance of the Ru/TiO2-R catalyst in the individual hydrogenolysis of LDPE and PP. As shown in Supplementary Fig. 13, when the reaction time was extended to 3 h, LDPE was completely converted, and further prolonging the reaction did not lead to excessive hydrogenolysis to methane. The gas yield remains low at 5.2%, while the liquid product distribution gradually shifts toward lower carbon numbers, centering around C13, indicating the catalyst’s good resistance to methanation during LDPE hydrogenolysis. In the case of PP, the Ru/TiO2-R catalyst enables near-complete conversion of the polymer within just 1 h. As the reaction proceeded, the heavy liquid fractions were progressively converted into lighter oil fractions (Supplementary Fig. 14), while the formation of low-value gaseous byproducts was negligible throughout the process. This could be attributed to the side methyl groups in the polypropylene backbone, which restrict the rotation of molecular chains, making the cleavage of 3C–2C bonds kinetically preferred and thereby suppressing methane formation associated with 3C–1C bond scission58. These results suggest that the Ru/TiO2-R catalyst holds great promise for the hydrogenolysis of mixed PE-PP polyolefin waste, enabling simultaneous depolymerization and controlled conversion into valuable liquid products.
In sharp contrast, the Ru/TiO2-A catalyst, which features a similar particle size but a different structure, exhibits significantly reduced catalytic performance. The surface-deposited metallic Ru nanoparticles achieve solid conversions of only 11.9% for LDPE and less than 5% for PP, highlighting the critical role of epitaxially grown RuOx structures in enabling efficient polymer hydrogenolysis. Moreover, we also evaluated the hydrogenolysis behavior of a commercial Ru/C catalyst possessing Ru nanoparticles with a size of approximately 3.45 nm (Supplementary Fig. 15). Owing to the larger particle size, which provides more surface adsorption and activation sites, Ru/C achieves an LDPE conversion of 80.0%, significantly outperforming Ru/TiO2-A. However, under identical conditions, Ru/C converted only 30.3% of PP solid, indicating that simply increasing the size of Ru nanoparticles may enhance PE hydrogenolysis efficiency, but is insufficient to promote the conversion of PP. For comparison, a Ru/TiO2-R-IMP catalyst with the same Ru loading was also prepared by the conventional wet impregnation method. Unlike the Ru/TiO2-R catalyst obtained via freeze-drying, the impregnated sample exhibited larger and unevenly distributed Ru nanoparticles with an average size of approximately 3.36 nm (Supplementary Fig. 16). Correspondingly, its hydrogenolysis activity was significantly lower than that of Ru/TiO2-R (Supplementary Fig. 17), highlighting the superior catalytic performance and structural advantages of the epitaxially grown RuOx architecture achieved through the freeze-drying approach.
Additionally, the size of Ru nanoparticles had a pronounced effect on product selectivity. As shown in Fig. 4a, although Ru/C (~3.45 nm Ru NPs) achieved a slightly lower LDPE conversion (80.0%) compared to Ru/TiO2-R (89.7%), the yield toward gaseous products (C1–C4) was markedly higher (8.7% vs. 3.5%), which suggests that larger Ru nanoparticles not only offer more surface coordination sites for adsorption and activation, but also facilitate continuous C–C bond cleavage, leading to excessive hydrogenolysis and the formation of low-value methane. Above all, we propose a mechanistic framework to elucidate the hydrogenolysis behavior of different Ru structures (Fig. 4d): Small-sized RuOx sites with epitaxial growth structures provide abundant adsorption and activation sites, enabling the efficient and selective co-conversion of PE and PP into light and heavy hydrocarbon oils. In contrast, small Ru metal nanoparticles lack the extended activation interfaces necessary for effective C–H and C–C bond cleavage, resulting in low activity for both polymers. Meanwhile, large Ru nanoparticles, although rich in surface active sites, tend to over-hydrogenolysis of PE, leading to excessive methane formation and significantly diminished product value.
To eliminate the influence of the support on hydrogenolysis performance and more accurately evaluate the intrinsic behavior of different Ru structures toward PE and PP hydrogenolysis, Ru nanoparticles were synthesized via an alcohol reduction method59 and subsequently deposited onto TiO2-R and TiO2-A supports by impregnation. To induce structural variations under different environments, the resulting precursors were treated under either reducing (5% H2/Ar) or oxidizing (air) atmospheres and were designated as Ru-NP/TiO2-R-H2, Ru-NP/TiO2-A-H2, Ru-NP/TiO2-R-Air, and Ru-NP/TiO2-A-Air, respectively. TEM images (Supplementary Fig. 18) confirmed that all Ru nanoparticles were uniformly dispersed across the support surfaces. As shown in Supplementary Fig. 19, after air and hydrogen treatments, the Ru species on TiO2 existed predominantly in oxidized and metallic states, respectively. The hydrogenolysis performance of the pre-synthesized Ru catalysts toward polyolefins was then evaluated at 240 °C (Supplementary Fig. 20). For LDPE hydrogenolysis, Ru-NP/TiO2-A-H2 and Ru-NP/TiO2-R-H2 achieved solid conversion rates of 20.9% and 49.1%, respectively, while for PP hydrogenolysis, the conversions were 5.3% and 10.5%. These results indicate that Ru nanoparticles treated under H2 are less favorable for PP hydrogenolysis, and confirm that the TiO2-R support itself contributes negligibly to the catalytic activity for PP hydrogenolysis. The relatively low hydrogenolysis activity of Ru-NP/TiO2-A-H2 may be attributed to the pronounced SMSI effect36.
Notably, Ru-NP/TiO2-A-Air exhibited nearly identical hydrogenolysis properties for both PE and PP compared with Ru-NP/TiO2-A-H2. This observation suggests that, despite the oxidative pretreatment, Ru oxide species supported on TiO2-A are unstable under hydrogenolysis conditions and are readily reduced to metallic Ru nanoparticles, leading to similar catalytic behavior as Ru-NP/TiO2-A-H2. Importantly, Ru-NP/TiO2-R-Air displayed a markedly higher solid conversion rate for PP (86.8%) than for PE (75.8%), consistent with the catalytic characteristics observed for Ru/TiO2-R (Fig. 4a), indicating that the RuOx active sites formed on Ru-NP/TiO2-R after air treatment are strongly coupled with the TiO2-R support, conferring exceptional structural stability and high intrinsic activity toward PP hydrogenolysis. Collectively, these results demonstrate that epitaxially grown RuOx sites effectively overcome the intrinsic hydrogenolysis kinetic mismatch between PE and PP, enabling efficient one-pot co-hydrogenolysis of mixed PE–PP feedstocks.
Temperature-programmed surface reaction (TPSR) experiments using n-hexane and 3-methylpentane as model molecules were conducted to clarify the activity differences. The onset temperature of CH4 formation was defined as the activation temperature. As displayed in Supplementary Fig. 21a, b, Ru/TiO2-R and Ru/C exhibited comparable activation temperatures for n-hexane hydrogenolysis, with Ru/C showing a slightly lower onset temperature, suggesting that metallic Ru nanoparticles are more favorable for the hydrogenolysis of linear alkanes. Importantly, Ru/TiO2-R exhibited a much lower activation temperature (~113.7 °C, Supplementary Fig. 21c) than Ru/C ( ~ 187.3 °C, Supplementary Fig. 21d) for 3-methylpentane hydrogenolysis, demonstrating superior activity for branched alkane hydrogenolysis. These results demonstrate that the epitaxial RuOx overlayer on the TiO2-R surface effectively promotes the activation of branched alkane structures.
Subsequently, we explored the performance of the Ru/TiO2-R catalyst in the co-hydrogenolysis of LDPE-PP mixtures. LDPE and PP powders were blended at varying mass ratios, while keeping the total polymer mass constant, and subjected to hydrogenolysis under identical conditions. As shown in Fig. 4e, Ru/TiO2-R can efficiently convert the mixed polyolefins into liquid fuels, with gas yields consistently remaining below 3%, demonstrating that Ru/TiO2-R serves as an efficient and selective catalyst for the co-hydrogenolysis of mixed polyolefins. With increasing PP content, the gas formation decreases to 0.62%, while the liquid yield increases to 95.02%, accompanied by a higher proportion of heavy oil fractions in the liquid products (Supplementary Fig. 22), which indicates the composition of the resulting liquid products can be continuously tuned by adjusting the type and ratio of the polymer substrates (Supplementary Fig. 23, Supplementary Table 4), offering a promising strategy for the selective conversion of mixed polyolefin waste into liquid fuels of varying quality.
To systematically investigate the catalytic behavior differences associated with various Ru structures in the PP hydrogenolysis and to elucidate their abilities to activate different types of C–C bonds, we employed squalane as a model compound for hydrogenolysis evaluation60. The molecular structure of squalane contains clearly defined 3C–2C bonds (tertiary–secondary carbon bonds) and 2C–2C bonds (secondary–secondary carbon bonds). By analyzing the distribution of hydrogenolysis products, the selective cleavage behavior can be quantitatively assessed. Specifically, the C24 and C27 products result from the cleavage of 3C–2C bonds, while the C25 and C26 products are generated from 2C–2C cleavage (Fig. 4f). We excluded small-molecule products because they may originate from the over-hydrogenolysis of high-carbon alkanes, making it difficult to unambiguously attribute them to specific cleavage events. Accordingly, we define a selectivity index as the yield ratio of (C25 + C26) to (C24 + C27), where a lower value indicates a stronger selectivity of the catalyst toward 3C–2C cleavage.
All three catalysts were evaluated under identical conditions at squalane conversions below 40% (Supplementary Fig. 24) to assess the intrinsic site selectivity of the hydrogenolysis reaction. As exhibited in Fig. 4g, h, the Ru/TiO2-R catalyst offers a selectivity index of 0.95, close to a 1:1 ratio, indicating comparable activation capabilities for both 3C–2C and 2C–2C bonds, which enables the simultaneous depolymerization of PE and PP. In contrast, catalysts composed of metallic Ru nanoparticles (Ru/TiO2-A and Ru/C) show significantly different selectivity characteristics, with selectivity indices of 1.53 and 1.39, respectively, indicating an intrinsically stronger activation ability toward 2C–2C bonds, leading to lower reactivity of Ru nanoparticles toward PP molecules bearing branched methyl groups, thereby hindering the cooperative depolymerization of mixed polyolefins. Furthermore, the slow-growth molecular dynamics method was employed to calculate the activation of C–H bond, with the bond length of the C–H bond used as the collective variable (CV) (Supplementary Fig. 25). The calculated adsorption energy of isopentane on Ru/TiO2-R is 0.43 eV, which is lower than that on Ru/TiO2-A (0.83 eV), indicating that the isomeric molecule is more easily adsorbed on the layered RuO2 surface. Moreover, the C–H activation of isopentane on Ru/TiO2-R (0.90 eV) is also lower than that on Ru/TiO2-A (1.13 eV). These findings demonstrate that epitaxial RuOx layers offer a clear advantage over Ru nanoparticles in promoting the hydrogenolysis of structurally distinct molecules. On epitaxial RuOx, the kinetic disparity between 3C–2C and 2C–2C bond cleavages is effectively eliminated, leading to a markedly enhanced synergistic hydrogenolysis performance for mixed polyolefins.
We further explore the stability and reusability of the optimal Ru/TiO2-R catalyst. In the hydrogenolysis stability tests of LDPE (Supplementary Fig. 26) and LDPE/PP mixtures (Supplementary Fig. 27), the optimal Ru/TiO2-R catalyst maintained stable performance over five consecutive reaction cycles. The solid conversions, liquid yields, and product distributions show negligible variation, indicating good catalytic stability and catalyst recyclability. Moreover, the TEM image (Supplementary Fig. 28a) confirmed that after repeated use, the catalyst retained a highly dispersed RuOx epitaxial structure with a consistent Ru nanoparticle size of approximately 1.4 nm (Supplementary Fig. 28b), further validating the outstanding structural stability of the Ru/TiO2-R catalyst.
Integrated Co-hydrogenolysis of Real-life Mixed Polyolefins
To expand the applicability of the mixed polyolefin co-upcycling system, we evaluated the hydrogenolysis performance of the optimal Ru/TiO2-R catalyst toward various polyolefin structures, post-consumer polyolefin waste, and mixed polyolefin waste streams. As seen in Fig. 5a–c, six representative waste-derived polyolefin products with distinct structures were selected as model substrates, which includes discarded LDPE bottle (PE1), mulch film (PE2, primarily composed of PE), LDPE packaging film (PE3), high-density polyethylene (HDPE) bottle (PE4), and PP box (PP1) and centrifuge tube (PP2). Prior to hydrogenolysis, all samples were mechanically processed by crushing and sieving into particles with dimensions ≤5 mm × 5 mm, and then uniformly mixed with the catalyst for subsequent hydrogenolysis experiments.

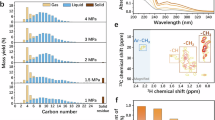
a Photographs of waste polyethylene and polypropylene. b Hydrogenolysis performance of various chemicals, single and mixed wastes over Ru/TiO2-R catalyst. The detailed catalytic conditions are listed in Supplementary Table 5. c Photographs of HDPE bottle and PP centrifuge tube and their co-hydrogenolysis performance within different reaction times. Reaction conditions: T = 260 °C, mRu/TiO2-R = 100 mg, mHDPE = 0.5 g, mPP = 0.5 g, PH2 = 2 MPa. d GPC results, e PDI and branched ratios and f non-solid product distributions of the HDPE/PP co-hydrogenolysis experiments.
We first evaluated the hydrogenolysis performance of the Ru/TiO2-R catalyst toward polyolefins with different molecular structures, including linear low-density polyethylene (LLDPE), HDPE, and PP with higher molecular weight (Mn = 8000 ± 500). As shown in Fig. 5b, Supplementary Figs. 29a–c, under reaction conditions of 240 °C, and 2 MPa H2, the catalyst efficiently and selectively converts these structurally diverse polyolefins into liquid alkane fuels, demonstrating that the small-sized epitaxially grown RuOx structure exhibits superior hydrogenolysis universality across various types of polyolefins. Considering that post-consumer plastic products typically contain various additives and impurities, and that mass transfer limitations under reaction conditions may further hinder performance, the hydrogenolysis of commercial polyolefin waste was conducted under more demanding conditions. The Ru/TiO2-R catalyst maintains high hydrogenolysis activity and selectivity, with the yields of liquid fuel consistently exceeding 80%, highlighting the catalyst’s superior potential for application in the co-hydrogenolysis of real-world mixed plastic wastes.
It is worth noting that in polyolefin hydrogenolysis, the composition of the resulting oil products determines their potential applications and economic value. Although we have preliminarily achieved one-pot co-hydrogenolysis of PE and PP polymers, further optimization is still required to precisely control the composition of the liquid products. Benefiting from the unique nanoscale epitaxial structure of the optimized Ru/TiO2-R catalyst, the methanation of PE-derived intermediates is effectively suppressed, which enables the liquid products from PE hydrogenolysis to remain in the form of low-carbon alkanes during the co-hydrogenolysis process, allowing for the continued hydrogenolysis of heavy oil fractions generated from PP. Consequently, the mixture of light and heavy hydrocarbons is transformed into high-quality liquid products with a simplified composition and an increased abundance of branched-chain structures. Taking the co-hydrogenolysis of PE4 and PP2 as a representative example, under reaction conditions of 260 °C, 2 MPa H2, a PE4/PP2 mass ratio of 1:1, and a reaction time of 5 h, the mixed polyolefin waste was efficiently converted into liquid alkanes (Fig. 5c). The yields of heavy oil (C35+) and light oil (C5–C35) are 49.5% and 36.1%, respectively. The resulting liquid products exhibit a Mw of 3460 Da (Fig. 5d, Supplementary Table 6) and a PDI of 2.28 (Fig. 5e). Upon further extension of the reaction time, the heavy oil fractions underwent continued hydrogenolysis, resulting in a progressive decrease in both Mw and PDI of the liquid products, along with a reduction in branched ratio due to the cleavage of 3C–2C bonds (calculated from the CH ratio in the 1H NMR spectra, Fig. 5e, Supplementary Fig. 30 and Supplementary Table 7). Notably, this transformation occurred with only a slight increase in gas formation. After 20 h of reaction, the heavy oil components were almost completely converted, yielding a liquid product with an Mw of 391 Da and a PDI of 1.21. The overall light oil yield reached 84.6%, comprising 40.7% gasoline-range (C5–C12) and 43.8% diesel-range (C13–C22) alkanes, while methane yield remains low at 10.5% (Fig. 5f). These results highlight the superior performance and robustness of the Ru/TiO2-R catalyst in converting real-world mixed polyolefins into high-quality liquid fuels and provide a solid foundation for controlling product quality in co-hydrogenolysis process.
Discussion
In summary, by constructing nanoscale epitaxial RuOx structures with an increased density of activation sites for PP hydrogenolysis, we successfully matched the hydrogenolysis rate of PP to that of PE, enabling the one-pot co-upcycling of polyolefin mixtures into liquid alkane fuels. Unlike conventional Ru metal nanoparticles, the epitaxially grown RuOx structures exhibit comparable activation capabilities toward various C–C bonds within the complex branched structures of PP, and the small particle size can effectively stabilize the intermediates from PE hydrogenolysis to suppress the over-hydrogenolysis. Experimentally, the optimal Ru/TiO2-R catalyst demonstrates good solid conversion and high liquid fuel selectivity across a range of mixed polyolefin compositions. For example, in a realistic co-upcycling system involving post-consumer HDPE bottles and PP centrifuge tubes, fine-tuning the reaction conditions enables continuous modulation of the hydrogenolysis products from these structurally distinct polyolefins, which can yield a high proportion (~84.6%) of low-carbon liquid alkanes, with valuable gasoline- and diesel-range fractions reaching 40.7% and 43.8%, respectively. These results highlight the exceptional versatility and robustness of the co-hydrogenolysis catalyst, providing an efficient, controllable, and industrially promising strategy for the upcycling of mixed polyolefin wastes.
Methods
Chemicals
Ruthenium (III) Chloride Trihydrate (RuCl3·3H2O, 99.0%) was purchased from Bide. Commercial rutile-phase TiO2 (99.9%, 20–30 nm) and anatase-phase TiO2 (99.9%, 20–30 nm) were purchased from XuShuo and DiBai, respectively. PVP (Mw = 40000) and ethylene glycol were purchased from Sigma-Aldrich. Squalane (95%) was purchased from Aladdin and used without further purification. Low-density polyethylene (LDPE, Mn = 1700, Mw = 4000) was provided by Sigma-Aldrich. Commercial Ru/C (5 wt.%), high-density polyethylene (HDPE, melt index = 6 ~ 9 g/10 min), linear low-density polyethylene (LLDPE, melt index = 2 g/10 min), polypropylene (PP, Mn = 4000 ± 500 and Mn = 8000 ± 500) were purchased from Macklin. LDPE bottle, mulch film, LDPE film, PP box, HDPE bottle and PP centrifuge tube were brought from Alibaba Group.
Synthesis of Ru/TiO2-R(A)
The Ru/TiO2 catalysts were prepared in the same method, except for a change in the crystal structure of TiO2. Typically, 1 g TiO2 powder was placed in a 50 mL beaker and dispersed in 10 mL of distilled water under sonication and stirring for 15 min. Then, 1.97 mL of a prepared 0.1 mmol/mL RuCl3 solution was added to the above suspension, while sonication and stirring were continued for another 30 min. The mixture was rapidly frozen with liquid nitrogen and placed in a -80 °C fridge overnight, followed by further drying in a freeze-drier. The dried catalyst was reduced at 300 °C for 2 h in a 5% H2/Ar atmosphere with a heating rate of 5 °C/min and a gas flow rate of 60 mL/min. The catalysts were named Ru/TiO2-R and Ru/TiO2-A according to the phase of TiO2.
Synthesis of Ru-NP/TiO2-R(A)
The Ru nanoparticle was prepared by polyol reduction59. Typically, 15 mg PVP and 39 mg RuCl3·3H2O were dissolved in 30 mL ethylene glycol in a 50 mL flask. The mixture was first heated to 60 °C under vacuum, and then maintained at 180 °C for 3 h under N2 atmosphere with vigorous stirring. The as-prepared Ru cluster was precipitated with acetone and centrifuged at 15557 g. The precipitates were washed 5 times to remove excess ethylene glycol. Then, the precipitates were dried under 70 °C oven and then re-dispersed into ethanol prior to impregnation. The quantitative TiO2 power was added into the ethanol dispersions of Ru nanoparticle, heated at 60 °C with stirring and then dried in an oven at 70 °C, named Ru-NP/TiO2-R(A). Subsequently, Ru-NP/TiO2-R(A) were reduced at 300 °C for 2 h in a 5% H2/Ar atmosphere with a heating rate of 5 °C/min and a gas flow rate of 60 mL/min, named Ru-NP/TiO2-R(A)-H2. The Ru-NP/TiO2-R(A)-Air were obtained by Ru-NP/TiO2-R(A) calcined in air with the same temperature program.
Synthesis of Ru/TiO2-R-IMP
The Ru/TiO2-R-IMP were prepared via the conventional impregnation method. Typically, 1 g rutile TiO2 power was placed in a 20 mL glass bottle and dispersed with 10 mL of distilled water. Then, 1.97 mL of RuCl3 solution (0.1 mmol/mL) was added under stirring at 400 rpm. The slurry product was obtained by evaporating the solvent at 80 °C and then dried in an oven at 70 °C. The dried solid was ground and subsequently reduced in a 5% H2/Ar atmosphere at 300 °C for 2 h with a heating rate of 5 °C/min. The obtained catalyst was denoted as Ru/TiO2-R-IMP.
Catalytic test of polyolefin hydrogenolysis
Typically, 50 mg of catalyst and 1 g of polyolefin were physically mixed and placed into the bottom of a 100 mL stainless-steel batch autoclave (Shanghai Yanzheng Experimental Instrument Co., Ltd.). The reactor was then sealed and purged with H2 (1 MPa) at least three times to remove residual air, after which the pressure was adjusted to 2 MPa. During the reaction, the stirring rate was maintained at 400 rpm. Upon completion of the reaction and after the system cooled to room temperature, the gaseous products were collected using a gas sampling bag, and the final pressure was recorded. The solid residue and liquid products were collected by rinsing the reactor with 30 mL of toluene into a centrifuge tube. The mixture was then refrigerated at 4–6 °C for 30 min to solidify the high molecular weight wax fractions. Subsequently, the mixture was separated using a centrifuge, where the liquid contained the products dissolved in toluene, and the solid residue was dried in an oven at 80 °C. The gas and liquid products were analyzed by a gas chromatograph (GC) with a flame ionization detector (FID), and the models of capillary-column were TM-Al2O3 and HP-1, respectively. The components dissolved in toluene were defined as liquid products; however, due to the good solubility of polypropylene hydrogenolysis products in toluene, high-molecular-weight products (> C35) could not be detected by GC. To enable more accurate quantification of the liquid products from polypropylene hydrogenolysis, 100 μL of mesitylene was added as an internal standard for the determination of light liquid fractions (C5–C35). The solid conversion and products yield are calculated by the following equations:
Catalytic test of squalane hydrogenolysis
For squalane hydrogenolysis, 1 mg Ru and 1 g squalane were placed at the bottom of a 100 mL batch stainless-steel autoclave. The operating steps of the reaction were the same as those for polyolefin hydrogenolysis. In this reaction, the temperature was 240 °C and the stirring rate was 400 rpm. The gas and liquid products were collected by a gas sampling bag and toluene solvent, and then analyzed by GC-FID with a capillary-column of TM-Al2O3 and HP-1, respectively. The products were quantitatively analyzed using mesitylene as an internal standard.
Temperature-programmed surface reaction experiments (TPSR)
The TPSR experiments were conducted in a custom-built reactor system equipped with three independent gas lines. Gas line 1 supplied 5% H2/Ar, gas line 2 supplied pure H2, and gas line 3 supplied He. The flow rates of all three gases were precisely regulated by individual mass flow controllers (MFCs). The outlet of the MFC on gas line 3 was connected to a bubbler containing liquid n-hexane or 3-methyl pentane, allowing alkane vapors to be introduced into the reactor via He bubbling. Prior to the TPSR measurement, the catalyst was reduced in gas line 1 at 300 °C for 30 min. Subsequently, gas line 1 was closed, and gas lines 2 (30 mL/min) and 3 (20 mL/min) were opened simultaneously to initiate the temperature-programmed reaction, with the reactor outlet connected to a mass spectrometer for product detection.
Reusability test
For PE hydrogenolysis, the proportion of catalyst and alkanes in the solid residue after the reaction was analyzed through thermo gravimetric analysis. Based on the catalyst content in the residue, a certain amount of fresh catalyst was added before the next reaction to ensure the total catalyst mass was 50 mg. For mixed PE and PP hydrogenolysis, the solid residue was washed with hot toluene for 5 times to remove the un-reacted polyolefins. The loss catalyst was supplemented with fresh catalyst to a total amount of 50 mg before the next cycle.
Computational methods
Density functional theory (DFT) calculations were carried out on a Vienna Ab initio Simulation Package (vasp. 6.4.2)61,62,63. The generalized gradient approximation (GGA)64 of Perdew−Burke−Ernzerhof was applied as the exchange−correlation functional, and the projector augmented wave (PAW) method65 was used to describe the electron-ion interactions. The Kohn−Sham valence states were expanded using a planewave basis set with a kinetic energy cutoff of 520 eV. The van der Waals interaction between atoms was treated using the DFT-D3 method with Becke-Johnson damping66. Gaussian smearing of the electronic states was applied, with Fermi temperatures of 0.05 for condensed phase calculations.
The crystal lattice parameters of rutile TiO2 bulk are as follows: a = b = 4.6458 Å, c = 2.9667 Å (α = β = γ = 90o). A Monkhorst-Pack k-mesh of 6 × 6 × 10 k-points was used in the structural relaxation for the bulk. Electronic energies were computed with a tolerance of 1 × 10‒7 eV and a total force of 0.02 eV for each atom.
For the rutile TiO2 (110) slab model, we constructed a 3 × 3 supercell with a crystal thickness of 30 Å to avoid periodic effects. The bottom three layers of atoms were fixed to simulate the bulk structure. The energy and force convergence thresholds for each atom were set to 10−6 eV and 0.04 eV/Å, respectively. For structural optimizations, the Brillouin zone was sampled using 2 × 1 × 1 k-points according to the Monkhorst-Pack scheme. For the calculation of lattice mismatch, we used the following formula:
Where a2 is the lattice constant of rutile TiO2 (110) or anatase TiO2 (101), and a1 is the lattice constant of RuO2 (110).
For the calculation of the formation energy of RuO2 grown on a rutile TiO2 (110) slab, we used the following formula48:
where E[mRuO2/TiO2] or E[nRuO2/TiO2] is the energy of TiO2 (110) with (m or n) layers RuO2 covered, E[RuO2(slab)] is the energy of RuO2, and k is the number of RuO2 units from nRuO2/TiO2 growing to mRuO2/TiO2 on TiO2(110). The more negative energy means the more favored formation of RuO2 on TiO2 surfaces.
The adsorption energy Ead can be defined as Ead = Gmolecule on surface - Gsub - Gmolecule, where Gmolecule on surface stands for the energy of the slab models with the adsorbate, Gsub is the calculated energy of the clean substrate, and Gmolecule is the energy of the molecular adsorbate under vacuum. The Gibbs free energy change (G) of reaction steps was calculated by the followings:
where EDFT is the electronic energy calculated for specified geometrical structures, EZPE is the zero-point energy, S is the entropy.
To assess the kinetic barriers, we utilized a constrained molecular dynamics (MD) technique known as the “slow-growth” approach67,68,69, which allowed us to derive the free-energy profile. In this method, the value of the reaction coordinate (denoted as \(\xi\)) is linearly changed from the characteristic value for the initial state (IS) to that for the final state (FS) with a velocity of transformation \(\dot{\xi }\). The resulting work needed to perform a transformation from IS to FS can be computed as:
where F(q) is the free energy at general coordinate q, which is evolving with time t. In the limit of infinitesimally small \(\xi\), the work \({w}_{\text{IS}\to \text{FS}}\) corresponds to the free-energy difference between the final and initial states. The time step is set as 1.0 fs, and a \(\partial \xi\) value of 0.0008 Å was adopted for each molecular dynamics (MD) step. The Andersen thermostat70 was employed to maintain a constant temperature at 300 K. We have home-made codes/scripts to post-process the output data.
Characterizations
The transmission electron microscopy (TEM) images were tested by the field emission high-resolution transmission electron microscopy (FEI Talos F200X, Thermo Fisher). The HAADF images and electron energy-loss spectroscopy (EELS) data were acquired using an aberration-corrected STEM (HF5000, Hitachi) operated at an acceleration voltage of 200 kV. The EELS analysis was performed with a dispersion of 0.25 eV per channel, and the energy resolution, as determined by the full width at half-maximum of the zero-loss peak, was better than 1.0 eV. X-ray diffraction (XRD) patterns were collected on an analytical Empyrean machine. Inductivity coupled plasma optical emission spectrometry (ICP-OES) was used to determine the actual Ru loading of catalysts. Thermal Gravimetric Analyzer (TGA) measurement was performed by the machine from METTLER TOLEDO. Near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS) was measured on a SPECS NAP-XPS instrument with a temperature-controllable laser heating device and a thermocouple equipped. The photon source is the monochromatic X-ray source of Al Kα (1486.6 eV). The sample was pressed onto copper foam, and then the sample was fixed onto the XPS sample holder with tantalum strips. In a routine test, the data were collected under UHV at 25 °C, followed by the heat-treatment of the sample using the laser at 150, 300, 400 °C in a pure hydrogen atmosphere (0.2 mbar) for 5 min. The H2-temperature-programmed desorption (H2-TPR) was collected by an automatic chemical adsorption instrument (FINETEC/FINE-SORB-3010). Typically, 20 mg of catalysts were put into the middle of a U-shaped tube for H2-TPR experiments. The H2-TPR progress involved two steps: (1) Ar purging to remove physically adsorbed gases, and (2) heating from room temperature to 700 °C in a H2/Ar flow. The H2 signal was determined by a Thermal Conductivity Detector (TCD) with the current and temperature of 80 mA and 100 °C, respectively.The data of diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) was collected by a Thermo Nicolet iS50 instrument. For the CO-DRIFTS, the catalyst was pre-treated under H2/N2 flow at 250 °C for 30 min in the cell. After cooling down to room temperature, the background data were collected under N2 atmosphere, then the CO gas was pumped into the cell to undergo adsorption for about 40 min. Finally, the data were collected with N2 purging until the gaseous CO signal disappeared. X-ray Absorption Near Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) data were collected at Beamline 13SSW at Shanghai Synchrotron Radiation Facility (SSRF). The molecular weight of the samples was analyzed using gel permeation chromatography (GPC) at an Agilent PL-GPC220. NMR spectroscopy was performed on a Digital NMR Spectrometer (AVANCE NEO 400 MHz).
Data availability
The source data generated in this study are provided in the Source Data file. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Stegmann, P. et al. Plastic futures and their CO2 emissions. Nature 612, 272–276 (2022).
MacLeod, M. et al. The global threat from plastic pollution. Science 373, 61–65 (2021).
Sardon, H. & Dove, A. P. Plastics recycling with a difference. Science 360, 380–381 (2018).
Zheng, K. et al. Progress and perspective for the conversion of plastic wastes into valuable chemicals. Chem. Soc. Rev. 52, 8–29 (2023).
Geyer, R. et al. Production, use, and fate of all plastics ever made. Sci. Adv. 3, e1700782 (2017).
Stubbins, A. et al. Plastics in the Earth system. Science 373, 51–55 (2021).
Coates, G. W. & Getzler, Y. D. Y. L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 5, 501–516 (2020).
Conk, R. J. et al. Polyolefin waste to light olefins with ethylene and base-metal heterogeneous catalysts. Science 385, 1322–1327 (2024).
Hu, P. et al. Stable interfacial ruthenium species for highly efficient polyolefin upcycling. J. Am. Chem. Soc. 146, 7076–7087 (2024).
Ellis, L. D. et al. Chemical and biological catalysis for plastics recycling and upcycling. Nat. Catal. 4, 539–556 (2021).
Chu, M. Y. et al. Efficient polyolefin upcycling over a single-atom alloy catalyst. CCS Chem 7, 2451–2464 (2025).
Sun, J. et al. Catalytic upcycling of polyolefins. Chem. Rev. 124, 9457–9579 (2024).
Jehanno, C. et al. Critical advances and future opportunities in upcycling commodity polymers. Nature 603, 803–814 (2022).
Chen, S. et al. Ultrasmall amorphous zirconia nanoparticles catalyse polyolefin hydrogenolysis. Nat. Catal. 6, 161–173 (2023).
Zhao, M. et al. Enhancing the conversion efficiency of polyethylene to methane through codoping of Mn atoms into Ru centers and CeO2 supports. J. Am. Chem. Soc. 146, 33104–33111 (2024).
Wei, T. et al. Catalytic recycling of plastics into value-added products. Nano Res. 17, 9428–9445 (2024).
Chu, M. et al. Co-recycling of plastics and other waste materials. Nat. Rev. Clean Technol. 1, 320–332 (2025).
Rorrer, J. E. et al. Hydrogenolysis of polypropylene and mixed polyolefin plastic waste over Ru/C to produce liquid alkanes. ACS Sustain. Chem. Eng. 9, 11661–11666 (2021).
Xu, Z. et al. Chemical upcycling of polyethylene, polypropylene, and mixtures to high-value surfactants. Science 381, 666–671 (2023).
Sullivan, K. P. et al. Mixed plastics waste valorization through tandem chemical oxidation and biological funneling. Science 378, 207–211 (2022).
Wang, M. et al. Complete hydrogenolysis of mixed plastic wastes. Nat. Chem. Eng. 1, 376–384 (2024).
Shi, Y. et al. Advances and challenges for catalytic recycling and upgrading of real-world mixed plastic waste. ACS Catal. 15, 841–868 (2024).
Korley, L. T. J. et al. Toward polymer upcycling—adding value and tackling circularity. Science 373, 66–69 (2021).
Clarke, R. W. et al. Dynamic crosslinking compatibilizes immiscible mixed plastics. Nature 616, 731–739 (2023).
Zhang, Z. et al. Mixed plastics waste upcycling with a high-stability single-atom Ru catalyst. J. Am. Chem. Soc. 145, 22836–22844 (2023).
Wang, C. et al. Polyethylene hydrogenolysis at mild conditions over ruthenium on tungstated zirconia. JACS Au 1, 1422–1434 (2021).
Wang, X. et al. Enhancing waste plastic hydrogenolysis on Ru/CeO2 through concurrent incorporation of Fe single atoms and FeOx nanoclusters. Angew. Chem. Int. Ed. 64, e202506035 (2025).
Kots, P. A. et al. Electronic modulation of metal-support interactions improves polypropylene hydrogenolysis over ruthenium catalysts. Nat. Commun. 13, 5186 (2022).
Zhao, T. Y. et al. Mechanistic insights into processive polyethylene hydrogenolysis through in situ NMR. Macromolecules 56, 4287–4295 (2023).
Hu, Q. et al. Polyethylene hydrogenolysis by dilute RuPt alloy to achieve H2-pressure-independent low methane selectivity. Nat. Commun. 15, 10573 (2024).
Nakaji, Y. et al. Low-temperature catalytic upgrading of waste polyolefinic plastics into liquid fuels and waxes. Appl. Catal. B-Environ. 285, 119805 (2021).
Kang, Q. et al. Entropy confinement promotes hydrogenolysis activity for polyethylene upcycling. Angew. Chem. Int. Ed. 62, e202313174 (2023).
Chen, L. et al. Efficient and selective dual-pathway polyolefin hydro-conversion over unexpectedly bifunctional M/TiO2-anatase catalysts. Appl. Catal. B-Environ. 335, 122897 (2023).
Chen, L. et al. Effect of reaction conditions on the hydrogenolysis of polypropylene and polyethylene into gas and liquid alkanes. React. Chem. Eng. 7, 844–854 (2022).
Tamura, M. et al. Structure-activity relationship in hydrogenolysis of polyolefins over Ru/support catalysts. Appl. Catal. B-Environ. 318, 121870 (2022).
Tu, W. et al. SMSI-induced charge transfer for selective hydrogenolysis of polyolefins. Appl. Catal. B-Environ. 339, 123122 (2023).
Si, X. et al. Ni-catalyzed carbon–carbon bonds cleavage of mixed polyolefin plastic waste. J. Energy Chem. 85, 562–569 (2023).
Ma, W. et al. Catalytic hydrogenolysis of polypropylene and polyethylene mixtures: Effect of temperature on liquid alkane components. J. Energy Inst. 115, 101615 (2024).
Zichittella, G. et al. Hydrogenolysis of polyethylene and polypropylene into propane over cobalt-based catalysts. JACS Au 2, 2259–2268 (2022).
Hackler, R. A. et al. Effect of macro- and microstructures on catalytic hydrogenolysis of polyolefins. Macromolecules 55, 6801–6810 (2022).
Flaherty, D. W. et al. Metal-catalyzed C–C bond cleavage in alkanes: effects of methyl substitution on transition-state structures and stability. J. Am. Chem. Soc. 136, 9664–9676 (2014).
Hibbitts, D. D. et al. Role of branching on the rate and mechanism of C–C cleavage in alkanes on metal surfaces. ACS Catal. 6, 469–482 (2015).
Kim, S. et al. Ru-catalyzed polyethylene hydrogenolysis under quasi-supercritical conditions. JACS Au 5, 1760–1770 (2025).
Chu, M. et al. Site-selective polyolefin hydrogenolysis on atomic Ru for methanation suppression and liquid fuel production. Research 6, 0032 (2023).
Chen, L. et al. Disordered, sub-nanometer Ru structures on CeO2 are highly efficient and selective catalysts in polymer upcycling by hydrogenolysis. ACS Catal. 12, 4618–4627 (2022).
Zhou, J. et al. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 13, 327 (2022).
O’Sullivan, M. et al. Interface control by chemical and dimensional matching in an oxide heterostructure. Nat. Chem. 8, 347–353 (2016).
Tang, X. et al. Significance of epitaxial growth of PtO2 on rutile TiO2 for Pt/TiO2 catalysts. J. Am. Chem. Soc. 146, 3764–3772 (2024).
Chen, S. et al. Morphology-engineered highly active and stable Ru/TiO2 catalysts for selective CO methanation. Angew. Chem. Int. Ed. 58, 10732–10736 (2019).
Guo, Y. et al. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect. ACS Catal. 8, 6203–6215 (2018).
Wang, F. et al. Catalytic behavior of supported Ru nanoparticles on the {100}, {110}, and {111} facets of CeO2. J. Catal. 329, 177–186 (2015).
Sanz-Marco, A. et al. Continuous photothermal gas-phase CO2 hydrogenation over highly dispersed Ru-Ni on TiO2. Catal. Today 459, 115440 (2025).
Cisneros, S. et al. Oxygen vacancies in Ru/TiO2-drivers of low-temperature CO2 methanation assessed by multimodal operando spectroscopy. iScience 25, 103886 (2022).
Hadjiivanov, K. et al. FTIR study of CO interaction with Ru/TiO2 catalysts. J. Catal. 176, 415–425 (1998).
Solymosi, F. & Raskó, J. An infrared study of the influence of CO adsorption on the topology of supported ruthenium. J. Catal. 115, 107–119 (1989).
Zhang, Y. et al. Strong metal-support interaction of Ru on TiO2 derived from the CO-reduction mechanism of RuxTi1–xO2 interphase. ACS Catal. 12, 1697–1705 (2022).
Chen, L.-N. et al. Ru nanoparticles supported on partially reduced TiO2 as a highly efficient catalyst for hydrogen evolution. Nano Energy 88, 106211 (2021).
Jaydev, S. D. et al. Identifying selective catalysts in polypropylene hydrogenolysis by decoupling scission pathways. Chem Cataly. 3, 100564 (2023).
Yan, X. et al. Size control of polymer-stabilized ruthenium nanoparticles by polyol reduction. J. Mater. Chem. 11, 3387–3391 (2001).
Nakagawa, Y. et al. Regioselectivity and reaction mechanism of Ru-catalyzed hydrogenolysis of squalane and model alkanes. ChemSusChem 10, 189–198 (2017).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Woo, T. K. et al. A combined Car−Parrinello QM/MM implementation for ab initio molecular dynamics simulations of extended systems: Application to transition metal catalysis. J. Phys. Chem. B 101, 7877–7880 (1997).
Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 78, 2690–2693 (1997).
Oberhofer, H. et al. Biased sampling of nonequilibrium trajectories: Can fast switching simulations outperform conventional free energy calculation methods?. J. Phys. Chem. B 109, 6902–6915 (2005).
Andersen, H. C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 72, 2384–2393 (1980).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 22376152 to J.C. and No. 42377013 to M.X.Z.), Jiangsu Provincial Fund for Excellent Young Scholars (BK20240154 to J.C.), and Suzhou Frontier Technology Research Advanced Materials Project (SYG202305 to J.C.). M.Z. thanks the support from Zhejiang Provincial Key Research and Development Program (2025C02207). J.C. thanks the support from the Suzhou Key Laboratory of Advanced Photonic Materials, Collaborative Innovation Center of Suzhou Nano Science & Technology, and the 111 Project. We thank the BL13SSW beamline at the Shanghai Synchrotron Radiation Facility (https://cstr.cn/31124.02.SSRF. BL13SSW) for the XAFS experiments support. The authors also thank Dr. Xuefei Weng and Dr. Yifan Li for the technical support for Nano-X from Suzhou Institute of Nano-Tech and Nano-Bionics (SINANO), Chinese Academy of Sciences, and Dr. Xuchun Wang for data curation.
Author information
Authors and Affiliations
Contributions
Q.Z. and J.C. conceived and supervised the project. W.T. and M.Y.C. carried out the experiments and collected the date. T.Y. and L.Z. contributed to the XAFS measurement and provided analysis. M.X.Z., Q.Q.Z. and X.W. provided the DFT calculation. W.T., M.Y.C. and M.H.C. wrote the manuscript, and M.S.Z., M.X.Z., J.C., and Q.Z. revised it. All the authors contributed to the whole manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tu, W., Chu, M., Yan, T. et al. One-pot co-upcycling of mixed polyolefin waste. Nat Commun 17, 3358 (2026). https://doi.org/10.1038/s41467-026-70104-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70104-z
