
Abstract
Acetylene semihydrogenation is a critical process in the polyolefin industry by selectively removing trace acetylene from ethylene-rich reformate. However, this reaction is generally limited by the inherent activity-selectivity trade-off due to the thermodynamic advantage of overhydrogenation. Herein, we develop a facile and straightforward strategy to construct Pd2 dual-atom sites anchored on defect-rich surface-graphitized nanodiamond (ND@G) via the solvent-mediated dispersion of palladium carboxylate driven by the chelation of palladium precursors via carboxylate anion. Cs-corrected HAADF-STEM images coupled with XAS analysis unambiguously manifest the successful architecting of Pd2 dual-atom sites. Compared to Pd1 single-atom sites, the obtained Pd2/ND@G sample demonstrates superior catalytic performance in acetylene semihydrogenation, with the corresponding TOF values increased from 0.151 s−1 to 1.953 s−1, without the obvious decline of ethylene selectivity (93.2%, at full acetylene conversion). C2H2/C2H4-TPD, H2-D2 exchange reaction, isotope-labeled TPSR combined with DFT calculations confirm the effective co-activation of C2H2/H2 on Pd2 dual-atom sites while maintaining the weak adsorption of ethylene similar to that on its single-atom sites, which can break the activity-selectivity trade-off in acetylene semihydrogenation.
Similar content being viewed by others

Introduction
Ethylene, as a key bulk chemical, has a global annual production exceeding 200 million metric tons, with the majority of them being utilized in the polymer industry1,2. It is typically produced through the catalytic cracking of naphtha or light hydrocarbons3,4. Consequently, this ethylene-rich reformate inevitably contains a small amount of acetylene impurity (0.5–2%), which can poison the Ziegler-Natta catalysts used in the downstream polymerization of ethylene due to the very strong π coordinated bond between Ti3+ and C2H25,6,7,8,9. Therefore, it is crucial to remove acetylene from the ethylene-rich reformate below 1 ppm before it is applied in the polymer industry10,11. So far, acetylene semihydrogenation is critical for ethylene purification in polyethylene production via the selective removal of ethyne without C2H4 overhydrogenation12,13. However, architecting a cost-effective catalyst with high activity and ethylene selectivity in acetylene semihydrogenation poses an intractable challenge due to the thermodynamic advantage of overhydrogenation and the linear scaling relations constraints of adsorption energies between acetylene and ethylene5,14.
Currently, Cu-, Ni-, and Pd-based catalysts are extensively studied in the acetylene semihydrogenation reaction15,16,17,18. Among them, alumina-supported Pd-based catalysts, especially PdAg bimetallic nano-catalysts, have been industrially implemented in the acetylene selective hydrogenation because of their excellent hydrogenation activity19,20. Nevertheless, the ethylene selectivity is generally unsatisfied on Pd nanoparticles (NPs) or clusters owing to their relatively strong adsorption of ethylene (di-σ bond mode) and the presence of β-PdHx21,22,23. To improve the selectivity of ethylene in acetylene semihydrogenation, forming an intermetallic compound or alloy is a common strategy by suppressing the strength of ethylene adsorption and the formation of PdHx due to the diluted surface Pd dispersion24,25,26. However, the increase of ethylene selectivity in the above strategy comes at the expense of reduced Pd atom utilization efficiency due to the nonparticipation of many more bulk Pd atoms in acetylene semihydrogenation. In contrast, single-atomization of active sites is another typical strategy to optimize the ethylene selectivity in acetylene semihydrogenation because of only ethylene’s weak π-bond absorption and absence of β-PdHx on single-atom sites27,28. Nonetheless, the hydrogenation ability of Pd single-atom sites is significantly decreased compared to their NP counterparts due to the insufficient hydrogen dissociation resulting from the diminished metallicity27,29. Apart from the inadequate hydrogen activation, the activity of acetylene semihydrogenation on Pd single-atom sites is also suppressed by the competitive adsorption of acetylene and H2 due to its inability to simultaneously activate multiple reactants12,30. At present, dual-atom catalysts have attracted extensive research attention because they not only possess the advantages of single-atom catalysts, such as high atom utilization efficiency and highly uniform active sites, but also exhibit a synergistic effect in their dual-atom structure, optimizing the interactions between the active sites and multiple reactants or intermediates31,32,33. This may be conducive to alleviating the competitive adsorption/activation of multiple reactants and breaking the intrinsic linear scaling relations of adsorption energies among reactants, intermediates, and products. However, the reported method for dual-atom catalysts synthesis, including anion replacement deposition-precipitation, metal ion recognition, and precursor-preselected, generally concerns cost-prohibitive reagents and intricate operation procedures, remaining a huge challenge31,34,35,36,37.
Herein, we report a facile and straightforward protocol for architecting a defect-engineered graphene hybridized nanodiamond (ND@G) supported Pd2 dual-atom catalyst via the solvent-dependent dispersion of palladium carboxylate due to the effective chelation of carboxylate anion in specific solvents38,39,40. The structure of Pd2/ND@G was systematically investigated by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-ray absorption analysis (XAS), confirming the successful construction of Pd2 dual-atom sites. The obtained Pd2/ND@G sample shows excellent performance in acetylene semihydrogenation reaction compared to its single-atom counterpart (Pd1/ND@G), lowering the total conversion temperature of acetylene from 180 to 100 oC and increasing the corresponding TOF values from 0.151 s−1 to 1.953 s−1, showing dramatic improvement in acetylene semihydrogenation activity without the obvious decline of ethylene selectivity (93.2%, at full acetylene conversion). C2H2/C2H4-TPD, H2-D2 exchange reaction, and isotope-labeled TPSR confirmed the effective simultaneous-activation of C2H2/H2 on Pd2 dual-atom sites while maintaining the weak adsorption of ethylene similar to that on its single-atom sites, which can break the activity-selectivity trade-off in acetylene semihydrogenation. Density functional theory (DFT) calculations manifested the advantages of Pd2 dual-atom in optimizing the interaction between active sites and C2H2/C2H4, and breaking the intrinsic linear scaling constraints of their adsorption energies.
Results
Characterization
Pd1/ND@G and Pd2/ND@G samples were prepared via the impregnation method, utilizing the solvent-dependent dispersion of palladium carboxylate, followed by a sequential pretreatment of O2-H2 at 200 oC. It is reported that palladium acetate (Pd(OAc)2) exists as a monomer species in acetic acid (HAc) solution, whereas palladium trifluoroacetate (Pd(TFA)2) forms a dimer in hot acetone (CP) solution (Fig. 1a)38,39,40. The Pd cluster supported on ND@G was synthesized by a deposition-precipitation strategy41,42. The detailed procedures are listed in the supporting information. To remove surface ligands of the as-obtained samples, the Pd1/ND@G and Pd2/ND@G were subjected to sequential pretreatment with air and hydrogen at 200 oC, which is confirmed by the temperature programmed oxidation (TPO) of Pd(OAc)2 and Pd(TFA)2 (Supplementary Fig. 1). The complete removal of carboxylate ligands was confirmed by Fourier transform infrared spectroscopy, as evidenced by the absence of their characteristic stretching vibration frequencies (Supplementary Fig. 2)40. Hereafter, the notations of Pd1/ND@G, Pd2/ND@G, and PdC/ND@G refer to samples subjected to a sequential O2-H2 pretreatment. No obvious diffraction peaks for Pd are detected in the X-ray diffraction (XRD) patterns of Pd1/ND@G, Pd2/ND@G, and PdC/ND@G, implying the high dispersion of Pd active sites (Supplementary Fig. 3)41,43. The actual loading of Pd in the above three samples was determined by inductively coupled plasma atomic emission spectrometry, almost in line with their nominal loadings (Supplementary Table 1).

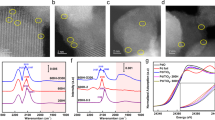
a The schematic illustration of Pd1/ND@G and Pd2/ND@G preparation. Cs-corrected HAADF-STEM images of (b, c) Pd1/ND@G (Scale bar: 2 nm) and (d, e) Pd2/ND@G (Scale bar: 5 nm). f, g Enlarged images of Pd2 dimers and their corresponding intensity profiles. Pd K-edge XANES profiles (h) and EXAFS spectra (i) for Pd1/ND@G and Pd2/ND@G (inset: expansion of the highlighted region). j The corresponding XAFS fitting curves for the Pd2/ND@G sample.
To further elucidate the geometric structure of Pd sites at the atomic level, Cs-corrected HAADF-STEM (Cs-HAADF-STEM) examination was performed with samples of Pd1/ND@G, Pd2/ND@G, and PdC/ND@G. With atomic resolution, it was found that only Pd single atoms exist in the Pd1/ND@G sample, without the presence of Pd clusters or NPs, indicating the single-atom dispersion of Pd active sites highlighted in yellow cycles (Fig. 1b, c and Supplementary Fig. 4). As regards Pd2/ND@G, a large number of Pd-Pd dual atom pairs are observed on the ND@G surface, as highlighted in the red rectangles. In contrast, some single-atom Pd sites are also detected due to the decomposition of palladium carboxylate during the ligand removal procedure (Fig. 1d, e and Supplementary Fig. 5). The Pd1:Pd2 site ratio was determined to be 51:49, based on the identification of 175 Pd2 dual-atom pairs and 179 Pd1 single-atom sites via directly counting their site numbers in Cs-corrected HAADF-STEM images, indicating approximately 33% of Pd2 carboxylate dimers in Pd2/ND@G-No treatment sample decomposing into Pd1 single-atom sites during calcination44. The mean distance between Pd-Pd in Pd2 dimers is about 2.75 Å, confirmed by their intensity profiles, demonstrating the formation of Pd2 dual-atom geometry (Fig. 1f, g and Supplementary Fig. 6-7)35,37. Similarly, no Pd clusters or NPs are present in Pd2/ND@G, manifesting the stable Pd2 sites without sintering under pretreatment. As a reference, the Cs-corrected HAADF-STEM images of PdC/ND@ and its size distribution are shown in Supplementary Figs. 8 and 9. Pd active sites, including single-atom, subnanometer clusters, as well as a small amount of Pd NPs, are present in the PdC/ND@G sample.
To obtain deep insight into the electronic structure and local coordination geometry of Pd species in the samples of Pd1/ND@G and Pd2/ND@G, the adsorption of Pd K-edge was collected for XAS analysis. Figure 1h shows the normalized X-ray absorption near-edge spectra (XANES) of Pd K-edge in Pd1/ND@G and Pd2/ND@G samples, located in the range of Pd foil and PdO, indicating the electron-deficient state of Pd species27,30. Notably, the adsorption threshold of Pd K-edge in Pd2/ND@G is lower than that of Pd1/ND@G, denoting the increased metallicity of Pd2 entity compared to Pd1 single-atom, namely enhanced electron state density35. This enhanced electron state density of Pd2 dimers is also evidenced by the negative binding energy shift of the Pd 3 d electron in the XPS spectra compared to that of Pd1/ND@G. (Supplementary Fig. 10). To further investigate the geometric construction of Pd species, extended X-ray absorption fine structure (EXAFS) spectroscopy was performed. For Pd1/ND@G sample, only a distinct peak centered at 1.6 Å assigned to the Pd-C/O first-shell scattering was observed, whereas no signal corresponding to Pd-Pd scattering at 2.5 Å was detected (Fig. 1i)28,45. According to the Pd K-edge fitting, Pd1/ND@G shows a coordination number (CN) of 2.67 ± 0.37 with a bond distance of 2.03 Å (Supplementary Fig. 11 and Supplementary Table 2). The above results confirmed the single-atom dispersed Pd sites. In contrast, a main peak located at 1.6 Å, accompanied by a weak band at 2.5 Å, is present in the Pd2/ND@G sample, implying the presence of both Pd-C/O and Pd-Pd scattering (Fig.1i)35,37. In addition to Pd-C/O oscillation, Pd2/ND@G also exhibits a Pd-Pd coordination with CN of 0.5 ± 0.19 based on the Pd K-edge fitting (Fig. 1j, Supplementary Fig. 11, and Supplementary Table 2). Given that the average CN of Pd-Pd in Pd2/ND@G sample represents the weighted average of the Pd-Pd coordination numbers in Pd1 and Pd2 sites (\({\mbox{C.N.}}\left({\mbox{Pd}}-{\mbox{Pd}}\right)\,=\,{\mbox{n}}\left({{\mbox{Pd}}}_{2}\right)/\left[{\mbox{n}}\left({{\mbox{Pd}}}_{1}\right)+{\mbox{n}}\left({{\mbox{Pd}}}_{2}\right)\right]\) Eq. 1, where Pd1 exhibits zero Pd-Pd coordination and Pd2 has a Pd-Pd coordination number of 1), it is concluded that the site number ratio of Pd1 to Pd2 is 1:1, consistent with the value derived from microscopic statistical analysis of Cs-corrected HAADF-STEM images. These consequences, combined with Cs-corrected HAADF-STEM images of Pd2/ND@G, unambiguously demonstrated the formation of Pd2 dual-atom pair sites (Fig. 1d-g and Supplementary Fig. 5-6).
Catalytic performances
Subsequently, the catalytic performance of the as-obtained Pd-based catalysts with single-atom, dual-atom, or cluster active sites was systematically evaluated for the semi-hydrogenation of acetylene under C2H4/H2-rich co-feed, which is an intractable challenge due to the facile overhydrogenation of ethylene46,47,48. The acetylene conversion over Pd1/ND@G is below 10% at 60 oC and reaches complete conversion only at high temperatures above 180 oC, showing inadequate catalysis of the Pd single-atom sites in acetylene semihydrogenation due to its inability to simultaneously activate C2H2 and H2 despite its superior ethylene selectivity (Fig. 2a, b)27,30. In contrast, the Pd cluster sites exhibit excellent activity in acetylene semihydrogenation with total conversion of C2H2 even at 60 oC (Fig. 2a). However, the ethylene selectivity over PdC/ND@G is even lower than –500% at 90 oC, implying consumption of much C2H4 in the co-feed gas due to overhydrogenation (Fig. 2b)49,50. Compared to Pd1/ND@G and PdC/ND@G, the complete conversion of acetylene over Pd2/ND@G decreased from 180 to 100 oC with the ethylene selectivity up to 93.2%, indicating significantly increased activity of Pd dual-atom sites in acetylene semi-hydrogenation owing to its’ increased electronic state density and synergistic effect in Pd2 geometry structures (Figs. 1 and 2a, b). Notably, the ethylene selectivity of Pd2 dual-atom sites at full C2H2 conversion is comparable to that of the Pd single-atom sites, which can also be indirectly verified by their activity in the ethylene hydrogenation reaction (Supplementary Fig. 12). Apart from the excellent activity and selectivity in acetylene semihydrogenation, the long-term stability of Pd2/ND@G is tested at 70% conversion or full conversion of acetylene (Fig. 2c and Supplementary Fig. 13). It can be observed that the Pd2/ND@G sample exhibits remarkable stability during acetylene semihydrogenation, without any obvious decline in acetylene conversion (Fig. 2c) or ethylene selectivity (Supplementary Fig. 13). Additionally, Pd2/ND@G sample after long-term stability test, labeled as Pd2/ND@G-used, was characterized with Cs-corrected HAADF-STEM. The Cs-corrected HAADF-STEM images revealed only Pd2 dual-atom sites and Pd1 single-atom sites, with no observation of Pd clusters or nanoparticles (Supplementary Fig. 14). Actually, the high ethylene selectivity during the long-term stability test also indirectly excluded the presence of Pd clusters or nanoparticles due to their tendency to over-hydrogenation (Supplementary Fig. 13). This is primarily attributed to the abundant carbon defects on the ND@G surface, which enable the robust anchoring of the Pd2 dual-atom site, evidenced by their change in the ID/IG ratio of the Raman spectra (Supplementary Fig. 15). Furthermore, to elucidate the role of the ND@G support in stabilizing Pd2 dual-atom sites, Pd1, Pd2, and PdC sites supported on silica and α-alumina (commonly used supports in industry) were evaluated in the acetylene semihydrogenation. The catalytic performance of Pd₁, Pd₂, and PdC sites on both supports was similar: complete acetylene conversion was achieved at 80 °C, but the selectivity remained below –150%, indicating over-hydrogenation of ethylene (Supplementary Fig. 16). This is primarily because the surfaces of silica and alumina supports lack specific sites capable of strongly anchoring Pd₁ or Pd₂, leading to the sintering of single-atom and dual-atom sites into clusters or particles.

The catalytic performance of acetylene semihydrogenation. a C2H2 conversion and b C2H4 selectivity over Pd1/ND@G, Pd2/ND@G, and PdC/ND@G. Reaction condition: Following ligand removal and reduction, each catalyst (amount normalized to a fixed Pd mass, diluted with 500 mg quartz sand) was tested under temperature-programmed reaction conditions, with triplicate sampling at each temperature and GHSVs of 60,000 (Pd1/ND@G, Pd2/ND@G) and 120,000 mL h⁻¹ g⁻¹ (PdC/ND@G). c The long-term stability of C2H2 semihydrogenation on Pd2/ND@G at 80 oC. d The TOF value of converted acetylene and space-time yield of obtained ethylene at 80 oC on Pd1/ND@G, Pd2/ND@G, and PdC/ND@G. e Comparison of the catalytic performance of various catalysts. The numbers in Fig. 2e correspond to the reference numbers in the supplementary information. f Arrhenius plots of the different catalysts. All conversions of acetylene for TOF calculations are controlled in the range of 7.5-15%. The catalytic data with error bars were replicated 3 times. The error bars in Fig. 2a, b, d, f represent the standard deviation.
To further quantitatively investigate the differences in catalytic performance of various Pd active sites in acetylene semihydrogenation, the TOF value of converted acetylene and ethylene space-time yield at 80 oC was calculated (Fig. 2d). The Pd2 dual-atom sites exhibit superior catalytic performance in acetylene semihydrogenation compared to the Pd single-atom sites with a 12.9-fold increase in the TOF values (1.953 s–1 vs 0.151 s–1), indicating distinguishable enhancement in catalytic activity. The specific activity of Pd2/ND@G is compared to the data reported in published literature, surpassing most results (Fig. 2e and Supplementary Table 3). Although PdC/ND@G shows a much higher TOF value than that of Pd2/ND@G, the space-time yield of ethylene on the PdC cluster is as low as –120.5 molethylenegPd–1h–1, which means a large amount of ethylene is consumed in the co-feed gas (Fig. 2d). Besides, the apparent kinetic activation energy barriers based on Arrhenius plots reveal that Pd2/NFD@G shows a lower activation energy compared to that of Pd1/ND@G (25.3 kJ/mol vs 43.3 kJ/mol), which further verifies the superior activity of acetylene semihydrogenation on Pd2 dual-atom sites (Fig. 2f).
Mechanism study
To elucidate the underlying reason for the notable difference in the catalytic performance among various Pd sites, the temperature-programmed experiments are conducted to investigate the adsorption, activation, and co-activation behaviors of the reactants/products (C2H2, C2H4, and H2) on Pd1, Pd2, and PdC sites. C2H2- and C2H4- TPD measurements are conducted to probe the adsorption/desorption ability of acetylene and ethylene on the obtained Pd-based catalysts (Fig. 3a, b). The main desorption peaks of acetylene on Pd1/ND@G, Pd2/ND@G, and PdC/ND@G samples are centered at 82, 88, and 88 oC, respectively, which may be assigned to the π-bonded C2H2 (Fig. 3a)30,51. Apart from the main desorption peak at 88 oC, a shoulder peak at 125 oC is also observed on Pd clusters, which may be attributed to the di-σ bond adsorption of acetylene (Fig. 3a)29,52. Compared to Pd1/ND@G, the higher desorption peak observed on Pd2 dual-atom sites indicates a stronger binding affinity. In contrast, ethylene remains physically adsorbed on both Pd1 and Pd2 sites due to its complete removal during the stabilization of the baseline at room temperature, which is confirmed by the lack of desorption peaks in the range of 40–200 oC (Fig. 3b). The C2H2/C2H4-TPD results reveal that Pd2 dual-atom sites exhibit enhanced acetylene adsorption while preserving the physical adsorption of ethylene, which may contribute to its high activity and ethylene selectivity in acetylene semihydrogenation.

a C2H2- and b C2H4-TPD profiles of Pd1/ND@G, Pd2/ND@G, and PdC/ND@G samples. c H2-D2 exchange reaction over Pd1/ND@G, Pd2/ND@G, and PdC/ND@G catalysts. d Isotope-labeled TPSR of acetylene semihydrogenation over Pd1/ND@G, Pd2/ND@G, and PdC/ND@G samples at 60 oC.
In addition to C2H2/C2H4 adsorption, the H2-D2 exchange reaction is conducted to probe the H2 activation, a factor influencing the catalytic performance of acetylene semihydrogenation (Fig. 3). The H2-D2 exchange reaction attains equilibrium on PdC/ND@G even at room temperature, verified by the constant concentration of HD, indicating the facile dissociation of H2, which accounts for its propensity for overhydrogenation (Figs. 2b, 3c, Supplementary Fig. 12, and Supplementary Fig. 17). In contrast, Pd single-atom sites show weak hydrogen dissociation ability, confirmed by the balanced conversion temperature of 120 oC, in line with its low activity in acetylene semihydrogenation (Figs. 2a, 3c, and Supplementary Fig. 17). For Pd2 dual-atom sites, the HD ion current at equilibrium is achieved at a temperature of 80 oC, indicating its increased H2 dissociative activation (Fig. 3c and Supplementary Fig. 17). Besides, the isotope-labeled TPSR measurements are performed to explore the competitive adsorption-activation between C2H2 and H2 (Fig. 3d and Supplementary Fig. 18). The isotopic tracer experiment is conducted by initially flowing a mixture of H2, D2, and He over a Pd-based sample, followed by substitution of He with the same acetylene flow to maintain the total feed rate, excluding the influence of gas flow rate on HD concentration. The intensity of competitive adsorption-activation between C2H2 and H2 can be quantitatively assessed by monitoring the evolution of HD concentration after acetylene introduction. The HD signal intensity over Pd1/ND@G is virtually eliminated upon switching the He flow to acetylene, demonstrating that the competitive adsorption-activation between C2H2 and H2 on Pd single-atom sites significantly inhibits its catalytic activity in acetylene semihydrogenation (Fig. 3d and Supplementary Fig. 18). In contrast, the intensity of the HD signal on the Pd2 dual-atom or PdC cluster still remains notably considerable despite a reduction due to the competitive adsorption of C2H2 and H2, indicating its significant alleviation. Additionally, the ethylene and ethane peak intensities observed in the isotope-labeled TPSR experiment on Pd-based samples align well with their corresponding catalytic performance in acetylene semihydrogenation (Supplementary Fig. 18).
DFT calculations
To investigate the underlying mechanism and reveal the catalytic origin of the Pd2 dual-atom sites in the semi-hydrogenation of acetylene, we conducted density functional theory (DFT) calculations. First, two atomically dispersed catalyst models (Pd1/ND@G and Pd2/ND@G) were constructed based on coordination number characteristics derived from EXAFS fitting. These correspond to a single Pd atom anchored at a dual-carbon-defect site (a defect region where three adjacent carbon atoms are missing) in graphene (Pd1/ND@G) and a Pd2 dual-atom stabilized at a tri-carbon-vacancy site (Pd2/ND@G), respectively. The optimized structure of Pd2/ND@G shows that the Pd atoms exhibit Pd-C bonds (coordination number, CN = 2.5; bond length, R = 1.96 Å) and an intradimer Pd-Pd bond (R = 2.55 Å). In addition, a Pd(111) unit cell comprising 36 Pd atoms was constructed to represent the PdC/ND@G sample based on the size distribution of the Pd cluster (Figs. 1i-1j, Supplementary Fig. 9, 11, 19, and Supplementary Table 2). Geometric optimization results show that the structural parameters, such as coordination numbers and bond lengths, of the constructed models agree well with experimental values (Supplementary Table 2 and Table 4). Bader charge analysis reveals that the charge transfer trend of Pd in Pd2/ND@G is consistent with the results of XPS and XANES, which further confirms the increased metallicity of the Pd2 site compared to that of Pd1 (Fig. 1h, Supplementary Fig. 10, and Table 4).
Figure 4 shows the reaction pathway of acetylene hydrogenation over Pd1/ND@G, Pd2/ND@G, and PdC/ND@G samples. The calculated adsorption energies of acetylene become increasingly negative across the order: Pd1/ND@G (–1.05 eV), Pd2/ND@G (–1.11 eV), and PdC/ND@G (–2.46 eV, Supplementary Fig. 20), indicating stronger adsorption and higher activation capability, consistent with the C2H2-TPD results (Fig. 3a). Accordingly, the energy barriers for the rate control step reveal a distinct activity trend in acetylene semihydrogenation activity. The highest transition state energy, corresponding to the competitive adsorption/activation of C2H2 and H2 (TS1), on PdC/ND@G is the lowest (–1.85 eV), followed by Pd2/ND@G (1.04 eV), and is highest on Pd1/ND@G (1.93 eV, Fig. 4). This difference in transition state energy between the above three samples indicates that the activity follows the order: PdC/ND@G > Pd2/ND@G > Pd1/ND@G, in line with their reaction activity (Figs. 2a, d, f). However, despite the superior activity, PdC/ND@G exhibits poor ethylene selectivity due to its higher desorption energy (–1.65 eV) than the energy barrier for further hydrogenation to C2H5 (–2.37 eV), implying that ethylene tends to undergo over-hydrogenation to ethane rather than desorb, leading to poor selectivity (Fig. 2b). This high desorption energy of ethylene on PdC/ND@G is consistent with its C2H2-TPD profile (Fig. 3b). In contrast, on both Pd2/ND@G and Pd1/ND@G, the ethylene desorption energy is lower than the barrier for C2H5 formation, in line with the physical adsorption of ethylene on both Pd sites, confirming the high selectivity toward ethylene (Figs. 2b and 3b). Furthermore, the H2-D2 exchange reaction pathway reveals a substantially lower energy barrier on Pd2/ND@G (0.96 eV) than on Pd1/ND@G (1.66 eV), further indicating the superior kinetic performance of the Pd₂ dual-atom site in hydrogen activation (Fig. 3c, Supplementary Fig. 14 and 21). DFT calculations demonstrated the advantages of Pd2 dual-atom in optimizing the interaction between active sites and C2H2/C2H4, and overcoming the intrinsic linear scaling constraints of their adsorption energies, which can break the activity-selectivity trade-off in acetylene semihydrogenation.

The free energy distribution of acetylene hydrogenation on Pd1/ND@G, Pd2/ND@G, and Pd(111) catalysts (the x-axis is the reaction coordinate, and the y-axis shows the free energy of C2H2, C2H4, C2H6, and H2 measured at 80 °C and 1 bar). Color coding: gray, blue, and yellow spheres represent carbon, palladium, and hydrogen, respectively.
Discussion
This work successfully constructed a Pd2 dual-atom site catalyst via the solvent-dependent dispersion of palladium carboxylate, which demonstrated superior catalytic performance in acetylene semihydrogenation with a TOF value as high as 117.1 molacetylene/molPd/min at 80 oC, outperforming the vast majority of reported catalysts. The XAS analysis, C2H2/C2H4-TPD, H2-D2 exchange reaction, isotope-labeled TPSR, and DFT calculations manifested that the enhanced metallicity of dual-atom sites and the synergistic effect between Pd atoms in the Pd2 entity enable the modulated interaction among reactants (C2H2 and H2), product (C2H4), and Pd active sites. This effectively breaks the inherent scaling relation constraints of C2H2/C2H4 adsorption energies and mitigates the competitive adsorption-activation between C2H2 and H2, thereby overcoming the activity-selectivity trade-off in acetylene semihydrogenation.
Methods
Materials
Nanodiamond (ND) powders were purchased from Beijing Grish Hitech Co. Ltd. China. Palladium acetate (Pd(OAc)2) and palladium trifluoroacetate (Pd(TFA)2) were purchased from Aladdin. Acetone (CP) and acetic acid (HAc) were purchased from Sinopharm Chemical Reagent Co. Ltd. China. All chemicals were analytical grade.
Preparation of ND@G
The ND powders were calcined at a temperature of 1300 K for 4 hrs under an argon flow with a rate of 100 mL/min. Upon cooling to room temperature, a graphene hybridized nanodiamond (ND@G) was obtained. This sample was successively purified by washing with concentrated hydrochloric acid and water. Ultimately, the purified ND@G powders were dried in a vacuum at 80 oC overnight.
Preparation of Pd1/ND@G and Pd2/ND@G
The Pd1/ND@G and Pd2/ND@G samples are prepared by the impregnation method. 200 mg of purified ND@G powder was impregnated into 2 mL of a certain concentration of Pd(OAc)2 in acetic acid solution and Pd(TFA)2 in a hot acetone solution (0.1 mgPd/mL). This slurry was stirred at room temperature for 24 h, followed by drying under vacuum at 80 °C overnight. Finally, the obtained samples were pretreated with air at 200 oC for 1 h, followed by reduction with 10 vol% H2/He at 200 oC for 1 h, denoted as Pd1/ND@G and Pd2/ND@G.
Preparation of PdC/ND@G
The PdC/ND@G sample was prepared by the deposition-precipitation method. 200 mg of ND@G powder was dispersed into 30 mL of deionized water in a 100 mL round-bottom flask by ultrasonic treatment for 30 min. The pH value of the above suspension was adjusted to 10 by dropping a certain amount of Na2CO3 solution (0.25 M). 12.5 μL of Pd(NO3)2 solution containing 16 mgPd/mL was added to 3 mL deionized water, followed by tuning its pH value with 3 drops of Na2CO3 solution (0.25 M). Subsequently, the Pd solution was added dropwise into the ND@G suspension under stirring at 100 oC for 1 h. After cooling to room temperature, the sample was filtered and washed several times with deionized water. Finally, the powder was dried in a vacuum oven at 80 oC overnight. The obtained Pd-based catalyst was pretreated 10% H2 at 250 oC for 1 h, denoted as PdC/ND@G.
Catalysts characterization
The Pd loading of Pd1/ND@G, Pd2/ND@G, and PdC/ND@G samples was determined by an inductively coupled plasma atomic emission spectrometer (ICP-AES). The powder X-ray diffraction (XRD) patterns of the catalysts were recorded on a X’ Pert PRO diffractometer operated at 40 kV and 40 mA using a Cu Kα radiation source (λ = 0.15432 nm), with a scanning rate of 10°/min. X-ray photoelectron spectroscopy (XPS) measurements were conducted on an ESCALAB 250 instrument equipped with an Al Kα X-ray source (1489.6 eV, 150 W, 50.0 eV pass energy). The binding energies in XPS were referenced to the C 1 s binding energy at 284.8 eV. Cs-corrected HAADF-STEM images were recorded by a JEOL JEM ARM 200CF cold-field-emission scanning transmission electron microscope operated at 200 kV. Temperature-programmed desorption (TPD) of C2H2/ C2H4 was performed in a quartz-bed flow reactor equipped with an online mass spectrometer (MS, Pfeiffer OMNIstar) for gas mixture analysis. The 150 mg of sample was first pretreated under a 10vol.% H2/He mixture at 200 °C for 1 h, followed by cooling to room temperature in a He flow. After cooling, a gas mixture of 1vol.% C2H2 or C2H4 balanced with He was introduced into the reactor for 3 h at 25 °C (flow rate = 20 mL/min) to saturated adsorption. Subsequently, the gas flow was switched to pure He flow (flow rate = 10 mL/min) for purging the physically adsorbed C2H2/C2H4. The mass spectrometer continuously monitored the signal intensities at m/z = 26 (C2H2), 28 (C2H4), 18 (H2O), and 4 (He). Once the MS signals stabilized, the sample was heated under a He flow (10 mL/min) to 350 oC at a rate of 2 °C/min while continuously recording the mass spectrum. H2-D2 exchange experiments were performed in a quartz-bed flow reactor with an online mass spectrometer (MS, Pfeiffer OMNIstarTM) to evaluate gas mixtures. Typically, a certain amount of sample (Pd1/ND@G and Pd2/ND@G 10 mg; PdC/ND@G using 5 mg) mixed with 500 mg of quartz sand was reduced with 10 vol.% H2 in He at 200 oC for 1 h (flow rate = 20 mL/min) and cooled to room temperature in He flow. Then, the mixture gas of 10 vol.% H2/He (20 mL/min), 5 vol.% D2/He (20 mL/min) in He was introduced into the reactor and kept for 30 mins until the baseline was stable (flow rate = 20 mL/min). The sample was heated under 10 vol.% H2, 5 vol.% D2 in He flow (flow rate = 20 mL/min) from room temperature to 200 oC. The intensities for the m/z values equaling 2, 3, and 4 were continuously recorded for H2, HD, and D2/He, respectively. Isotope-labeled TPSR experiments were also performed on a quartz-bed flow reactor with an online mass spectrometer (MS, Pfeiffer OMNIstarTM). Typically, a certain amount of samples (Pd1/ND@G and Pd2/ND@G 10 mg; PdC/ND@G using 5 mg) mixed with 500 mg of quartz sand were pretreated with 10 vol.% H2 balanced with He at 200 oC for 1 h. Then, the gas flow was switched to pure He during the cooling process. After cooling to 50 oC, the He flow was continuously introduced for some time, followed by switching to 10 vol.% H2 (20 mL/min) and 5 vol.% D2 (20 mL/min). After some time, the 10 mL/min pure He was also introduced to the reactor for a while. Subsequently, the pure He flow was replaced by a 1 vol.% C2H2 at 10 mL/min. The intensities of m/z values of 2 (H2), 3 (HD), 4 (D2+He), 26 (C2H2), 28 (C2H4), and 30 (C2H6) were continuously recorded by an online mass spectrometer (MS, Pfeiffer OMNIstarTM). X-ray absorption spectroscopy (XAS) measurements were performed at the Argonne Advanced Photon Source, 10-ID beamline under operating conditions of 3.5 GeV and 250 mA, using a Si(111) double-crystal monochromator. The X-ray beam size is 500 µm horizontal × 1000 µm vertical.
Catalytic test
The acetylene semihydrogenation performances of the Pd-based catalysts were evaluated in a quartz-bed flow reactor with a certain amount of samples (20 mg for Pd1/ND@G and Pd2/ND@G or 10 mg for PdC/ND@G to maintain a consistent content of palladium) mixed with 500 mg of quartz sand. Prior to testing, the prepared Pd-based samples were pretreated sequentially with oxygen (20% O2/N2) and hydrogen (10% H2/He) at 200 oC for 1 h, respectively. After cooling to room temperature, a mixture gas of 1 vol.% C2H2, 10 vol.% H2, and 20 vol.% C2H4 balanced with He was introduced with a flow rate of 20 mL/min, followed by temperature rise testing. The gas hour space velocity (GHSV) of Pd1/ND@G and Pd2/ND@G is 60,000 mL/g/h, while GHSV for PdC/ND@G is 120,000 mL/g/h/. The concentrations of reactants and products were periodically analyzed online using an Agilent 7890 A Gas Chromatograph equipped with a HP-PLOT AL/S capillary column and a flame ionization detector (FID).
Acetylene conversion, ethylene selectivity, TOF values of acetylene, space-time yield, and specific activity were calculated as the following formulas:
Where c(C2H2) is the concentration of acetylene; mCat., wt% are the weight of the catalyst tested and the loading of Pd.
Similarly, the ethylene hydrogenation performances of Pd1/ND@G and Pd2/ND@G were evaluated in the same reactor. Before testing, the prepared Pd-based samples were pretreated sequentially with oxygen (20% O2/N2) and hydrogen (10% H2/He) at 200 oC for 1 h, respectively. After cooling to room temperature, a mixture gas of 0.5 vol.% C2H4, 5 vol.% H2 balanced with He was introduced with a flow rate of 40 mL/min, followed by ascending temperature testing. The gas hour space velocity (GHSV) of Pd1/ND@G and Pd2/ND@G is 120,000 mL/g/h. The concentrations of reactants and products were periodically analyzed online using an Agilent 7890 A Gas Chromatograph equipped with a HP-PLOT AL/S capillary column and a flame ionization detector (FID). The ethylene conversion was calculated as the following formulas:
Computational details
All density functional theory calculations were performed using the VASP package(VASP.6.3.2)53. The projector augmented wave (PAW) pseudopotential was employed to describe the interactions between nuclei and valence electrons, and the exchange-correlation functional was treated within the generalized gradient approximation using the PBE functional54,55. A plane-wave energy cutoff of 400 eV was applied56. To accurately describe the electronic structures of different systems, differentiated spin settings were adopted: spin-restricted calculations were used for the metallic Pd(111) surface, while spin-polarized calculations were applied for the Pd1/ND@G and Pd2/ND@G catalyst models, which may possess localized magnetic moments57. The Brillouin zone was sampled with k-point grids of 4 × 4 × 1 for Pd(111) and 2 × 2 × 1 for Pd1/ND@G and Pd2/ND@G58. The energy convergence threshold for electronic self-consistent iteration was set to 10⁻⁵ eV, and van der Waals interactions were corrected using the DFT-D3 method59. During structural optimization, the convergence criterion for interatomic forces was set to 0.03 eV/Å. Transition states were located using the NEB and dimer methods60,61. To better reflect realistic reaction conditions (1 bar, 80 °C, 1% acetylene concentration), thermodynamic corrections were applied to the species along the reaction pathways, with the corresponding correction values provided in Supplementary Table 5.
The adsorption energy (\({E}_{{ads}}\)) was calculated as:
where \({E}_{{totoal}}\) is the total energy of the adsorbate on the substrate, \({E}_{{slab}}\) is the energy of the substrate, and \({E}_{{adsorbent}}\) is the energy of the isolated adsorbate.
The Gibbs free energy at finite temperature and pressure (\({G}_{(T,P)}\)) was calculated as:
where \({E}_{{DFT}}\) is the DFT-calculated energy at 0 K, and \(\Delta {G}_{{corrt}}\) is the thermodynamic correction term, which accounts for the effects of temperature and pressure on the adsorption energy, enabling accurate prediction of reaction free energies under experimental conditions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Chauhan, R., Sartape, R., Minocha, N., Goyal, I. & Singh, M. R. Advancements in environmentally sustainable technologies for ethylene production. Energ. Fuel. 37, 12589–12622 (2023).
Xu, J.-X., Yuan, Y., Wu, X.-F. Ethylene as a synthon in carbonylative synthesis. Coord. Chem. Rev. 477, 214947 (2023).
Zhou, H. et al. PdZn intermetallic nanostructure with Pd-Zn-Pd ensembles for highly active and chemoselective semi-hydrogenation of acetylene. ACS Catal. 6, 1054–1061 (2016).
Hyun, K., Yun, S. & Choi, M. Synergistic combination of inorganic and organic promoters on palladium catalysts for effective acetylene partial hydrogenation. ACS Catal. 14, 2938–2948 (2024).
Ayodele, O. B. et al. Synergistic computational-experimental discovery of highly selective PtCu nanocluster catalysts for acetylene semihydrogenation. ACS Catal. 10, 451–457 (2020).
Almisbaa, Z., Aljama, H. A., Almajnouni, K., Cavallo, L. & Sautet, P. Acetylene semi-hydrogenation on intermetallic Ni-In catalysts: Ni ensemble and acetylene coverage effects from a theoretical analysis. ACS Catal. 13, 7358–7370 (2023).
Hu, Y. et al. Bioinspired molecular catalyst for photocatalytic semihydrogenation of acetylene with water as a proton source. ACS Catal. 15, 1135–1146 (2025).
Morra, E. et al. Probing the coordinative unsaturation and local environment of Ti3+sites in an activated high-yield Ziegler–Natta catalyst. Angew. Chem. Int. Ed. 54, 4857–4860 (2015).
Groppo, E. et al. Activation and in situ ethylene polymerization on silica-supported Ziegler–Natta catalysts. ACS Catal. 5, 5586–5595 (2015).
Liu, F. et al. Integration of bimetallic electronic synergy with oxide site isolation improves the selective hydrogenation of acetylene. Angew. Chem. Int. Ed. 60, 19324–19330 (2021).
Liu, Y. et al. Polyoxometalate-based metal-organic framework as molecular sieve for highly selective semi-hydrogenation of acetylene on isolated single Pd atom sites. Angew. Chem. Int. Ed. 60, 22522–22528 (2021).
Ballesteros-Soberanas, J. et al. A MOF-supported Pd1-Au1 dimer catalyses the semihydrogenation reaction of acetylene in ethylene with a nearly barrierless activation energy. Nat. Catal. 7, 452–463 (2024).
Huang, F. et al. Anchoring Cu1 species over nanodiamond-graphene for semi-hydrogenation of acetylene. Nat. Commun. 10, 4431 (2019).
Zhu, C. et al. Molecule saturation boosts acetylene semihydrogenation activity and selectivity on a core-shell Ruthenium@Palladium Catalyst. Angew. Chem. Int. Ed. 62, e202300110 (2023).
Studt, F. et al. Identification of non-precious metal alloy catalysts for selective hydrogenation of acetylene. Science 320, 1320–1322 (2008).
Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209–1212 (2012).
Zheng, M. et al. Recent advances in electrocatalytic hydrogenation reactions on copper-based catalysts. Adv. Mater. 36, 2307913 (2024).
Zhang, W. et al. Ambient-condition acetylene hydrogenation to ethylene over WS2-confined atomic Pd sites. Nat. Commun. 15, 9457 (2024).
Wang, J., Xu, H., Che, C., Zhu, J. & Cheng, D. Rational design of PdAg catalysts for acetylene selective hydrogenation via structural descriptor-based screening strategy. ACS Catal. 13, 433–444 (2023).
Pei, G. X. et al. Ag alloyed Pd single-atom catalysts for efficient selective hydrogenation of acetylene to ethylene in excess ethylene. ACS Catal. 5, 3717–3725 (2015).
Teschner, D. et al. The roles of subsurface carbon and hydrogen in palladium-catalyzed alkyne hydrogenation. Science 320, 86–89 (2008).
Liu, H. et al. Unveiling atomic-scaled local chemical order of high-entropy intermetallic catalyst for alkyl-substitution-dependent alkyne semihydrogenation. J. Am. Chem. Soc. 146, 20193–20204 (2024).
Jiang, X. et al. Cu single-atom catalysts for high-selectivity electrocatalytic acetylene semihydrogenation. Angew. Chem. Int. Ed. 62, e202307848 (2023).
Ball, M. R. et al. AgPd and CuPd catalysts for selective hydrogenation of acetylene. ACS Catal. 10, 8567–8581 (2020).
Hu, M. et al. MOF-confined Sub-2 nm atomically ordered intermetallic PdZn nanoparticles as high-performance catalysts for selective hydrogenation of acetylene. Adv. Mater. 30, 1801878 (2018).
Chen, H. et al. Co-infiltration and dynamic formation of Pd3ZnCx intermetallic carbide by syngas boosting selective hydrogenation of acetylene. Nat. Commun. 15, 9850 (2024).
Huang, F. et al. Atomically Dispersed Pd on Nanodiamond/Graphene Hybrid for Selective Hydrogenation of Acetylene. J. Am. Chem. Soc. 140, 13142–13146 (2018).
Yang, B. et al. Incorporation of Pd single-atom sites in perovskite with an excellent selectivity toward photocatalytic semihydrogenation of alkynes. Angew. Chem. Int. Ed. 63, e202410394 (2024).
Ge, X. et al. Embedding single Pd atoms on NiGa intermetallic surfaces for efficient and selective alkyne hydrogenation. Angew. Chem. Int. Ed. 63, e202410979 (2024).
Huang, F. et al. Low-temperature acetylene semi-hydrogenation over the Pd1–Cu1 dual-atom catalyst. J. Am. Chem. Soc. 144, 18485–18493 (2022).
Tian, S. et al. Dual-atom Pt heterogeneous catalyst with excellent catalytic performances for the selective hydrogenation and epoxidation. Nat. Commun. 12, 3181 (2021).
Fang, C. et al. Synergy of dual-atom catalysts deviated from the scaling relationship for oxygen evolution reaction. Nat. Commun. 14, 4449 (2023).
Sun, J. et al. MOF-derived Ru1Zr1/Co dual-atomic-site catalyst with promoted performance for Fischer-Tropsch synthesis. J. Am. Chem. Soc. 145, 7113–7122 (2023).
Tian, S. et al. Carbon nitride supported Fe2 cluster catalysts with superior performance for alkene epoxidation. Nat. Commun. 9, 2353 (2018).
Zhang, N. et al. A supported Pd2 dual-atom site catalyst for efficient electrochemical CO2 reduction. Angew. Chem. Int. Ed. 60, 13388–13393 (2021).
Wang, B. et al. A general metal ion recognition strategy to mediate dual-atomic-site catalysts. J. Am. Chem. Soc. 146, 24945–24955 (2024).
Yun, Y. et al. Dynamically precise constructing dual-atom Pd2 catalyst:A monodisperse catalyst with high stability for semi-hydrogenation of alkyne. Adv. Mater. 36, 2409436 (2024).
Adrio, L. A., Nguyen, B. N., Guilera, G., Livingston, A. G. & Hii, K. K. Speciation of Pd(OAc)2 in ligandless Suzuki–Miyaura reactions. Catal. Sci. Technol. 2, 316–323 (2012).
Wang, C.-H., Gao, W.-Y., Ma, Q. & Powers, D. C. Templating metastable Pd2 carboxylate aggregates. Chem. Sci. 10, 1823–1830 (2019).
Stephenson, T. A., Morehouse, S. M., Powell, A. R., Heffer, J. P. & Wilkinson, G. Carboxylates of palladium, platinum, and rhodium, and their adducts. J. Chem. Soc. 667, 3632–3640 (1965).
Hong, F. et al. Strong metal-support interaction boosting the catalytic activity of Au/TiO2 in chemoselective hydrogenation. Chin. J. Catal. 42, 1530–1537 (2021).
Zhang, Y. et al. Boosting the catalysis of gold by O2 activation at Au-SiO2 interface. Nat. Commun. 11, 558 (2020).
Hong, F. et al. Selective and stable Au-Cu bimetallic catalyst for CO-PROX. Nano Res. 16, 9031–9038 (2023).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Zhou, S. et al. Pd single-atom catalysts on nitrogen-doped graphene for the highly selective photothermal hydrogenation of acetylene to ethylene. Adv. Mater. 31, 1900509 (2019).
Bai, L. et al. Efficient industrial-current-density acetylene to polymer-grade ethylene via hydrogen-localization transfer over fluorine-modified copper. Nat. Commun. 14, 8384 (2023).
Bai, R. et al. Encapsulation of palladium carbide subnanometric species in zeolite boosts highly selective semihydrogenation of alkynes. Angew. Chem. Int. Ed. 62, e202313101 (2023).
Gu, J. et al. Synergizing metal-support interactions and spatial confinement boosts dynamics of atomic nickel for hydrogenations. Nat. Nanotechnol. 16, 1141 (2021).
Zhang, J. et al. Tuning hydrogenation chemistry of Pd-based heterogeneous catalysts by introducing homogeneous-like ligands. Nat. Commun. 14, 3944 (2023).
Guo, Y. et al. Pd single-atom catalysts derived from strong metal-support interaction for selective hydrogenation of acetylene. Nano Res. 15, 10037–10043 (2022).
Luneau, M. et al. Achieving high selectivity for alkyne hydrogenation at high conversions with compositionally optimized PdAu nanoparticle catalysts in raspberry colloid-templated SiO2. ACS Catal. 10, 441–450 (2020).
Gao, Q. et al. Atomic layers of B2 CuPd on Cu nanocubes as catalysts for selective hydrogenation. J. Am. Chem. Soc. 145, 19961–19968 (2023).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens Matter 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Hammer, B., Hansen, L. B. & Norskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Perdew, J. P. & Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation-Energy. Phys. Rev. B 45, 13244–13249 (1992).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple (vol 77, pg 3865, 1996). Phys. Rev. Lett. 78, 1396–1396 (1997).
Alavi, A., Hu, P. J., Deutsch, T., Silvestrelli, P. L. & Hutter, J. CO oxidation on Pt(111): An ab initio density functional theory study. Phys. Rev. Lett. 80, 3650–3653 (1998).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Sheppard, D., Terrell, R. & Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 128, 134106 (2008).
Acknowledgements
This work was supported by the National Key R&D Program of China (grant numbers 2021YFA1502802), the National Natural Science Foundation of China (grant numbers 22502215, 22502214, 22579171, 22203012, U21B2092, 22572200, 22202213, and 22402210), the International Partnership Program of Chinese Academy of Sciences (grant number 172GJHZ2022028MI), the Natural Science Foundation of Liaoning Province (2025BS0153), Natural Science Foundation of Chongqing (CSTB2025NSCO-GPX0721), China Postdoctoral Science Foundation (grant number 2024M763338), Shenyang Bureau of science and technology (grant number 24-213-3-25), and Zhongke Technology Achievement Transfer and Transformation Center of Henan Province 2025119. D.M. acknowledges support from the Tencent Foundation through the XPLORER PRIZE and New Cornerstone Investigator Program.
Author information
Authors and Affiliations
Contributions
‡F.H., H.C., J.C., and Z. Y. contributed equally. H.L., D.M., and G.S. conceived the research. F.H. conducted catalyst synthesis and carried out most of the characterization and catalytic reaction tests. H.C. conducted the DFT calculations. J.C. conducted the XAS data analysis. Z.Y. conducted the HAADF-STEM experiments. F.H. wrote the article with edits and approval from all authors. M.P., J.Z., Z.Y., G.W., J.D., and B.S participated in the discussion of this manuscript. All authors contributed to the preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hong, F., Chen, H., Chen, J. et al. Breaking the activity-selectivity trade-off for acetylene semihydrogenation by Pd2 dual-atom site. Nat Commun 17, 4391 (2026). https://doi.org/10.1038/s41467-026-70107-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70107-w
