
Abstract
Capturing the structural changes that molecules undergo during chemical reactions in real space and time is a long-standing dream and an essential prerequisite for understanding and ultimately controlling femtochemistry. A key approach to tackle this challenging task is Coulomb explosion imaging, which has benefited decisively from recently emerging high-repetition-rate X-ray free-electron laser sources. With this technique, information on the molecular structure is inferred from the momentum distributions of the ions produced by the rapid Coulomb explosion of molecules. Retrieving molecular structures from these distributions poses a highly nonlinear inverse problem that remains unsolved for molecules consisting of more than a few atoms. Here, we address this challenge using a diffusion-based Transformer neural network. We show that the network reconstructs unknown molecular geometries from ion-momentum distributions with a mean absolute error below one Bohr radius, which is half the length of a typical chemical bond.
Similar content being viewed by others

Introduction
Imaging molecular structure and, in particular, its temporal evolution is fundamental to understanding and steering ultrafast processes, including chemical reactions1. Several experimental techniques have been developed during the last decades to study the evolution of molecular structure on picosecond and femtosecond time scales. Relying on a variety of measurement concepts, these techniques probe different aspects of molecular structure and dynamics with different levels of fidelity. Static molecular structures can be captured with the highest position-space resolution using electron microscopy2. Time-resolved measurements of evolving molecular geometries often rely on X-rays or high-energy electrons to infer molecular structure from recorded diffraction patterns3,4. Coulomb explosion imaging (CEI), which takes advantage of Coulomb repulsion of nuclei within molecules that are rapidly stripped of their electrons5,6,7, is a less mature technique that can also provide time-resolved information if combined with short laser pulses8. Since atomic motion typically unfolds on femtosecond time scales (as determined by molecular vibrations), CEI with intense femtosecond laser or X-ray pulses has been exploited for studying molecular structural changes9,10,11,12,13,14,15,16,17,18. These pulses rapidly ionize the target molecules, causing their atomic constituents to repel and fragment as a result of Coulombic forces. The resulting ion-momentum distributions contain information about the initial geometric configuration of the molecule before ionization19,20,21,22,23,24.
In all of the imaging methods discussed above, extracting molecular structure from experimental data requires computational algorithms of varying complexity. For diffraction-based imaging techniques, reliable inversion methods are available. In contrast, a corresponding inversion of measured momentum-space data to molecular geometry is not routinely available in the case of CEI. For CEI, geometry retrieval requires solving a highly nonlinear inverse problem, which is extremely challenging when dealing with molecules containing more than 3-4 atoms. In general, inverse problems involve the reconstruction of hidden causal factors from observable data25,26, which are connected by a forward process. If the forward process is trivial to calculate and the noise distribution of this process is known, inverse problems can be solved with the maximum likelihood estimation or the maximum a posteriori approach. Both approaches are typically implemented with an iterative solver, which requires the forward process to be calculated at each step of the iterations. This makes them unfeasible for solving the CEI inverse problem because, in this case, the forward process is driven by the time-dependent many-body interactions governed by quantum mechanics, which is computationally prohibitive to be integrated into an iterative solver. Consequently, direct reconstruction of molecular geometry from CEI has only been demonstrated in a few cases using a classical implementation of the forward process27,28. Most CEI studies5,10,11,13,14,15,17,18,19,20,21,23,24 have relied on a single-pass simulation of the forward process to compare with experimental measurements, leaving accurate, general reconstruction of molecular geometries an open and unresolved problem.
In this work, we address the molecular structure retrieval problem in CEI with a deep generative neural network designed to reconstruct molecular geometries from ion momentum measurements, which we termed MOLEXA (molecular structure reconstruction from Coulomb explosion imaging). It is built on the Transformer architecture29 and the diffusion generative modeling framework30,31,32,33,34,35, with a novel memory mechanism implemented in between the Transformer blocks. The complex forward process of CEI not only renders the classical iterative solvers inapplicable but also poses a severe challenge for deep learning techniques because it is computationally too demanding to generate adequate data for neural network training. To address the issue of data scarcity, MOLEXA uses a two-stage training approach. Stage 1 trains on a large dataset generated using a computationally inexpensive, approximate forward model, while stage 2 fine-tunes the model on a smaller, high-quality dataset derived from ab initio simulations. The dual-phase strategy reduces the mean absolute prediction error to less than one atomic unit, or half the length of a typical chemical bond. Our present work focuses on the reconstruction of the molecular structure from CEI measurements using X-ray pulses, but the demonstrated generative modeling approach can also be applied to building reconstruction models for CEI measurements using optical lasers19,36,37 and highly charged ion beams38.
Results
The MOLEXA network
The MOLEXA model takes the measurable quantities (i.e., the three-dimensional ion momenta measured in coincidence) from CEI as an input and predicts the initial structure of a molecule before its interaction with the X-ray pulse (Fig. 1a). It comprises four modules for input embedding, dynamics extraction, structure denoising, and uncertainty estimation, which will be briefly described in the following. Full network details can be found in Supplementary Note 1.

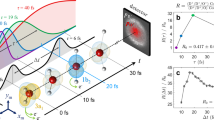
a Illustration of the Coulomb explosion imaging technique and the molecular structure retrieval from its momentum measurements using the MOLEXA neural network. The main architectural details of MOLEXA are displayed in panels (b) and (c). The ball-and-stick models in this and the subsequent figures represent the scaled spatial arrangement of the atomic constituents in the molecules. b Dynamics Extraction Module. c Structure Denoising Module. Images in this figure were created with Blender and Inkscape.
The input to the Embedding Module (Fig. 1a) contains the atomic number, charge state, and molecular-frame momentum of each atomic fragment. The embeddings of the atomic number and charge state are concatenated with the linear projection of the momentum to form atom-wise features. The atomic features are concatenated to create pairwise features, which are then processed by a residual block before being sent as input to the Dynamics Extraction Module.
The Dynamics Extraction Module (Fig. 1b) generates conditioning information used in the Structure Denoising Module. The basic Transformer block, which includes multi-head self-attention, is implemented and accounts for the majority of the computational load for both this and subsequent modules. Instead of directly stacking the Transformer blocks on top of one another, we found that adding memory operations at the end of each block enhances model performance. Similar to the long short-term memory mechanism39, the memory operations (on the right side of Fig. 1b) include a forget gate that regulates what information to discard from the previous state, an update gate that decides which information in the Transformer output should be added to the memory, and an output gate that selectively sends the current state of the memory to the next Transformer block. In comparison to using skip connections, the memory mechanism is found to suppress the mean (maximum) atomic distance and angle errors by 3.6% (4%) and 1.3% (4.4%), respectively. We refer to the combination of the Transformer and memory operations as the “Transformer with Memory” (TM) block. There are six TM blocks in the Dynamics Extraction Module.
The Structure Denoising Module, illustrated in Fig. 1c, reconstructs the molecular structure using a reverse diffusion process. It starts with a noisy molecular structure. Its atomic positions are encoded on the basis of the output of the Dynamics Extraction Module and the current noise level. Pairwise features derived from this encoding are processed by two TM blocks. The output is projected to obtain atom-wise features that are further processed through a self-attention block. The Position Decoder takes the transformed atomic features and outputs a less noisy molecular structure. During inference, this structure is iteratively refined by a diffusion sampler with a noise schedule adapted from Ref. 34. As shown in Fig. 1c, five iterations, corresponding to four intermediate structures, are performed to obtain the final molecular structure. The smaller atom sizes shown for the earlier iterations reflect larger interatomic distances.
The Uncertainty Estimation Module is trained to match the predicted uncertainty with the absolute error between the predicted and ground-truth structures. It can provide uncertainty estimations for the structure predictions. Using pairwise features from the Dynamics Extraction Module and the predicted molecular structure, the Uncertainty Estimation Module estimates the errors of the predicted atomic positions using two TM blocks, followed by an uncertainty decoder. It pre-defines the uncertainty bins r = [0, 0.05, 0.1, …, 9.95] and estimates the probability that the prediction error falls within each of these bins. For each coordinate of the predicted atomic positions, the uncertainty is then calculated as the probability-weighted sum of the bin values.
Training
Unlike text or image generation models, for which there exists an enormous amount of training data, deep learning models in physical sciences often face the data scarcity issue, which is one of the main obstacles preventing the widespread adoption of deep learning techniques for solving physics-related problems. For the molecular structure retrieval problem, we created two training datasets by performing Coulomb explosion simulations at two levels of theory. One level involves the ab initio calculation of the XFEL-induced Coulomb explosion of molecules, which tracks the quantum transition probabilities across all participating electronic configurations while treating the nuclei as moving in a classical force field40. It has been shown to produce results that agree with experiments18. A similar level of theory was used, for example, in Refs. 21,23. These high-level simulations are computationally expensive. Thus, we created only a small dataset containing 76,000 samples, using a thousand CPUs for more than a month. A portion of this dataset was kept for validation (10%) and testing (10%) purposes. Since the computation time scales roughly exponentially with the number of atoms, these simulations were limited to molecules with fewer than ten atoms, which was a compromise to balance the dataset size with computational constraints. The second level of theory is a much cheaper, classical Coulomb explosion model with crude approximations22. It was used to generate a dataset that is about a hundred times larger. MOLEXA was first trained on this large but inaccurate dataset and then on the small dataset, which is more accurate and best reflects the reality of a CEI experiment. We found that the two-stage training approach reduced the structure prediction error by a factor of two compared to training solely on the smaller but more accurate dataset.
Before training in each stage, both the ion-momentum and ground-truth position distributions were centered and aligned to a common molecular frame. Specifically, for each molecule, the emission direction of the heaviest ion fragment is defined as the x-axis. Then the cosine similarities of the momenta of the other ion fragments relative to the x direction are calculated. The ion momentum with the smallest cosine similarity is chosen to define the y-axis through Gram-Schmidt orthonormalization so that this momentum vector would fall within the first quadrant of the x-y plane. Both the momenta of all ion fragments and the initial molecular structure, after being centered at the origin, are transformed into this coordinate frame. For diatomics, the coordinate system is fully determined once the emission direction of the heavier ion fragment is defined as the x-axis. This pre-alignment procedure, applied consistently across all molecules, transforms the structure retrieval task from one with arbitrary coordinate frames to one within a fixed molecular frame, thereby eliminating the need to explicitly incorporate translational and rotational invariance into the model.
The loss function consists of two parts: a weighted mean squared error for the predicted molecular structures, and a cross-entropy loss for uncertainty estimations. Both parts were used throughout the two training stages. The Uncertainty Estimation Module was further fine-tuned using the validation dataset while keeping the other modules frozen, during which only the second part of the loss function was used. All reported results were generated from the test dataset. The training and validation datasets only contain molecules with fewer than eight atoms, while the eight- or nine-atom molecules were set aside to test the generalization capability of the model. Additional details on loss function, training, and testing are provided in the Methods section.
Model performance
Using the test dataset of molecules with less than eight atoms, the mean absolute error (MAE) is 0.52 a.u. (atomic units), the mean (maximum) distance error (DE) of all atomic pairs in a molecule is 0.98 (2.11) a.u., and the mean (maximum) angle error (AE) of all directional triplets in a molecule is 13.97 (38.39) degrees. MOLEXA, which was trained on only molecules with up to seven atoms, is also capable of reconstructing the structure of molecules containing eight or nine atoms. For these molecules larger than those included in the training dataset, the MAE is 0.66 a.u., the mean (maximum) DE is 1.16 (3.43) a.u., and the mean (maximum) AE is 14.12 (57.91) degrees. The inference time distribution evaluated on all test molecules is plotted in Supplementary Fig. 1 and has a mean of 59.8 ms. Each of the reconstructed molecular structures is from a single model prediction. Based on the standard deviations of the model predictions plotted in Supplementary Fig. 2, the expected discrepancy between the corresponding atomic coordinates of two independent reconstructions for a single molecule is about 0.07 a.u.
Figure 2 provides an overview of the structure retrieval performance of the model. In each column of the figure, the results for molecules consisting of N atoms are presented, showing exemplary structures predicted with low (top row) and high (second row) reconstruction uncertainties. The two bottom rows indicate how the general performance of MOLEXA (in terms of mean DE and AE) behaves as a function of the predicted uncertainty for molecules of different sizes. The Uncertainty Estimation Module produces uncertainties for all coordinates of all atoms in a molecule. The overall uncertainty displayed here was obtained by taking an average of the uncertainties of all atomic positions of a molecule. The heatmaps in Fig. 2, as well as those in Supplementary Figs. 4–5 for MAE, maximum DE and AE, show that there is a strong correlation between the predicted uncertainty and the errors of the reconstructed molecular structures. This indicates that the former can serve as a reliable metric for assessing whether a MOLEXA reconstruction is trustworthy or not. It can be observed from these heatmaps that the predicted uncertainties can underestimate the reconstruction errors, especially for larger molecules with larger prediction errors. We attribute this to a training bias caused by the Uncertainty Estimation Module being trained on reconstructions, most of which have relatively low errors. Such bias can potentially be mitigated with a weighted cross-entropy loss in future developments.

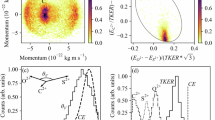
The columns from left to right represent molecules with an increasing number of atoms. Top row: Exemplary structure predictions with low predicted uncertainties. The predicted and ground-truth structures are plotted as opaque and semi-transparent ball-and-stick models, respectively. The corresponding uncertainty, mean DE, and AE are listed below each molecular structure. The color coding of the elements is as follows—H: white, C: gray, N: blue, O: red, F: cyan, Si: brown, P: orange, S: yellow, and Cl: green. The ball-and-stick models were plotted with PyMOL, which considers two atoms bonded if their distance is smaller than a tolerance-expanded sum of their covalent sizes. Second row: Exemplary structure predictions with high predicted uncertainties. The maximum DE and AE values for these molecules are summarized in Supplementary Tables 2 and 3. The two bottom rows depict the dependence of the mean DE and AE on the predicted uncertainty. The density plots use the same color scale. The dash-dotted line in the bottom left corner marks the mean DE (1.27 a.u.) of reconstructions with the classical \(\frac{1}{KER}\) model, and the dashed line marks the error (0.49 a.u.) for the optimized empirical model \(\frac{0.797}{KER}\). The corresponding MOLEXA input data, including the ion charge states and momentum distributions, are shown in Supplementary Fig. 3. Source data are provided as a Source Data file.
With MOLEXA being a diffusion model, an uncertainty quantification for each sample is also obtainable by calculating the standard deviation of an ensemble of its structure predictions. The uncertainties calculated with these two approaches are shown in Supplementary Fig. 2 to have an approximately linear relationship. The former method is used here because it provides an uncertainty estimate for each prediction without requiring an ensemble of predictions. Note that although an uncertainty estimate can assist the assessment of the trustworthiness of a prediction, it is not effective in selecting the most accurate reconstruction out of many predictions for a molecule. This is indicated by the small correlation coefficients between the uncertainty estimate and prediction errors plotted in Supplementary Fig. 6.
Figure 2 also depicts the dependency of prediction errors on the size of the molecules examined. For diatomics, the mean DE is 0.155 a.u. As a reference, the mean DEs from calculating the bond length of the same set of diatomic molecules with the classical \(\frac{1}{KER}\) model and the optimized empirical \(\frac{0.797}{KER}\) model are 1.27 and 0.49 a.u., respectively. (KER stands for the kinetic energy release from Coulomb explosion. The empirical factor 0.797 was determined by minimizing the error of the \(\frac{c}{KER}\) model on the test data.) For larger molecules, the average DE and AE distributions gradually shift upwards, implying that it becomes more difficult for the model to learn the underlying X-ray-induced dynamics in molecules containing more atoms. Although the increased prediction error for molecules with eight or nine atoms is still acceptable, it is expected to increase even more for larger molecules. Corresponding example predictions for the 1,3-cyclohexadiene molecule containing fourteen atoms are shown in Supplementary Fig. 7. As expected, they show large discrepancies from the ground-truth structures. In addition to the dependence on the number of atoms, predictions for molecules with larger atomic distances tend to result in larger errors, as indicated by Supplementary Fig. 8. Training with a more diverse dataset that includes larger molecules with larger atomic distances would be needed to break these limitations and further extend the applicability of MOLEXA in the future.
The model has two other major limitations. One originates from the non-unique mapping between ion momentum vectors and real-space molecular geometries as revealed by Ref. 41, which can increase the overall prediction error. In future developments, the non-uniqueness issue can be mitigated by a model that makes use of multiple coincidence channels as input data, in contrast to a single channel used by the current model. The other limitation is that the input data must include all ions produced by the Coulomb explosion of the molecule. But in a CEI experiment, the complete coincident ion detection from each explosion is a relatively low-probability event, especially for larger systems. Our current way to circumvent this limitation is by distilling the required full-coincidence momentum vectors from the data accumulated over many explosion events21,22,23,24. This approach is used to prepare the input data of the experimentally studied molecules discussed in the next section. Further development of the neural network is needed to make it capable of molecular structure prediction from partial coincidence sets, which would be particularly beneficial for single-event reconstruction.
Application of MOLEXA
In this subsection, we first demonstrate the ability of MOLEXA to perform the inversion of experimental data into real-space molecular geometries. For this, we used the data acquired during several experiments carried out at the European X-ray Free-Electron Laser facility to reconstruct the equilibrium geometry of ground-state molecules, including water, tetrafluoromethane, and ethanol. No further molecule-specific input was provided to the model for retrieving the structures. The experiments were performed in multiple CEI beamtimes using the COLTRIMS (Cold Target Recoil Ion Momentum Spectroscopy) Reaction Microscope42 at the Small Quantum Systems (SQS) instrument. The beamtimes and X-ray pulse parameters are summarized in Supplementary Table 4. In all experiments, the molecular samples were delivered to the interaction region through a supersonic expansion followed by three skimmers and an adjustable collimator. The distance from the nozzle to the interaction region is about 54 cm. The pressure in the main chamber was maintained at 1 × 10−11 mbar. The focal spot size of the X-ray beam was about 1.5–3 μm, and the X-ray pulse duration was less than 25 fs based on the 250 pC electron bunch charge. The ion fragments produced in the interaction region were guided by a homogeneous electric field to a time- and position-sensitive detector. The lab-frame momentum vectors of the ion fragments were then reconstructed from the detector readouts, which were subsequently transformed into the molecular frame with the alignment procedure discussed in the Training subsection.
In real molecules, the nuclear ground state exhibits a spatial distribution due to the uncertainty principle, such that different nuclear geometries are sampled even in the absence of excitation. Because the mapping between ion momentum and molecular geometry is nonlinear, averaging in momentum space does not strictly correspond to averaging in real space. In principle, the equilibrium molecular structure is best estimated by reconstructing geometries from individual single-shot, full-coincidence Coulomb explosion events and averaging the predicted structures in real space. But single-shot full-coincidence detection remains challenging for today’s CEI experiments. In the following, the structures are instead predicted from the coincident momentum vectors (Supplementary Figs. 9–14) obtained by averaging over the single-shot data. This procedure yields an approximate equilibrium geometry from the ensemble-averaged observables, rather than the full nuclear probability distribution or any single instantaneous molecular configuration.
As a first example, Fig. 3a shows the reconstruction of the molecular structure of water molecules. The employed dataset43 used in this analysis is identical to the one used in Ref. 16. The 2D heatmap in the left-most part of the panel displays the experimentally measured molecular-frame momentum distribution of two protons detected in coincidence with a singly charged oxygen ion. Next to it in the middle, we show an illustration of the centroids of the momentum distributions of the three ions, which serve as the input for MOLEXA. The results obtained from the reconstruction are shown on the right. The reconstructed molecular geometry (opaque) is plotted on top of the ground truth (semi-transparent), with the corresponding MAE being 0.296 a.u. The mean (maximum) DE and AE are 0.674 (1.199) a.u. and 18.459 (27.689) degrees, respectively. Next, we test the model on tetrafluoromethane, a molecule consisting of five atoms. The corresponding reconstruction is shown in Fig. 3b. The left-most panel depicts the momenta of three of the four fluorine ions in a molecular frame spanned by the fourth fluorine ion and one of the three. The reconstructed position-space geometry has an MAE of 0.238 a.u., a mean (maximum) DE of 0.66 (1.117) a.u., and a mean (maximum) AE of 5.943 (17.173) degrees. The data was recorded during the commissioning of the SQS reaction microscope44.

a Molecular structure reconstruction of water. The measured 2D momentum map is shown on the left. To its right is the illustration of the averaged momentum distribution of the three ion fragments, which is used as the input to MOLEXA. In the real space, the reconstructed and ground-truth structures are plotted as opaque and semi-transparent ball-and-stick models, respectively. b Molecular structure reconstruction of tetrafluoromethane. c Molecular structure reconstruction of ethanol. The corresponding orientations of the pre-explosion molecule are displayed at the top of the 2D momentum maps. The color coding of the elements is as follows—H: white, C: gray, O: red, and F: cyan. The ground-truth structures are from the NIST Computational Chemistry Comparison and Benchmark Database55. Source data are provided as a Source Data file.
As a benchmark for an application to molecules with up to nine atoms, we applied MOLEXA to a CEI dataset recorded for ethanol molecules45. The results are shown in Fig. 3c. The 2D maps show the molecular-frame momentum distributions of protons in the coincidence channel O+/C+/C+/H+, viewed from three different perspectives. We added the corresponding orientation of the real-space molecule to the top of each graph to aid the identification of the six protons in the momentum maps. The input to the model is again obtained by taking the centroids of the momentum distributions of the nine ions. The retrieved molecular geometry plotted together with the ground truth at the right has an MAE of 0.429 a.u. The mean (maximum) DE and AE are 1.024 (2.007) a.u. and 9.011 (32.319) degrees, respectively. More details on the reconstruction from experimental data, including the momentum distributions of other ions not displayed in Fig. 3, the momentum centroid data, and the reconstructed atomic coordinates as well as the predicted error estimates, can be found in Supplementary Figs. 9–14 and Supplementary Tables 5–7.
The ultimate aim of CEI is to directly observe molecular dynamics during a chemical reaction in a time-resolved manner. In order to achieve this, coincident momentum-space fragmentation patterns are measured at different instants during the chemical reaction, thus allowing us to study the molecular structural changes as the chemical reaction unfolds on femtosecond or longer time scales. In the following example, we exploit MOLEXA to reconstruct the different geometries of cyclobutene as predicted by ab initio simulations46. The electrocyclic reactions of cyclobutene represent a textbook example of pericyclic reactions that are among the important classes of chemical reactions in organic chemistry. Figure 4a shows that MOLEXA is capable of reconstructing different possible geometrical changes, including ring opening, twisting, and proton migration, after cyclobutene is excited from the ground state (S0) to the S1 state. In Fig. 4b, MOLEXA is used to reconstruct position-space “snapshots” of cyclobutene as it undergoes a ring-opening reaction. The reconstructions show that the model can provide insight about the overall structure and identify gross structural rearrangements such as proton migration and ring opening. Further details on the reconstruction of cyclobutene geometries can be found in Supplementary Tables 8–14. More examples demonstrating the model’s capability to reconstruct varying structures of molecules are displayed in Supplementary Fig. 15. It is worth noting that the reconstructions in Fig. 4 are idealized scenarios where the input momentum vectors correspond to a single well-defined molecular geometry. This can work if the input coincidence data is assembled from a single explosion event. For time-resolved CEI experiments, the accumulated data for each time delay typically results from a mixture of geometries corresponding to multiple quantum states. Classification of the experimental data into individual states, based on, e.g., ion charge-state characteristics and kinetic energies18, is hence required prior to applying MOLEXA for reconstruction.

a Reconstructed geometries of cyclobutene in its S0 and S1 states. b Reconstructed “snapshots” of cyclobutene during a chemical reaction. The color coding of the elements is as follows—H: white and C: gray. Source data are provided as a Source Data file.
Discussion
MOLEXA is a powerful neural network designed for molecular structure reconstruction with the CEI technique. It allows for inverting momentum-space datasets to position space, providing the structure of a molecule right before its explosion by an X-ray pulse and showing particular effectiveness in reconstructing the overall structure of molecules. In addition, it is capable of providing an uncertainty estimate for its reconstructed molecular geometries. By employing time-resolved CEI datasets, MOLEXA has the potential to provide “snapshots” of a molecule at different instants during a chemical reaction, which can enable the use of the CEI technique for direct reconstruction of molecular dynamics in position space as they unfold on their natural time scales.
Apart from taking advantage of recent advances in deep learning, such as the Transformer framework and diffusion-based generative modeling, MOLEXA utilized the “Transformer with Memory” architecture and went through a two-stage training, both of which were important for achieving its effectiveness in molecular structure predictions. It demonstrates the potential of generative modeling in solving inverse problems that classical approaches cannot address due to the excessive complexity of their forward models, which prevents integration into an iterative procedure. Even with deep learning techniques, solving such problems poses a challenge because of training data scarcity. The two-stage modeling can be applied as a general approach to addressing this issue when a complicated forward process can be approximated as a simple model.
Methods
Dataset creation
For the ab initio simulation, we used a theoretical model that combines Monte Carlo/Molecular Dynamics simulations (MC/MD)40 with a classical over-the-barrier (COB) model47,48,49 to track inner-shell photoionization, Auger-Meitner cascades, valence electron redistribution, and nuclear dynamics. Photoabsorption and inner-shell cascade processes were modeled using a Monte Carlo method to calculate quantum electron transition probabilities across all participating electronic configurations (ECs), including ground, core-excited, and valence-excited states. The electronic-structure calculations were based on the relativistic Hartree-Fock-Slater (HFS) method, which provided bound-state and continuum wavefunctions for computing cross sections of photoionization, shake-off, electron-impact ionization, and electron-ion recombination, as well as Auger-Meitner and fluorescence decay rates. The molecular-dynamics component tracked the motion of atoms, ions, and delocalized ionized electrons. The COB model simulates electron-transfer dynamics in the valence shell. In this model, an electron fills a vacancy in the valence shell of a neighboring atom when its binding energy is higher than the Coulomb barrier. When the atoms are far apart, the resulting Coulomb barrier suppresses electron transfer. Electron transfer takes place instantaneously when the electron orbital energy is higher than the Coulomb barrier.
With this ab initio model, the Coulomb explosion of three hundred different molecules with fewer than ten atoms was first simulated in their equilibrium geometry. The X-ray pulses have a photon energy of 2 keV, pulse energy of 1 mJ, pulse duration of 15 fs, and focal spot size of 1 μm. In order to expand the dataset, the simulations were additionally performed tens of times on each of these molecules after randomly varying their structures. Because of the stochastic nature of the X-ray interaction with molecules, the atomic charge-state combination of the resulting ion fragments from a molecule can vary from one simulation trajectory to another. With a hundred thousand trajectories simulated for each molecule at a fixed structure, the number of trajectories ending at each of the possible charge-state combinations was enumerated. Only combinations with a count greater than three hundred were considered. The momentum of each ion fragment was obtained by averaging all trajectories. For every such charge-state combination, the atomic number, charge state, and momentum of all ion fragments, as well as the initial atomic coordinates of the molecule, were included in the dataset as a single entry. With an average of ten structures simulated for each of the three hundred molecules and an average of about ten charge-state combinations produced from a molecule at a particular structure, the dataset contains 76,000 entries. It was further split into training (80%), validation (10%), and test (10%) datasets. The training dataset was used in the second step of the two-stage modeling process. Exemplary samples from the test dataset are shown in Supplementary Fig. 17 together with the corresponding predictions.
Because the ab initio simulation was computationally expensive and could only be used to generate a small dataset, an approximate forward Coulomb explosion model22 was used to create a dataset about two orders of magnitude larger. The model describes the charge-up of each atom in a molecule with a modified error function that increases from zero to the final charge number within a time window controlled by the constant τ. For the simulation, τ was set to be 45 fs, which was determined by minimizing the discrepancy of the results of the approximate model with respect to those of the ab initio simulations. Using the time-dependent charge states given by the modified error function, the Coulomb explosion dynamics were simulated with the Runge-Kutta approach that propagates the time-dependent positions and velocities of ion fragments according to classical mechanics. The “molecules” used for this approximate simulation were generated by enumerating all possible combinations of the 9 elements (H, C, N, O, F, Si, P, S, and Cl), with the number of atoms in each combination less than 10. The positions of the atoms in a “molecule” were sampled from a uniform distribution ranging from −10 a.u. to 10 a.u. The dataset produced from the simulation with these “molecules” consists of six million entries and was used for the first step of the two-stage modeling process.
Training details
During training, the reverse diffusion process (Structure Denoising Module) was run only once for each training step. Instead of taking a random structure as input, it starts with a noisified ground-truth structure with the noise level controlled by σi. The Structure Denoising Module was trained to denoise this input and generate a geometry \({{{{\bf{G}}}}}_{i}^{prediction}\) that is a reconstruction of the ground truth \({{{{\bf{G}}}}}_{i}^{ground\_truth}\). The corresponding loss function is
where the weight wi is set according to Ref. 34 and given by
with σdata determined by the standard deviation of the molecular structures in the dataset. In addition to structure reconstruction, MOLEXA was trained to estimate the uncertainty of its predicted structures. As already mentioned in the main text, it first gets the probability \({s}_{n}^{i}\) that the uncertainty of the ith predicted coordinate \({x}_{i}^{prediction}\) falls into the nth bin of the pre-defined uncertainty list [r0, …, r200]. The absolute error \(| {x}_{i}^{prediction}-{x}_{i}^{ground\_truth}|\) is classified according to this list as a one-hot encoded vector qi. The loss function for the uncertainty estimate is then calculated as the averaged cross entropy
The combined loss function used during training is given by
where cx and cu are the weights of the structure retrieval and uncertainty estimation loss functions, respectively.
The neural network was trained through two stages. The weights were initialized using the orthogonal Glorot initialization50,51 with a scale of 2 for the linear layers and sampled from the uniform distribution with a range from \(-\sqrt{3}\) to \(\sqrt{3}\) for the embedding layers. In the first stage, it was trained on the large dataset generated by the approximate forward model. The weight cx in the loss function was set to 400. And cu was set to 0.1 for the first seven epochs and 1 afterwards. For the second stage, the training was performed with the dataset, which is about a hundred times smaller and generated by the ab initio forward model. The weights cx and cu in the loss function were set to 400 and 0.01, respectively. In order to improve the accuracy of uncertainty predictions, after the two-stage training, the network was further trained on the validation dataset. Only the Uncertainty Estimation Module was trained while the other modules were kept frozen. The weight cx was set to 0 and cu to 0.01. During all training phases, the Adam optimizer52 was used for optimization. Its parameters β1, β2, and ϵ were fixed at 0.9, 0.99, and 10−5, respectively. The learning rate was kept at 0.001. Training with learning rate decay was tested, but it did not improve the prediction errors. More details on the two-stage training are summarized in Supplementary Table 1.
Coordinate frame transformations for experimentally studied molecules
The molecular frames used by the 2D maps in Fig. 3 were defined with a procedure similar to that described in the main text. The flying direction of a reference ion (O+ for water and ethanol, and F+ for tetrafluoromethane, as indicated by the arrows in the 2D maps of Fig. 3) was set as x-axis. The y-axis was then defined such that the momentum vector of a second reference ion (H+ for water, and F+ for tetrafluoromethane) falls within the positive x-z plane. For ethanol, the second reference ion was chosen to be the C+ ion that has a smaller cosine similarity with respect to the first reference ion (O+). The y-axis was defined such that the momentum vector of this second reference ion falls within the positive x-y plane. The molecular-frame momentum distributions and their centroids are shown for the four molecules in Supplementary Figs. 9–14. The atomic coordinates reconstructed by MOLEXA together with the ground truth are listed in Supplementary Tables 5–7.
Data availability
The datasets and model weights used in this study have been deposited in the Zenodo database under accession code 1579447053. The raw data recorded for the water, tetrafluoromethane, and ethanol experiments at the European XFEL are available at https://doi.org/10.22003/XFEL.EU-DATA-002150-00, https://doi.org/10.22003/XFEL.EU-DATA-002181-00, and https://doi.org/10.22003/XFEL.EU-DATA-002926-00, respectively. Source data are provided with this paper.
Code availability
The source code is publicly available at https://github.com/xli025/molexa54.
References
Zewail, A. H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond. J. Phys. Chem. A 104, 5660–5694 (2000).
Egerton, R. F. Physical Principles of Electron Microscopy. Vol. 56, (Springer US, New York, NY, 2005).
Centurion, M., Wolf, T. J. A. & Yang, J. Ultrafast Imaging of Molecules with Electron Diffraction. Annu. Rev. Phys. Chem. 73, 21–42 (2022).
Odate, A., Kirrander, A., Weber, P. M. & Minitti, M. P. Brighter, faster, stronger: ultrafast scattering of free molecules. Adv. Phys. X 8, 2126796 (2023).
Vager, Z., Naaman, R. & Kanter, E. P. Coulomb Explosion Imaging of Small Molecules. Science 244, 426–431 (1989).
Levin, J. et al. Study of Unimolecular Reactions by Coulomb Explosion Imaging: The Nondecaying Vinylidene. Phys. Rev. Lett. 81, 3347 (1998).
Herwig, P. et al. Imaging the Absolute Configuration of a Chiral Epoxide in the Gas Phase. Science 342, 1084–1086 (2013).
Stapelfeldt, H., Constant, E., Sakai, H. & Corkum, P. B. Time-resolved Coulomb explosion imaging: A method to measure structure and dynamics of molecular nuclear wave packets. Phys. Rev. A 58, 426 (1998).
Ergler, T. et al. Spatiotemporal Imaging of Ultrafast Molecular Motion: Collapse and Revival of the \({{{{{\rm{D}}}}}_{2}}^{+}\) Nuclear Wave Packet. Phys. Rev. Lett. 97, 193001 (2006).
Hishikawa, A., Matsuda, A., Fushitani, M. & Takahashi, E. J. Visualizing Recurrently Migrating Hydrogen in Acetylene Dication by Intense Ultrashort Laser Pulses. Phys. Rev. Lett. 99, 258302 (2007).
Jiang, Y. H. et al. Ultrafast Extreme Ultraviolet Induced Isomerization of Acetylene Cations. Phys. Rev. Lett. 105, 263002 (2010).
Hansen, J. L. et al. Control and femtosecond time-resolved imaging of torsion in a chiral molecule. J. Chem. Phys. 136, 204310 (2012).
Ibrahim, H. et al. Tabletop imaging of structural evolutions in chemical reactions demonstrated for the acetylene cation. Nat. Commun. 5, 4422 (2014).
Liekhus-Schmaltz, C. E. et al. Ultrafast isomerization initiated by X-ray core ionization. Nat. Commun. 6, 8199 (2015).
Endo, T. et al. Capturing roaming molecular fragments in real time. Science 370, 1072–1077 (2020).
Jahnke, T. et al. Inner-Shell-Ionization-Induced Femtosecond Structural Dynamics of Water Molecules Imaged at an X-Ray Free-Electron Laser. Phys. Rev. X 11, 041044 (2021).
Jahnke, T. et al. Direct observation of ultrafast symmetry reduction during internal conversion of 2-thiouracil using Coulomb explosion imaging. Nat. Commun. 16, 2074 (2025).
Li, X. et al. Imaging a light-induced molecular elimination reaction with an X-ray free-electron laser. Nat. Commun. 16, 7006 (2025).
Pitzer, M. et al. Direct Determination of Absolute Molecular Stereochemistry in Gas Phase by Coulomb Explosion Imaging. Science 341, 1096–1100 (2013).
Pitzer, M. et al. Absolute Configuration from Different Multifragmentation Pathways in Light-Induced Coulomb Explosion Imaging. ChemPhysChem 17, 2465–2472 (2016).
Boll, R. et al. X-ray multiphoton-induced Coulomb explosion images complex single molecules. Nat. Phys. 18, 423–428 (2022).
Li, X. et al. Coulomb explosion imaging of small polyatomic molecules with ultrashort x-ray pulses. Phys. Rev. Res. 4, 013029 (2022).
Richard, B. et al. Imaging collective quantum fluctuations of the structure of a complex molecule. Science 389, 650–654 (2025).
Green, A. E. et al. Visualizing the Three-Dimensional Arrangement of Hydrogen Atoms in Organic Molecules by Coulomb Explosion Imaging. J. Am. Chem. Soc. 147, 37133–37143 (2025).
Ongie, G. et al. Deep Learning Techniques for Inverse Problems in Imaging. JSAIT 1, 39–56 (2020).
Zhao, Z., Ye, J. C. & Bresler, Y. Generative Models for Inverse Imaging Problems: From mathematical foundations to physics-driven applications. IEEE Signal Process. Mag. 40, 148–163 (2023).
Légaré, F. et al. in Ultrafast Phenomena XIV (eds. Kobayashi, T., Okada, T., Kobayashi, T., Nelson, K. A. & De Silvestri, S.) 888–890 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2005).
Kunitski, M. et al. Observation of the Efimov state of the helium trimer. Science 348, 551–555 (2015).
Vaswani, A. et al. Attention Is All You Need. Adv. Neural Inf. Process. Syst. 5998–6008 (2017).
Sohl-Dickstein, J., Weiss, E., Maheswaranathan, N. & Ganguli, S. Deep Unsupervised Learning Using Nonequilibrium Thermodynamics. Proc. 2256–2265 (ICML, 2015).
Ho, J., Jain, A. & Abbeel, P. Denoising Diffusion Probabilistic Models. Proc. (NeurIPS, 2020).
Song, Y. et al. Score-Based Generative Modeling through Stochastic Differential Equations. Proc. (ICLR, 2021).
Song, J., Meng, C. & Ermon, S. Denoising Diffusion Implicit Models. Proc. (ICLR, 2021).
Karras, T., Aittala, M., Aila, T. & Laine, S. Elucidating the Design Space of Diffusion-Based Generative Models. Adv. Neural Inf. Process. Syst. 35, 26565–26577 (2022).
Xu, M. et al. Geodiff: A Geometric Diffusion Model for Molecular Conformation Generation. Proc. (ICLR, 2022).
Bhattacharyya, S. et al. Strong-Field-Induced Coulomb Explosion Imaging of Tribromomethane. J. Phys. Chem. Lett. 13, 5845–5853 (2022).
Lam, H. V. S. et al. Differentiating Three-Dimensional Molecular Structures Using Laser-Induced Coulomb Explosion Imaging. Phys. Rev. Lett. 132, 123201 (2024).
Yuan, H. et al. Coulomb Explosion Imaging of Complex Molecules Using Highly Charged Ions. Phys. Rev. Lett. 133, 193002 (2024).
Hochreiter, S. & Schmidhuber, J. Long Short-Term Memory. Neural Comput. 9, 1735–1780 (1997).
Ho, P. J. & Knight, C. Large-scale atomistic calculations of clusters in intense x-ray pulses. J. Phys. B: At. Mol. Opt. Phys. 50, 104003 (2017).
Sayler, A. M. et al. Nonunique and nonuniform mapping in few-body Coulomb-explosion imaging. Phys. Rev. A 97, 033412 (2018).
Dörner, R. et al. Cold Target Recoil Ion Momentum Spectroscopy: a ‘momentum microscope’ to view atomic collision dynamics. Phys. Rep. 330, 95–192 (2000).
Piancastelli, M. N. et al. Dynamic response of water molecules to a ultra-intense X-ray beam. https://doi.org/10.22003/XFEL.EU-DATA-002150-00 (2019).
Jahnke, T. et al. Multi-photon ionization of atoms and small molecules. https://doi.org/10.22003/XFEL.EU-DATA-002181-00 (2019).
Boll, R. et al. REMI commissioning. https://doi.org/10.22003/XFEL.EU-DATA-002926-00 (2021).
Ong, M. T. The Photochemical and Mechanochemical Ring Opening of Cyclobutene from First Principles. (University of Illinois at Urbana-Champaign, 2010).
Ryufuku, H., Sasaki, K. & Watanabe, T. Oscillatory behavior of charge transfer cross sections as a function of the charge of projectiles in low-energy collisions. Phys. Rev. A 21, 745 (1980).
Niehaus, A. A classical model for multiple-electron capture in slow collisions of highly charged ions with atoms. J. Phys. B: Atom. Mol. Phys. 19, 2925 (1986).
Ho, P. J. et al. X-ray induced electron and ion fragmentation dynamics in IBr. J. Chem. Phys. 158, 134304 (2023).
Glorot, X. & Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. Proc. AISTATS 9, 249–256 (2010).
Saxe, A. M., McClelland, J. L. & Ganguli, S. Exact solutions to the nonlinear dynamics of learning in deep linear neural networks. https://arxiv.org/abs/1312.6120 (2014).
Kingma, D. P. & Ba, J. Adam: A Method for Stochastic Optimization. Proc. (ICLR, 2015).
Li, X. Datasets and model weights for paper “Generative modeling enables molecular structure retrieval from coulomb explosion imaging”, Zenodo. https://doi.org/10.5281/zenodo.15794470 (2025).
Li, X. xli025/molexa: v1.0.0, Zenodo. https://doi.org/10.5281/zenodo.18510545 (2026).
NIST Computational Chemistry Comparison and Benchmark Database. NIST Standard Reference Database Number 101. http://cccbdb.nist.gov/. Release 22 May (2022).
Acknowledgements
We acknowledge the LCLS data team and the SLAC Shared Science Data Facility (S3DF) for providing the computing and data storage used in model development. We acknowledge the teams of the three European XFEL experiments (2150, 2181, and 2926) for sharing the associated data. X.L. would like to thank Patricia Vindel Zandbergen for her help related to the model testing and Philipp Schmidt for his support in experimental data processing. This work is supported by the Linac Coherent Light Source, SLAC National Accelerator Laboratory, which is funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. P.J.H. is supported by the U.S. DOE BES Chemical Sciences, Geosciences, and Biosciences Division under Contract No. DE-AC02-06CH11357. D.R. and A.R. are supported by grant no. DE-FG02-86ER13491 from the same funding agency, and also acknowledge dedicated support for ML/AI developments through the GRIPex program at Kansas State University. F.T. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project 509471550, Emmy Noether Program. T.J.A.W. was supported by the Atomic, Molecular, and Optical Sciences Program of the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, through Contract No. DE-AC0276SF00515.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology: X.L. with support from J.B.T., J.P.C. and P.J.H.; Dataset creation and curation: X.L. and P.J.H.; Generative model development: X.L., J.H., M.X. and S.E.; CEI experiments: X.L., T.J., R.B., M.M., M.N.P., D.R., A.R. and F.T.; CEI experiment data analysis: X.L. and T.J.; Original draft: X.L., T.J., R.B., J.H., M.X., D.R., F.T., T.J.A.W., S.E. and P.J.H.; Final draft: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xinwen Ma, Henrik Stapelfeldt, Daniel Strasser, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Jahnke, T., Boll, R. et al. Generative modeling enables molecular structure retrieval from Coulomb explosion imaging. Nat Commun 17, 3430 (2026). https://doi.org/10.1038/s41467-026-70160-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70160-5
