
Abstract
Influenza B virus (IBV) has circulated in the human population for a long time, yet the evolutionary mechanisms responsible for host adaptation remain poorly understood. Here we show that recent IBV strains exhibit an enhanced ability to evade the innate immune response and an increased replication efficiency compared with earlier strains. Our data indicate that the nonstructural protein 1 (NS1) of recent IBV strains interacts with TUFM and LC3B to induce mitophagy, leading to degradation of MAVS, suppression of interferon production and enhanced viral replication. In contrast, NS1 of earlier strains displays minimal ability to trigger mitophagy-mediated MAVS degradation. Sequence analyses show that, over the past two decades, IBV has acquired a phenylalanine (F)-to-leucine (L) substitution at residue 247 of NS1, altering its interaction with LC3B. A rescued recent IBV strain carrying the NS1-L247F mutation exhibits diminished NS1-LC3B binding, impaired mitophagy, and attenuated replication. Our study shows that adaptive evolution involving a single mutation in NS1 enables mitophagy-mediated innate immune evasion, contributing to IBV adaptation to the host.
Similar content being viewed by others


Introduction
Influenza A virus (IAV) and influenza B virus (IBV) cause significant morbidity and mortality each year during seasonal epidemics. IBVs have circulated in humans since at least 1940 (B/Lee/1940)1. In the 1980s, two antigenically and genetically distinct lineages of IBV, B/Victoria and B/Yamagata, were detected2. On average, IBV accounts for approximately 25% of influenza cases; however, this proportion can be significantly higher in certain seasons and geographic regions3,4, reaching up to 50–80% of infections in some seasons5,6. Notably, between 2001 and 2009 in the United States, IBV was responsible for as much as 66% of all influenza-related healthcare costs7. This disease burden is typically more severe in school-aged children and adolescents, who can develop multiple systemic complications6,8,9.
Under host immune pressure, influenza viruses undergo adaptive evolution through mutation, antigenic shift, and recombination, leading to annual global epidemics and occasional pandemics. Compared with IAV, the adaptive evolution of IBV is poorly understood. The analysis of the relative rates of nonsynonymous and synonymous substitutions per site (dN/dS) provides direct evidence for adaptive evolution. It has been suggested that IBV hemagglutinin (HA, dN/dS = 0.22) and neuraminidase (NA, dN/dS = 0.20) display relatively high dN/dS values compared with most other segments, indicating that IBV HA and NA are under strong selection pressure from host adaptive immunity. Notably, the nonstructural protein 1 (NS1) of IBV also has a high average dN/dS ratio (0.29), reflecting its potential role in evading innate immune responses10,11. Although several positively selected sites have been predicted in the HA, NA, and NS1 datasets10, their functional significance remains unknown.
IBV counters the host innate immune response, largely driven by interferons (IFNs) and interferon-stimulated genes (ISGs), through various mechanisms involving NS16. The C-terminal domain of IBV NS1 can block RIG-I activation12, prevent IRF3 nuclear translocation, and inhibit IFN-β promoter activation13. IBV NS1 also blocks the covalent linkage of ISG15 to its target proteins14. Additionally, IBV PB1 inhibits IRF3 and STAT1 activation, with a specific residue (A523) in PB1 being critical for its IFN-antagonistic function15. However, which amino acid residues of IBV NS1 are crucial for innate immune evasion and adaptive evolution is still unclear. Elucidating how IBV adapts to the host by evading the innate immune responses is critical for understanding the epidemiology of IBV and informing influenza surveillance and control.
In this study, we found that the amino acid at position 247 of IBV NS1 gradually changed from phenylalanine (F) to leucine (L) during IBV evolution. This mutation triggers TUFM-dependent mitophagy, promoting MAVS degradation and allowing IBV to evade the host innate immune response.
Results
Recent IBV strains exhibit an enhanced ability to evade innate immune responses and increased replication efficiency compared with earlier strains
To investigate the host adaptation of IBV, we selected three representative epidemic strains from distinct periods: B/Lee/1940 (BLee-1940), B/Yamagata/16/88 (BYam-1988), and B/Shanghai/PD114/2018 (BYam-2018). A dendrogram of the full-length amino acid sequences of their respective HA genes illustrated their evolutionary relationships (Fig. 1a). Next, we evaluated the IFN-antagonistic functions and replication capacities of these IBV strains in A549 cells. BYam-2018 showed the strongest suppression of IFN and ISG production (Fig. 1b), yielded higher viral titers (Fig. 1c), and exhibited elevated nucleoprotein (NP) expression (Fig. 1d–f) compared with BLee-1940 and BYam-1988. We further examined viral replication in mice. As expected, BYam-2018-infected mice had the strongest suppression of IFN and ISG production (Fig. 1g), higher viral titers (Fig. 1h) and NP mRNA levels (Figure S1a) in their lungs than mice infected with BLee-1940 or BYam-1988. Accordingly, BYam-2018 infection caused greater body weight loss (Fig. 1i), higher mortality (Fig. 1j), more severe lung injury (Fig. 1k), and an increased lung index (Fig. S1b). These results demonstrate that recent IBV strains have a strong capacity to suppress innate immune responses, thereby promoting their replication and adaptation within the host.

a Dendrogram of all non-redundant, full-length influenza B HA sequences from the GISAID Flu database (https://gisaid.org/). b The mRNA levels of IFN and interferon-stimulated genes (ISGs) in A549 cells infected with B/Lee/1940 (BLee-1940), B/Yamagata/16/88 (BYam-1988), and B/Shanghai/PD114/2018 (BYam-2018) were detected by RT-qPCR at 12 h post-infection. c Virus titers were determined by plaque assay at the indicated time points in A549 cells infected with different IBV strains (MOI = 0.05). d Immunoblot analysis of IBV NP in A549 cells infected with different IBV strains (MOI = 0.1) for 12 h (left). Band intensities were determined using ImageJ (right). e Immunofluorescence analysis of NP in A549 cells infected with different IBV strains (MOI = 0.1) for 24 h (left). Viral nucleoprotein (NP) and cell nuclei were stained using an anti-NP antibody (red) and DAPI (blue), respectively. The quantitative analysis of NP-positive cells is shown on the right. Scale bar: 10 µm. f The mRNA levels of NP in IBV-infected A549 cells were detected by RT-qPCR at 12 h post-infection. g The mRNA levels of IFN and ISGs in the lungs of IBV-infected mice were detected by RT-qPCR at 1 d post-infection. h Virus titers in the lungs of IBV-infected mice were determined by plaque assay at day 4 post-infection. i, j Body weight changes (i) and survival rate (j) of mice after IBV infection. k H&E staining image of mouse lungs from IBV-infected mice at day 4 post-infection (left). The severity of pathological manifestations was scored (right). Scale bar: 30 µm. Data are shown as mean ± SD. ns: not significant. One-way analysis of variance (ANOVA) followed by Tukey’s test (c–f, h, k). n = 3 biological replicates (b–f). Data are representative of three independent experiments (g–k). n = 3 mice per group (g), n = 7 mice per group (h, k), n = 10 mice per group (i, j). Source data are provided as a Source Data file.
BYam-2018-NS1 protein suppresses the innate immune response by inducing autophagy-mediated MAVS degradation
The NS1 protein of IAV and IBV facilitates viral replication by inhibiting the host innate immune response through multiple mechanisms16. In this study, we compared the innate immune suppression abilities of NS1 proteins from earlier and recent IBV strains. The results showed that BYam-2018-NS1 most effectively inhibited the production of IFN and ISGs induced by Sendai virus (SeV) compared with BLee-1940-NS1 and BYam-1988-NS1 (Fig. 2a). Next, we investigated which key protein in the IFN production signaling pathway was affected by BYam-2018-NS1. We observed that the relative luciferase activity (Fig. 2b–e) and mRNA levels of IFNB1 (Fig. S2) were significantly suppressed by BYam-2018-NS1 in RIG-I- and MAVS-transfected HEK293T cells, respectively, indicating that BYam-2018-NS1 inhibited the pathway at or upstream of MAVS. Further analysis showed that the overexpression of BYam-2018-NS1 reduced MAVS expression but did not affect the levels of RIG-I, TBK1, and IRF3 (Fig. 2f). Additionally, BYam-2018-NS1 overexpression led to dose-dependent MAVS degradation (Fig. 2g). Consistently, infection with BYam-2018 reduced MAVS expression (Fig. 2h). In contrast, BYam-1988-NS1 showed no impact on the mRNA levels of IFNB1 (Fig. S2) and MAVS expression (Fig. 2f, h). These data suggest that BYam-2018-NS1 inhibits IFN production by promoting the degradation of MAVS.

a The mRNA levels of IFN and ISGs were determined by RT-qPCR in HEK293T cells transfected with the indicated plasmids for 24 h, and then stimulated with SeV (MOI = 0.1) for 12 h (top). Corresponding protein expression levels were analyzed by immunoblotting (bottom). b–e The relative luciferase activity of IFNB1 in HEK293T cells transfected with the indicated plasmids, along with Myc-tagged RIG-I (b), MAVS (c), TBK1 (d), or IRF3/5D (e) for 24 h (top). Corresponding protein expression levels were analyzed by immunoblotting (bottom). f The expression of proteins was detected by immunoblotting in HEK293T cells transfected with the indicated plasmids. g The expression of endogenous MAVS was determined by immunoblotting in HEK293T cells transfected with different doses of Flag-BYam-2018-NS1 for 24 h. h The expression of proteins was detected by immunoblotting in A549 cells infected with IBV (MOI = 0.1) for 12 h. i The expression of endogenous MAVS was evaluated by immunoblotting in HEK293T cells transfected with the indicated plasmids for 24 h, followed by the treatment of DMSO, MG132, CQ, or SBI, along with CHX (left). Band intensities were determined by ImageJ (right). j The expression of proteins was detected by immunoblotting in A549 cells infected with IBV for 12 h and treated with DMSO or inhibitors. k Virus titers were determined by plaque assay in A549 cells infected with IBV (MOI = 0.1) for 24 h and treated with DMSO or inhibitors. l mRNA levels of IFNB1 in HEK293T cells co-transfected with Myc-MAVS together with the indicated plasmids for 24 h, followed by the treatment of DMSO or inhibitors, along with CHX (top). Corresponding protein expression levels were analyzed by immunoblotting (bottom). m IFNB1 luciferase activity in HEK293T cells co-transfected with IFNB1 Luc reporter together with the indicated plasmids for 24 h, followed by the treatment of DMSO or inhibitors, along with CHX (top). Corresponding protein expression levels were analyzed by immunoblotting (bottom). Data are shown as mean ± SD. ns: not significant. Unpaired two-tailed Student’s t-test (b–e) and one-way ANOVA followed by Tukey’s test (i, k–m). n = 3 biological replicates. Source data are provided as a Source Data file.
Protein degradation in eukaryotic cells is primarily controlled by two major systems: the ubiquitin-proteasome and autophagy-lysosomal pathways17. To determine which pathway is involved in BYam-2018-NS1-mediated MAVS degradation, HEK293T cells were transfected with BYam-2018-NS1, followed by treatment with inhibitors targeting different protein degradation pathways. Immunoblot analysis showed that the degradation of MAVS by BYam-2018-NS1 was rescued by the autophagy inhibitors chloroquine (CQ) and SBI, but not by the proteasome inhibitor MG132 (Fig. 2i), indicating that BYam-2018-NS1-mediated MAVS degradation depends on autophagy-lysosomal pathways. Meanwhile, BYam-2018-NS1 increased LC3B-II expression and promoted sequestosome 1 (SQSTM1) degradation. SBI reversed the effects on LC3B-II expression and SQSTM1 degradation. CQ led to the accumulation of LC3B-II and SQSTM1 due to its role in inhibiting autophagic flux by decreasing the fusion of autophagosomes and lysosomes18. Furthermore, treatment with CQ and SBI blocked MAVS degradation during BYam-2018 infection (Fig. 2j) and subsequently attenuated viral replication (Fig. 2k). In addition, autophagy inhibitors abolished the inhibitory effect of NS1 on MAVS-mediated IFNB1 expression (Fig. 2l, m). To further examine whether autophagy induction is essential for BYam-2018-NS1-mediated MAVS degradation, MAVS expression was assessed in wild-type (WT) and ATG5−/− A549 cells. ATG5 deficiency significantly impaired BYam-2018-NS1-induced MAVS degradation (Fig. S3a) and abolished the inhibitory effect of BYam-2018-NS1 on MAVS-stimulated IFNB1 luciferase activity (Fig. S3b) and IFNB1 mRNA expression (Fig. S3c). Moreover, ATG5 knockout reduced BYam-2018 titers in A549 cells (Fig. S3d). In contrast, neither autophagy inhibitors nor ATG5 deficiency affected MAVS expression (Figs. 2j, S3a), viral replication (Figs. 2k, S3d), or IFN production (Figs. 2l, m, Figs. S3b, c) during BYam-1988-NS1 overexpression or BYam-1988 infection. Collectively, BYam-2018-NS1 inhibits host innate immune responses by inducing autophagy-mediated MAVS degradation.
BYam-2018-NS1 demonstrates an enhanced ability to induce autophagy compared with NS1 proteins of earlier strains
We further compared the ability of recent and earlier IBV strains, as well as their NS1 proteins, to induce autophagy. As expected, infection with the recent strains BYam-2018 (Fig. 3a) and B/Guangxi/JZ1352/2018 (BVic-2018) (Fig. S4a), or transfection with their NS1 proteins (Figs. 3b, S4b), led to puncta formation of LC3B in A549 cells. However, infection with the earlier strains BLee-1940, BYam-1988, and BVic-1987, or transfection with their NS1 proteins, hardly triggered autophagy. In addition, infection with the recent strains (Fig. 3c) or overexpression of their NS1 proteins (Fig. 3d) significantly increased LC3B-II expression and promoted SQSTM1 and MAVS degradation, whereas the earlier strains did not. Moreover, BVic-2018-NS1 overexpression led to more MAVS degradation than BVic-1987-NS1 transfection (Fig. S4c). These data indicate that the recent IBV strain BYam-2018-NS1 has a stronger capacity to induce autophagy than earlier strains.

a, b The formation of GFP-LC3 puncta was evaluated by fluorescence microscopy in A549 cells transfected with GFP-LC3B and then infected with different IBV strains (MOI = 0.1) for 24 h (left) (a), or in A549 cells transfected with mCherry-GFP-LC3B along with BLee-1940-NS1, BYam-1988-NS1, or BYam-2018-NS1 for 24 h (left) (b). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) in blue. Scale bars: 5 µm. LC3 puncta per cell were quantified in at least 30 cells (right). c MAVS and LC3B expression was detected by immunoblotting in A549 cells infected with BYam-1988 or BYam-2018 (MOI = 0.1) for 24 h (top). Band intensities were determined by ImageJ (bottom). d MAVS, LC3B, and SQSTM1 expression was detected by immunoblotting in A549 cells transfected with BLee-1940-NS1, BYam-1988-NS1, or BYam-2018-NS1 for 24 h (left). Band intensities were determined by ImageJ (right). Data are shown as mean ± SD. ns: not significant. One-way ANOVA followed by Tukey’s test. n = 3 biological replicates. Source data are provided as a Source Data file.
BYam-2018-NS1 localizes to mitochondria to induce mitophagy
Next, we investigated how BYam-2018-NS1 induces autophagy. Western blot results showed that BYam-2018 infection (Fig. 4a) or its NS1 protein transfection (Fig. 4b) significantly reduced the expression of the mitochondrial membrane proteins TIM23 and TOM20 compared with BLee-1940 or BYam-1988 infection or their NS1 transfections. Meanwhile, autophagosomes and damaged mitochondria were observed in BYam-2018-infected A549 cells via TEM imaging (Fig. 4c), suggesting that BYam-2018 can induce mitophagy. We then examined whether BYam-2018-NS1 localizes to mitochondria. Immunofluorescence colocalization showed that not only did NS1 in BYam-2018- or BYam-1988-infected A549 cells colocalize with the mitochondria (Fig. 4d), but also the transfected NS1 from both strains showed similar co-localization (Fig. 4e). Immunogold labeling further revealed the presence of overexpressed BYam-2018-NS1 in the mitochondria of HEK293T cells (Fig. 4f). Using double-label immunoelectron microscopy, we observed BYam-2018-NS1 and LC3 on the double-membrane structure of autophagosomes in HEK293T cells (Fig. 4g). Colocalization of LC3 and NS1 was further confirmed in Fig. S4a, b. These results demonstrate that BYam-2018-NS1 localizes to mitochondria to induce mitophagy.

a TOM20 and TIM23 expression in A549 cells infected with BLee-1940, BYam-1988, or BYam-2018 (MOI = 0.1) was detected by immunoblotting at 24 h post-infection (left). Band intensities were determined by ImageJ (right). b TOM20 and TIM23 expression in HEK293T cells transfected with Flag-BLee-1940-NS1, Flag-BYam-1988-NS1, Flag-BYam-2018-NS1, or Flag-Vector was detected by immunoblotting at 24 h post-transfection (left). Band intensities were determined by ImageJ (right). c Autophagosomes (yellow arrows) and damaged mitochondria (red arrows) were visualized by transmission electron microscopy in IBV-infected A549 cells (MOI = 0.1) at 24 h post-infection. d Immunofluorescence assay (IFA) showing the colocalization of NS1 and mitochondria in A549 cells transfected with mCherry-Mito and infected with BYam-1988 or BYam-2018 (MOI = 0.1) for 24 h, fixed with 4% paraformaldehyde, and stained with anti-NS1 (green). Nuclei were stained with DAPI in blue. Scale bar: 3 µm. e IFA showing the colocalization of NS1 and mitochondria in A549 cells transfected with Flag-BYam-1988-NS1 or Flag-BYam-2018-NS1 and mCherry-Mito for 24 h, fixed with 4% paraformaldehyde, and stained with anti-NS1 (red). Nuclei were stained with DAPI in blue. Scale bar: 3 µm. f The localization of IBV NS1 was detected by immunoelectron microscopy in Flag-BYam-2018-NS1-transfected HEK293T cells for 24 h, where gold particles against Flag (10 nm, red arrows) were observed in the mitochondria. Scale bars: 60 nm. g The colocalization of IBV NS1 and LC3 was detected by immunoelectron microscopy with 15 nm gold particles against LC3B (red arrows) and 5 nm gold particles against Flag (blue arrows) in HEK293T cells transfected with Flag-BYam-2018-NS1 and Myc-LC3. Scale bars: 150 nm. Data are shown as mean ± SD. One-way ANOVA followed by Tukey’s test. n = 3 biological replicates (a, b). Data are representative of 3 biological replicates (c–g). Source data are provided as a Source Data file.
BYam-2018-NS1 protein mediates TUFM-dependent mitophagy to inhibit the host innate immune responses
Mass spectrometry was used to identify potential cargo receptors participating in BYam-2018-NS1-mediated mitophagy in HEK293T cells (Fig. 5a). Among the candidate proteins, TUFM and SQSTM1 emerged as proteins of interest due to their pivotal roles in mitophagy (Fig. 5b). Glutathione S-transferase (GST) pull-down assays demonstrated that BYam-2018-NS1 interacted with TUFM but not SQSTM1 (Fig. 5c). Co-IP assays confirmed that NS1 from BYam-1988, BYam-2018, BVic-1987, and BVic-2018 were able to interact with TUFM under both transfection (Fig. 5d) or infection (Fig. 5e) conditions. These results corroborate the mitochondrial localization of NS1 from BYam-1988 and BYam-2018 observed in Fig. 4d, e. To further investigate TUFM’s role in BYam-2018-NS1-induced mitophagy, siRNAs were used to knock down TUFM (Fig. 5f). Western blot analysis showed that TUFM knockdown reversed the BYam-2018-NS1-induced reduction in MAVS degradation (Fig. 5g). Consistently, TUFM knockdown restored the mRNA levels of IFNB1 (Fig. 5h), ISG20 (Fig. 5i), IFIT1 (Fig. 5j), and IFITM1 (Fig. 5k), which were reduced by BYam-2018-NS1 in response to SeV infection. These results demonstrate that BYam-2018-NS1 mediates TUFM-dependent mitophagy to inhibit host innate immune responses.

a Schematic diagram of the screen for target autophagy receptors interacting with BYam-2018-NS1 by immunoprecipitation-mass spectrometry (IP-MS). b LC-MS/MS analysis of host proteins that interacted with BYam-2018-NS1 by mass spectrometry. c The interaction between BYam-2018-NS1 and TUFM was determined by a GST pull-down assay. Escherichia coli (BL21) expressed His-tagged BYam-2018-NS1 and GST-tagged TUFM, or GST-tagged SQSTM1 recombinant proteins were purified, and protein-protein interactions were examined by GST-mediated pull-down, followed by immunoblotting with anti-His and GST antibodies. d Immunoblot analysis of lysates from HEK293T cells co-transfected with Myc-BYam-1988-NS1, Myc-BYam-2018-NS1, Myc-BVic-1987-NS1, or Myc-BVic-2018-NS1 and Flag-TUFM for 24 h, followed by immunoprecipitation with the indicated antibodies. e A549 were infected with BYam-1988 or BYam-2018 (MOI = 0.1) for 24 h, and then co-immunoprecipitation was performed with the indicated antibody. f HEK293T cells were transfected with TUFM siRNA, and the extracts were subjected to immunoblotting with the indicated antibodies (left). Band intensities were determined by ImageJ (right). g The expression of MAVS was detected by immunoblotting in HEK293T cells co-transfected with Flag-BYam-2018-NS1 along with siRNA-TUFM or siRNA-NC. h–k RT-qPCR analysis of IFNB1 (h), ISG20 (i), IFIT1 (j), and IFITM1 (k) expression in HEK293T cells transfected with Flag-BYam-2018-NS1 or Flag-Vector, along with either siRNA-TUFM or siRNA-NC for 24 h, followed by SeV treatment for 8 h. Data are shown as mean ± SD. Unpaired two-tailed Student’s t-test. Data are representative of 3 biological replicates (c–f). n = 3 biological replicates (g–k). Source data are provided as a Source Data file.
A single mutation in BYam-1988-NS1 switches its interaction with LC3B and induces mitophagy
NS1 of IBV is composed of three structural domains: an N-terminal RNA-binding domain (RBD) (aa 1–90), a flexible polypeptide linker (aa 91–120), and a C-terminal effector domain (TED) (aa 121–281). Interestingly, a potential autophagy-related LC3-interacting region (LIR) motif, FERL, was identified at residues 244–247 of BYam-2018 NS1. This motif is predicted to adopt the α-helical conformation essential for LC3 binding (Figs. 6a, S5). Furthermore, we discovered that the amino acid at position 247 is phenylalanine (F) in the early IBV strains (BLee-1940, BYam-1988, and BVic-1987) but switches to leucine (L) in the recent IBV strains (BYam-2018 and BVic-2018) and vaccine strains (B/Yamagata/Phuket/3073/2013 and B/Victoria/Washington/02/2019), suggesting that this mutation may affect IBV’s ability to induce autophagy.

a Schematic representation of the full-length NS1 protein and separate domains of Influenza BYam-2018-NS1. Autophagy-related LC3-interacting region (LIR) motif predicted using the iLIR web server (https://ilir.warwick.ac.uk). b The interaction between BYam-2018-NS1 and LC3B was analyzed by co-IP assay in 293 T cells co-transfected with the indicated plasmids for 24 h. c The interaction between BYam-1988-NS1 and TUFM was analyzed by co-IP assay in 293 T cells co-transfected with the indicated plasmids for 24 h. d–f Structure prediction of BYam-2018-NS1 (aa 121-281) using AlphaFold2 and SWISS-MODEL (https://swissmodel.expasy.org) and visualized with PyMOL (https://pymol.org/). Molecular docking was conducted to analyze the interaction between NS1 (red) and LC3B (silver, PDB code: 1UGM). g The expression of LC3B and MAVS was analyzed by immunoblotting in HEK293T cells transfected with the indicated plasmids for 24 h (left). Band intensities were determined by ImageJ (right). h LC3B puncta were investigated by fluorescence microscopy in HEK293T cells co-transfected with GFP-LC3B along with Flag-BYam-2018-WT, Flag-BYam-2018-NS1-F247L, or Flag-Vector (left). LC3 puncta per cell were quantified in at least 30 cells (right). Scale bar: 3 µm. i Analysis of the L247 percentage of NS1 in all IBV strains, retrieved from the Global Initiative on Sharing All Influenza Data (GISAID) database, from 1964 to 2024 (top). For each mutation, the sequence numbers were given (bottom). Data are shown as mean ± SD. ns: not significant. One-way ANOVA followed by Tukey’s test (g) and unpaired two-tailed Student’s t-test (h). Data are representative of 3 biological replicates (b, c). n = 3 biological replicates (g, h). Source data are provided as a Source Data file.
We next investigated whether L at residue 247 of NS1 is critical for IBV-induced mitophagy. A co-immunoprecipitation assay showed that BYam-1988-NS1(Fig. 6b) or BVic-1987-NS1 (Fig. S4d) could not interact with LC3. However, the F247L mutant acquired the ability to interact with LC3, and this mutation did not affect their interactions with TUFM (Figs. 6c, S4e). We further explored whether the L247 of BYam-2018-NS1 was responsible for binding to the previously defined LIR-docking site19,20,21,22 in LC3B using AlphaFold2 and molecular docking analysis. The results revealed that the hydrophobic side chain of L247, the third residue of the BYam-2018-NS1 LIR, was inserted into the HP1 region of LC3 and formed hydrophobic interactions with the side chain of residue K49 in HP1(Figs. 6d–f, S6). We then assessed how this site affects autophagy. The results showed that BYam-1988-NS1-F247L promoted LC3B conversion, whereas BYam-1988-NS1-WT did not (Fig. 6g). Consistently, BYam-1988-NS1-F247L overexpression increased the number of LC3B puncta (Fig. 6h), indicating that substituting F with L at residue 247 of BYam-1988-NS1 enhanced autophagy. To investigate whether L at residue 247 of NS1 is an adaptive site in IBV, we analyzed IBV sequences in the GISAID database (https://www.gisaid.org/). The results showed that as IBV prevalence increased, the amino acid at residue 247 of NS1 was gradually replaced from F to L over the past two decades (Fig. 6i). These data indicate that L at residue 247 of IBV NS1 is critical for its interaction with LC3B and is indispensable for activating mitophagy.
BYam-2018 with an NS1-L247F mutation exhibits diminished innate immune evasion due to impaired mitophagy-mediated degradation of MAVS
To investigate the role of L247 of NS1 during infection, a recombinant IBV containing the NS1-L247F mutation (named BYam-2018-L247F) was constructed by reverse genetics based on the recent IBV strain BYam-2018. BYam-2018-L247F induced fewer LC3 puncta (Fig. 7a) and enhanced MAVS expression (Fig. 7b) compared with BYam-2018-WT in A549 cells. Consequently, both viral titer (Fig. 7c) and protein levels of NP (Fig. 7d) were reduced with this mutation, and BYam-2018-L247F exhibited a weaker ability to suppress host immunity than BYam-2018-WT (Fig. 7e–g). Furthermore, mouse infection experiments showed that infection with BYam-2018-L247F led to reduced weight loss (Fig. 7h), increased survival (Fig. 7i), decreased lung viral titers (Fig. 7j), and attenuated lung injury (Fig. 7k). These results suggested that the amino acid at position 247 of IBV NS1 is critical for regulating the autophagy-mediated innate immune response.

a The formation of GFP-LC3 puncta was evaluated by fluorescence microscopy in A549 cells infected with BYam-2018-WT and BYam-2018-L247F (MOI = 0.1) for 24 h (left). Nuclei were stained with DAPI in blue. Scale bars: 10 µm. LC3 puncta per cell were quantified in at least 30 cells (right). b MAVS and LC3B expression was detected by immunoblotting in A549 cells infected with BYam-2018-WT and BYam-2018-L247F (MOI = 0.1). c Virus titers were determined by plaque assay at the indicated time points in A549 cells infected with BYam-2018-WT and BYam-2018-L247F (MOI = 0.05). d Western blot analysis of NP in A549 cells infected with BYam-2018-WT and BYam-2018-L247F (MOI = 0.1) for 24 h (left). e–g The mRNA levels of IFN and ISGs in A549 cells infected with BYam-2018-WT and BYam-2018-L247F were detected by RT-qPCR at 12 h post-infection. h, i Body weight changes (h) and survival rate (i) of mice after BYam-2018-WT or BYam-2018-L247F infection. j Virus titers in the lungs of BYam-2018-WT or BYam-2018-L247F mice were determined by plaque assay at day 4 post-infection. k H&E staining image of mouse lungs from BYam-2018-WT or BYam-2018-L247F-infected mice at day 4 post-infection (left). The severity of pathological manifestations was scored from 0 to 4, with 0 for no damage and 4 for maximum damage (right). Scale bar: 30 µm. Data are shown as mean ± SD. Unpaired two-tailed Student’s t-test (a, d–g, j, k), one-way ANOVA followed by Tukey’s test (b, c). n = 3 biological replicates (a–g). Data are representative of three independent experiments (h–k). n = 10 mice per group (h, i), n = 7 mice per group (j, k). Source data are provided as a Source Data file.
Discussion
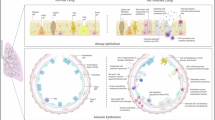
Host innate and adaptive immune responses act as the primary defense against influenza virus infection23. Concurrently, influenza viruses, particularly their HA and NA genes, undergo adaptive evolution driven by selective pressure from the host adaptive immune system24,25,26,27. Nevertheless, the relationship between influenza virus adaptive evolution and the host innate immune response remains unclear. In this study, we found that the amino acid at residue 247 of IBV NS1 was progressively replaced from F to L, culminating in complete substitution within the last twenty years. NS1 with L247 in recent IBV strains interacts with TUFM and LC3B, inducing mitophagy that degrades MAVS and thereby suppresses IFN production. In contrast, NS1 with F247 in earlier strains scarcely triggers autophagy-mediated MAVS degradation. These findings provide insight into how IBV adapts to its host by evading antiviral innate immunity (Fig. 8), thereby establishing a link between innate immunity and viral adaptive evolution.

A phenylalanine (F)-to-leucine (L) substitution at residue 247 of NS1 has emerged during the past two decades, switching to mitophagy-mediated innate immune evasion. Upon IBV infection, RIG-I/MAVS-mediated antiviral signals are activated, triggering IFN responses. The NS1 of recent IBV interacts with TUFM and LC3B, inducing TUFM-dependent mitophagy that degrades MAVS, thereby suppressing IFN production. However, NS1 of earlier strains could hardly trigger autophagy-mediated MAVS degradation.
Viruses undergo adaptive evolution to enhance their ability to infect, replicate, and transmit across species, involving receptor-binding proteins and polymerases. For example, the deletion of V483 in the spike protein grants SARS-CoV-2 an evolutionary advantage for human adaptation and host-range expansion following a prolonged pandemic28. Likewise, the mutations H110Y, T160A, Q226L, and G228S of hemagglutinin enable H5N1 influenza viruses to cross the species barrier29,30,31,32,33. Individual mutations in the HA proteins, R137M or N144E, in H2N2 pandemic influenza virus shift receptor binding from dual specificity for human and avian receptors to predominantly binding human receptors34. The PB2-E627K substitution in influenza H7N9 signifies in vivo genetic tuning and rapid host adaptation35. In this study, we demonstrate that recent IBV strains are better equipped to evade the innate immune response and replicate with greater pathogenicity in mice than earlier isolates. This advantage appears to stem from an F-to-L substitution at NS1 residue 247, since reversion to NS1-L247F in a recent strain markedly attenuated its replication and pathogenicity in mice. Similarly, the N342K substitution in IBV neuraminidase, whose prevalence rose after 2016, enhances viral pathogenicity in mice36, and the PA K338R mutation, selected through serial lung-to-lung passages, increases IBV virulence in mice and promotes replication in the ferret respiratory tract37. Collectively, these data support the hypothesis that, since the early 2000s, IBV has undergone adaptive evolution toward greater pathogenicity by improving its ability to evade innate immune responses, although direct clinical comparisons of IBV pathogenicity before and after that period remain limited.
Autophagy is a conserved, tightly regulated intracellular catabolic process in which mammalian cells degrade and recycle damaged or dysfunctional macromolecules and organelles, playing an important regulatory role during viral infection38. Although IAV-induced autophagy is well documented, whether IBV triggers this pathway has remained unresolved, largely because most studies have relied on early IBV strains. For instance, the prototypic B/Lee/1940 strain failed to induce autophagosome accumulation39. In this study, we demonstrate that while early IBV strains cannot activate autophagy, more recent IBV isolates do so efficiently. Consistent with this result, our previous work showed that the expression of autophagy-related genes was significantly upregulated in A549 cells infected with the recent IBV strain BYam-201840. Therefore, comparing the immune responses to different virus strains, especially those from various time periods, will enhance our understanding of virus adaptive evolution.
Mitophagy is a selective autophagic defense that clears damaged or excess mitochondria to maintain mitochondrial homeostasis41. Increasing evidence shows that various viruses use TUFM-dependent mitophagy to facilitate their replication via different strategies. In this study, we show that NS1 from the current BYam-2018 strain binds TUFM to localize to mitochondria and then recruits LC3 to trigger mitophagy, thereby suppressing the host innate immune response. In contrast, although NS1 from the early BYam-1988 strain still interacts with TUFM and targets the mitochondria, it fails to engage LC3 and so cannot induce mitophagy. Likewise, SARS-CoV-2 ORF10 activates BNIP3L-mediated mitophagy to enhance viral replication42. IAV PB1-F2 and NP proteins each hijack their respective cargo receptors, TUFM and TOLLIP, to trigger full mitophagy, resulting in suppression of IFN-I production43,44. Human parainfluenza virus type 3 matrix protein and Hantaan virus Gn protein localize to mitochondria by interacting with TUFM and subsequently recruit LC3B to mitochondria to mediate mitophagy44,45. Bunyavirus SFTSV nucleoprotein exploits TUFM-mediated mitophagy to impair antiviral innate immunity46. Together, these findings highlight the essential role of mitophagy in viral innate immune evasion.
In conclusion, our results demonstrate that adaptive evolution involving a single mutation at NS1 residue 247 promotes mitophagy-mediated MAVS degradation, thereby enhancing the capacity of IBV to evade the host innate immune response. This mutation may be one of the key factors contributing to IBV’s successful adaptation to the host and its continuous circulation in nature. Thus, monitoring the adaptive evolution of NS1 is critical and will inform strategies for influenza prevention and control.
Methods
Reagents and antibodies
Rabbit monoclonal antibodies against RIG-I (D14G6) (Cat #3743), MAVS (D5A9E) (Cat #24930), TBK1 (E8I3G) (Cat #38066), IRF3 (D6I4C) XP® (Cat #11904), Flag (Cat #14793), GST-Tag (91G1) (Cat #2625), SQSTM1 (D6M5X) (Cat #23214), ATG5 (D5F5U) (Cat #12994), Tom20 (D8T4N) (Cat #42406), TIM23 (E1Q7L) (Cat #34822), His-Tag (D3I1O) XP® (Cat #12698), and mouse monoclonal antibodies against Myc-Tag (9B11) (Cat #2276), GAPDH (D4C6R) (Cat #97166), LC3B (Cat #3868) were purchased from Cell Signaling Technology (USA). GST-BYam-2018-NS1 and His-TUFM fusion proteins were prepared in Escherichia coli BL21 and purified using standard procedures. Anti-NP and anti-BNS1 antibodies were generated by immunizing rabbits with E. coli–expressed recombinant proteins. HRP-conjugated anti-mouse secondary antibody (Cat #A0216) and HRP-conjugated anti-rabbit secondary antibody (Cat #A0208) were from Beyotime. Fluorescein (FITC) donkey anti-mouse IgG (Cat #715-096-151), Rhodamine (TRITC) donkey anti-mouse IgG (Cat #715-025-151), and Fluorescein (FITC) donkey anti-rabbit IgG (Cat #711-095-152) were purchased from the Jackson Laboratory. The plasmid prep purification kit was from Genemark (China). Transcriptase kits were from Tiangen Biotech (Beijing, China). ANTI-FLAG M2 Affinity Agarose Gel (Cat #A4596), ivermectin (Cat #1354309), and importazole (Cat #401105) were purchased from Sigma (USA). GST Magnetic Beads (MG-103) were from Biomed (Beijing, China). Primary antibodies were used at a 1:1000 dilution, and HRP-coupled secondary antibodies were used at a 1:10,000 dilution for immunoblotting. For immunofluorescence, primary antibodies were used at a 1:200 dilution.
Cells and cell culture
HEK293T (CRL-11268), human lung epithelial carcinoma (A549) (CCL-185), and Madin-Darby canine kidney (MDCK) (CCL-34) cells were obtained from the American Type Culture Collection (ATCC). ATG5-knockout A549 cells (ATG5−/−A549) were generated by CRISPR/Cas9-mediated genome editing47. HEK293T, A549, and ATG5−/−A549 cells were maintained in Dulbecco’s Modified Eagle Medium (Gibco, c11965500BT) supplemented with 10% fetal bovine serum (Gibco) and 1% (w/v) penicillin-streptomycin at 37 °C with 5% CO₂.
Viruses
B/Lee/1940 (BLee-1940), B/Yamagata/16/88 (BYam-1988), and B/Victoria/1987 (BVic-1987), kindly provided by Xianzhu Xia (Institute of Military Veterinary Medicine), were propagated in the allantoic cavities of 10-day-old SPF embryonated chicken eggs. Each strain (1000 PFU) was inoculated into embryonated chicken eggs maintained at 37 °C, and viruses were harvested from allantoic fluid at 48 h post inoculation48. B/Shanghai/PD114/2018 (BYam-2018) and B/Guangxi/JZ1352/2018 (BVic-2018) were provided by Dayang Wang (Chinese Center for Disease Control and Prevention), also amplified and quantified per the same procedures48. These IBV strains have undergone more than 15 passages and exhibit strong adaptability to mice12,40,48,49. Sequencing results indicate that there has been no change at residue 247 of NS1 during passaging. The IBV-L247F mutant was generated using a 12-plasmid reverse-genetics system.
For cell infection, the medium was removed, cells were washed twice with PBS (Gibco, 10010072), and serum-free medium containing IBV was added for 2 h. The medium was then replaced with 2% FBS medium and incubated until a full cytopathic effect was observed or until the designated time.
All viruses were titrated using plaque assays on MDCK cells50. Confluent MDCK monolayers (5 × 10^6 cells/well in 12-well plates) were washed with PBS and infected with serial virus dilutions for 1 h at 37 °C. After removing the inoculum and washing, cells were overlaid with DMEM containing 1% low-melting-point agarose and 1 µg/ml TPCK-trypsin, then incubated at 37 °C. Plaques were counted at 3 days post-infection to calculate viral titers. Data are the means of three independent experiments.
Mice
Female BALB/c mice (6–8 weeks old) were obtained from Vital River, China. All animals were housed in a barrier facility under standard conditions, with a 12 h light/12 h dark cycle, an ambient temperature of 22 ± 2°C, and a relative humidity of 40–70%. For mouse infection, animals were intranasally inoculated with IBV (3000 PFU) or PBS as a control. Body weight was recorded immediately before virus challenge (defined as day 0), and the mice were monitored daily for weight changes and survival for up to day 11 or 15 post-challenge. Mice that lost more than 25% of their initial body weight were humanely euthanized. To assess viral replication in the lungs, mice in each group were sacrificed at 4 days post-infection, and the lungs were collected. Lung tissues were homogenized in PBS, and viral titers were quantified by plaque assay using MDCK cells.
All animal experiments were approved by, and conducted in strict accordance with, the Guidelines for the Care and Use of Laboratory Animals of the Ethics Committee of the Institute of Microbiology, Chinese Academy of Sciences (Permit Number APIMCAS2021126).
Plasmids
The IFNB1 promoter-luciferase reporter (IFNB1-Luc) was kindly provided by Dr. Chunfu Zheng (Fujian Medical University, Fuzhou, China). Blee-1940-NS1, BYam-1988-NS1, BYam-2018-NS1, BVic-1987-NS1, BVic-2018-NS1, and BYam-1988-NS1-F247L were synthesized by GENEWIZ and cloned into the pcDNA3.0-Flag vector (MiaoLingBio, P6921). Gene encoding human TUFM and LC3B were synthesized and cloned into the PCAGGS-Myc vector. GST-tagged BYam-2018-NS1 and His-TUFM were synthesized by TSINGKE (Tsingke Biotechnology Co., Ltd) and cloned into the PGEX-4T-1 and pET30a-His vectors, respectively.
siRNA interference assay
All siRNA fragments were purchased from Sangon Biotech (Shanghai, China). TUFM siRNA sequences were as follows: TUFM siRNA-1 sense: 5’- GGUCAUGCAGAUUAUGUUA-3’ and TUFM siRNA-1 antisense: 5’- UAACAUAAUCUGCAUGACC-3’. TUFM siRNA-2 sense: 5’- GGGCUAAGUUCAAGAAGUA-3’ and TUFM siRNA-2 antisense: 5’- UACUUCUUGAACUUAGCCC-3’. TUFM siRNA-3 sense: 5’- GAUUAUGUUAAGAAUAUGA-3’ and TUFM siRNA-3 antisense: 5’- UCAUAUUCUUAACAUAAUC-3’. A scrambled siRNA (Invitrogen) was used as a negative control. Plasmids or siRNA were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen, 11668019). After 24 h, proteins were isolated, and expression levels were analyzed by immunoblotting.
Reverse transcription and qPCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, 15596026CN), following the manufacturer’s instructions. Each sample (2 μg) was digested with DNase I and reverse-transcribed into cDNA using oligonucleotide (dT) primers. Mock reactions were performed in the absence of reverse transcriptase. Relative real-time PCR was carried out to measure gene expression in HEK293T cells. SYBR Green (Takara, RR420A)-based real-time PCR was run on an ABI 7500 instrument. The reaction conditions included an initial step of 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 31 s. Each sample was amplified in triplicate. GAPDH served as an internal control. Primer sequences used in this study are listed in Table S1.
Immunofluorescence microscopy
HEK293T or A549 cells were seeded into 24-well plates (5.0 × 104 cells per well) one day before transfection or infection. When the cells reached approximately 70% confluency, 1 μg of plasmid was transfected per well. Before transfection, the medium was replaced with pre-warmed Opti-MEM without phenol red (Life Technologies). Plasmid transfection was performed using a transfection reagent (Cat#40802, YEASEN, China) according to the manufacturer’s instructions. At the specified time post-transfection or infection, cells were washed three times with PBS, fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized with 0.5% Triton X-100 in PBS (PBST) for 20 min, and stained with the appropriate antibodies. Nuclei were counterstained with 5 μg/mL DAPI (Sigma). Coverslips were examined using a Leica SP8 confocal microscope.
LC3 puncta per cell were quantified on confocal images using ImageJ 1.51 as follows: cell boundaries were defined by DAPI staining, background was subtracted (Rolling Ball radius 50 px), and LC3 puncta were detected by thresholding and “Analyze Particles” (size 0.2–2 μm2, circularity 0.3–1.0). A minimum of 30 cells per condition were analyzed, and data are presented as mean ± SD.
Coimmunoprecipitation and immunoblotting
Cells were cultured in 10-cm or 6-well plates and transfected (Cat#40802, YEASEN, China) for 24 h. They were then lysed in buffer containing 0.5% NP40, 150 mM NaCl, 20 mM HEPES (pH 7.4), 10% glycerol, and 1 mM EDTA, followed by centrifugation at 14,000 × g for 10 min to remove debris. The supernatant was incubated with ANTI-FLAG® M2 affinity gel (Sigma-Aldrich) at 4 °C for 6 h. After five washes with buffer containing 0.5% NP40, 300 mM NaCl, 20 mM HEPES (pH 7.4), 10% glycerol, and 1 mM EDTA, bound proteins were eluted by boiling in SDS loading buffer for 10 min and analyzed by immunoblotting. Representative immunoblot images from at least three independent replicates are shown in the figures.
Mass spectrometry (MS)
HEK293T cells were transfected with Flag-BYam-2018-NS1 or Flag-vector for 24 h, and were lysed in lysis buffer supplemented with a complete protease inhibitor cocktail (Roche Diagnostics) (n = 1 represents three pooled technical replicates). The target proteins were immunoprecipitated with ANTI-FLAG M2 Affinity Agarose Gel (Sigma, USA, Cat #A4596) and subjected to SDS-PAGE electrophoresis. The proteins on the lanes of SDS-PAGE gels were digested with trypsin and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis at the Institutional Center for Shared Technologies and Facilities of the Institute of Microbiology, Chinese Academy of Sciences.
LC separation was conducted using an EASY-nLC™ 1200 HPLC system interfaced via a Nanospray Flex ion source to an Orbitrap Exploris 240 mass spectrometer (Thermo Fisher Scientific). The peptides were loaded onto a trap column (C18, 1.9 μm particles, 150 μm ID, 2 cm length, Dr. Maisch GmbH) and separated using an analytical column (C18, 1.9 μm particles, 150 μm ID, 15 cm length, Dr. Maisch GmbH) at a flow rate of 500 nL/min with a liner 60 min LC gradient composed of Solvent A (0.1% formic acid (v/v)) and Solvent B (80% acetonitrile, 0.1% formic acid (v/v)). The gradient was 8–12% B for 2 min, 12–30% B for 38 min, 30–45% B for 11 min, 45–90% B for 3 min, and finally 90% B for 6 min. A data-dependent acquisition mode was used, the MS data was acquired in the Orbitrap at a resolution setting of 120,000 (m/z 200) across the mass range of 350–1500 m/z, the target value was 2.00E + 06 with a maximum injection time of 50 ms. Data-dependent mode was selected as the cycle time mode setting at 2 s. The precursor ions were selected from each MS full scan with an isolation width of 1.6 m/z for fragmentation in the Ion Routing Multipole with a normalized collision energy of 30%. Subsequently, MS/MS spectra were acquired at a resolution of 15,000 at m/z 200. The target value was 7.50E + 04 with a maximum injection time setting of Auto. The dynamic exclusion time was 45 s. For nano electrospray ion source setting, the spray voltage was 2.0 kV; no sheath gas flow; the heated capillary temperature was 320 °C.
MS/MS data were processed using the Mascot search engine (v.3.0.0, 2024, http://www.matrixscience.com; Matrix Science Ltd., London, UK). Tandem mass spectra were searched against the Homo Sapiens Uniprot database. For precursor ions, the mass error was set to 10 ppm, and for fragment ions, the mass error was set to 0.02 Da. The max missed cleavage was set to 1. False discovery rate (FDR) thresholds for peptides were adjusted to <1%. p-value < 0.05 was considered statistically significant. All the other parameters in Mascot were set to default values.
Glutathione S-transferase (GST) pull-down assay
GST pull-down assays were performed using a GST Magnetic Beads kit (Beijing Biomed Gen Technology Co., Ltd., China). GST-BYam-2018-NS1 and His-TUFM were expressed in Escherichia coli Rosetta (DE3) for 6 h at 37 °C after induction with 0.5 mM IPTG. The resulting protein complexes were visualized by immunoblotting using the appropriate antibody.
Protein modeling and molecular docking
The BYam-2018-NS1 (aa 121–281) protein model was constructed using the Robetta prediction server (http://robetta.bakerlab.org) and AlphaFold. LC3B (PDB code: 1UGM) protein crystal structures were obtained from the PDB database (https://www.rcsb.org/pdb) in PDB format. Protein docking of BYam-2018-NS1 (aa 121–281)–LC3B was performed using the ZDOCK tool (https://zdock.umassmed.edu/). Two-dimensional and three-dimensional structural interaction diagrams for BYam-2018-NS1 (aa 121–281)–LC3B were generated using LigPlot (https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/) and PyMol software (http://www.pymol.org), displaying details of the interacting amino acids, bridging water molecules, and hydrogen bonds.
Luciferase reporter assay
HEK293T cells were seeded in 24-well plates. The following day, cells were transfected with 200 ng of a luciferase reporter plasmid and 100 ng of a β-Gal plasmid, along with other required expression plasmids as needed for each experiment. After 24 h, cells were lysed, and the supernatant was stored at −80 °C. Fluorophore assays were performed using a Fluorophore Assay Kit (TransGen, FR201).
Transmission electron microscopy
HEK293T cells were infected with IBV for 24 h. A subset of the cells was harvested for western blot analysis of viral infection in Fig. 4a. The remaining cells were trypsinized and pelleted by centrifugation at 1200 × g for 10 min at 4 °C. A fixative solution (2.5% glutaraldehyde, 4% paraformaldehyde, 0.2 M phosphate buffer, pH 7.5) was added to the cell pellet and incubated at room temperature for 2 h. Cells were then fixed in 2% glutaraldehyde for 24 h at room temperature. After treatment with 1% osmium tetroxide for 2 h at 4 °C, cells were dehydrated in a graded ethanol series and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and examined under a transmission electron microscope (Hitachi H-7650, Japan) (TEM).
Immunogold staining and electron microscopy
HEK293T cells were transfected with Flag-BYam-2018-NS1 or Flag-Vector. At 24 h post-transfection, cells were fixed with 4% paraformaldehyde–0.5% glutaraldehyde for 12 h at 4 °C, washed three times with PBS, and post-fixed in 1% osmium tetroxide in PBS for 1 h at 4 °C. After three more washes in PBS, cells were dehydrated in a graded ethanol series and treated with acetone. Infiltration was performed using Spurr resin in gelatin capsules. Ultrathin sections were immunostained with anti-Flag (1:20) and anti-LC3B (1:20) antibodies, followed by goat anti-mouse IgG conjugated to 5 nm gold particles and goat anti-rabbit IgG conjugated to 15 nm gold particles (1:50). Samples were observed with a FEI Tecnai G2 TEM at a 75-kV acceleration voltage. All sample preparation and immunocolloidal gold TEM were conducted at the Center for Biological Imaging (CBI, http://www.ibp.cas.cn/cbi/) of the Institute of Biophysics, Chinese Academy of Sciences.
CHX, MG132, chloroquine (CQ), and SBI treatment
HEK293T cells were seeded in 12-well plates (1.0 × 105 cells/well) one day before transfection. When they reached ~70% confluency, 1.5 μg of plasmid was transfected per well. At 24 h post-transfection, cells were treated with 100 μg/mL cycloheximide (CHX; YEASEN, 40325ES03) for 9 h. Cells were then harvested and analyzed by immunoblotting. In experiments using MG132 (10 μM; Sigma, C2211) and CQ (20 µM), SBI (25 mM), these reagents were added alongside CHX, and cells were collected 6 h later.
Western blot quantification
Western blot images were analyzed using ImageJ 1.51 (National Institutes of Health, https://imagej.net/ij/). TIFF files were opened without alteration to bit depth. For each lane, a fixed rectangular region of interest (ROI) was drawn around the band to record the Integrated Density. A same-sized adjacent ROI was used to measure local background, and the corrected band intensity was calculated as: Corrected intensity = Integrated Density_band – (Area_ROI × Mean background intensity). Target protein signals were normalized to the corresponding GAPDH loading control from the same lane and expressed relative to the control sample (set to 1). Any brightness or contrast adjustments were applied uniformly to the entire image. Quantification was performed on at least three independent biological replicates, and data are presented as mean ± SD.
Statistical analysis
All results are presented as the mean ± SD. Two-tailed Student’s t-test and one-way ANOVA were used to evaluate statistical significance. All statistical analyses were performed using GraphPad Prism 9.3.1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the findings of this study are available in the article and its Supplementary Information files. Source data are provided with this paper. The mass spectrometry proteomics data that support the findings of this study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD074269. Source data are provided with this paper.
References
Francis, T. Jr a new type of virus from epidemic influenza. Science 92, 405–408 (1940).
Rota, P. A. et al. Cocirculation of two distinct evolutionary lineages of the influenza type B virus since 1983. Virology 175, 59–68 (1990).
Caini, S. et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21st century. PLoS ONE 14, e0222381 (2019).
Schwab, L. S. U., Do, T. H. T., Pilapitiya, D. & Koutsakos, M. Dissemination of influenza B virus to the lower respiratory tract of mice is restricted by the interferon response. J. Virol. 98, e0160423 (2024).
Ambrose, C. S. & Levin, M. J. The rationale for quadrivalent influenza vaccines. Hum. Vaccin. Immunother. 8, 81–88 (2012).
Hensen, L., Kedzierska, K. & Koutsakos, M. Innate and adaptive immunity toward influenza B viruses. Future Microbiol. 15, 1045–1058 (2020).
Yan, S., Weycker, D. & Sokolowski, S. US healthcare costs attributable to type A and type B influenza. Hum. Vaccin. Immunother. 13, 2041–2047 (2017).
Koutsakos, M., Nguyen, T. H., Barclay, W. S. & Kedzierska, K. Knowns and unknowns of influenza B viruses. Future Microbiol. 11, 119–135 (2016).
Zaraket, H., Hurt, A. C., Clinch, B., Barr, I. & Lee, N. Burden of influenza B virus infection and considerations for clinical management. Antivir. Res. 185, 104970 (2021).
Chen, R. & Holmes, E. C. The evolutionary dynamics of the human influenza B virus. J. Mol. Evol. 66, 655–663 (2008).
Vijaykrishna, D. et al. The contrasting phylodynamics of human influenza B viruses. eLife 4, e05055 (2015).
Jiang, J. et al. Robust Lys63-linked ubiquitination of RIG-I promotes cytokine eruption in early influenza B virus infection. J. Virol. 90, 6263–6275 (2016).
Donelan, N. R. et al. The N- and C-terminal domains of the NS1 protein of the influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 78, 11574–11582 (2004).
Yuan, W. & Krug, R. M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 20, 362–371 (2001).
Schreiber, A. et al. Type I interferon antagonistic properties of influenza B virus polymerase proteins. Cell. Microbiol. 22, e13143 (2020).
Hale, B. G., Randall, R. E., Ortín, J. & Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89, 2359–2376 (2008).
Jiao, P. et al. SARS-CoV-2 nonstructural protein 6 triggers endoplasmic reticulum stress-induced autophagy to degrade STING1. Autophagy 19, 3113–3131 (2023).
Mauthe, M. et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455 (2018).
Kuang, Y. et al. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12, 2363–2373 (2016).
Wirth, M. et al. Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins. Nat. Commun. 10, 2055 (2019).
Johansen, T. & Lamark, T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 432, 80–103 (2020).
Alcober-Boquet, L. et al. The PB1 and the ZZ domain of the autophagy receptor p62/SQSTM1 regulate the interaction of p62/SQSTM1 with the autophagosome protein LC3B. Protein Sci. 33, e4840 (2024).
Liu, Y. et al. Uncompromised NK cell activation is essential for virus-specific CTL activity during acute influenza virus infection. Cell. Mol. Immunol. 15, 827–837 (2018).
Smith, D. J. et al. Mapping the antigenic and genetic evolution of the influenza virus. Science 305, 371–376 (2004).
Fitch, W. M., Bush, R. M., Bender, C. A. & Cox, N. J. Long-term trends in the evolution of H(3) HA1 human influenza type. A. Proc. Natl. Acad. Sci. USA 94, 7712–7718 (1997).
Koel, B. F. et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science 342, 976–979 (2013).
Hensley, S. E. et al. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 326, 734–736 (2009).
Yue, C. et al. Deletion of V483 in the spike confers evolutionary advantage on SARS-CoV-2 for human adaptation and host-range expansion after a prolonged pandemic. Cell Res. 34, 739–742 (2024).
Lin, T. H. et al. A single mutation in bovine influenza H5N1 hemagglutinin switches specificity to human receptors. Science 386, 1128–1134 (2024).
Herfst, S. et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336, 1534–1541 (2012).
Imai, M. et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486, 420–428 (2012).
Xiong, X. et al. Receptor binding by a ferret-transmissible H5 avian influenza virus. Nature 497, 392–396 (2013).
Zhang, W. et al. An airborne transmissible avian influenza H5 hemagglutinin seen at the atomic level. Science 340, 1463–1467 (2013).
Sun, J. et al. Dual receptor-binding, infectivity, and transmissibility of an emerging H2N2 low pathogenicity avian influenza virus. Nat. Commun. 15, 10012 (2024).
Liu, W. J. et al. Dynamic PB2-E627K substitution of influenza H7N9 virus indicates the in vivo genetic tuning and rapid host adaptation. Proc. Natl. Acad. Sci. USA 117, 23807–23814 (2020).
Qi Chen, X. X. a. M. T. e. a. NAN342K mutation enhances the pathogenicity of influenza B virus in mice. Zoonoses https://doi.org/10.15212/ZOONOSES-2023-0013 (2023).
Bae, J. Y. et al. A single amino acid in the polymerase acidic protein determines the pathogenicity of influenza B viruses. J. Virol. https://doi.org/10.1128/jvi.00259-18 (2018).
Luo, T. et al. PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy 12, 1355–1371 (2016).
Gannagé, M. et al. Matrix protein 2 of the influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380 (2009).
Jiao, P. et al. Robust induction of interferon and interferon-stimulated gene expression by influenza B/Yamagata lineage virus infection of A549 cells. PLoS ONE 15, e0231039 (2020).
Zhang, Q. et al. The kinase Jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor ARF for degradation. Nat. Immunol. 16, 458–466 (2015).
Li, X. et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell. Mol. Immunol. 19, 67–78 (2022).
Zhang, B. et al. The nucleoprotein of the influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 19, 1916–1933 (2023).
Wang, R. et al. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 17, 496–511 (2021).
Wang, K. et al. The glycoprotein and nucleocapsid protein of hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep. 27, 2075–2091.e2075 (2019).
Zhang, W. K. et al. Bunyavirus SFTSV nucleoprotein exploits TUFM-mediated mitophagy to impair antiviral innate immunity. Autophagy 21, 102–119 (2025).
Newman, A. C., Kemp, A. J., Drabsch, Y., Behrends, C. & Wilkinson, S. Autophagy acts through TRAF3 and RELB to regulate gene expression via antagonism of SMAD proteins. Nat. Commun. 8, 1537 (2017).
Zheng, W. et al. Naproxen exhibits broad anti-influenza virus activity in mice by impeding viral nucleoprotein nuclear export. Cell Rep. 27, 1875–1885.e1875 (2019).
Meng, Q. et al. [Phylogenetic and pathogenicity analysis of influenza B virus strain B/Guangxi-Jiangzhou/1352/2018]. Sheng Wu Gong. Cheng Xue Bao 38, 3390–3405 (2022).
Wang, S. et al. Tyrosine 132 phosphorylation of influenza A virus M1 protein is crucial for virus replication by controlling the nuclear import of M1. J. Virol. 87, 6182–6191 (2013).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32061123001 to W.L.), the National Key Research and Development Program of China (2023YFC2308600 to L.S.), and Shenzhen Bay Laboratory. We thank Jingfang Liu (the Technological Platform of Mass Spectrum Centre of Institute of Microbiology, Chinese Academy of Sciences) for assistance with LC-MS/MS analysis.
Author information
Authors and Affiliations
Contributions
L.S. initiated and supervised the project. L.S., W.L., and P.J. designed the experiments, analyzed the data. P.J., X.J., X.B., and Y.Z. performed the experiments. R.L., Z.F., W.F., H.Z., and H.L. helped with some experiments. P.J. wrote the original draft of the paper. L.S., W.L., D.W., and Y.B. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiao, P., Jia, X., Bai, X. et al. A single mutation in nonstructural protein 1 is critical for the adaptive evolution of influenza B virus. Nat Commun 17, 3353 (2026). https://doi.org/10.1038/s41467-026-70211-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70211-x
