
Abstract
The conversion of CO2 into high-value syngas (CO and H2) using Cu-based catalysts has garnered considerable interest. However, these catalysts deactivate rapidly due to Ostwald-ripening and thermal aggregation. Herein, we propose to encapsulate Cu-cluster into mMOR zeolite for engineering a separation-site catalyst Cu@mMOR. In the reverse water-gas shift reaction (RWGS), mMOR zeolite of Cu@mMOR directly captures and then activates CO2, and simultaneously serves as a molecular fence to prevent CO2 contact with Cu-clusters surrounding with small pore of mMOR (<3.0 Å) channels. These encapsulated Cu-clusters in mMOR possess considerable Cu vacancies, which significantly facilitate H2 heterolytic dissociation, transferring H* species towards activated CO2 on mMOR and converting them into CO. This separation-site strategy efficiently increases catalytic activation while intelligently altering the CO2 reaction pathway, preventing Cu from thermal agglomeration and Oswald ripening in the catalyst. Consequently, Cu@mMOR attains a space-time yield of CO as high as 3290 mmol gcat-1 h-1, with catalytic stability extending up to 788 hours.
Similar content being viewed by others

Introduction
The ongoing and excessive emissions of CO2 have led to global climate change, which is currently receiving considerable attention. The conversion of CO2 into high-value chemicals is essential for mitigating the greenhouse effect and maintaining a sustainable environment and energy supply1. Among these, syngas (CO and H2 mixture) is reported to be an essential chemical used in the large-scale production of alcohols, esters, and liquid fuels2. Recently, the insufficient provision stemming from reliance solely on fossil resources has spurred growing interest in catalytic CO2 hydrogenation-a promising strategy for converting greenhouse gases into valuable syngas3. Despite its apparent appeal, this approach presents formidable obstacles.
The reverse water-gas shift reaction (RWGS) is a desirable and promising CCU technology that catalytically converts CO2 into syngas under a H2 environment. Operating the RWGS at atmospheric pressure and temperatures above 500 °C enables direct conversion of CO2 into syngas with the ideal H2/CO ratio for Fischer-Tropsch synthesis, requiring only moderate energy input and avoiding extra downstream separation4. For this reaction, Cu-based catalysts are promising non-noble metal candidates for RWGS because of their high hydrogenation activity and high CO selectivity5. In general, high temperature during the RWGS reaction usually tends to lead to the sintering of Cu, which can result in catalyst deactivation via the aggregation of Cu NPs. Encapsulating the active metal inside thermally robust zeolites is an effective way to curb high-temperature aggregation. For instance, Xiao et al. embedded Pt NPs within Beta zeolite via a seed-mediated growth route, constructing a rigid “protective shell” around the Pt that markedly suppressed thermal migration and aggregation6. Likewise, Liu et al. confined sub-nanometer Pt clusters in the straight channels of MEL zeolite, firmly anchoring the naked Pt species and achieving excellent stability in methyl-cyclohexane dehydrogenation7. These reports confirm that zeolite-confinement can efficiently mitigate thermally induced particle aggregation. However, high-temperature sintering can also proceed via an Ostwald-ripening path, where volatile metal species migrate from interior pores to the outer surface and aggregate. For example, Julian et al. reported that confined Mo zeolite still diffuses outward during high-temperature methane dehydrogenation, causing agglomeration and coking8. Analogous to the above catalysts, Cu species are susceptible to forming volatile Cu-carbonyl complexes under CO-rich conditions, leading to significant catalyst deactivation9. Therefore, the simultaneous inhibition of Ostwald-ripening alongside the migration and aggregation of Cu species in the RWGS reaction remains a crucial challenge for achieving high-performance catalysts that exhibit both efficient activation and enduring stability.
Herein, we proposed to construct Cu@mMOR (copper-encapsulated modified mordenite zeolite) that encapsulate Cu clusters within mMOR (modified mordenite zeolite) crystals to realize catalytic site separation for CO2 and H2 activation in RWGS reaction. For our designed catalyst, mMOR was triggered to crystallize around Cu clusters and encapsulate Cu species insides via its Na+-induced crystallized small pore channels (<3.0 Å) linking with Cu-O bonds. CO2 was efficiently captured and activated on outside mMOR, which served as the molecular fence to isolated CO2 from Cu clusters depended on its small channels. On the other hand, H2 was dissociated into H* on internal Cu clusters and spillover toward activated CO2 via Cu-O interface of Cu@mMOR, and finally produced CO with high selectivity. The catalytic site separation strategy significantly enhanced catalytic activation for both CO2 and H2, and rationally changed the CO2 reaction pathway, thereby effectively preventing Cu Oswald ripening and thermal aggregation in catalysts. Therefore, a space-time yield of 3290 mmol gcat-1 h-1 was attained in Cu@mMOR at high-temperature conditions (600 °C), showcasing the long-term stability of catalyst performance over a duration of 788 h (49.1% CO2-conversion-rate and 99.6% CO-selectivity).
Results and discussion
Design idea and synthesis strategy for Cu@mMOR
Figure 1 showed our design idea and the corresponding synthesis strategy for mMOR-confined Cu catalyst (denoted as Cu@mMOR) having separated catalytic sites for H2 and CO2. Firstly, SiO2 nanospheres were used to encapsulate Cu-based precursor via a reversed-phase micro-emulsion method (see Fig. 1a–b). Next, Cu-based precursor were calcined and reduced into Cu particles surrounding with SiO2 (named Cu@SiO2, Fig. 1c). In this process, Na+ was used as a directing agent to induce the growth of small Si-O cages (<3.0 Å) in mMOR to isolate CO2 from Cu species. Next, the whole part of SiO2 of Cu@SiO2 was gradually converted into mMOR zeolite under the induction of N-methyl-pyrrolidine and followed by the second calcination and H2 reduction (seen Fig. 1d–f and Supplementary Fig. 1). Finally, a portion of Na+ was selectively retained while the remainder was converted to NH4+ via low-concentration NH4Cl exchange, followed by calcination to ultimately form H⁺, resulting in the formation of Cu@mMOR.

a Model illustration of the inverse microemulsion step. b Model illustration of the inverse microemulsion after adding TEOS and NH3·H2O. c Model illustration of Cu@SiO2. d Model illustration of Cu@SiO2 with Na+-rich layer. e Model illustration of Cu@mMOR. f Atomic-scale illustration of Cu@mMOR.
Morphology of different Cu-based catalysts
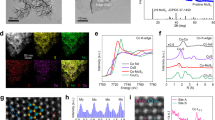
It is evident from Fig. 2a–b that Cu@mMOR contained numerous Cu black dots (1.5 ± 0.3 nm) that were homogenously dispersed throughout the nanosheets. These amorphous Cu nanodots were encapsulated in mMOR, which was identified by the lattice fringes of (110) plane10 (Fig. 2c). According to the elemental energy-dispersive X-ray spectroscopy (EDS)-mapping images depicted in Fig. 2d, the Cu element was homogeneously distributed throughout the mMOR zeolite. However, the particle size of Cu in spherical Cu@SiO2 (~20 nm, Fig. 2e) was about ~2.1 nm (Fig. 2f), which was obviously larger than that in Cu@mMOR. It can be inferred that Cu was further reduced into smaller clusters in a confined space during the crystal transformation from SiO2 to mMOR. In contrast, Cu-impregnated mMOR (denoted as Cu/mMOR) contained nano-sized particles of approximately 4.4 nm on its outer surface of mMOR (Fig. 2g), and these nanoparticles displayed distinct lattice patterns corresponding to the metallic Cu (111) crystal plane11 (Fig. 2h). It can be seen that the copper acetylacetonate impregnation method proved challenging for incorporating Cu nanoparticles into mMOR crystals and effectively controlling their particle size within mMOR.

a–d TEM images and elemental distribution of Cu@mMOR. e–f TEM images of Cu@SiO2. g–h TEM images of Cu/mMOR.
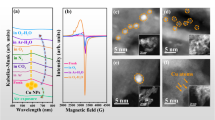
X-ray diffraction (XRD) and X-ray fluorescence (XRF) analyses (Supplementary Fig. 2 and Supplementary Table 1) revealed that Cu/mMOR and Cu@mMOR had high crystallinity of MOR type zeolite, whereas Cu@SiO2 showed a diffuse X-ray peak of amorphous silica phase. This indicated that the amorphous silica nanosphere was completely transformed into a highly crystalline MOR zeolite structure in Cu@mMOR12. In addition, none of the three catalysts (Cu@SiO2, Cu/mMOR, and Cu@mMOR) showed clear diffraction peaks of Cu crystals. However, the Cu element detected in XRF (Supplementary Table 1) had a very similar percentage composition in the range of 2.6–4.1 wt%, suggesting that Cu species were highly dispersed in these catalysts. Additionally, the XPS survey (Supplementary Fig. 3) showed the slight presence of Na element in Cu@mMOR but not in Cu/mMOR, implying that the former remains some Na+ sites. Moreover, the Cu 2p XPS patterns of Cu@mMOR and Cu@SiO2 exhibited much lower peak area than that of impregnated Cu/mMOR (Supplementary Fig. 4), suggesting the extensive encapsulation of Cu nanoparticles rather than their exposure on the MOR external surface13. Next, the pore texture (Supplementary Fig. 5 and Supplementary Table 2) of Cu@mMOR showed a slight reduction in N2 adsorption uptake (427.1 m2 g−1) of type I isotherm compared to that of pure mMOR (530.1 m2 g−1). This meant that most of the micropore structure of mMOR maintained upon encapsulating Cu nanoparticles in mMOR. In contrast, Cu/mMOR exhibited a lower surface area of 310.7 m2 g−1, indicating a partial loss of the mMOR porous structure due to the obstruction caused by the impregnated Cu nanoparticles14. As anticipated, Cu@SiO2 had minimal porous structures attributed to the low porosity of SiO2. The textural structure analysis inferred that Cu@mMOR maintained the mMOR porous property after encapsulating Cu species with homogeneous distribution, which would favor the adsorption and transport of reactants and active species during the reaction. Further, positron annihilation lifetime spectroscopy (PALS) was conducted to determine the pore size distribution of the ultra-microporous region in MOR, mMOR, and their Cu-based catalysts15. The deconvoluted PALS spectra display four distinct peaks corresponding to different positron annihilation lifetimes (τ1-τ4; Supplementary Fig. 6a). An increase in τ value indicates larger pore sizes within the MOR framework. Here, τ1 denotes the bulk lifetime, which is attributed to direct annihilation within the non-porous regions16. The remaining lifetimes τ2-τ4 correspond to positron annihilation within the 5-membered, 8-membered, and 12-membered ring channels of MOR, respectively (Supplementary Fig. 6b–c). In comparison to the support, the τ3 components associated with 8-MRs exhibited distinctly opposite trends in Cu@mMOR and Cu/mMOR. Specifically, the calculated pore size for Cu/mMOR increased to 3.6 Å. This observation aligns with the pore-blocking effect identified via N2 adsorption isotherms (Supplementary Fig. 5), ascribed to the obstruction of pore mouths by external Cu particles. Thus, it compels positrons to annihilate within larger inter-particle voids on the surface17. In contrast, the encapsulation strategy yielded a shrunken pore size of 2.8 Å for Cu@mMOR, directly confirming the effective occupation of the 8-MR channels by Cu species. Furthermore, coincidence Doppler broadening (CDB) analysis revealed a higher electron density for Cu@mMOR in the low-momentum valence electron region (<4 × 10−3 m0c; Supplementary Fig. 7), corroborating the tight confinement of copper clusters within the unique micro-environment constructed by the 5-MRs and 8-MRs18. Notably, this shrunken pore mouths of Cu@mMOR (2.8 Å) precisely matches the kinetic diameter of H2 (2.8 Å) while remains smaller than that of CO2 (3.3 Å)19. Conceptually, this double-ring encapsulation architecture could act as a “molecular fence” for suppressing thermal aggregation and Ostwald ripening via the spatial separation of CO2 and H2.
Structural characterization of different Cu-based catalysts
To better understand the local structure of Cu species in different micro-environments, X-ray absorption spectroscopy (XAS) was performed in order to evaluate the Cu chemical state and coordination status in various catalysts. The edge energy of the two catalysts (Cu@mMOR and Cu/mMOR) at the Cu K-edge (Fig. 3a) was positioned between the Cu foil and Cu2O standards, with no pre-edge band of Cu2+ detected in the rising edge. This implied that Cu species were in the form of Cu0 and Cu+ rather than Cu2+ in the two catalysts20. Compared to Cu/mMOR, Cu@mMOR exhibited a much higher Cu adsorption edge energy, attributing to a higher proportion of Cu+ species in Cu@mMOR21. To quantify the Cu+ species in the catalyst, a mathematical relationship between the absorption edge energy position and the valence state was established by fitting data from the corresponding standards22,23. As shown in Supplementary Fig. 8, the Cu+ content in Cu@mMOR was calculated to be ∼ 38.1 at.%. In contrast, the Cu+ proportions in Cu/mMOR and Cu@SiO2 were determined to be only 9.0 and 6.9 at.%, respectively. The significantly higher Cu+ content in Cu@mMOR is indicative of enhanced interfacial charge transfer from Cu0 to mMOR, a process facilitated by the abundant interfaces inherent to the encapsulation structure.

a XANES spectra at Cu K-edge. b Fourier transform EXAFS plot of the Cu K-edge. c Wavelet-transformed two-dimensional spectra for c1 Cu@mMOR, c2 Cu2O standard, c3 Cu-foil standard and c4 Cu/mMOR. d Schematic illustration of the formation of Cu clusters in Cu@mMOR by lattice pressure. e UV-Raman spectra for Cu@mMOR and Cu/mMOR. f TPR-MS profile for Cu@mMOR and Cu/mMOR.
The local coordination environment of Cu species in various catalysts was identified by fitting the Fourier transform extended X-ray absorption fine structure (FT-EXAFS) of the various catalysts, as illustrated in Fig. 3b. As shown, Cu@mMOR exhibited a notable Cu-O peak at 1.46 Å, as well as minor Cu-O-Cu and Cu-Cu peaks at 2.57 and 2.08 Å, respectively, indicating that most of its Cu species were coordinated with O within Cu@mMOR24. Furthermore, the wavelet transform (WT) plot of Cu@mMOR in Figs. 3c-1 showed an obvious Cu-O bond (k = 3.2–5.6 Å−1, R = 1.8-1.9 Å) and a weak Cu-Cu bond (k = 6.2-7.4 Å−1, R = 2.1-2.7 Å) when compared to Cu2O (Figs. 3c-2) and Cu foil (Figs. 3c-3) counterparts25, corroborating the results from the FT-EXAFS spectra. The shell fitting analysis presented in Supplementary Table 3 depicts that most of the copper in Cu@mMOR coordinated with oxygen (O) and formed Cu-O clusters, with only a small amount of Cu-Cu bonding remaining26. The XANES, FT-EXAFS, and WT plot results proved that Cu-O clusters dominated the main position in the Cu@mMOR, with a small portion of metallic Cu clusters in mMOR. Differently, Cu/mMOR and Cu@SiO2 (Supplementary Fig. 9) displayed significant similarities to Cu-foil in their EXAFS and WT plot features, reflecting the predominant presence of metallic Cu0 with larger nanoparticle sizes in Cu/mMOR and Cu@SiO2. From the above results, the schematic representation of Cu@mMOR formation is illustrated in Fig. 3d. Initially, the SiO2 in Cu@SiO2 began to crystallize into mMOR. Then, Cu grains confined in mMOR underwent mMOR compression and tore into smaller Cu nanoclusters. During this process, confined space continuously generated strong interactions between Cu and silanol, resulting in enrichment of the Cu-O coordination within mMOR. Thus, Cu@mMOR contained abundant Cu-O clusters with atomic distribution in the mMOR crystal. Conversely, this character was not observed for Cu/mMOR due to the lack of the in situ mMOR crystallization process.
Moreover, the types and contents of defects in Cu clusters within Cu@mMOR were investigated via UV-Raman spectroscopy, with the results shown in Fig. 3e. As seen, the confined Cu@mMOR exhibited a sharp peak at 221 cm−1 and a broad peak around 400–463 cm-1, corresponding to the defective Cu-O of Cu vacancies (the missing Cu atom in Cu-O-Cu motifs) and Si-O of MOR framework, respectively27,28. In comparison, Cu/mMOR displayed a negligible peak of defective Cu-O around Cu vacancies at 218 cm-1 and a minor peak of Si-O of MOR at 397 cm-1. The intensities of these peaks indicated that Cu@mMOR had a much higher concentration of Cu vacancies than Cu/mMOR, which was attributed to the Cu-O defective coordination during the crystallization of mMOR from SiO2. Additionally, Raman spectra (Supplementary Fig. 10) were also measured for Cu@mMOR and Cu@SiO2. As observed, the defective Cu-O around Cu vacancies had a significantly higher peak intensity with a blue shift from 215 to 221 cm-1 in Cu@mMOR compared to Cu@SiO2. This once again demonstrated that the lattice compression of crystal transformation induced the contraction of Cu-O bonds in Cu clusters, resulting in the generation of additional Cu vacancies29.
It has been reported that Cu vacancies can significantly influence the redox properties of catalysts30,31. Furthermore, H2 temperature-programmed reduction-mass spectrometry (H2-TPR-MS) analyses of various catalysts were carried out to investigate the redox properties of Cu. Initially, all catalysts underwent complete oxidation to CuO prior to measurement. Figure 3f showed the TPR-MS profiles of Cu@mMOR and Cu/mMOR. As shown, hydrogen reduction profile of Cu@mMOR exhibited a single hydrogen reduction peak at 262 °C, whereas the corresponding peak for Cu/mMOR appeared at a higher temperature (ca. 300 °C). This significant shift to a lower temperature suggested that the copper species in Cu@mMOR possessed smaller sizes and higher reducibility32. To quantitatively assess the hydrogen spillover effect, the hydrogen spillover capacity was determined by calculating the additional H2 consumption beyond that used for water formation. The results showed that each Cu atom in Cu@mMOR consumed an extra 0.45 H atoms, while the Cu/mMOR exhibited almost no additional H2 consumption (0.01 H/Cu). These results suggest that a significant hydrogen spillover phenomenon occurred in Cu@mMOR, thereby demonstrating that the synergy between its confined microenvironment and copper vacancies effectively promotes H2 activation and spillover.
Importantly, the electron transfer induced by metal vacancies can alter the surface acidic properties of support33. In order to better understand this effect, NH3-TPD (Supplementary Fig. 12) was performed to explore the acidic concentration and acidity of mMOR in various catalysts. As displayed, pristine mMOR had a total acid content of 580 μmol g-1, mainly consisting of strong (peak > 350 °C) and weak (peak <250 °C) acids34. Compared to pristine mMOR, Cu@mMOR showed a notable reduction in total acid content to 197 μmol g-1, with the retained portion almost composed of strong acids. Conversely, Cu/mMOR exhibited a slight reduction in all acids, yielding a total value of 398 μmol g-1. Furthermore, pyridine-IR spectra were utilized to gain insight into the effect of Cu loading mode on the acid distribution of catalysts, as shown in Supplementary Fig. 13. Compared to Cu/mMOR, Cu@mMOR demonstrated a much larger abatement in content of Lewis acid sites (LAS), owing to the numerous coordination-saturated Cu clusters in confined microenvironment35. Interestingly, the remaining Brønsted acid sites (BAS) in Cu@mMOR almost belonged to strong acid sites36, aligning well with our NH3-TPD curves (Supplementary Fig. 12). As calculated, its strong BAS ratio in total BAS was up to 81.7%, far higher than those of mMOR (62.3%) and Cu/mMOR (57.0%). This increase was mainly contributed to the enriched Cu-acid interactions in mMOR confined spaces, which induced massive electron transfer from Cu to the O of BAS (as evidenced by Cu XANES in Fig. 3a and O 1 s XPS in Supplementary Fig. 14). Meanwhile, this charge transfer could further enable Cu@mMOR to retain more Na+ sites (mentioned in Supplementary Fig. 3), which are stabilized by stronger electrostatic interactions towards O of Cu@mMOR37.
Reaction performance of Cu@mMOR and related catalysts
The effects of Cu diverse microenvironment within prepared Cu-based catalysts on their catalytic activity for CO2 hydrogenation to syngas were evaluated, and the results are presented in Fig. 4. As seen in Fig. 4a, there was an upward tendency in the CO2 conversion of each catalyst when the reaction temperature increased from 450 to 650 °C. By comparison, Cu@mMOR exhibited a superior RWGS activity and was very close to the thermodynamic equilibrium (calculated using HSC6.0 software)38, which could reach 49.1% of CO2 conversion at 600 °C. In addition, it had the lowest CO apparent activation energies (34.09 ± 6.88 kJ mol-1) among the three catalysts (Supplementary Fig. 15), signifying its superior catalytic activity for CO2 conversion.

a–c CO2 conversion rate of Cu-based catalysts as a function of a reaction temperature, b H2/CO2 ratio and c space velocity. d Durability test for Cu@mMOR and Cu/mMOR. e RWGS performance comparison between Cu@mMOR and other reported catalysts. f XRD patterns of spent Cu@mMOR and Cu/mMOR. g–h TEM images of Cu@mMOR and Cu/mMOR before and after reaction of 120 h.
Additionally, a higher H2 ratio in the feed gas was beneficial in improving CO2 conversion for all three catalysts (Fig. 4b). Notably, Cu@mMOR and Cu@SiO2 demonstrated a similar variation trend in CO2 conversion with H2/CO2 ratios to the thermodynamic equilibrium curve, with the former one approaching close to the dashed line of reaction thermodynamic equilibrium, demonstrating much higher catalytic activity of Cu@mMOR than that of Cu@SiO2. On the contrary, the trend of Cu/mMOR shifted away from the dashed line of reaction thermodynamic equilibrium with an increase in H2/CO2 ratios. The discrepant RWGS trends in encapsulated catalysts and exposed catalysts mainly presented different activation mechanisms. For the encapsulated catalyst Cu@mMOR, the reaction likely followed an H-assisted mechanism39. In this process, H2 facilitates CO2 activation through active species, which is a typical feature in dual-site catalysts40. However, as an exposed catalyst, Cu/mMOR may require H2 and CO2 to compete for the same site for activation (the Redox mechanism)41. Therefore, the excessive dosage of H2 would pre-occupy Cu active sites and suppress the trend of H2 promoting CO2 conversion. Figure 4c depicts the variation trend in CO2 conversion over various catalysts as space velocity increased. As seen, it showed a plateau and then a slight descend of CO2 conversion in Cu@mMOR with the increase of space velocity. By contrast, Cu@SiO2 and Cu/mMOR displayed obvious descend trend, indicating Cu@mMOR possessed an ultra-rapid pathway from CO2 into CO compared to Cu/mMOR and Cu@SiO242.
Based on the above results, H2/CO2 ratios of 2, space velocities of 270 L gcat−1 h−1, and a reaction temperature of 600 °C were chosen as the plausible conditions for the subsequent durability test. As presented in Fig. 4d, Cu@mMOR exhibited a steady period of up to 788 hours with its corresponding CO2 conversion remaining similar at around 49.1%, which consistently approached the thermodynamic limit of CO2 conversion (50.2%). On the contrary, Cu/mMOR showed much faster catalytic deactivation with markedly reduced CO2 conversion. Moreover, it demonstrated a significant decline in catalytic stability, with its CO2 conversion plummeting from 30.8% to 13.5% after 158 h of reaction. Obviously, Cu@mMOR with the unique MOR-confined microenvironment could withstand the ordeal of a long-term RWGS reaction. Through calculation, the highest space-time yield (STY) of CO over Cu@mMOR was 3290 mmol gcat−1 h−1 and the max stability duration reached up to 788 h, displaying the highest STY and notable durability, surpassing most of the reported Cu-based catalysts under a severe condition of high temperature and high flow43,44,45,46,47,48,49,50,51,52 (Fig. 4e and Supplementary Table 4).
To further understand the structural evolution of Cu species during the long-term reaction process, the structure and morphology of fresh catalysts and spent catalysts after 120 h reaction were determined by using XRD and TEM, as shown in Fig. 4f–h. Spent Cu/mMOR displayed obvious diffraction peaks at 43° and 51° assigned to metallic Cu bulks53, which did not appear in the fresh samples. This suggested that the local aggregation of Cu increased with reaction time. On the other hand, no distinct Cu diffraction peaks were found in spent Cu@mMOR, implying a high dispersion of Cu species within mMOR. Additionally, TEM images revealed that Cu species in fresh and spent Cu@mMOR had nearly the same particle size of approximately 2 nm. In the case of Cu/mMOR, the Cu nanoparticles increased in size from an initial range of 3–6 nm in the fresh sample to a range of 15–33 nm in the spent sample. Taking account of these above characterizations, the sintering of Cu species was the main cause of the Cu/mMOR deactivation.
RWGS reaction mechanism of Cu@mMOR
To validate the sites separated effect of Cu@mMOR for CO2 and H2, N2O was employed to replace CO2 for touching with Cu species in Cu@mMOR and Cu/mMOR owing their similar molecular configuration and diffusion properties54. At medium temperature, N2O selectively oxidized the surface and subsurface Cu0 and Cu+ species of the catalysts55. The resulting oxidized Cu species were subsequently quantified via H2-temperature programmed reduction (H2-TPR). As depicted in Fig. 5a and Supplementary Fig. 16, the Cu@mMOR sample exhibited only trace H2 reduction peak around 281 °C, whereas Cu/mMOR displayed a pronounced reduction peak at 294 °C. Correspondingly, the H2O signal generated during the reduction of Cu@mMOR was significantly weaker than that of Cu/mMOR. These results confirm that most Cu sites in Cu@mMOR were effectively shielded from N2O oxidation, demonstrating the ‘molecular fence’ function of the tight encapsulation. In contrast, the absence of such restriction in Cu/mMOR led to substantial exposure of Cu species to the N2O atmosphere. Finally, CO2-DRIFTS was conducted at elevated temperatures to further validate the molecular fence effect under realistic reaction conditions. As demonstrated in Fig. 5b, Cu@mMOR exclusively showed absorbed CO2 and carbonate species upon CO2 introduction without other reacted species, while the conventional Cu/mMOR catalyst immediately exhibited characteristic CO vibrational bands. These results demonstrated that the molecular fence structure of Cu@mMOR effectively prevented CO2 from accessing Cu clusters even under actual reaction conditions, thereby maintaining spatial isolation of the active sites.

a TPR-MS of Cu@mMOR and Cu/mMOR after N2O treatment. b CO2-DRIFTS of Cu@mMOR and Cu/mMOR. c HD-exchange of Cu@mMOR and Cu/mMOR. d Gibbs free energy profiles of the H2 dissociation for Cu@mMOR and Cu/mMOR. CO2/H2-DRIFTS of Cu@mMOR (e) and Cu/mMOR (f). g Gibbs free energy profiles of the CO2 hydrogenation in Cu@mMOR.
Next, CO2-TPD was employed to analyze the influence of Na+ alkaline sites on mMOR for CO2 adsorption and active sites. As displayed in Supplementary Fig. 17, Cu@mMOR demonstrated strong CO2 chemisorption at 231 °C, whereas Cu/mMOR without Na+ displayed an upward-trending peak above 450 °C. This implied that Na+ may serve as the CO2 chemisorption site in Cu@mMOR56. Furthermore, CO2-TPD, NH3-TPD, and CO2 hydrogenation performance of the Cu@mMOR with varying Na contents were systematically studied to investigate the influence of Na+ on the CO2 chemisorption, acidic site density, and CO2 hydrogenation activity. As shown in Supplementary Fig. 18a–b, CO2 chemisorption capacity of Cu@mMOR exhibited a well-defined linear increase with rising Na+ content, while the protonation degree (acidic site density) decreased progressively with Na+ incorporation. Meanwhile, CO2 hydrogenation rate (Supplementary Fig. 18c) displayed a characteristic volcano-shaped trend with increasing Na+ content, which initially rose to a maximum and then declined. This volcano-type behavior suggested that Na+ played an auxiliary role in activating CO2. Specifically, Na+ enhanced the adsorption of CO2 on adjacent Si-O--Al sites via electrostatic interactions, thereby increasing the surface coverage and residence time of CO256. This enhancement ultimately contributed to the superior CO2 hydrogenation performance of Cu@mMOR.
Subsequently, the CO2 diffusion process in Cu@mMOR was validated using ab initio molecular dynamics simulations (AIMD). As seen in Supplementary Movie 1, CO2 was only prone to diffuse in the channels of mMOR at a reaction temperature of 600 °C. It is noteworthy that the distance between CO2 and Na+ site gradually transitioned from a fluctuating state to a stabilizing one at 3.4 Å (Supplementary Fig. 20). This indicated that CO2 was rapidly adsorbed by the Na+ site during diffusion, occurring within approximately 10 ps. Constrained ab initio molecular dynamics (cAIMD) was further employed to probe the CO2 adsorption-activation mode at 600 °C (Supplementary Movie 3-7). As illustrated in Supplementary Fig. 21, the formed carbonate involved the lowest energy barrier (0.7 eV) to bend O–C–O bond. This demonstrated that Na+ markedly reduced the energy barriers for both CO2 adsorption and activation. Therefore, it was proved that CO2 rapidly bonded with Na+ and then formed carbonate during its diffusion through the channels of Cu@mMOR, effectively triggering the reaction57.
H2 adsorption behaviors of Cu@mMOR and Cu/mMOR were carried out through H2-TPD. As seen in Supplementary Fig. 22, Cu@mMOR had a higher desorption temperature than Cu/mMOR, suggesting that the Cu-O clusters in Cu@mMOR are more favorable for adsorbing H2. Next, H2 dissociation behaviors of various catalysts were evaluated by using H-D exchanged tests, and the obtained results were shown in Fig. 5c. The results clearly revealed that Cu@mMOR exhibited varying HD production rates compared to Cu/mMOR as the temperature increased (Eq. 5, SI). In contrast, the HD curve of Cu/mMOR grew slowly until it significantly rose at 435 °C. Its formation rate of HD was only 78 μmol gcat-1 s-1 at 500 °C with an H2 dissociation rate of 3.9%. By comparison, Cu@mMOR showed about 11.5 times the H* formation rate than Cu/mMOR, attributing to abundant Cu vacancies in Cu@mMOR boosting H2 dissociation capacity58. To further validate this hypothesis, Fig. 5d illustrated the free energy profiles of the H2 dissociation process on various models (the detailed models see Supplementary Figs. 34–35). These models include Cu@mMOR with Cu vacancies (denoted as Cu@mMOR-VCu), Cu@mMOR without Cu vacancies (denoted as Cu@mMOR-absent-VCu), and Cu/mMOR. As shown, the energy barrier of H2 dissociation of Cu@mMOR-VCu, Cu@mMOR-absent-VCu and Cu/mMOR were 0.37, 0.70, and 1.23 eV, respectively. It indicated that Cu vacancies promote accelerated H-H bond cleavage. Notably, in Cu@mMOR systems, the dissociated H* species preferentially coordinate with both Si-O--Al groups and Cu+ sites, leading to the formation of H+/H- pairs (Supplementary Fig. 34). Consequently, Cu@mMOR utilized Cu-O clusters to adsorb and activate H2, promoting its heterolytic dissociation through its Cu vacancies. This phenomenon boosted CO2 reduction by providing a steady supply of highly reductive H* species.
To unravel the chemical nature and dynamic behavior of hydrogen spillover at the molecular level, the evolution of hydrogen-containing species on Cu@mMOR were monitored using steady-state isotopic transient kinetic analysis coupled with FTIR (SSITKA-FTIR) during H2/D2 switching. As depicted in Supplementary Fig. 36, under a H2 atmosphere, distinct signals assigned to hydride species (Al-H and Si-H) and characteristic peaks for protic species (Si-OH-Al and Si-OH) were clearly observed on Cu@mMOR59,60. The coexistence of these H+ and H- species provided direct evidence for the heterolytic cleavage of H2 on Cu@mMOR and identifies the specific nature of the resulting fragments. Upon switching the feed to D2, the intensities of these hydrogenated species gradually decayed, with a concomitant growth of deuterated species peaks at the lower wavenumbers59,60. This rapid H/D isotopic exchange demonstrated that the generated H+/H- pairs exhibited high mobility and dynamic exchange activity over the framework acid-base sites61,62.
Next, in situ EPR was carried out to investigate the electronic mechanism driving H2 heterolytic activation. As shown in Supplementary Fig. 37, Cu@mMOR displayed a strong signal associated with unpaired electrons on the Cu clusters under H2 feeding63. When the atmosphere was switched to CO2/H2, this EPR signal decreased markedly. This indicated that the electrons were transferred to adsorbed hydrogen to generate H- species, which subsequently migrate into the channels and participate in the RWGS reaction. Furthermore, XPS analysis was conducted to elucidate the migration and consumption dynamics of hydrogen species by monitoring valence shifts of Cu and Al in Cu@mMOR under H2 and CO2/H2 atmospheres. As depicted in Supplementary Fig. 38, H2 treatment decreased the binding energies of both Cu and Al to indicate an electron-rich state, whereas the subsequent introduction of CO2/H2 triggered a synchronous positive shift in these values64,65. These findings corroborated the spillover of H- species from Cu surfaces onto the framework, where they were consumed by the reaction with CO2 within the pores. Collectively, this unique H+/H- pair migration mechanism facilitated an efficient hydrogen supply. Furthermore, it modulated the hydrogenation pathways of formate intermediates via electrostatic interactions to enhance both the sustainability and selectivity of the RWGS reaction.
Furthermore, the spillover kinetics of the catalyst were evaluated using the kinetic isotope effect (KIE). In this context, the CO formation rate under CO2/H2 and CO2/D2 atmosphere was respectively measured and the corresponding KIE values with reaction time increasing were shown and calculated in Supplementary Fig. 39. As shown, the KIE value (rH/rD) of Cu@mMOR increased progressively from 1.03 at 300 °C to 1.43 at 600 °C, revealing a pronounced temperature-dependent discrepancy. Generally, the magnitude of KIE is governed by the zero-point energy (ZPE) and the tunneling effect66. The observed rise in KIE with temperature indicated that the hydrogen spillover process on the Cu@mMOR catalyst was accompanied by a significant quantum tunneling effect, contrasting with the temperature-diminishing ZPE contribution67. This tunneling process facilitated the rapid spillover of active hydrogen species from Cu sites into the mMOR channels, thereby promoting CO2 reduction.
Notably, the increasing KIE value suggested that more hydrogen-containing species were involved in the H* spillover process of Cu@mMOR68. Additionally, the DRIFTS of Cu@mMOR revealed the presentation of H(H2O)n*, which initially increased before reaching a steady state (Supplementary Fig. 40). These findings implied that H(H2O)n* may function as a transporter of H* during the spillover process in Cu@mMOR69,70. To deeply investigate the spillover progress of H(H2O)n*, an AIMD simulation was carried out and displayed in Supplementary Movie 2. The video shows that H* on the Cu-O cluster was prone to coordinate with H2O and subsequently migrated within the mMOR channel. As calculated in Supplementary Fig. 42, the energy barrier of H2O-assisted spillover was only 0.29 eV, lower than most of the reported values (0.4 ~ 0.8 eV)71,72,73. In this system, [H(H2O)*] had a higher tendency to migrate through the channels of Cu@mMOR owing to its delocalization. Thus, the transfer efficiency of H* in Cu@mMOR was significantly improved, allowing adsorbed CO2 to be rapidly reduced. Furthermore, the independent gradient model based on Hirshfeld partition (IGMH, Supplementary Fig. 43a) indicated that within the Cu@mMOR channels, H- could form H(H2O)n- clusters with water molecules via strong hydrogen bonds74. This interaction facilitated the dispersion of the negative charge on H-, thereby enhancing thermodynamic stability. Simultaneously, the stability of H(H2O)n- was found to increase with the number of water molecules (Supplementary Fig. 43b–c), implying that H- was thermodynamically driven to approach the CO2 reaction sites where water was produced75. To further investigate the water effects on hydrogen spillover, H2/D2 and H2 + H2O/D2 + D2O switching SSITKA experiments were performed on Cu@mMOR (Supplementary Fig. 44). At which, water-assisted hydrogen spillover exhibited an extremely long residence time of 199.4 s under H2 + H2O/D2 + D2O switching conditions (Supplementary Fig. 44b), significantly exceeding the 46.6 s observed during H2/D2 switching (Supplementary Fig. 44a). The markedly extended residence time indicates that water enables hydrogen spillover to occur over a significantly much longer distance on Cu@mMOR via facile the O-H bond cleavage and the pronounced quantum tunneling effects of H(H2O)n+/H(H2O)n-76. Thereby, it established a highly efficient cycle for the spatially separated RWGS reaction by maximizing the active hydrogen and CO2 contact.
In order to elucidate the reaction species of RWGS over Cu@mMOR, DRIFTS tests under mixed CO2/H2 atmospheres (CO2/H2-DRIFTS) were respectively conducted to analyze the variation of RWGS intermediates from CO2 and H2, and the obtained spectra are illustrated in Fig. 5e. As observed, Cu@mMOR displayed distinct peaks of CO2* (carbonate species) and Al-H around 1310-1450 cm-1, which demonstrated an upward trend with increasing reaction time. Meanwhile, the peaks of HCOO* species (1603 cm−1), H2O (3363 cm−1), B-acids (3588 cm−1), and CO* (2152 cm−1) appeared and maintained a dynamic equilibrium77,78,79,80. These intermediates confirmed that the RWGS reaction over Cu@mMOR likely followed exclusively the formate pathway, and H2O molecules actively participated in H+ transfer during H* spillover. Furthermore, steady-state isotope transient kinetic analysis (SSITKA) experiments were performed to the Cu@mMOR system to gain deeper kinetic insight. The procedure involved equilibrating the catalyst under a CO2/H2 atmosphere, followed by a rapid switch to an isotopic gas mixture (13CO2/H2 or CO2/D2) supplemented with an inert Kr internal standard at an identical flow rate. The dynamic evolution of surface species and effluent products were simultaneously monitored in real-time via infrared spectroscopy and mass spectrometry, respectively. As shown in Supplementary Figs. 46–52, the residence times τ of key reaction species over Cu@mMOR followed the order: 13CO2* (3.4 s, CO2 chemisorption) <H13COO* (11.4 s, CO2 hydrogenation) <H12COO* (46.4 s, formate dissociation) <Al-H (57.5 s, hydrogen spillover). Among which, Al-H species exhibited the longest isotopic residence time, indicating that hydrogen spillover was kinetically limited and thus identified as the reaction’s rate-determining step (RDS)81,82,83,84.
Density functional theory (DFT) calculations were performed to investigate the CO2 hydrogenation mechanism over the Cu@mMOR at the atomic level. As shown in Fig. 5g, H2 heterolytically dissociated on the Cu cluster, generating active H+ and H- that migrate to the Si-ONa-Al site. Meanwhile, CO2 was chemisorbed to carbonate at the same site. In this chemisorption configuration, CO2* (carbonate species) could be hydrogenated to COOH+ via the carboxyl pathway or to HCOO- via the formate pathway. A comparison of energy barriers exhibited that forming COOH+ needed 0.98 eV, whereas producing HCOO- required only 0.64 eV. Due to the lower barrier, CO2* preferred to follow formate pathway, which was consistent with the DRIFTS observation that detected only the formate intermediate (Fig. 5e). Next, HCOO- may either become protonated or directly dissociate into CO. Protonation is kinetically much more favorable, with a very low barrier of only 0.43 eV, leading easily to HCOOH formation. In contrast, direct dissociation pathways face much higher barriers (1.72 eV for HCOO⁻ and 2.04 eV for COOH⁺), making them far less likely. These substantial barriers (particularly for COOH+) were attributed to the necessity of rupturing an additional Al-H bond to facilitate electron transfer, strong antibonding orbital repulsion, and the requirement to overcome hydrogen bonding interactions (Supplementary Figs. 53–54 and Supplementary Movies 11–14)74,85,86,87,88. Subsequently, the generated HCOOH could either dissociate directly or undergo further hydrogenation. By comparing both energy barrier, direct dissociation of HCOOH required merely 0.92 eV, whereas successive hydrogenation must overcome higher barriers of 2.04 eV and 0.93 eV. In addition, AIMD further exhibited that the dissociation energy barrier of HCOOH inside the mMOR channel was 0.24 eV lower than that outside the channel (Supplementary Fig. 55). Therefore, HCOOH rapidly decomposed to CO and H2O within the Cu@mMOR channels, which explained why Cu@mMOR achieved nearly 100 % CO selectivity. Notably, DFT calculations supported that Cu@mMOR followed the formate pathway and identified H+/H- migration from Cu to the mMOR as the highest-energy step, establishing it as the rate-determining step in line with our SSITKA analysis (Supplementary Fig. 52).
By comparison, Cu/mMOR showed no apparent vibration peaks of hydrogenated intermediates in CO2/H2-DRIFTS (Fig. 5f), signifying the absence of H* spillover for the reduction of CO2 to CO. In addition, its KIE curve displayed that the KIE value was almost similar in CO2/H2 and CO2/D2 (Supplementary Fig. 39). In addition, the intensities of typical vibration peaks (1900 to 2100 cm-1) of metal carbonyl compounds increased with reaction time, indicating a gradual rise of Cu(CO)n species in Cu/mMOR89. These species were typically linked to the Oswald-ripening phenomenon resulting from excessive CO adsorption90. Therefore, unencapsulated Cu was prone to form highly mobile species in the absence of an effective barrier to prevent CO adsorption (Fig. 6). It triggered Ostwald ripening, which eventually led to the deactivation of Cu/mMOR.

The schematic diagrams of Cu deactivation in Cu/mMOR and the sites-separation system in Cu@mMOR.
In summary, confined Cu-O clusters in MOR zeolite (Cu@mMOR) were designed for a highly efficient CO2 RWGS reaction through a separation strategy. We induced the growth of small cages (<3 Å) tightly encapsulating Cu-O clusters in order to separate catalysis sites of Cu@mMOR for H2 and CO2, respectively. At which, confined Cu-O clusters generated abundant Cu vacancies and robust Cu-O bonds within the mMOR shell, thereby promoting the heterolytic dissociation of H2 by an 11.5-fold enhancement compared to unencapsulated Cu/mMOR. Additionally, mMOR zeolite captured and activated CO2 with fast kinetics (a rapid progress of ~10 ps), effectively forming a molecular fence to isolate CO2 from the Cu-O clusters. In the continuous RWGS reaction, the generated H* species migrated from Cu-O clusters to mMOR and contacted CO2, realizing the reduction of CO2 to CO via the formate pathway. Interestingly, H2O was proved to serve as an effective H* carrier, which significantly promoted H* delocalization and its reduction efficiency by a 20.5-fold increase. The established synergistic interactions endowed Cu@mMOR with high catalytic activity in the RWGS reaction. During the long-term reaction, Cu@mMOR prevented thermal aggregation owing to its strong Cu-O interactions. Moreover, the spatial separation of CO prevented the Ostwald ripening of Cu-O clusters. Eventually, Cu@mMOR exhibited remarkable RWGS activity of 3290 mmol CO gcat-1 h-1 under high space velocity conditions, along with 788 hours of high-temperature stability, surpassing the majority of the latest reported Cu-based catalysts. This work offers valuable insights for the development of catalysts that can overcome the challenges associated with Ostwald ripening and agglomeration.
Methods
Catalyst preparation
Cu@SiO2 was prepared by a modified reported method from the literature91. Briefly, 3.37 g of Brij-58 was dissolved in 15 mL of cyclohexane in a 50 mL sample bottle at 50 °C under stirring. Then, 0.4 mL of copper nitrate solution (2 M) was added to form a reverse microemulsion system. After 1 h of stirring, 1 mL of ammonium hydroxide was added dropwise, followed by 2 g of tetraethyl orthosilicate after 30 minutes. The silica coating proceeded for 2 h, after which 15 mL of isopropanol was added to demulsify the microemulsion. The mixture was centrifuged to obtain the precipitate (8000 rpm, with a relative centrifugal force of 5800 g), which was dried in a vacuum oven at 70 °C for 12 h. The dried powder was then calcined at 500 °C in an air stream to remove organic species. During calcination, the temperature was increased slowly at a rate of 1 °C per minute to avoid the formation of quartz phases and to prevent the rapid diffusion of decomposition gases, which could create excessive meso-porosity. Eventually, the product Cu@SiO2 was reduced at 773 K for 3 h under a 30 mL/min flow of 5% H2 in N2.
Cu@mMOR was prepared via a 1-methylpyrrolidine (NMP)-assisted crystallization method. In a typical preparation, 500 mg of Cu@SiO2 was mixed with sodium aluminate (0.513 g), sodium bromide (1.28 g), aqueous sodium hydroxide (8.6 g, 30 wt%), and deionized water (66.37 g) in a homogenizer at a high speed of 20000 rpm to form a uniform solution. The purpose of this step was to enable the pre-adsorption of Na+ on the Cu@SiO2 interface. Then, 23.3 g of silica sol and 6 g of the structure-directing agent NMP were slowly added dropwise to this uniform solution. The purpose of adding NMP was to accelerate the crystallization of the MOR framework. Next, the mixture was kept at 60 °C and stirred for 6 hours to form a uniform gel. Subsequently, the gel was transferred to a 150 mL Teflon-lined autoclave for hydrothermal crystallization at 170 °C. After 4 days of heating, the solid product was separated by centrifugation (8000 rpm, with a relative centrifugal force of 5800 g), washed with deionized water, and then dried overnight at 120 °C. The sample was then calcined at 550 °C for 8 hours to remove the organic components. To retain the Na as CO2 adsorption sites and avoid the damage of framework by Al leaching, a “low-concentration, multi-step” exchange was adopted to precisely adjust the Na content. The calcined sample was subjected to ion exchange with 0.1 M ammonium chloride solution three times for 2 hours each, washed with deionized water, and dried overnight at 60 °C. Eventually, the dried sample was followed by air calcination and H2 reduction at 500 °C to obtain Cu@mMOR.
Typically, the preparation of mMOR was similar to that of Cu@mMOR. The main difference was that Cu@SiO2 was not added, and the amount of silica sol was adjusted according to the XRF result of Cu@SiO2.
As a comparison material, Cu/mMOR was prepared via a modified impregnation method. To increase the presence of more Cu species on the catalyst surface, copper (II) acetylacetonate with a larger molecular size was used as the Cu source. In a typical procedure, copper (II) acetylacetonate was first dissolved in an appropriate amount of ethanol to prepare a uniform solution. Subsequently, mMOR powder was impregnated with this solution and ultrasonically dispersed in a sealed glass bottle for 30 minutes. The excess solvent in the mixture was then removed by rotary evaporation. Finally, the solid powder was calcined at 500 °C for 4 hours and subsequently reduced by H2 at 500 °C to obtain the Cu/mMOR sample.
Catalytic reaction test
The catalytic reaction tests for the reverse water-gas shift (RWGS) reaction were conducted in a mixed bed reactor with an internal diameter of 14 mm and a length of 300 mm in downflow mode. To prevent temperature fluctuations due to exothermic and endothermic reactions, a mixture of 20 mg of catalyst (120 mesh) and 300 mg of quartz sand (100 mesh) was placed in the reactor and supported between quartz wool. The catalyst was exposed to a CO2/H2 flow. The reaction was carried out at 600 °C, 0.1 MPa, CO2/H2 = 1/2, and a GHSV of 270 L gcat−1 h−1, unless otherwise specified. The outlet light gases (CO, CH4, and CO2) were analyzed online via an Agilent 7820 A gas chromatograph (GC) equipped with a TCD and a PoraPak Q column (Agilent G3591-70010).
The conversion of CO2 (\({X}_{{{CO}}_{2}}\)) and selectivity to CO (SCO) were calculated as follows:
At which, the \({N}_{{{CO}}_{2}{{\rm{in}}}}\), is the molar quantity of CO2 fed into the reactor, while NCO,out and \({N}_{{{\rm{C}}}{{{\rm{O}}}}_{2},{{\rm{out}}}}\) are the molar quantities of CO and CO2 in the outlet gas, respectively.
All results were obtained with a carbon mole balance greater than 95%.
Characterizations
Various characterization methods, including HRTEM, EDS mapping, XRD, XPS, XRF, N2 adsorption-desorption isotherms, temperature-programmed desorption (TPD), XANES, EXAFS, isotope analysis, the in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS), and positron annihilation spectroscopy (PAS), were adopted to reveal the structure, physical and chemical properties and reaction mechanism over Cu@mMOR and related catalysts. Specifically, the positron annihilation lifetime (PAL) spectra were recorded using a PAL spectrometer (DPLS4000, Anhui Nuclear and Electronics Technology Co., Ltd., China) with a time resolution of approximately 180 ps and a source intensity of 20 µCi. The coincidence Doppler-broadening measurements were performed using a DCDB3000 spectrometer (Anhui Nuclear and Electronics Technology Co., Ltd., China). The detailed processes are provided in the Supplementary Information.
Theory calculations
Various theoretical calculation methods—including transition states calculation, charge density difference contour plots, work function, crystal orbital Hamilton population (COHP), independent gradient model based on Hirshfeld partition (IGMH) and Ab initio molecular dynamics (AIMD)—were adopted to gain a deep insight for reaction mechanism of Cu@mMOR. The details of theory calculations are provided in the Supplementary Information.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information. The data generated in this study are provided in the Source Data file. All data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Velty, A. & Corma, A. Advanced zeolite and ordered mesoporous silica-based catalysts for the conversion of CO2 to chemicals and fuels. Chem. Soc. Rev. 52, 1773–1946 (2023).
Weber, J. L., Mejía, C. H., de Jong, K. P. & de Jongh, P. E. Recent advances in bifunctional synthesis gas conversion to chemicals and fuels with a comparison to monofunctional processes. Catal. Sci. Technol. 14, 4799–4842 (2024).
Zhang, W., Sun, J., Wang, H. & Cui, X. Recent advances in hydrogenation of CO2 to CO with heterogeneous catalysts through the RWGS reaction. Chem. Asian J. 19, e202300971 (2024).
Portillo, E., Gandara-Loe, J., Reina, T. R. & Pastor-Pérez, L. Is the RWGS a viable route for CO2 conversion to added value products? A techno-economic study to understand the optimal RWGS conditions. Sci. Total Environ. 857, 159394 (2023).
Bahmanpour, A. M., Signorile, M. & Kröcher, O. Recent progress in syngas production via catalytic CO2 hydrogenation reaction. Appl. Catal. B 295, 120319 (2021).
Zhang, J. et al. Sinter-resistant metal nanoparticle catalysts achieved by immobilization within zeolite crystals via seed-directed growth. Nat. Catal. 1, 540–546 (2018).
Wang, H. et al. Subnanometer Pt catalysts encapsulated in MEL zeolite mesocrystals for H2 production from methylcyclohexane dehydrogenation. J. Am. Chem. Soc. 147, 21789–21802 (2025).
Julian, I. et al. Supercritical solvothermal synthesis under reducing conditions to increase stability and durability of Mo/ZSM-5 catalysts in methane dehydroaromatization. Appl. Catal., B 263, 118360 (2020).
Hou, P., Yu, Q., Luo, F. & Liu, J.-C. Reactant-induced dynamic active sites on Cu catalysts during the water gas shift reaction. ACS Catal. 15, 352–360 (2025).
Chen, F. et al. Tailor-made the ultrathin nanosheet and acid site accessibility of mordenite zeolite for carbonylation of dimethyl ether. Chem. Eng. J. 498, 155451 (2024).
Nishimura, S., Takagaki, A., Maenosono, S. & Ebitani, K. In situ time-resolved XAFS study on the formation mechanism of Cu nanoparticles using poly(N-vinyl-2-pyrrolidone) as a capping agent. Langmuir 26, 4473–4479 (2010).
Gao, W. et al. Insight into solvent-free synthesis of MOR zeolite and its laboratory scale production. Micropor. Mesopor. Mat. 280, 187–194 (2019).
Wang, S. et al. Activation and spillover of hydrogen on sub-1 nm palladium nanoclusters confined within sodalite zeolite for the semi-hydrogenation of alkynes. Angew. Chem. Int. Ed. 58, 7668–7672 (2019).
Zhai, P., Zhang, L., Cullen, D. A., Aireddy, D. R. & Ding, K. Construction of inverse metal-zeolite interfaces via area-selective atomic layer deposition. ACS Appl. Mater. Interfaces 13, 51759–51766 (2021).
Bosnar, S., Vrankic, M., Bosnar, D., Ren, N. & Saric, A. Positron annihilation lifetime spectroscopy (PALS) study of the as prepared and calcined MFI zeolites. J. Phys. Chem. Solids 110, 227–233 (2017).
Huang, S. J. et al. Anylasis and comparison of several methods for calculation of positron bulk lifetime in perfect crystals. Acta Phys. Sin. 63, 217804 (2014).
Cízek, J. Characterization of lattice defects in metallic materials by positron annihilation spectroscopy: A review. J. Mater. Sci. Technol. 34, 577–598 (2018).
Taylor, C. N. et al. Catalyst deactivation probed by positron annihilation spectroscopy. ACS Catal. 11, 14967–14976 (2021).
Mehio, N., Dai, S. & Jiang, D. E. Quantum mechanical basis for kinetic diameters of small gaseous molecules. J. Phys. Chem. A 118, 1150–1154 (2014).
Matsuoka, M., Ju, W.-S., Takahashi, K., Yamashita, H. & Anpo, M. Photocatalytic decomposition of N2O into N2 and O2 at 298 K on Cu(I) ion catalysts anchored onto various oxides. The effect of the coordination state of the Cu(I) ions on the photocatalytic reactivity. J. Phys. Chem. B 104, 4911–4915 (2000).
Jiang, Y., Li, H., Chen, C., Zheng, Y. & Qiao, S.-Z. Dynamic Cu0/Cu+ interface promotes acidic CO2 electroreduction. ACS Catal. 14, 8310–8316 (2024).
Caballero, A. et al. An in situ XAS study of Cu/ZrO2 catalysts under de-NOx reaction conditions. J. Catal. 235, 295–301 (2005).
Vitova, T., Hormes, J., Peithmann, K. & Woike, T. X-ray absorption spectroscopy study of valence and site occupation of copper in LiNbO3: Cu. Phys. Rev. B 77, 144103 (2008).
Cao, P. et al. Zeolite-encapsulated Cu nanoparticles for the selective hydrogenation of furfural to furfuryl alcohol. ACS Catal. 11, 10246–10256 (2021).
Pankin, I. A. et al. Identifying Cu-oxo species in Cu-zeolites by XAS: A theoretical survey by DFT-assisted XANES simulation and EXAFS wavelet transform. Catal. Today 345, 125–135 (2020).
Sokol, H. J., Ebrahim, A. M., Caratzoulas, S., Frenkel, A. I. & Valla, J. A. In situ XAFS, XRD, and DFT characterization of the sulfur adsorption sites on Cu and Ce exchanged Y zeolites. J. Phys. Chem. C. 126, 1496–1512 (2022).
Sander, T. et al. Correlation of intrinsic point defects and the raman modes of cuprous oxide. Phys. Rev. B 90, 045203 (2014).
Zhang, W. et al. One-pot template-free synthesis of Cu-MOR zeolite toward efficient catalyst support for aerobic oxidation of 5-hydroxymethylfurfural under ambient pressure. ACS Appl. Mater. Interfaces 8, 23122–23132 (2016).
Brinzari, T. V. et al. Local lattice distortions in Mn[N(CN)2]2 under pressure. Inorg. Chem. 55, 1956–1961 (2016).
Liu, Z. S. et al. Cation concavities induced d-band electronic modulation on Co/FeOx nanostructure to activate molecular and interfacial oxygen for CO oxidation. Environ. Sci. Technol. 57, 21272–21283 (2023).
Dong, Q. H. et al. Ultrahigh mass activity for the hydrogen evolution reaction by anchoring platinum single atoms on active {100} facets of TiC via cation defect engineering. Adv. Funct. Mater. 33, 2210665 (2023).
Wang, B. et al. Nature of active sites on Cu-CeO2 catalysts activated by high-temperature thermal aging. ACS Catal. 10, 12385–12392 (2020).
Yi, H. et al. Frustrated Lewis pairs on metal-cation vacancy catalysts enhanced the electroreduction of NO to NH3. J. Mater. Chem. A 12, 24237–24247 (2024).
Cai, K. et al. Influence of acid strength on the reactivity of dimethyl ether carbonylation over H-MOR. ACS Sustain. Chem. Eng. 7, 2027–2034 (2019).
Chanquía, C. M. et al. Speciation of copper in spherical mesoporous silicates: from the microscale to angstrom. J. Phys. Chem. C. 114, 12221–12229 (2010).
Corma, A., Martinez-Triguero, J., Valencia, S., Benazzi, E. & Lacombe, S. IM-5: a highly thermal and hydrothermal shape-selective cracking zeolite. J. Catal. 206, 125–133 (2002).
Beerdsen, E., Smit, B. & Calero, S. The influence of non-framework sodium cations on the adsorption of alkanes in MFI- and MOR-type zeolites. J. Phys. Chem. B 106, 10659–10667 (2002).
Smith, W. R. HSC chemistry for windows, 2.0. J. Chem. Inf. Computer Sci. 36, 151–152 (1996).
Ocampo-Restrepo, V. K., Verga, L. G. & Da Silva, J. L. F. Ab initio study of the C-O bond dissociation in CO2 reduction by redox and carboxyl routes on 3d transition metal systems. J. Phys. Chem. C. 125, 26296–26306 (2021).
Wang, H. et al. Synergistic interactions of neighboring platinum and iron atoms enhance reverse water-gas shift reaction performance. J. Am. Chem. Soc. 145, 2264–2270 (2023).
Jing, H., Li, Q., Wang, J., Liu, D. & Wu, K. Theoretical study of the reverse water gas shift reaction on copper modified β-Mo2C(001) surfaces. J. Phys. Chem. C. 123, 1235–1251 (2019).
Zhang, C. et al. Blocking methanation during reverse water gas shift reaction on Ni/SiO2 catalysts by surface Ag. ChemCatChem 15, e202201284 (2023).
Zhang, X. et al. Highly dispersed copper over beta-Mo2C as an efficient and stable catalyst for the reverse water gas shift (RWGS) reaction. ACS Catal. 7, 912–918 (2017).
Liu, H. X. et al. Partially sintered copper-ceria as excellent catalyst for the high-temperature reverse water gas shift reaction. Nat. Commun. 13, 867 (2022).
Jin, R., Easa, J. & O’Brien, C. P. Highly active CuOx/SiO2 dot core/rod shell catalysts with enhanced stability for the reverse water gas shift reaction. ACS Appl. Mater. Interfaces 13, 38213–38220 (2021).
Yu, J. et al. Ultra-high thermal stability of sputtering reconstructed Cu-based catalysts. Nat. Commun. 12, 7209 (2021).
Bahmanpour, A. M. et al. Cu-Al spinel as a highly active and stable catalyst for the reverse water gas shift reaction. ACS Catal. 9, 6243–6251 (2019).
Pahija, E. et al. FeOx nanoparticle doping on Cu/Al2O3 catalysts for the reverse water gas shift. J. CO2 Util. 64, (2022).
Tarifa, P., González-Castaño, M., Cazaña, F., Monzón, A. & Arellano-García, H. Development of one-pot Cu/cellulose derived carbon catalysts for RWGS reaction. Fuel 319, 123707 (2022).
Xu, N. et al. Structure and activity comparison of Cu/CeyZr1–yOx series catalysts in the reverse water–gas shift reaction. Energy Fuels 38, 7158–7166 (2024).
Zhang, J., Wang, Y., Tian, J. & Yan, B. Cu/LaFeO3 as an efficient and stable catalyst for CO2 reduction: exploring synergistic effect between Cu and LaFeO3. AlChE J. 68, e17640 (2022).
Li, Z.-X., Xu, K., Wang, W.-W., Fu, X.-P. & Jia, C. -j Stabilized inverse Y2O3/Cu interfaces boost the performance of the reverse water gas shift reaction. Catal. Sci. Technol. 14, 3483–3492 (2024).
Liang, Y. et al. Decorating of Ag and CuO on Cu nanoparticles for enhanced high catalytic activity to the degradation of organic pollutants. Langmuir 33, 7606–7614 (2017).
Chen, Q. et al. Validation of the CO2/N2O analogy using molecular simulation. Ind. Eng. Chem. Res. 53, 18081–18090 (2014).
Jensen, J. R., Johannessen, T. & Livbjerg, H. An improved N2O-method for measuring Cu-dispersion. Appl. Catal. a-Gen. 266, 117–122 (2004).
Delgado, J. A. & Gómez, J. M. Estimation of adsorption parameters from temperature-programed-desorption thermograms: application to the adsorption of carbon dioxide onto Na- and H-mordenite. Langmuir 21, 9555–9561 (2005).
Mir, S. H., Yadav, V. K. & Singh, J. K. Efficient CO2 capture and activation on novel two-dimensional transition metal borides. ACS Appl. Mater. Interfaces 14, 29703–29710 (2022).
Sljivancanin, Z. & Hammer, B. H2 dissociation at defected Cu: preference for reaction at vacancy and kink sites. Phys. Rev. B 65, 085414 (2002).
Liu, R. et al. Increasing the number of aluminum atoms in T3 sites of a mordenite zeolite by low-pressure SiCl4 treatment to catalyze dimethyl ether carbonylation. Angew. Chem. Int. Ed. 61, e202116990 (2022).
Chen, G., Fang, L., Li, T. & Xiang, Y. Ultralow-loading Pt/Zn hybrid cluster in zeolite HZSM-5 for efficient dehydroaromatization. J. Am. Chem. Soc. 144, 11831–11839 (2022).
Bettahar, M. M. The hydrogen spillover effect. A misunderstanding story. Catal. Rev.-Sci. Eng. 64, 87–125 (2022).
Bettahar, M. M. The hydrogen spillover effect-a misunderstanding study II: Single oxide and zeolite supports. Catalysts 14, 458–477 (2024).
Yang, X. et al. Understanding the synergistic catalysis of balanced Cu0-Cu+ sites and oxygen vacancies in Cu/ZrO2 catalysts for the efficient hydrogenation of furfural. Chem. Eng. J. 501, 157796 (2024).
Halder, A. et al. CO2 methanation on Cu-cluster decorated zirconia supports with different morphology: A combined experimental in situ GIXANES/GISAXS, ex situ xps and theoretical dft study. ACS Catal. 11, 6210–6224 (2021).
Liu, C. et al. In situ reconstruction of Cu-N coordinated MOFs to generate dispersive Cu/Cu2O nanoclusters for selective electroreduction of CO2 to C2H4. ACS Catal. 12, 15230–15240 (2022).
Hammes-Schiffer, S. Explaining kinetic isotope effects in proton-coupled electron transfer reactions. Acc. Chem. Res. 58, 1335–1344 (2025).
An, B. et al. Molecular iridium complexes in metal-organic frameworks catalyze CO2 hydrogenation via concerted proton and hydride transfer. J. Am. Chem. Soc. 139, 17747–17750 (2017).
Ojeda, M. et al. Kinetically relevant steps and H2/D2 isotope effects in Fischer-Tropsch synthesis on Fe and Co catalysts. J. Phys. Chem. C. 114, 19761–19770 (2010).
Gu, Z. et al. Water-assisted hydrogen spillover in Pt nanoparticle-based metal-organic framework composites. Nat. Commun. 14, 5836 (2023).
Luo, J.-Y. & Epling, W. S. New insights into the promoting effect of H2O on a model Pt/Ba/Al2O3 NSR catalyst. Appl. Catal. B 97, 236–247 (2010).
Hülsey, M. J., Fung, V., Hou, X., Wu, J. & Yan, N. Hydrogen spillover and its relation to hydrogenation: Observations on structurally defined single-atom sites. Angew. Chem. Int. Ed. 61, e202208237 (2022).
Spreafico, C., Karim, W., Ekinci, Y., van Bokhoven, J. A. & VandeVondele, J. Hydrogen adsorption on nanosized platinum and dynamics of spillover onto alumina and titania. J. Phys. Chem. C. 121, 17862–17872 (2017).
Karim, W. et al. Catalyst support effects on hydrogen spillover. Nature 541, 68 (2017).
Lu, T. & Chen, Q. X. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 43, 539–555 (2022).
Lee, H. M., Anick, D. J. & Kim, K. S. Structures of tri-, tetra-, and hexahydrated hydride anion clusters. Int. J. Quantum Chem. 109, 1820–1826 (2009).
Ceriotti, M. et al. Nuclear quantum effects in water and aqueous systems: Experiment, theory, and current challenges. Chem. Rev. 116, 7529–7550 (2016).
Sun, Q. et al. Zeolite encaged Pd-Mn nanocatalysts for CO2 hydrogenation and formic acid dehydrogenation. Angew. Chem. Int. Ed. 59, 20183–20191 (2020).
Rabee, A. I. M. et al. Uncovering the synergy between gold and sodium on ZrO2 for boosting the reverse water gas shift reaction: in situ spectroscopic investigations. Appl. Catal. B 345, 123685 (2024).
Kondo, J. N., Iizuka, M., Domen, K. & Wakabayashi, F. IR study of H2O adsorbed on H-ZSM-5. Langmuir 13, 747–750 (1997).
Osuga, R., Yokoi, T. & Kondo, J. N. IR observation of activated ether species on acidic OH groups on H-ZSM-5 zeolites. Mol. Catal. 477, 110535 (2019).
Otroshchenko, T., Kondratenko, V. A., Zanina, A., Zhang, Q. Y. & Kondratenko, E. V. Progress through temporal analysis of products and steady-state isotopic transient kinetic analysis to elucidate oxidation, CO2 hydrogenation and lower olefins production reactions. ChemCatChem 16, e202301447 (2024).
Janssens, P., Poissonnier, J., Chakkingal, A., Bos, R. & Thybaut, J. W. Recent advances in the use of steady-state isotopic transient kinetic analysis data in (micro)kinetic modeling for catalyst and process design. Catal. Commun. 179, 106688 (2023).
Wang, K. et al. DRIFTS-SSITKA-MS investigations on the mechanism of plasmon preferentially enhanced CO2 hydrogenation over Au/γ-Al2O3. Appl. Catal., B 328, 122531 (2023).
Wang, X. Y. et al. The nature of interfacial catalysis over Pt/NiAl2O4 for hydrogen production from methanol reforming reaction. J. Am. Chem. Soc. 145, 905–918 (2023).
Dronskowski, R. & Blochl, P. E. Crystal orbital hamilton populations (COHP) - energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617–8624 (1993).
Lu, T. Visualization analysis of covalent and noncovalent interactions in real space. Angew. Chem.-Int. Ed. 64, e202504895 (2025).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 161, 082503 (2024).
Qin, Y. et al. CO intermediate-assisted dynamic Cu sintering during electrocatalytic CO2 reduction on Cu-N-C catalysts. Angew. Chem. Int. Ed. 63, e202404763 (2024).
Dietze, E. M., Abild-Pedersen, F. & Plessow, P. N. Comparison of sintering by particle migration and ripening through first-principles-based simulations. J. Phys. Chem. C. 122, 26563–26569 (2018).
Ge, Y. et al. Highly efficient Pt decorated CoCu bimetallic nanoparticles protected in silica for hydrogen production from ammonia–borane. ACS Sustain. Chem. Eng. 5, 1675–1684 (2017).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. U23A20120 to Zhen. Z.), Natural Science Foundation of Guangxi Province (No. AD24010026 to Zhen. Z.). The authors would like to thank Jianhua Chen for providing the calculation software support. The simulation calculations in this work are supported by the high-performance computing platform of Guangxi University. The authors would like to thank Jiabao Wang from Scientific Compass (www.shiyanjia.com) for the XPS analysis. The authors would like to thank Jiaqi Li from SCI-GO (www.sci-go.com) for the Raman and DRIFTS analysis. The authors would like to thank Tieying Zhao and Xiaoge Bai from Tianjin Xianquan Industry and Trade Development Co., Ltd. for assisting with the SSITKA experiments and analysis.
Author information
Authors and Affiliations
Contributions
W.S., Zhong. Z., and Zhen. Z. conceived the research idea and designed the experiments. W.S. performed the majority of the experiments and analyzed the data. X.J., X.F., C.L., J.Z., and Y.L. took part in the synthesis of samples and characterizations. X.D. performed the positron annihilation spectroscopy analysis and assisted in the corresponding manuscript writing. W.S. and J.C. performed the DFT calculations. W.S., Zhongxing Zhao (Z.Z.), and Zhenxia Zhao (Z.Z.) wrote the paper. S.J.S. revised the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, W., Jia, X., Deng, X. et al. Molecular fence Cu-based catalyst for CO2 hydrogenation to CO with high activity and durability. Nat Commun 17, 3552 (2026). https://doi.org/10.1038/s41467-026-70333-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70333-2
