
Abstract
Copper catalysts feature unique superiorities for the electrochemical conversion of CO2 to C2+ fuels and chemicals. Their surface oxidation states dominantly determine the reaction pathways to various products. However, most Cu-based catalysts inevitably undergo electroreduction from Cu2+ to Cu1+ or Cu0 species during the electrochemical CO2 reduction. Herein, we propose a straightforward strategy to stabilize Cu2+ ions by coordinating them with benzobistriazole (H2BBTA), producing a metal-organic polymer (CuBBTA) with periodically adjacent copper atoms. Remarkably, CuBBTA delivers a high Faradaic efficiency of 62.0 ± 1.9% for CO2-to-C2H4 conversion and a half-cell power conversion efficiency of 34.4% in a flow cell. It also maintains stable operation for over 50 hours in a zero-gap electrolyzer, sustaining a FE > 55% at ≈ 1 A total current density. Operando X-ray absorption, Raman, and attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) reveal that the catalyst remains structurally stable with no dynamic transformation during the reaction. Online differential electrochemical mass spectrometry (DEMS), operando ATR-SEIRAS and theoretical calculations show that neighboring Cu2+ ions in the polymer provide suitably-distanced dual sites that enable the energetically favorable formation of an *COCHO intermediate. This study presents a strategic method for developing stable catalysts for efficient CO2-to-ethylene electroconversion.
Similar content being viewed by others
Introduction
Electrochemical CO2 reduction reaction (CO2RR) driven by renewable electric energy presents a promising strategy for mitigating atmospheric CO2 levels while enabling sustainable carbon recycling through the production of value-added chemicals1,2,3,4,5,6. Cu-based catalysts have garnered significant attention due to their high selectivity toward C2+ products, such as ethylene (C2H4), ethanol (EtOH), acetic acid and n-propanol7,8,9. Among various carbon dioxide reduction products, ethylene stands out as a focal point in this realm, given its essential role as an important building block in the chemical industry10,11. However, the efficient conversion of CO2 to ethylene remains several interconnected scientific and engineering challenges. Achieving high ethylene selectivity requires precise control over C–C coupling and protonation steps. For instance, *OH adsorption strength on copper defect sites determines whether EtOH or C2H4 dominates: weak *OH adsorption favors EtOH, while strong *OH adsorption promotes C2H412. Although catalysts like Cu2O nano-homojunctions achieve 73.7% ethylene Faraday efficiency (FE) in H-cells, maintaining selectivity under industrial current densities remains challenging13.
The uncontrolled restructuring of Cu-based catalysts during electrochemical CO2RR also remains a major challenge for stability14,15. In addition to structural reconstruction, reduction of Cu oxidation states has been identified as another critical factor leading to catalyst deactivation during CO2RR. Recent studies have revealed that maintaining partially oxidized Cu species is essential for stable C-C coupling activity16,17. Reported work focused primarily on strategies such as incorporating secondary metals18,19,20,21, optimizing coordination environment22,23,24, integrating active ligands25,26,27, employing carbon/nitrogen confinement28,29, and enhancing metal–support interactions to stabilize the valence state of Cu in electrocatalysts30,31,32,33. In the case of Cu2+ ions, experimental studies demonstrated that their higher oxidation state facilitates *CO hydrogenation due to stronger electrostatic interactions compared to their Cu1+ or Cu0 counterparts, thereby promoting the formation of deeply reduced hydrocarbon products. As a result, catalysts with isolated, stable Cu2+ sites tend to favor the electroreduction of CO2 to CH434,35,36. However, achieving efficient, selective and long-term C-C coupling in catalysts with single, stable Cu2+ sites under CO2RR conditions still remains a significant challenge. The C-C coupling process involves the interaction of surface-bound *CO and/or *CHO intermediates, which requires an appropriate spatial arrangement of at least two adjacent binding sites37,38. Therefore, the rational design and preparation of electrocatalysts featuring stable and neighboring Cu2+ sites are of critical importance for enhancing their sustained activity and selectivity, as well as for fundamentally elucidating their structure–activity relationships in CO2RR.
Metal–organic polymers (MOPs) have attracted growing interest as atomically precise single-site catalysts for the electrochemical CO2RR, owing to their well-defined structures and tunable functionalities22,39. Through strong ligand interactions, MOPs can stabilize Cu⁺ species, thereby preserving active sites for CO2 reduction40. By modulating their electronic structures, MOPs can lower the dimerization energy of CO intermediates, facilitating the formation of ethylene24. The uniform metal–organic frameworks with open channels enhance the transport and diffusion of reactants and products41. Furthermore, their intrinsic structures provide abundant catalytic sites, including unsaturated metal centers, metal clusters, and functional groups on the organic linkers42. Despite these advantages, a fundamental understanding at the molecular level remains lacking, hindering the rational design of highly active and stable MOP-based catalysts.
Here, we develop Cu-based quasi-two-dimensional (2D) metal-organic polymer catalysts for electrocatalytic CO2RR. Strong coordination between Cu2+ and benzobistriazole (H2BBTA) stabilizes Cu2+ during CO2RR. Electrochemical tests and spectroscopic analysis demonstrated that the CuBBTA electrocatalyst remained chemically and structurally stable during long-term electrocatalytic CO2RR without dynamic deactivation due to the robust Cu-BBTA coordination. Neighboring Cu sites in the polymer provide suitably distanced dual Cu sites that enable the energetically favorable formation of an *COCHO intermediate. As a result, CuBBTA delivers a half-cell C2H4 power conversion efficiency (PCE) of 34.4%, which is 1.8 times that of the Cu control, together with a C2H4 FE of 62.0 ± 1.9%. Assembling the catalyst into a membrane electrode assembly (MEA) system enables stable full-device CO2 electrolysis at a total current of ~ 1 A for 50 h.
Results and discussion
Structural characterizations of CuBBTA
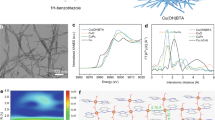
The CuBBTA catalyst – consisting of repeated Cu-BBTA coordination and bridged transversely via hydroxyl groups – was synthesized using a one-pot synthesis approach. The H2BBTA ligand and cupric chloride were stirred in an alkaline environment at 25 °C for 10 h (Fig. 1a, details in “Methods”). Powder X-ray diffraction (PXRD) patterns exhibited a prominent peak at 10.1°, which can be attributed to a periodic chain or interlayer spacing, implying a partially ordered polymeric stacking structure (Fig. 1b)35. The product presents a cross-linked network structure, as evidenced by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images(Fig. 1c, d and Supplementary Fig. 1). Isolate and dense Cu atoms were clearly observed via high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1e and Supplementary Fig. 2). Scanning transmission electron microscopy (STEM) elemental mapping shows that C, N, O and Cu disperse uniformly (Fig. 1f). Actual Cu loadings were quantified by inductively coupled plasma atomic emission spectrometry (ICP-AES) (Supplementary Table 1). The Cu loading in CuBBTA was determined to be 39.4 wt%, which is comparable to values reported for Cu single-atom catalysts (SACs) (Supplementary Table 2).

a Schematic of the formation process of CuBBTA catalyst and its structure at the mesoscopic and microscopic scales, showcasing its wireframe model. b Experimental (blue) and simulated (red) PXRD patterns of CuBBTA. The calculated refinement is based on the structure simulated by DFT calculations. c SEM and (d) TEM images of CuBBTA. e HAADF-STEM image of CuBBTA. The white dots are isolated and dense Cu atoms. f EDS mapping of CuBBTA (scale bar: 200 nm). Source data are provided as a Source Data file.
We performed a series of spectroscopic analyses to characterize the structure of CuBBTA. The N 1 s X-ray photoelectron spectroscopic (XPS) spectrum of CuBBTA was deconvoluted into two peaks, which were attributed to Cu-N and pyridine-N, respectively (Fig. 2a)43,44. The binding energy of N in CuBBTA was slightly shifted to a lower, single-peak value of 399.6 eV. This suggested the electrons of the N atoms were delocalized and the charge was distributed evenly over the three N atoms after coordinating with Cu22. The N − H in CuBBTA was deprotonated, revealed by the Fourier-transform infrared (FT-IR) spectroscopy (Supplementary Fig. 3a). The solid-state 13C NMR spectrum indicated that the N1, N3, N4 and N6 positions of the triazole ring coordinated with Cu, resulting in a symmetric benzene environment in CuBBTA (Fig. 2b). For H2BBTA, two distinct peaks at 140 ppm and 131 ppm were observed, corresponding to two types of carbon atoms in the benzene ring. In CuBBTA, the 13C signals shifted slightly to 142 ppm and 132 ppm, still representing two distinct carbon environments, which suggests retention of symmetry upon coordination. These results, together with elemental analysis, support that Cu is coordinated with N1, N3, N4, and N6 of the BBTA ligand. Cu sites were also bridged by a hydroxyl group, evidenced by FT-IR spectroscopy and XPS (Supplementary Figs. 3b and 4). No Cl signal was detected in CuBBTA via XPS (Supplementary Fig. 5), suggesting that Cl- did not participate in the coordination process.

a N 1 s XPS spectra of the benzobistriazole (H2BBTA) molecule and CuBBTA. b 13C solid-state NMR spectra of the H2BBTA molecule and CuBBTA. Inset: corresponding H2BBTA structure before and after coordination with Cu. c Cu K-edge XANES spectrum of CuBBTA. The inset displays a magnification of the pre-edge peak. d Cu K-edge EXAFS spectrum of CuBBTA. The Cu K-edge XANES spectra of Cu foil, CuO, and copper(II) phthalocyanine (CuPc) were used as references. e XAFS wavelet transform (WT) of CuBBTA. f Cu K-edge EXAFS fitting of CuBBTA. Inset: an illustration of CuBBTA local structure near the Cu atom. Source data are provided as a Source Data file.
We further employed X-ray absorption fine structure (XAFS) spectroscopy to probe the local coordination environment of copper in CuBBTA. Figure 2c shows that the X-ray absorption near-edge structure (XANES) spectrum of CuBBTA was close to that of CuO and copper phthalocyanine (CuPc), indicating a Cu(II) oxidation state in CuBBTA. Besides, the XANES of CuBBTA displayed the K-pre-edge peak, which is characteristic for Cu2+. These results are consistent with Cu 2p X-ray photoelectron spectroscopy (XPS) and LMM spectra (Supplementary Fig. 6)45,46,47. The extended X-ray absorption fine structure (EXAFS) spectra revealed the absence of Cu-Cu bonding in CuBBTA (Fig. 2d). The peaks observed at 1.45 Å and 1.5 Å were attributed to Cu-O bonding in CuO and Cu-N bonding in CuPc, respectively. The wavelet transforms of the Cu K-edge further confirmed the involvement of both nitrogen (N) and oxygen (O) in the coordination of Cu (Fig. 2e and Supplementary Fig. 7).
The structural model of CuBBTA shown in Supplementary Fig. 8 was established based on Pawley refinement of the PXRD data (Fig. 1b), supported by density functional theory (DFT) calculations and spectroscopic analyses. The optimized structural models of CuBBTA are provided in Supplementary Data 1. The CuBBTA structure extends longitudinally through repeated Cu-BBTA coordination and is transversely bridged by hydroxyl groups. The EXAFS spectrum calculated based on this model demonstrated a satisfactory fit with the experimental data, yielding coordination numbers (CN) of 2 for both Cu-N and Cu-O (Fig. 2f and Supplementary Table 3). The peak at ≈ 3.15 Å can be attributed to the Cu-Cu coordination in the second shell, consistent with the DFT-optimized structure and EXAFS fitting results (Cu-Cu ≈ 3.15 ± 0.08 Å). The simulated structure of CuBBTA was also in good agreement with the elemental composition and mass percentages derived from elemental analysis and inductively coupled plasma atomic emission spectroscopy (ICP-AES) (Supplementary Table 1, 4).
CO2 electroreduction performances
The electrochemical CO2RR performance of CuBBTA was initially assessed in 1.0 M KOH solution within a flow cell configuration (Supplementary Fig. 9). Linear sweep voltammetry (LSV) curves (Fig. 3a) show that CuBBTA exhibits a lower onset potential and faster reaction kinetics than CuBBTA-derived Cu (D-Cu), particularly under CO2. D-Cu was prepared by in situ electroreduction at − 300 mA cm−2 for 10 min under Ar. Figure 3b illustrates the CO2RR product selectivity of CuBBTA and comparative samples, quantified using both gas chromatography and nuclear magnetic resonance (NMR) spectroscopy (see “Methods” and Supplementary Fig. 10 for details). CuBBTA showed a prominent selectivity for C2H4 (FEC2H4 > 40%) over the whole measured potentials from − 0.69 to − 0.94 V vs. RHE (All potentials are referenced to RHE unless otherwise noted). The FEC2H4 reached a maximum of 62.0 ± 1.9% at − 0.84 V (Fig. 3b), corresponding to a partial current density for C2H4 (jC2H4) of − 305 mA cm−2 (Fig. 3c). The rest C2+ products included EtOH (5.8%), acetate (2.4%), and n-PrOH (1.9%), totaling a C2+ FE of 72.2% and partial current density of 351 mA cm−2 (Supplementary Fig. 11). In contrast, the D-Cu exhibited lower selectivity, yielding nearly equal amounts of C1 and C2 products, with FEC2H4 in the range of 20–40% (Fig. 3b). Subsequently, electrochemical impedance tests were conducted to analyze the electron transfer kinetics in CO2RR (Supplementary Fig. 12). The charge-transfer resistance (Rct) of CuBBTA is minimal, indicating efficient charge transport. Notably, although the total catalytic current density of CuBBTA is lower than that of derived Cu in the potential of − 0.94 V, the jC2H4 on CuBBTA is considerably more advantageous due to its higher FEC2H4 (Fig. 3c). CuBBTA demonstrates a half-cell C2H4 power conversion efficiency (PCE) of 34.4% at − 0.84 V, which is 1.8 times higher than that of the D-Cu (Fig. 3d and Supplementary Table 5). By comparing the PCE of CO2 to C2H4 and the corresponding jC2H4 of CuBBTA with the state of the art reported Cu-based electrocatalysts (Fig. 3e and Supplementary Table 6), the CO2RR performance of CuBBTA is competitive with that of previously reported Cu-based catalysts.

a LSV curves of CuBBTA and D-Cu in a flow cell under CO2 or Ar atmospheres. b FE of various products from CuBBTA and D-Cu at different potentials in a flow cell. c Ethylene partial current densities of CuBBTA and D-Cu at various potentials in a flow cell. d Half-cell PCE of C2H4 on CuBBTA and D-Cu electrodes versus different applied potentials. e Comparison of the PCEethylene and reduction current of CuBBTA with recently reported catalysts. f FEC2H4 of CuBBTA at various potentials in a biphasic electrode MEA system. g Stability performance of CuBBTA within the MEA to produce ethylene. a–d No iR correction was applied to calculate the applied potential. The error bars represent standard deviations from at least three independent measurements. Source data are provided as a Source Data file.
We further evaluated the CO2 electrolysis performance in a full-cell configuration utilizing a compact, commercialization-relevant membrane electrode assembly (MEA) system with an electrode geometric area of 4 cm2. CuBBTA catalysts were employed as the cathode, while iridium oxide supported on titanium mesh (IrO2/Ti) served as the anode, with an anion exchange membrane acting as the separator (Supplementary Fig. 13). The trends of C2H4 selectivity over the applied full-cell voltage range of 3.0–3.8 V (without iR compensation) were consistent with those observed in the flow cell. A maximum Faradaic efficiency of 60.1% for C2H4 was achieved at 3.6 V, corresponding to a partial current density of almost 150 mA cm−2 (Fig. 3f and Supplementary Fig. 14). Initial stability assessments demonstrated sustained operation for 50 hours at a total current of almost 1 A, maintaining an average FE of 60% for C2H4 (Fig. 3g). Post-electrolysis characterization via XRD, SEM, XPS, STEM and HAADF-STEM revealed no discernible changes in the morphology or atom distribution of the CuBBTA catalyst (Supplementary Figs. 15–19).
Investigation of catalyst stability and origin of the stability
Studying the atomic structure-activity relationship of the catalyst during the CO2RR process is crucial to reveal the intrinsic catalytic mechanism. To assess the chemical state of Cu in the CuBBTA under CO2RR, operando XAS was performed (Supplementary Fig. 20). The cross-sectional SEM images and energy-dispersive spectroscopy (EDS) mappings reveal that the thickness of the catalytic layer in the table-top XAS spectrometer is approximately 42 μm (Supplementary Fig. 21). The operando XANES spectra reveal that the oxidation states of Cu under all applied potentials are close to + 2 from − 0.6 to − 1.1 V (Fig. 4a). No Cu-Cu bond is discerned in the EXAFS (Fig. 4b). In parallel, the FT-EXAFS fitting results of Cu show that the coordination number of Cu-N/O in the first shell stays relatively stable at around 4 (Fig. 4c, Supplementary Figs. 22 and 23 and Supplementary Table 7), demonstrating that Cu remains in a coordinated, atomically dispersed state under CO2RR condition. Additional operando XAS measurements collected at a synchrotron source provide consistent evidence, further supporting our conclusions (Supplementary Figs. 24 and 25). CuBBTA was reduced to metallic Cu after electroreduction at −300 mA cm-2 for 10 minutes under an Ar atmosphere, with the Cu oxidation state transitioning from + 2 to 0 (Supplementary Fig. 26).

Operando Cu K-edge XANES spectra (a) and Fouriertransformed k2-weighted EXAFS signals of the Cu K-edge spectra (b) recorded at different potentials on CuBBTA. c Changes of coordination number for the Cu–N/O coordination shells. d Operando Raman spectra of CuBBTA under CO2 atmosphere at applied potentials from − 0.6 to − 1 V. e Operando ATR-SEIRAS on CuBBTA at the applied potential of − 0.5 to − 1.2 V. f Operando ATR-SEIRAS on D-Cu at the applied potential of − 0.5 to − 1.2 V. Source data are provided as a Source Data file.
We further verified the structural stability of CuBBTA under electrochemical CO2RR conditions using operando Raman spectroscopy (Supplementary Fig. 27). At open-circuit potential (OCP), CuBBTA exhibited typical Raman shifts that correspond to the vibrational modes of molecular structure under CO2 and Ar conditions (Supplementary Table 8). When the potential was elevated every 0.1 V from the potential range of − 0.6 to − 1 V, a drastic change took place in the Ar atmosphere (Supplementary Fig. 28). The intensity of the Raman shifts decreased and completely disappeared, suggesting no residual BBTA molecule staying on or coordinating with the surface of the as-reduced Cu nanoparticle. In contrast, the CuBBTA displayed a slight change under CO2 conditions (Fig. 4d). This observation indicated high stability of the coordination structure of CuBBTA under electrochemical CO2RR conditions.
We also investigated the catalyst and its adsorption behavior toward CO2RR intermediates using operando ATR-SEIRAS to further validate the structural stability of CuBBTA under electroreduction conditions (Supplementary Fig. 29). Due to the single-site nature of the Cu centers in CuBBTA, only a single adsorption mode is expected for this specific CO2RR intermediate. As shown in Fig. 4e, a vibrational band appearing in the range of 2108–2077 cm−1 is assigned to electrogenerated atop-adsorbed CO (*COatop) on CuBBTA (Supplementary Fig. 30a). The redshift of this band position under increasingly negative potentials is attributed to the Stark effect, confirming the operando formation of *CO during CO2RR48,49. Upon reversing the applied potentials to − 0.6 V and − 0.7 V, followed by measurement at − 1.2 V, the CO peak intensities recover and blue-shift to 2115 and 2090 cm−1, respectively-matching the positions observed in the initial scans. These results indicate that no Cu agglomeration occurred to an extent detectable by IR spectroscopy during the CO2RR process. In addition, distinct absorption bands are observed at approximately 1750 and 1580 cm-1, which are assigned to surface-bound *CHO and *COCHO intermediates, respectively. These assignments are consistent with previously reported ATR-SEIRAS analyses of Cu-based CO2RR systems50. In contrast, D-Cu exhibits two distinct vibrational bands: one within 2115-2095 cm−1, corresponding to the asymmetric stretching vibration of *COatop, and another in the range of 1840–1815 cm−1, associated with bridge-adsorbed CO (*CObridge) (Fig. 4f and Supplementary Fig. 30b)51. Previous studies have demonstrated that the presence of *CObridge, which requires at least two adjacent adsorption sites, serves as an indicator of Cu agglomeration52,53.
We further explore the origin of CuBBTA stability in CO2RR. To probe the reaction mechanism, we initially performed electrochemical CO reduction (CORR), given its similarity to CO2RR54. Unexpectedly, applying a constant current density of − 300 mA cm−2 in 1 M KOH led to the complete reduction of CuBBTA to metallic Cu within 10 minutes (Supplementary Fig. 31). Substituting CO with Ar resulted in similar decomposition of CuBBTA (Supplementary Figs. 31 and 32). Comparison of CO2RR gas products before and after Ar purging revealed that, at a constant current density of − 300 mA cm−2, the ethylene Faradaic efficiency decreased from approximately 60% to 40% when CO2 was replaced with Ar (Supplementary Fig. 33). Cyclic voltammetry (CV) studies further revealed the instability of CuBBTA under an Ar atmosphere, with Cu2+/Cu+/Cu0 redox features emerging. In contrast, no such redox processes were observed under a CO2 atmosphere in the same flow cell using 1 M KOH as the electrolyte (Supplementary Fig. 34).
The instability of CuBBTA under CORR suggests that the highly alkaline local environment (local pH » 14) generated during these processes may compromise its structural stability55,56. In contrast, such high local pH conditions are mitigated during CO2RR, as CO2 reacts with OH⁻ to form carbonate species, thereby lowering the local pH. Indeed, previous modeling and experimental studies have indicated that the local pH remains below 14 in flow cells operating with 1 M KOH electrolyte52. To test this hypothesis, CO2RR was conducted using 10 M KOH as the electrolyte, which resulted in a similar decomposition of CuBBTA (Supplementary Fig. 31). These results collectively indicate that the CO2RR process creates a favorable local environment that prevents CuBBTA from being exposed to the harsh alkaline conditions, thereby maintaining the stability of its molecular structure.
Overall, operando XAS spectroscopy indicates that the oxidation state of Cu in CuBBTA remains consistently at +2, while operando Raman spectroscopy shows no significant changes across different applied potentials. In addition, the absence of *CObridge species in operando FT-IR spectroscopy further supports the conclusion that CuBBTA maintains structural integrity without undergoing substantial structural transformations. Compared to CORR, CO2RR provides a more favorable local environment that prevents CuBBTA from being exposed to harsh alkaline conditions, thereby preserving its molecular structure.
Density functional theory calculations
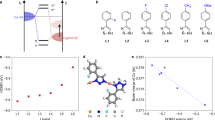
Density functional theory (DFT) calculations were further conducted to study the formation mechanism of the CuBBTA. For the formation of the CuBBTA, the BBTA first associates with the Cu2+ ion by forming a triazole-Cu coordination bond and one H2O is released with a decrease of free energy of 4.8 eV (Fig. 5a and Supplementary Fig. 35a). Subsequently, the second BBTA group substitutes at the trans position of the Cu(H2O)3BBTA intermediate with a decrease of free energy of 5.5 eV (for cis-isomer, the decrease of free energy is only 5.3 eV, Supplementary Fig. 35b). The third substitution of a H2O by a hydroxyl group shows lower free energy of 4.8 eV (Supplementary Fig. 35c). However, the fourth substitution leads to a further release of free energy by 4.1 eV, yielding a fairly stable coordination complex. Obviously, the abundant Pyrrolic N groups in the micellar brush play an important role in anchoring and stabilizing the Cu atoms.

a DFT calculation of the energy pathway and possible intermediates for the formation of CuBBTA. b Online DEMS spectra of CuBBTA during four consecutive LSV scans in the potential range of − 0.4 to − 1 V vs RHE. c The linear relationship between reduction potential and Operando ATR-SEIRAS transmittance wavelength for CuBBTA. The wavelength is the peak value of each absorption spectrum. d Energy barriers of *CO-*CHO coupling on the CuBBTA surface (Cu2+ site) and D-Cu surface (Cu0 site). The corresponding transition state structures are shown in the insets. Source data are provided as a Source Data file.
We herein used online differential electrochemical mass spectrometry (DEMS, Supplementary Fig. 36) to study the dynamic evolution of products on CuBBTA during the electrocatalytic reduction of CO2. DEMS brings a mass spectrometer into close contact with an electrochemical interface, allowing for rapid product analysis during the course of potential sweeps57. As observed in the mass spectrometry of Fig. 5b, the C2H4 and CO product signals were clearly resolved, the amounts of ethylene and *CO exhibit a negative correlation at the potential sweep. This behavior is consistent with the electrochemical data in Fig. 3b, where the Faradaic efficiency of C2H4 increases while that of CO decreases with increasingly negative potentials. By combing the results of operando ATR-SEIRAS analysis (Fig. 4e), the peaks of *COatop and *CHO decrease with increasing voltage, while the peak of *COCHO increases with increasing voltage (Fig. 5c), suggesting the dimerization of *CO and *CHO to *COCHO on adjacent copper atoms53,58. The formed *COCHO dimer exhibited a thermodynamically favorable adsorption on two adjacent Cu sites, with a Cu-Cu distance of 3.25 Å, closely aligning with that in the pristine CuBBTA coordination framework (3.12-3.41 Å, Supplementary Fig. 37a, b). The structural configuration of CuBBTA was verified through EXAFS analysis (Fig. 2f and Supplementary Table 3). This structural similarity reduces the conformational energy required for reconstruction, thereby minimizing damage to the molecular scaffold. The charge density difference plot of the C2 intermediate, *COCHO, in CuBBTA slab showed that electrons, once accepted by Cu, tended to transfer to adsorbates instead of reducing the Cu sites (Supplementary Fig. 37c), avoiding Cu leaching from its coordination environment during CO2RR.
To understand the structure-function relationship, a density functional theory (DFT) calculation was performed. The rate-determining step (RDS) along the CO2-to-C2+ path was calculated to be *CO2 hydrogenation by H2O to *COOH. This step on CuBBTA slab has an energy barrier of 0.91 V, which was 0.12 V lower than that on Cu(111) slab (Fig. 5d). H/D KIE experiment using H2O and D2O as the electrolyte (i.e., proton source) showed a ratio of ethylene production rates (KH:KD) close to 2:1 (Supplementary Fig. 38), supporting a RDS involving the hydrogenation by H2O59,60,61. The small energy difference indicates that the Cu0 and Cu2+ surfaces exhibit comparable overall reaction kinetics, in line with the experimentally observed LSV results showing similar current densities over a wide potential range (Fig. 3a). Subsequently, the energy barrier of the C-C coupling transition state plays the dominant role in determining the product selectivity.
We further investigated the dimerization kinetics of *CO and *CHO to *COCHO on Cu surfaces with different oxidation states. As shown in Fig. 5d and Supplementary Figs. 39 and 40, when the catalyst surface is entirely composed of Cu0 (CuBBTA is electrochemically reduced to Cu(111) under an Ar atmosphere), the dimerization of *CO and *CHO on the surface requires overcoming a high activation energy barrier (1.02 eV) to form the transient state. When the catalyst surface is entirely composed of Cu2+ (CuBBTA), the barrier for the transient state is reduced to 0.88 eV, indicating the formation of the *COCHO in Cu2+ site is more favorable than Cu0 site in kinetics, consistent with our experimental results. We further studied the free energies (ΔG) of this step. On the basis of reaction ΔG calculated at constant potential of − 0.8 V, the Cu2+ site representing the formation of the *COCHO was supports to be more energetically favorable with a free energy change of 0.44 eV, thermodynamically modestly more favorable than that for the Cu0 site (ΔG = 0.58 eV). This clearly demonstrates the importance of the Cu2+ environment maintained by CuBBTA for the efficient C–C coupling34,35,36,62.
In summary, operando XAS, in combination with operando Raman and ATR-SEIRAS analyses, revealed that CuBBTA remains in a stable Cu2+ state with no dynamic structural transformations during CO2RR. The structural stability originates from a suitable local environment during CO2RR. Online DEMS, operando ATR-SEIRAS and theoretical investigations suggest that the CuBBTA metal-organic polymer, featuring neighboring Cu sites, retains its stability throughout CO2RR, thereby facilitating C–C coupling. The thermodynamically favorable formation of the *COCHO intermediate underscores the critical role of the preserved Cu2+ environment in CuBBTA for the efficient generation of C2+ products. Our work not only proposes a strategy for stabilizing the valence state of Cu to achieve precise product selectivity in CO2RR but also highlights the significance of tailored coordination structures in the design of CO2RR electrocatalysts for the efficient synthesis of high-value-added products.
Methods
Chemicals and materials
Copper(II) chloride dihydrate (CuCl2·2H2O, AR), sodium hydroxide (NaOH, AR), potassium bicarbonate (KHCO3, AR), potassium hydroxide (KOH, AR), hydrochloric acid (HCl, CP, 36–38%), nitric acid (HNO3, AR, 65.0–68.0%) were all purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Benzobistriazole (H2BBTA) was purchased from Macklin. Copper(II) phthalocyanine (CuPc), Copper(II) hydroxide (Cu(OH)2), Cupric oxide (CuO), phenol (C6H5SO2C6H5), Dimethyl sulfoxide (DMSO), (dimethyl sulfoxide)-d6 (DMSO-d6), and Nafion solution (~5 wt%) were purchased from Sigma-Aldrich. N-Propanol (n-PrOH), acetate, formate, ethanol (EtOH) were purchased from Fisher Scientific. Proton exchange membrane (Nafion 117) was purchased from Dupont. The anion exchange membrane (Sustainion X37- 50 RT) was received from Dioxide Materials. Carbon paper-based gas diffusion layers (GDLs, Sigracet 28 BB) were purchased from the Fuel Cell Store. The aqueous solutions were prepared using deionized water with a resistivity of 18.25 MΩ cm.
Synthesis of Cu Metal-organic polymer (CuBBTA)
1.28 g of CuCl2·2H2O was dissolved in 37.5 mL of ultrapure water to form a blue solution. Separately, 0.60 g of Benzobistriazole (H2BBTA), 1.20 g of NaOH, and 75 mL of ultrapure water were mixed to yield a clear, colorless solution. The CuCl2 solution was rapidly added to the mixture and stirred at room temperature for 10 h. The mixture was centrifuged, washed with deionized water, centrifuged again, and the final product was obtained by drying.
Synthesis of CuBBTA-derived Cu (D-Cu)
CuBBTA-derived Cu was prepared by operando electroreduction at − 300 mA cm−2 for 10 min under an Ar atmosphere.
Electrochemical reduction of CO2
The CO2 electroreduction reaction was conducted in a flow cell or a membrane electrode assembly (MEA) system. Within the flow cell measurements, an electrolytic solution of 1 M KOH (pH = 13.7 ± 0.2) was utilized as both the anolyte and catholyte. The gaseous environments perfusing the cathodic compartment were composed of CO2 and Argon, tailored to the specific exigencies of the reaction conditions. A microporous gas diffusion layer (GDLs, Sigracet 28 BB, 1.5 × 3.5 cm2) with the catalyst loading of 1 mg cm–2 was employed as the working electrode, and its actual working area is 0.5 × 2 cm2. A platinum foil (1.5 × 3.5 cm2) and Hg/HgO (with 1 M KOH) were used as counter electrode and reference electrode, respectively. An anion exchange membrane of fumasep FAB-PK130 provided a discrete partition between the cathode and anode chambers. The flow rate of electrolyte was set at 2.5 mL min−1 by controlling a peristaltic pump. The regulation of gaseous flow was achieved with a mass flowmeter, maintaining a rate of 20 mL min−1. The calibration of potentialities was scrupulously performed in reference to the reversible hydrogen electrode (RHE), utilizing the equation: E (vs. RHE) = E (vs Hg/HgO) + 0.098 + pH × 0.059 V. All potentials reported in this work are not iR corrected unless otherwise specified. Linear sweep voltammetry (LSV) was executed within the gas diffusion cell at a scanning velocity of 10 mV s−1, traversing a potential range from 0.1 to − 1.3 V versus RHE. Cyclic voltammetry (CV) curves of the catalysts were recorded at a scan rate of 100 mV s−1 in the aforementioned flow cell.
The half-cell power conversion efficiency (PCE) of ethylene in the flow cell was calculated by the following equation:
The overpotential of oxygen evolution is assumed to be zero. Eethylene = 0.08 V (vs RHE) for CO2RR63.
In the MEA system, the anodic and cathodic flow fields were constructed from titanium and stainless steel, respectively, each with an exposed area of 4 cm2. A two-electrode configuration was employed, with gas diffusion electrodes (GDEs), an anion exchange membrane, and an IrO2/Ti mesh assembled between the flow fields. A 3 mm thick PTFE gasket was inserted between the membrane and anodic flow field to ensure proper sealing. The anolyte is 0.1 M KHCO3 (pH = 8.3 ± 0.2) with a flow rate controlled at 20 milliliters per minute, while the cathode gas is humidified with deionized water at 50 °C before entering the cathode chamber, with its flow rate controlled at 30 standard cubic centimeters per minute (sccm). For the stability test, the MEA was operated at a constant full-cell potential of 3.6 V. To maintain electrolyte composition during prolonged electrolysis, 10 L of 0.1 M KHCO3 anolyte was prepared. Electrolyte renewal was carried out at approximately 20 h and 43 h during the long-term electrochemical stability test. Gas products were analyzed at intervals of approximately 4 h. The electrochemical workstation was a CorrTest CS Studio electrochemical workstation at room temperature.
Products analysis
Electrochemical CO2 reduction was carried out at ambient temperature in a saturated 1 M KOH electrolyte over a potential range of − 0.69 V to − 0.94 V versus the reversible hydrogen electrode (RHE). Cathodic electrolysis was conducted for 10 min at each potential. Simultaneously, oxygen generated at the anode was removed along with the electrolyte using a peristaltic pump. Gaseous products from the cathodic compartment were continuously monitored using online gas chromatography (Shimadzu GC-2014) equipped with both a flame ionization detector (FID) and a thermal conductivity detector (TCD), with analyses performed at twenty-five-minute intervals.
Liquid-phase CO2RR products were quantified using a 400 MHz Inova NMR spectrometer with a pre-saturation pulse sequence for water suppression. Typically, 600 μL of the post-electrolysis electrolyte was mixed with 70 μL of D2O and 30 μL of an internal standard solution containing 10 mM DMSO and 50 mM phenol in H2O. Formate was quantified by comparing its peak area (singlet, δ = 8.33 ppm) to that of phenol (triplet, δ = 7.03 ppm). Other products, including ethanol (triplet, δ = 1.06 ppm), acetate (singlet, δ = 1.80 ppm), and n-propanol (triplet, δ = 0.77 ppm), were quantified by referencing their peak areas to that of DMSO (singlet, δ = 2.60 ppm). Calibration curves for each product were constructed using standard solutions of known concentrations (Supplementary Fig. 10). FE of the CO2RR products were computed from:
where F is the Faraday constant (96485 C mol−1), n is the total product (in mole), e is the number of transferred electrons for each product, and Q is the current time integral the amount of charge obtained.
Material characterizations
All 1H NMR spectroscopy spectra were recorded on a Bruker AVANCE NEO 400 MHz spectrometer at 295 K. The 13 C solid-state NMR was tested on an AVANCE NEO 600 MHz instrument. All spectra were obtained from deuterated chloroform. Powder X-ray diffraction (PXRD) was carried out on a Bruker D8-Focus Bragg-Brentano X-ray Powder Diffractometer equipped with a Cu sealed tube (λ = 1.54178 Å) at 40 kV and 40 mA. The chemical properties of surface elements in materials were analyzed using X-ray photoelectron spectroscopy (XPS) with an Al Kα source on a Thermo Electron Corporation ESCALAB 250 spectrometer. The transmitted infrared spectra were carried out by a Fourier transform-infrared absorption (FT-IR) spectrometer (Bruker vertex 80 v). Raman spectra were conducted using a Thermo Scientific DXR3 Raman spectrometer with an excitation of 532 nm laser light. The morphology and element distribution of the catalysts were examined using scanning electron microscopy (SEM, JSM-6510) and transmission electron microscopy (TEM, Philips-FEI Tecnai G 2S-Twin F20) that were equipped with energy dispersive X-ray spectroscopy (EDX). HAADF-STEM imaging was performed using a JEOL ARM-200F field-emission transmission electron microscope operated at an accelerating voltage of 200 kV. The concentrations of Cu present in the catalysts were determined through the use of inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis, using the Perkin-Elmer Optima 3300 DV. Gas analysis during electrolysis was conducted using a Shimadzu GC-2014 gas chromatography (GC) system 8 equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID).
Online differential electrochemical mass spectrometry
Operando mass spectrometric measurements were performed on a QAS-100 system (Linglu, China) to monitor volatile intermediates and products. The electrolyte consisted of 0.5 M KHCO3 saturated with either Ar or CO2 and was continuously purged throughout the experiment. Catalyst inks were uniformly drop-cast onto gold film or hydrophobic carbon paper substrates to serve as the working electrode. An Ag/AgCl electrode and a platinum wire were used as the reference and counter electrodes, respectively. Prior to each linear sweep voltammetry (LSV) scan (5 mV s−1), the mass spectrometry (MS) signal was allowed to stabilize. MS signals were recorded during the potential sweep, and subsequent scans were conducted only after the MS signal returned to a stable baseline.
Operando Raman spectroscopy
Operando Raman spectroscopy was conducted using a Thermo Scientific DXR3. The electrode configuration was consistent with that used in prior electrochemical experiments, except that the electrolyte was replaced with 0.1 M KHCO3 to minimize CO2 absorption by KOH. Spectra were acquired under 532 nm laser excitation at 10% laser power, with an exposure time of 20 s. Raman spectra at open-circuit potential (OCP) were obtained by immersing the sample directly in 0.1 M KHCO3. Operando Raman spectra were collected under chronoamperometric conditions at potentials ranging from −0.6 to − 1 V vs. RHE.
Operando ATR-SEIRAS spectroscopy
The catalyst layer was deposited onto a chemically prepared Au film on a Si ATR prism for subsequent ATR-SEIRAS measurements, which were performed using a Bruker vertex 80 v Spectrum FTIR spectrometer equipped with an MCT detector. Spectra were recorded at a resolution of 4 cm-1 over a wavenumber range of 600–4000 cm−1, with four scans averaged per acquisition. The Au film functioned as the working electrode, onto which the catalyst ink was drop-cast and dried prior to testing. A platinum sheet and an Ag/AgCl electrode served as the counter and reference electrodes, respectively, in a three-electrode configuration. Electrochemical measurements were carried out in 0.1 M KHCO3 using chronoamperometry across a potential range from − 0.5 V to − 1.2 V vs. RHE. Spectra were collected twice after 30 seconds of reaction at each potential. The Operando ATR-SEIRAS spectra were recorded in transmission mode. The negative features in the spectra arise from the differential subtraction process between spectra obtained under potential and at open-circuit potential.
Operando XAFS on the easyXAFS300 +
Cu K-edge X-ray absorption spectra were collected using an easyXAFS300 + benchtop instrument (easyXAFS, LLC), using a Si (553) ball-bending crystal analyzer and an Mo anode X-ray tube. Spectra were deadtime corrected and energy was calibrated using a standard Cu metal foil. Operando XAS measurements were performed in transmittance mode using a custom-designed flow cell with a Kapton-sealed window (Supplementary Fig. 20). The CO2RR product distribution and selectivity observed in this operando cell closely matched those obtained from the standard flow cell used for CO2RR performance evaluation. Operando XAFS spectra were scanned over the Cu K-edge with an energy step of 0.5–2.0 eV. The acquisition time for a single scan was approximately 10–20 min, and repeated scans were averaged to improve the signal-to-noise ratio.
Operando XAFS at the BL17B1 beamline
The Cu K-edge XAFS spectra were measured at the BL17B1 beamline of Shanghai Synchrotron Radiation Facility (SSRF), China. The storage ring of the SSRF was operated at 2.5 GeV with a maximum electron current of 250 mA. Operando XAFS measurements were performed with catalyst-coated carbon cloth using a home-built cell. The catalyst powders were dispersed in ethanol with 20 μl Nafion solution (5%, Sigma-Aldrich), and then sonicated for 30 min. The catalyst ink was drop-cast onto carbon paper as the working electrode (~ 1 cm × 1 cm) to ensure all of the electrocatalyst reacted with KOH electrolyte at a geometric metal loading of ~ 1 mg cm−2. The XAFS spectra were collected through the fluorescence mode. To monitor the changes during the CO2RR process, cathodic voltages from − 0.6 V to − 1.1 V were applied for 10 min in 1 M KOH as the conditioning step.
XAFS analysis and results
During operando XANES measurements, occasional glitches caused by transient gas bubbles in the operando cell were observed. Isolated spike points were removed prior to normalization, and the spectra were averaged over repeated scans to minimize bubble-induced fluctuations. Importantly, this treatment does not affect the absorption edge position or overall spectral evolution.
XAFS data were processed using the Demeter software package. Energy calibration was performed in Athena using a Cu foil standard, followed by spectral normalization and linear combination fitting, with Cu K-edge spectra of CuBBTA and metallic Cu foil serving as reference standards. The normalized EXAFS function, χ(E), was subsequently converted to χ(k), where k represents the photoelectron wave vector. To enhance the EXAFS oscillations in the mid-k region and evaluate the local coordination environment of Cu, χ(k) was multiplied by k². Fourier transformation of the k²-weighted χ(k) was conducted over a k range of 2.5–12 Å−1 to obtain radial structure functions in R space, allowing differentiation of coordination shells. EXAFS fitting was carried out in R space using Artemis within the Demeter package, employing the FEFF6 code to generate theoretical phase and amplitude functions for Cu–O, Cu–N, and Cu–Cu paths. Structural parameters, including coordination number (CN) and bond distance (R), were extracted through standard fitting procedures.
Density functional theory computation
All the density functional theory (DFT) calculations were performed with a periodic slab model using the Vienna ab initio simulation program (VASP)64,65. We used the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional66. We utilized the projectoraugment wave (PAW) method to describe the electron-ion interactions67. We set the cut-off energy for the plane-wave basis at 450 eV. The D3 correction method, by Grimme et al., was utilized to illustrate the long-range dispersion interactions between the adsorbates and catalysts68. The implicit solvation model implemented in VASPsol was used to consider the effect of solvent69. The entropies of free molecules at standard conditions were taken from the NIST database, while the vibrational entropy is considered for the adsorbed species.
To expose under-coordinated Cu sites, an -OH vacancy was introduced into the van der Waals-stacked MA-1 bulk phase. For comparison, a 3 × 3 Cu(111) supercell with 36 atoms was built to simulate C–C coupling under high CO coverage. The Brillouin zone was sampled by a 2 × 2 × 1 k points mesh for the monolayer slab and 3 × 3 × 1 for Cu(111) slab.
The Gibbs free energy change (ΔG) is calculated as: ΔG = ΔE + ΔZPE − TΔS where ΔE is the reaction energy obtained from DFT calculations, ΔZPE denotes the zero-point energy correction, and ΔS represents the entropy change. The entropy of all adsorbed species is assumed to be zero (S = 0), while the entropy values of gaseous molecules at standard conditions are taken from the NIST database. According to the computational hydrogen electrode (CHE) model, the free energy change of a proton–electron transfer process at U = 0 V is equivalent to the formation of hydrogen gas, described by the equation: H+ + e− = 1/2 H270. The adsorption energy of the *OCCHO intermediate is thus calculated using the following expression: Eads = E*OCCHO + slab – Eslab – 2 E*CO – 0.5 E*H2. Charge transfer between the *OCCHO species and the CuBBTA surface was analyzed using Bader charge analysis.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information. All other relevant data supporting the findings of this study are available from the corresponding authors upon request. Source data are provided in this paper.
References
Liu, M. et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 537, 382–386 (2016).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes?. Science 364, eaav3506 (2019).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Yang, H. B. et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 3, 140–147 (2018).
Li, P. et al. Manipulation of oxygen species on an antimony-modified copper surface to tune the product selectivity in CO2 electroreduction. J. Am. Chem. Soc. 146, 26525–26533 (2024).
Crandall, B. S. et al. Kilowatt-scale tandem CO2 electrolysis for enhanced acetate and ethylene production. Nat. Chem. Eng. 1, 421–429 (2024).
Li, F. et al. Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces. Nat. Catal. 3, 75–82 (2020).
Han, C. et al. Steering the product selectivity of CO2 electroreduction by single atom switching in isostructural copper nanocluster catalysts. Angew. Chem. Int. Ed. 137, e202503417 (2025).
Li, Q. et al. Ultralow coordination copper sites compartmentalized within ordered pores for highly efficient electrosynthesis of n-propanol from CO2. J. Am. Chem. Soc. 147, 6688–6697 (2025).
Li, W. et al. Bifunctional ionomers for efficient co-electrolysis of CO2 and pure water towards ethylene production at industrial-scale current densities. Nat. Energy 7, 835–843 (2022).
Chen, X. et al. Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20–27 (2021).
Zhong, D. et al. Selective electrochemical CO2 reduction to ethylene or ethanol via tuning *OH adsorption. Angew. Chem. Int. Ed. 64, e202501773 (2025).
Zhang, R. et al. Cu2O Nano-homojunction for high-efficiency electrocatalytic CO2-to-ethylene conversion. Angew. Chem. Int. Ed. 64, e202501554 (2025).
Ma, X. et al. Carbonate shell regulates CuO surface reconstruction for enhanced CO2 electroreduction. Nat. Synthesis 4, 53–66 (2025).
Lei, Q. et al. Structural evolution and strain generation of derived-Cu catalysts during CO2 electroreduction. Nat. Commun. 13, 4857 (2022).
Li, Y. et al. Strong electronic interactions of the abundant Cu/Ce interfaces stabilized Cu2O for efficient CO2 electroreduction to C2+ products under large current density. Adv. Funct. Mater. 35, 2509899 (2025).
Li, S. et al. Operando reconstruction of porous carbon supported copper selenide promotes the C2 production from CO2RR. Adv. Funct. Mater. 34, 2311989 (2024).
Chen, X. et al. Metal variance in multivariate metal–organic frameworks for boosting catalytic conversion of CO2. J. Am. Chem. Soc. 146, 19271–19278 (2024).
Li, J. et al. Cascade dual sites modulate local CO coverage and hydrogen-binding strength to boost CO2 electroreduction to ethylene. J. Am. Chem. Soc. 146, 5693–5701 (2024).
Cai, H. et al. Combining the active site construction and microenvironment regulation via a bio-inspired strategy boosts CO2 electroreduction under ampere-level current densities. Angew. Chem. Int. Ed. 137, e202425325 (2025).
Xie, G. et al. Dual-metal sites drive tandem electrocatalytic CO2 to C2+ products. Angew. Chem. Int. Ed. 136, e202412568 (2024).
Liang, Y. et al. Stabilizing copper sites in coordination polymers toward efficient electrochemical C-C coupling. Nat. Commun. 14, 474 (2023).
Yue, K. et al. Stabilized Cu0-Cu1+ dual sites in a cyanamide framework for selective CO2 electroreduction to ethylene. Nat. Commun. 15, 7820 (2024).
Yang, Y. et al. Ligand-tuning copper in coordination polymers for efficient electrochemical C–C coupling. Nat. Commun. 15, 6316 (2024).
Wu, H. et al. Selective and energy-efficient electrosynthesis of ethylene from CO2 by tuning the valence of Cu catalysts through aryl diazonium functionalization. Nat. Energy 9, 422–433 (2024).
Wang, Y. et al. Enhancing CO2 Electroreduction to Multicarbon Products by Modulating the Surface Microenvironment of Electrode with Polyethylene Glycol. Angew. Chem. Int. Ed. 137, e202420661 (2025).
Liu, Z. et al. Switching CO2 electroreduction toward ethanol by delocalization state-tuned bond cleavage. J. Am. Chem. Soc. 146, 14260–14266 (2024).
Zhang, L. et al. Elucidating the structure-stability relationship of Cu single-atom catalysts using operando surface-enhanced infrared absorption spectroscopy. Nat. Commun. 14, 8311 (2023).
Wu, H. et al. Pressure-dependent CO2 electroreduction to methane over asymmetric Cu–N2 single-atom sites. J. Am. Chem. Soc. 146, 22266–22275 (2024).
Zhang, Y. et al. Electronic metal-support interaction modulates Cu electronic structures for CO2 electroreduction to desired products. Nat. Commun. 16, 1956 (2025).
Zhu, J. et al. Superexchange-stabilized long-distance Cu sites in rock-salt-ordered double perovskite oxides for CO2 electromethanation. Nat. Commun. 15, 1565 (2024).
Zhang, R. et al. Dynamically stable Cu0Cuδ+ pair sites based on in situ-exsolved Cu nanoclusters on CaCO3 for efficient CO2 electroreduction. Angew. Chem. Int. Ed. 137, e202421860 (2025).
Chen, R. et al. Visualizing catalytic dynamics of Single-Cu-Atom-Modified SnS2 in CO2 electroreduction via rapid freeze-quench mossbauer spectroscopy. J. Am. Chem. Soc. 146, 24368–24376 (2024).
Tan, X. et al. Stabilizing copper by a reconstruction-resistant atomic Cu–O–Si interface for electrochemical CO2 reduction. J. Am. Chem. Soc. 145, 8656–8664 (2023).
Chen, X. et al. Highly stable layered coordination polymer electrocatalyst toward efficient CO2-to-CH4 conversion. Adv. Mater. 36, 2310273 (2024).
Zhou, X. et al. Stabilizing Cu2+ ions by solid solutions to promote CO2 electroreduction to methane. J. Am. Chem. Soc. 144, 2079–2084 (2022).
Xu, F. et al. Oxygen-bridged Cu binuclear sites for efficient electrocatalytic CO2 reduction to ethanol at ultralow overpotential. J. Am. Chem. Soc. 146, 9365–9374 (2024).
Zhao, Z.-H., Huang, J.-R., Liao, P.-Q. & Chen, X.-M. Highly efficient electroreduction of CO2 to ethanol via asymmetric C–C coupling by a metal–organic framework with heterodimetal dual sites. J. Am. Chem. Soc. 145, 26783–26790 (2023).
Chen, Y. et al. N-Heterocyclic carbene polymer-stabilized Au nanowires for selective and stable reduction of CO2. J. Am. Chem. Soc. 147, 14845–14855 (2025).
Heng, J.-M. et al. Dicopper(I) sites confined in a single metal–organic layer boosting the electroreduction of CO2 to CH4 in a neutral electrolyte. J. Am. Chem. Soc. 145, 21672–21678 (2023).
Jiao, L., Wang, Y., Jiang, H.-L. & Xu, Q. Metal–organic frameworks as platforms for catalytic applications. Adv. Mater. 30, 1703663 (2018).
Zhang, W. et al. Dynamic restructuring of coordinatively unsaturated copper paddle wheel clusters to boost electrochemical CO2 reduction to hydrocarbons. Angew. Chem. Int. Ed. 61, e202112116 (2022).
Zhang, Y. et al. Hydrogen-bond regulation of the microenvironment of Ni(II)-porphyrin bifunctional electrocatalysts for efficient overall water splitting. Adv. Mater. 35, 2210727 (2023).
Liu, Y. et al. A highly efficient metal-free electrocatalyst of F-doped porous carbon toward N2 electroreduction. Adv. Mater. 32, 1907690 (2020).
Qu, Y. et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat. Catal. 1, 781–786 (2018).
Yang, T. et al. Coordination tailoring of Cu single sites on C3N4 realizes selective CO2 hydrogenation at low temperature. Nat. Commun. 12, 6022 (2021).
Wang, Y. et al. CO electroreduction on single-atom copper. Sci. Adv. 9, eade3557 (2023).
Fang, M. et al. Anionic ionomer: released surface-immobilized cations and an established hydrophobic microenvironment for efficient and durable CO2-to-ethylene electrosynthesis at high current over one month. J. Am. Chem. Soc. 146, 27060–27069 (2024).
Gunathunge, C. M., Ovalle, V. J., Li, Y., Janik, M. J. & Waegele, M. M. Existence of an electrochemically inert CO population on Cu electrodes in alkaline pH. ACS Catal. 8, 7507–7516 (2018).
Yao, Y. et al. A surface strategy boosting the ethylene selectivity for CO2 reduction and in situ mechanistic insights. Nat. Commun. 15, 1257 (2024).
Ding, J. et al. Unraveling dynamic structural evolution of single atom catalyst via in situ surface-enhanced infrared absorption spectroscopy. J. Am. Chem. Soc. 147, 9601–9609 (2025).
Chou, T.-C. et al. Controlling the oxidation state of the Cu electrode and reaction intermediates for electrochemical CO2 reduction to ethylene. J. Am. Chem. Soc. 142, 2857–2867 (2020).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical CO2 reduction and C–C coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935–1945 (2024).
Xu, Z. et al. Electroreduction of CO2 to methane with triazole molecular catalysts. Nat. Energy 9, 1397–1406 (2024).
Sun, Q. et al. Probing inside the catalyst layer on Gas diffusion electrodes in electrochemical reduction of CO and CO2. Angew. Chem. Int. Ed. 64, e202504715 (2025).
Luo, W., Zhang, J., Li, M. & Zütte, A. Boosting CO production in electrocatalytic CO2 reduction on highly porous Zn catalysts. ACS Catal. 9, 3783–3791 (2019).
Ren, H. et al. Operando proton-transfer-reaction time-of-flight mass spectrometry of carbon dioxide reduction electrocatalysis. Nat. Catal. 5, 1169–1179 (2022).
Huang, P. et al. Balancing *CHO/*CO intermediate flux via carbonyl-hydroxyl motif synergy enables high-selectivity ethanol electrosynthesis from dilute CO2. J. Am. Chem. Soc. 147, 22062–22071 (2025).
Liu, W. et al. Efficient ammonia synthesis from the air using tandem non-thermal plasma and electrocatalysis at ambient conditions. Nat. Commun. 15, 3524 (2024).
Zhu, C. et al. Engineering the coordination environment of metal centers for selective and high-current CO2 electromethanation. J. Am. Chem. Soc. 147, 26185–26194 (2025).
Li, J. et al. Copper-catalysed electrochemical CO2 methanation via the alloying of single cobalt atoms. Angew. Chem. Int. Ed. 64, e202417008 (2025).
Albertini, P. P. et al. Hybrid oxide coatings generate stable Cu catalysts for CO2 electroreduction. Nat. Mater. 23, 680–687 (2024).
Lai, W., Qiao, Y., Zhang, J., Lin, Z. & Huang, H. Design strategies for markedly enhancing energy efficiency in the electrocatalytic CO2 reduction reaction. Energy Environ. Sci. 15, 3603–3629 (2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Acknowledgements
The authors are grateful to the Natural Science Foundation of Jilin Province (No. SKL202302017), the National Natural Science Foundation of China (NO. 22279041), the National Key Research and Development Program of China (No. 2022YFC2105800) and the 111 Center (B17020). We also thank Prof. Ya Tang and Dr. Zixian Song of Shanghai University for their assistance with the operando XANES experiments at the BL17B1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF).
Author information
Authors and Affiliations
Contributions
H.R. and J.S.Q. conceived the idea of the project. Z.H.Z. carried out the chemical synthesis and electrochemical experiments, and Operando experimental investigations. Y.M.H. and H.S. participated in the characterization. Q.X. and R.Z. provided a helpful discussion. Z.H.Z., J.W.H., and K.R. performed the DFT calculations. All authors discussed the results, drew conclusions and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ke Xie, Jun Yu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Xu, Q., Han, J. et al. Operando insights on stable Cu2+ active sites for efficient electrochemical CO2-to-C2H4 conversion. Nat Commun 17, 2654 (2026). https://doi.org/10.1038/s41467-026-70442-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70442-y
