
Abstract
Transcriptional regulation, critical for cellular differentiation and adaptation to environmental changes, involves coordinated interactions among DNA sequences, regulatory proteins, and chromatin architecture. Despite extensive chromatin profiles and gene expression data from consortia, understanding the dynamics of cis-regulatory elements in gene expression remains challenging. Deep learning is a powerful tool for learning gene expression and epigenomic profiles from DNA sequences, exhibiting superior performance compared to conventional machine learning approaches. However, even the most advanced deep learning-based methods may fall short in capturing the regulatory effects of distal elements such as enhancers, limiting their predictive accuracy. In addition, these methods may require significant resources to train or adapt to newly generated data. To address these challenges, we present EPInformer, a scalable deep-learning framework for predicting gene expression by integrating promoter-enhancer interactions with their sequences, epigenomic profiles, and chromatin contacts. Our model outperforms existing gene expression prediction models in rigorous cross-chromosome validation, accurately recapitulates enhancer-gene interactions validated by genome editing experiments, and identifies crucial transcription factor motifs within regulatory sequences.
Similar content being viewed by others


Introduction
Transcriptional regulation is intricately governed by the complex interplay of DNA sequences, epigenomic signals, and three-dimensional (3D) chromatin contacts1,2,3. This process shapes gene expression and plays a crucial role in cell differentiation and environmental response. The DNA sequence interacts with various epigenetic modifications and chromatin structures to fine-tune gene expression4. Epigenomic signals, including DNA methylation and histone modifications, add a dynamic layer to gene regulation, influencing transcriptional activity without altering the underlying DNA sequence. Additionally, the spatial organization of chromatin, evidenced by chromatin contacts and looping, further orchestrates transcriptional regulation, bringing distant regulatory elements into proximity with gene promoters. Together, these factors constitute a multifaceted system that drives the precise and context-dependent expression of genes in living organisms.
The collaborative work and data generation efforts of consortia like ENCODE5,6, FANTOM7,8, and 4D Nucleome9,10 have significantly enhanced our understanding of gene regulation through epigenomics and chromatin interactions. The rich and large dataset generated by these consortia has been crucial for training powerful deep-learning methods11, furthering our ability to dissect and understand gene regulatory mechanisms12,13,14,15. These models excel by learning to predict genomic and epigenomic signals—such as transcription factor binding, chromatin contacts and accessibility, DNA methylation, and histone modifications—to improve gene expression predictions and identify regulatory elements16,17,18,19. This underscores deep learning’s transformative impact on computational biology and genomics13,20,21. However, fully understanding the complexity of cis-regulatory elements (CREs), such as enhancers and repressors, remains a significant challenge.
Transformer-based deep learning models have shown remarkable proficiency in predicting gene expression22,23,24,25. Their architecture effectively captures interactions across genomic elements, with the attention mechanism offering an advantage over traditional convolutional neural networks (CNNs) by better handling long-range genomic interactions. Among sequence-based models, Enformer is the most widely used. It excels at predicting gene expression, transcription factor binding, and chromatin states directly from DNA sequence. More recently, an updated architecture, Borzoi, was introduced to model substantially longer genomic contexts (up to 524 kb), improving predictions of gene expression and diverse epigenomic signals. However, both Enformer and Borzoi’s extensive training requirements limit their adaptability to unseen cell types, and their ability to capture regulatory effects from very distal enhancers remains constrained, particularly for elements located more than 10 kb from a gene’s transcription start site (TSS), as previously reported26,27. Another model, CREaTor28 comprises two transformer encoders that integrate cCREs from ENCODE open chromatin regions with ChIP-seq data on transcription factors and histone modifications to predict gene expression levels. However, like Enformer, Borzoi, and CREaTor do not account for 3D chromatin interactions, which are essential for understanding spatial regulatory relationships. To incorporate this missing dimension, Seq-GraphReg29 integrates chromatin contact data using a graph attention network for expression prediction. However, its effectiveness is restricted by the limited availability of Hi-C data across various cell lines.
Notably, large genomic language models have emerged as an important approach for deciphering regulatory grammar from sequence in a self-supervised manner, inspired by the success of large language models. DNABERT‑230, Nucleotide Transformer31, and HyenaDNA32 are among the most popular models and have been extensively benchmarked33,34,35. Although they show promising performance in identifying cell-type-agnostic regulatory elements and genes directly from DNA sequence, they are not designed for cell-type-specific gene expression modeling, which limits their utility for understanding gene regulation in specific cellular contexts. Moreover, genomic language models typically require substantial computational resources for fine-tuning on downstream tasks, which is not feasible for most experimental laboratories.
Hence, there’s a pressing need for a more flexible framework for combining DNA sequences, epigenomic states, and chromatin contact data to refine predictive accuracy in cell-type-specific gene expression modeling. To achieve this, we introduce EPInformer (a portmanteau of Enhancer–Promoter Interaction and Transformer), a scalable and efficient deep-learning framework based on the transformer architecture.
EPInformer offers several key advantages over existing approaches for predicting gene expression: (i) flexible integration of heterogeneous multi-omic data (DNA sequence, epigenomic signals, chromatin contacts); (ii) explicit modeling of long-range promoter–enhancer interactions via dedicated attention module, improving enhancer–gene link prediction and interpretability, as validated by CRISPR perturbations; and (iii) compact, efficient training (0.4 M parameters; around 1 hour on a single GPU for 18,377 protein-coding genes). These capabilities enable EPInformer to outperform state-of-the-art methods while remaining computationally efficient.
Unlike other sequence-based models, EPInformer uses multi-head attention modules to directly model interactions between promoters and the potential enhancers (Supplementary Table 1). It integrates epigenomic signals (e.g., H3K27ac and DNase) with DNA sequences and, if available, chromatin contact data such as Hi-C to significantly enhance prediction accuracy. Notably, EPInformer’s streamlined architecture models gene expression in a single cell type with just 0.2% (447,149 total parameters) of Enformer’s requirements, facilitating rapid training and deployment for new cell types and reducing computational demands, a point especially important for researchers with modest computing resources. Our study rigorously tested EPInformer through a 12-fold cross-chromosome validation, confirming its superiority over existing models in predicting Cap Analysis Gene Expression (CAGE) and RNA-seq gene expression. EPInformer excels in its adaptability to various multimodal inputs. It can be trained on DNase-seq data alone or by integrating DNase-seq, H3K27ac ChIP-seq, and Hi-C contacts for a more comprehensive analysis. Its interaction encoder effectively identifies crucial distal enhancer information, validated through CRISPR perturbation experiments. Additionally, to explore and provide interpretability of the sequence features learned by the model, we utilized TF-MoDISco-lite36 and Tangermeme37 to uncover important transcription factor motifs within cell-type-specific enhancer sequences.
Results
Overview of the EPInformer framework
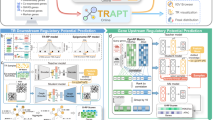
EPInformer is a transformer-based framework for predicting gene expression by explicitly modeling promoter and enhancer interactions. The model integrates genomic sequences, epigenomic signals (e.g., DNase-seq, H3K27ac ChIP-seq), and chromatin contacts through a flexible architecture to capture their interactions. EPInformer consists of four key modules (Fig. 1a and Supplementary Fig. 1): a sequence encoder (EPInformer-seq), a feature fusion layer, a promoter-enhancer interaction encoder, and a predictor module. Given a gene locus, EPInformer-seq learns DNA sequence embeddings of the promoter region (2-kb sequence around the Transcription Start Site) and potential enhancers in open chromatin regions within 100 kb of the TSS. Sequences shorter than 2 kb are padded with “N” to reach a uniform length. Residual convolutional layers learn DNA motifs in promoter and enhancer sequences, whereas dilated convolutional layers learn motif cooperation by extracting distal sequence patterns, facilitated by the dilated convolution operator12. Convolutional and pooling operations in the sequence encoder work together to learn a comprehensive sequence embedding, preserving key features of the DNA sequence as shown by several past approaches38,39,40,41. EPInformer-seq can also be pre-trained with fully connected layers to predict epigenomic signals (e.g., H3K27ac ChIP-seq) from potential enhancer regions (“Methods” and Supplementary Fig. 1). This pre-training process accelerates the optimization of EPInformer and equips it with an understanding of enhancer sequence patterns prior to full training for gene expression prediction. Moreover, this pretrained sequence encoder enhances interpretability and helps to uncover the key motifs at the putative enhancer of the target gene.

a EPInformer is trained on multimodal epigenomic data and promoter–enhancer sequences to predict CAGE or RNA-seq expression in specific cell types. It first derives promoter and candidate enhancer embeddings using residual and dilated convolutions in EPInformer-seq, which can be pre-trained on cell-type-specific enhancer signals to initialize the convolutional filters. The fusion layer optionally merges the sequence embeddings with distance, chromatin contacts, or epigenomic signals (e.g., H3K27ac and DNase). The interaction encoder employs a series of transformer encoders with multi-head attention modules designed to capture promoter-enhancer interactions. Finally, the prediction module integrates the resulting embeddings with mRNA half-life features and the promoter signal through fully connected layers to predict the gene expression. The EPInformer model is versatile for multiple tasks: b predicting gene expression from promoter and enhancer sequences with multimodal epigenomic signals; c prioritizing enhancers that may drive expression using the attention module of the interaction encoder, with scores derived from the average attention weights of the attention heads and layers; and d identifying regulatory sequence features and transcription factor binding motifs at enhancers pinpointed by attention score for the target gene through the sequence encoder with downstream interpretation tools (e.g., TF-MoDISco-lite36 and Tangermeme37).
The fusion layer is designed to merge sequence embeddings with information such as distance to the target gene, epigenomic signals (e.g., H3K27ac ChIP-seq and DNase-seq), and chromatin contact data (e.g., Hi-C) between a promoter and candidate enhancer regions. It starts by concatenating the epigenomic signals of candidate enhancers with their sequence embeddings, followed by a 1 × 1 convolution block to refine the combined embedding dimension for the following interaction encoder. The fusion layer can integrate any number and type of genomic or epigenomic signals with sequence embedding for subsequent interaction modeling. This versatility enhances the model’s capability to incorporate diverse data types available to the users, boosting its performance and flexibility.
The interaction encoder, comprising transformer layers with multi-head attention, is designed to learn the interplay between promoters and potential enhancers. It derives a weighted sum from their embeddings, with attention weights based on fused sequence and epigenomic signal embeddings. Notably, the interaction encoder focuses solely on enhancer-promoter interactions, ignoring enhancer-enhancer interactions through attention masking. In addition, only the promoter representation after the final layer of the transformer encoder is passed directly to the prediction module. This reduces the space of interactions to learn, focusing on promoter-enhancer interactions and increasing the computational efficiency of the model. This particular promoter representation encapsulates comprehensive relationships between a promoter and all candidate enhancers for the final predictor module, analogous to the CLS token functionality in BERT42,43. Subsequently, the predictor module, a feed-forward neural network, utilizes the promoter representation and genomic features like mRNA half-life44 and H3K27ac signals at the promoter region (500-bp around the transcription start site) to predict gene expression levels accurately. Importantly, EPInformer-trained models can be combined with TF-MoDISco-lite36 and Tangermeme37 to identify transcription factor binding motifs at the putative enhancer region, incorporating the attention score of promoter-enhancer pairs to elucidate their impact on gene expression prediction.
EPInformer was trained to minimize the discrepancy between predicted and observed gene expression levels, as measured by RNA-seq or CAGE using different feature sets. EPInformer excels in three key applications: (1) Accurately predicting gene expression levels using promoter-enhancer sequences, epigenomic signals, and chromatin contacts (Fig. 1b); (2) Efficiently identifying cell-type-specific enhancer-gene interactions, validated by CRISPR perturbation experiments (Fig. 1c); (3) Precisely predicting enhancer activity and identifying transcription factor binding motifs from sequences (Fig. 1d).
EPInformer improves gene expression prediction by explicitly modeling promoter–enhancer epigenomic profiles and chromatin contacts
To develop and evaluate EPInformer models for gene expression prediction, we initially used the ABC pipeline45 (Supplementary Fig. 2; “Methods”) to identify candidate promoter-enhancer pairs for coding genes in six well-characterized cell lines, K562, GM12878, HepG2, H1, NHEK, and HUVEC. In brief, we extracted promoter sequences from the 2-kb region surrounding the transcription start site (TSS) and candidate enhancer sequences from DNase I hypersensitive (DHS) sites, prioritizing up to 60 nearby enhancers per gene. This threshold covers 95% of potential regulatory elements within a 200-kb region centered on the TSS. We validated this distance choice by evaluating model performance across different genomic ranges (50 kb to 500 kb from the TSS), finding optimal performance at 100–250 kb with substantial degradation at 500 kb (Supplementary Fig. 6). For pre-training EPInformer-seq, we collected H3K27ac ChIP-seq peaks from ENCODE for six cell lines. We targeted 256 bp regions centered on the H3K27ac peak summits and included two additional 256 bp regions flanking each side with a 100 bp overlap. The ABC pipeline was used to calculate enhancer activity from DNase-seq and H3K27ac signals for these regions in all cell lines (see “Methods”). To further enrich our dataset, we included the reverse complement of each sequence, retaining the same activity level, resulting in datasets of an average 419,344 sequences across six cell lines. Additionally, chromatin contacts of promoter and candidate enhancer pairs were obtained from KR-normalized Hi-C contact maps using the ABC pipeline45,46.
Two gene expression datasets were curated for model training: protein-coding mRNA RNA-seq and Cap Analysis Gene Expression (CAGE) sequencing. For the CAGE dataset, expression values were quantified by aggregating read counts within a 384-bp window centered on each gene’s unique transcription start site (TSS), following the protocol established by Enformer22. RNA-seq expression data were obtained from the Xpresso dataset44, which utilizes pre-processed data from the Roadmap Epigenomics Consortium47. To address the large dynamic range inherent in raw read counts, a log transformation was applied to the expression values.
To evaluate model performance under varying data availability scenarios, we evaluated several EPInformer variants in predicting gene expression: EPInformer-PE takes in input promoter-enhancer sequences and the distance between the candidate enhancer and its target gene (TSS). EPInformer-PE-Activity extended this by incorporating H3K27ac and DNase signals of each enhancer element. The most comprehensive model, EPInformer-PE-Activity-HiC, in addition to including promoter-enhancer sequences and enhancer signals, can also leverage Hi-C contacts. To improve interpretability, the sequence encoders of EPInformer-PE-Activity and EPInformer-PE-Activity-HiC were pre-trained on cell-type-specific H3K27ac signals covering all peak regions (Supplementary Fig. 3).
To rigorously evaluate the EPInformer models, we conducted separate benchmarking for CAGE and RNA-seq expression. For RNA-seq prediction, we compared Xpresso44 and CREaTor against three EPInformer models using 12-fold cross-chromosome validation (Fig. 2a). In each fold, two chromosomes were designated for testing, two for validation, and the remainder for training, following protocols established in previous studies19,29. Regarding CAGE expression benchmarking, retraining seq-graphReg, Enformer, and Borzoi requires substantial computational resources and complex data preprocessing. Therefore, we evaluated their officially released pre-trained models using the original hold-out test sets defined in their respective studies22,48 (Fig. 2b–d). To ensure a fair comparison, the EPInformer models were trained and tested using these identical splits. Performance across all genes was assessed using the Pearson Correlation Coefficient between predicted and observed expression levels.

a Pearson correlation between observed and predicted RNA-seq expression levels for 18,377 protein-coding genes across six cell lines. Left: Spider plot showing correlation between observed and predicted expression levels for six cell lines. Middle: Spider plot showing correlation between observed and predicted expression levels for each test chromosome across six cell lines. Right: Scatter plot showing relationship between EPInformer-PE-Activity-HiC predicted and observed RNA-seq expression levels in K562 cells, with each point representing an individual gene. Genes with high expression in K562 are highlighted in red, and those with low expression are highlighted in green. All evaluations were conducted using 12-fold cross-chromosome validation. b Comparison of EPInformer models with Enformer for predicting CAGE gene expression levels using Enformer's hold-out test set in K562 (top) and GM12878 (bottom) cells. c Comparison of EPInformer models with Seq-GraphReg and CREaTor for CAGE gene expression prediction in K562 (top) and GM12878 (bottom) cells using 12-fold cross-chromosome validation. d Comparison of EPInformer models with Borzoi and CREaTor for predicting CAGE gene expression levels using Borzoi's hold-out test set in K562 (top) and GM12878 (bottom) cells. Source data are provided as a Source Data file.
For RNA-seq expression prediction, EPInformer-PE achieved a mean Pearson correlation of 0.78 across six cell lines, outperforming the sequence-based Xpresso model (0.67; Fig. 2a). This performance gap is likely driven by the difference in input scope: whereas Xpresso relies on a 20-kb sequence surrounding the target gene, EPInformer-PE utilizes a 200-kb window. This extended context enables the capture of distal cis-regulatory elements, underscoring the critical role of long-range regulatory information in accurate expression prediction.
Next, we evaluated two multimodal models: EPInformer-PE-Activity and CREaTor, both of which integrate H3K27ac, DNase, and DNA sequence data from open chromatin regions. Using 12-fold cross-chromosome validation across six cell lines, EPInformer-PE-Activity yielded a mean Pearson correlation of 0.83 (range: 0.79–0.86), surpassing the 0.78 mean achieved by CREaTor. Further enhancements were achieved by incorporating Hi-C chromatin contact data into EPInformer via a fusion layer, resulting in EPInformer-PE-Activity-HiC. This model delivered the highest performance, with a mean Pearson correlation of 0.84, surpassing EPInformer-PE-Activity. EPInformer-PE-Activity-HiC consistently outperformed Xpresso and CREaTor across all test chromosomes (Fig. 2a) and accurately predicted gene expression levels driven by strong enhancers in K562 cells. Across six cell lines using 12-fold leave-two-chromosomes-out cross-validation, EPInformer-PE-Activity-HiC achieved the highest median Pearson correlation (0.69), exceeding EPInformer-PE-Activity (0.65) and CREaTor (0.51; Supplementary Fig. 4), highlighting its superior capacity to capture gene dynamics across diverse cell types.
For CAGE expression prediction, EPInformer‑PE‑Activity‑HiC demonstrated superior performance compared to all EPInformer variants and external benchmarks in both K562 and GM12878 (Fig. 2b–d). Across two held‑out test sets, it surpassed Enformer (Fig. 2b) and Borzoi (Fig. 2d) in both K562 and GM12878 cell lines. Notably, even without Hi‑C data, EPInformer‑PE‑Activity exceeded Borzoi, yielding higher Pearson correlations in K562 (r = 0.841 vs. 0.792) and GM12878 (r = 0.847 vs. 0.800). Under 12‑fold cross‑chromosome validation in K562 and GM12878 (Fig. 2c), EPInformer‑PE‑Activity‑HiC achieved the highest average Pearson correlation (r = 0.883), outperforming Seq‑GraphReg (r = 0.744) and CREaTor (r = 0.810). Given that the original Xpresso was designed to predict RNA-seq expression, we retrained and evaluated it on the CAGE dataset to enable a comparison with the EPInformer models across the three benchmarks in Fig. 2b–d. As expected, all three EPInformer models significantly outperformed Xpresso in CAGE expression prediction, consistent with our findings in RNA-seq prediction (Supplementary Fig. 9).
While the above results demonstrate EPInformer’s superior performance on gene-level expression prediction, it is important to note that most existing models are evaluated only on canonical transcripts. As many genes produce isoforms with distinct functions and tissue-specific expression, we further assessed EPInformer‑PE‑Activity at the isoform level. To evaluate performance, we compiled all alternative TSSs for protein-coding genes, yielding 46,616 isoforms with distinct TSSs (median two per gene; Supplementary Fig. 7a). Isoform-specific expression was quantified using 128-bp CAGE read counts centered on each TSS. Under the same 12-fold cross-chromosomal validation, EPInformer-PE-Activity (using DNase and H3K27ac signals) accurately predicted K562 CAGE expression (Pearson r = 0.814; Supplementary Fig. 7b). As an example, predictions closely recapitulated the CAGE signal at GATA2, a key transcription factor gene expressed in K562 cells (Supplementary Fig 7c). We also compared our models with MENTR49 using a larger CAGE dataset comprising 92,473 mRNA coding transcripts. EPInformer-PE-Activity-HiC significantly outperformed MENTR, achieving a Pearson correlation of 0.792 compared to MENTR’s 0.675 when predicting average CAGE signals across four K562 samples (Supplementary Fig. 8).
In addition, we evaluated EPInformer’s ability to leverage ATAC-seq data for modeling gene expression, since ATAC-seq data are widely available across cell types. We adapted the ABC pipeline to obtain potential enhancer regions and their corresponding ATAC-seq signals to train EPInformer-PE-Activity on both CAGE and RNA-seq expression. Under 12-fold cross-validation (Supplementary Fig. 5), ATAC-based EPInformer-PE-Activity achieved an average Pearson correlation of 0.843 for K562 and 0.854 for GM12878, which is comparable to DNase+H3K27ac-based EPInformer-PE-Activity (average Pearson correlation: 0.867 for K562 and 0.874 for GM12878). These results demonstrate that EPInformer can effectively use ATAC-seq for gene expression modeling and is practical for cell types where only ATAC-seq data is available, highlighting our framework’s flexibility.
In summary, the EPInformer framework is effective and scalable for modeling gene expression. Our most advanced model, EPInformer-PE-Activity-HiC, significantly outperforms existing methods in predicting CAGE and RNA-seq expression across diverse cell lines, offering a robust approach for inferring gene expression from DNA sequence and multimodal epigenomic data.
EPInformer accurately prioritizes cell-type-specific enhancer–gene interactions
Linking candidate enhancers to their target genes using biochemical annotations remains a central challenge in genomics. To evaluate whether EPInformer can accurately learn from gene expression and multimodal epigenomic data to identify enhancer–gene interactions, we normalized the attention weights in the final layer of EPInformer interaction encoder to generate an attention score quantifying each candidate enhancer’s contribution to a given target gene (Fig. 3a; “Methods”). As a baseline, we used the Activity-by-Contact (ABC) score45, a widely adopted method for linking enhancers to target genes22,50. ABC score quantifies enhancer-gene links using either Hi-C contact maps (ABC.score.Hi-C) or a 1/distance-to-TSS contact proxy (ABC.score.distance). To align comparisons, we derived two attention-based scores from EPInformer: Attention.score.distance (from EPInformer-PE-Activity) and Attention.score.Hi-C (from EPInformer-PE-Activity-HiC).

a Schematic of attention score calculation in EPInformer-PE-Activity(-HiC). The red and blue boxes represent three candidate enhancers (E1, E2, and E3) located near the promoter (P) of the gene (gray box). The dashed box shows the simplified calculation of the attention score for promoter-enhancer pairs, incorporating DNase, H3K27ac, and Hi-C signals (expressed in arbitrary units, not to scale). b Precision– recall curves for predicting enhancer–gene (E–G) pairs. Positive E–G pairs are defined as cases where perturbing the candidate enhancer significantly decreases expression of the target gene. The curves compare attention-based scores (Attention.score.Hi-C from EPInformer-PE-Activity-HiC and Attention.score.distance from EPInformer-PE-Activity) with two ABC scores in classifying 1575 E–G pairs across 244 genes assayed by CRISPR perturbations. c Enrichment–recall curves for enhancer–gene interaction scores in GM12878 cells. The y-axis shows enrichment—the ratio of fine-mapped distal noncoding eQTLs (PIP > 70% in GM12878) that fall within predicted enhancers relative to all distal noncoding common variants. The x-axis shows recall—the fraction of eQTL variants overlapping enhancers linked to the correct gene. Curves represent varying score thresholds for enhancers predicted by different models. d Area under the precision-recall curve (AUPRC) values for E-G score performance shown in panel b when classifying enhancer-gene pairs at three different distance ranges. e Enrichment of putative causal variants (PIP > 70%) within predicted enhancers across different distance ranges. f Top three genes with the largest per-gene F1 score gains when comparing Attention.score.Hi-C to ABC.score.Hi-C. The first two rows display enhancer activity and Hi-C contact values computed by the ABC pipeline for each candidate enhancer. Light blue boxes indicate enhancers predicted by ABC.score.Hi-C, orange boxes indicate enhancers predicted by Attention.score.Hi-C. Red boxes indicate CRISPR-validated enhancers. Stars indicate correctly predicted enhancers. Source data are provided as a Source Data file.
To evaluate this approach, we compiled genetic perturbation datasets inspired by a recent study51: K562 CRISPR enhancer perturbations and fine-mapped eQTL variants from lymphoblastoid cells in GTEx52. Following the ABC pipeline45, we restricted candidate enhancers to those within 100 kb of a gene’s transcriptional start site (TSS) and computed enhancer activity and Hi-C contact frequencies for K562 and GM12878.
We first evaluated K562 enhancer-gene predictions by assembling 1,575 candidate enhancer-gene links (within 100 kb of their target genes) from the largest K562 CRISPR perturbation dataset51, of which 370 were experimentally validated. We compared attention scores from EPInformer-PE-Activity (Attention.score.distance) and EPInformer-PE-Activity-HiC (Attention.score.Hi-C) against two ABC scores
(ABC.score.Hi-C and ABC.score.distance) using precision–recall plots (Fig. 3b), reporting the area under the precision-recall curve (AUPRC). Attention.score.Hi-C achieved the highest AUPRC (0.732) versus the ABC.score.Hi-C (0.698). Without Hi-C, Attention.score.distance also outperformed the ABC.score.distance (0.600 vs. 0.579). Additionally, across varying distance ranges, Attention.score.Hi-C consistently exceeded ABC.score.Hi-C, with the largest gains for distal enhancers (60–100 kb: AUPRC 0.510 vs. 0.429; Fig. 3d). Notably, Attention.score.distance also surpassed ABC.score.distance for predicting distal enhancers (5–100 kb), indicating that EPInformer’s attention score better captures distal enhancer–gene links than ABC score.
To further assess functional relevance, we set a 70% recall threshold for all scores and compared per-gene F1 scores. Remarkably, Attention.score.Hi-C outperformed or matched ABC.score.Hi-C for 212 of 244 genes (89%). We highlight PRDX2, FUT1, and LYL1, where Attention.score.Hi-C achieved the largest F1 score gains over ABC.score.Hi-C (Fig. 3f), attributable to more precise identification of distal enhancers, further supports the effectiveness of the attention score.
Next, we evaluated how well attention scores can link eQTL variants to target genes in GM12878 lymphoblastoid cells. We collected distal noncoding eQTL variants with fine-mapping posterior inclusion probability (PIP) > 70% (n = 166). For each score, we quantified (1) recall for eQTL variant-gene links (the fraction of eQTLs that fall within predicted regulatory elements and are assigned to the correct gene), and (2) enrichment of eQTLs within predicted enhancers relative to all distal noncoding variants, across score thresholds. Attention.score.Hi-C showed significantly higher enrichment than ABC.score.Hi-C at recall <10% and comparable enrichment at ≥10% (Fig. 3c). Using a fixed 70% recall threshold (as in K562), Attention.score.Hi-C nominated enhancers showed higher enrichment for putative causal variants (PIP > 70%) across distance ranges than ABC.score.Hi-C (Fig. 3e), consistent with the K562 results and reinforcing the attention score’s precision in identifying true enhancer–gene interactions.
EPInformer recapitulates transcription factor motifs required for enhancer activities
Having established that EPInformer’s attention scores effectively prioritize enhancers relevant to a target gene, we next examined sequence patterns underlying the functionality of these predicted enhancers. We implemented a base-resolution motif-discovery workflow using EPInformer‑seq, integrating TF-MoDISco-lite36 and Tangermeme37 to analyze transcription factor motifs within genome-wide putative enhancers (Fig. 4a). For each cell line, we first trained EPInformer-seq to predict enhancer activity from 256-bp sequences centered on cell-type-specific H3K27ac peaks, along with two flanking sequences (100-bp overlap), using the same 12-fold leave-two-chromosomes-out cross-validation as EPInformer. The enhancer activity is computed as the geometric mean of H3K27ac and DNase RPM, following the ABC score protocol. EPInformer‑seq achieved an average Pearson correlation of 0.727 across six cell lines for enhancer activity prediction spanning all H3K27ac peak regions (Supplementary Fig. 3).

a The workflow of using EPInformer-seq with TF-MoDISco-lite and Tangermeme to discover transcription factor binding motifs. b Representative motifs enriched in K562 (left) and GM12878 (right) discovered by TF-MoDISco-lite from recurring sequence patterns learned by EPInformer-seq at cell-type-specific H3K27ac peaks. c Bar plots showing the number of TF-MoDISco-discovered motifs matching JASPAR 2024 CORE vertebrate non-redundant database (q < 0.05) in K562 (left) and GM12878 (right). d EPInformer discovers several important transcription factor binding motifs at a putative enhancer of KLF1. The orange dashed line indicates the transcription start site (TSS) of KLF1. The black box with the red arc represents a CRISPRi-validated enhancer, while the gray box denotes an EPInformer-PE-Activity predicted enhancer. The bar plot displays predicted enhancer activities (geometric mean of H3K27ac and DNase signals) for 256-bp sequences tiling the putative enhancer locus (chr19:12,977,587-12,979,123). The bottom panel shows insilico mutagenesis (ISM) attribution scores for the region (chr19:12,978,099-12,978,355) with the highest predicted enhancer activity. High-scoring sequence motifs are highlighted with dashed boxes, with labels and arrows indicating matching TF motifs from the JASPAR 2024 CORE vertebrate non-redundant database. Source data are provided as a Source Data file.
We next employed the TF-MoDISco-lite pipeline to interpret the sequence encoder at base resolution. Tangermeme was used to calculate DeepLIFT53 attribution scores for 256-bp sequences centered on H3K27ac summits of enhancers prioritized by our model. TF‑MoDISco‑lite (default settings) was applied to identify genome‑wide motifs associated with H3K27ac ChIP-seq peaks. Using Tomtom54,55 to match discovered motifs against the JASPAR 2024 CORE vertebrate non‑redundant database, we identified shared TF motifs including JUN, ELF1, ELK1, BACH1, and NFYA, as well as K562‑specific motifs (GATA1, GATA2, GATA1::TAL1) and GM12878‑specific TF motifs (SPI1, FOSL1; Fig. 4b, c). These results suggest that the pre-trained sequence encoder can help with model interpretation and discover cell-type-specific motifs important for enhancer activity.
Additionally, we aimed to uncover transcription motifs that may be required for KLF1’s enhancers in K562 cells. KLF1 is an erythroid-specific transcription factor essential for β-globin locus activation, erythroid differentiation, and red blood cell maturation56,57. Focusing on one of the distal predicted enhancers at chr19:12,977,587-12,979,123 (hg19) located 19,662 bp from KLF1, we first identified the window (256-bp resolution) with the highest predicted enhancer activity. Subsequently, we used Tangermeme within this window to obtain attribution scores at base resolution using the in-silico mutagenesis (ISM) approach. We observed four seqlets with high ISM scores (Fig. 4d), which were matched against the JASPAR 2024 CORE vertebrate non-redundant database using FIMO58. We found three matching motifs: GATA1, SP4, and ETV6. In K562 cells, GATA1 is a well-known master regulator necessary for erythroid differentiation, playing a crucial role in the activation and repression of various genes involved in hematopoiesis59,60; ETV6 is known for its involvement in hematopoiesis and oncogenesis, contributing to the regulation of genes essential for blood cell development and differentiation61,62; The SP4 transcription factor is a member of the SP/KLF family of zinc finger transcription factors, and is known to bind to GC-rich promoter elements and influence the transcription of target genes59,63. While these insights are derived from predictive analysis, they lay the groundwork for empirical validation. Future experiments, such as motif perturbations using CRISPR deletion, base editing, or prime editing, could directly assess these motifs’ influence on gene expression.
Discussion
Identifying promoter-enhancer interactions and decoding the cis-regulatory code remains a significant challenge in gene regulation. EPInformer, a novel transformer-based framework, significantly improves gene expression prediction by modeling promoter and enhancer sequences alongside multimodal epigenomic data. EPInformer excels by holistically integrating DNA sequence, epigenomic features and chromatin contact data, offering a refined understanding of gene regulatory patterns. The CNN-based sequence encoder learns sequence patterns of promoters and enhancers, providing insight into the cis-regulatory code. The feature fusion layer integrates epigenomic signals and chromatin contacts with sequence embeddings, enhancing the prediction power and the model’s flexibility to include additional data types. The interaction encoder explicitly models promoter-enhancer interactions, while the predictor, a feed-forward neural network, harmonizes multimodal data representations to predict gene expression levels. This approach resulted in a substantial performance increase as compared to state-of-the-art tools like Enformer, GraphReg, and Xpresso and achieved Pearson correlation coefficients of 0.875 in K562 cells and 0.891 in GM12878 cells for predicting CAGE expression in a 12-fold cross-chromosome validation.
EPInformer stands out from other gene expression prediction methods due to its lightweight design and versatility. Its architecture, requiring only 0.4 million parameters compared to Enformer’s 250 million and Borzoi’s 186 million, allows for faster training speeds without sacrificing efficacy. The model completes training in just one hour on an A100 GPU (Supplementary Table 2), making sophisticated gene expression modeling more accessible and user-friendly for the scientific community. Importantly, the model can be trained using only DNase-seq data if necessary. However, EPInformer’s structure can easily integrate DNA sequences with multiple types of epigenomic information and chromatin interactions, enhancing its ability to predict gene expression from diverse assays like CAGE and RNA-seq. This versatility ensures broad applicability and superior performance compared to models like Enformer, GraphReg, CREaTor and Xpresso.
We demonstrated that EPInformer attention scores can effectively identify relevant enhancer-promoter interactions. Importantly, this approach demonstrates higher accuracy in predicting CRISPRi-validated enhancers than state-of-the-art ABC scores. Additionally, applying downstream model interpretation tools to attention score-predicted enhancers can uncover key transcription factor motifs important for cell identity. We have also shown how EPInformer can aid in fine-mapping eQTL variants by accurately linking them to their target genes through enhanced enhancer-gene predictions. Future studies could extend this approach to systematically evaluate EPInformer’s utility for fine-mapping GWAS variants, potentially providing insights into the regulatory mechanisms underlying complex traits and diseases.
Future enhancements to EPInformer will focus on several key areas to further improve its performance and applicability. First, a current limitation is that EPInformer predicts gene-level expression using the canonical TSS, rather than modeling isoform-specific expression. Given that many genes produce multiple isoforms with distinct functions and tissue-specific expression patterns, extending EPInformer to predict isoform-level expression would enhance its biological relevance and utility for interpreting genetic variants. Future versions could incorporate isoform-specific TSSs from databases like FANTOM, utilize isoform-quantified RNA-seq data, and integrate isoform-specific regulatory features to achieve transcript-level resolution.
We also plan to develop more sophisticated methods for identifying and defining candidate enhancer regions, potentially incorporating additional epigenomic markers and evolutionary conservation data. While the current implementation focuses on activating epigenomic marks (H3K27ac and DNase-seq), the framework can capture some aspects of negative regulation indirectly through the absence of these signals. Future versions could explicitly incorporate repressive histone modifications (e.g., H3K9me3, H3K27me3) to distinguish between actively repressed and simply inactive regions, potentially improving predictions for genes under active repression. Extending the model to train on and predict gene expression across multiple cell types simultaneously will improve its generalizability and ability to capture cell-type-specific regulatory mechanisms. Given the importance of CTCF in chromatin organization, we aim to integrate CTCF binding site information to better model long-range interactions and chromatin domain boundaries. Implementing relative positional encoding schemes may improve the model’s ability to capture spatial relationships between regulatory elements. Incorporating reverse complement sequences of enhancers in the model architecture could capture additional regulatory information and improve prediction accuracy. Integrating pre-trained DNA foundation models as sequence embeddings may enhance EPInformer’s performance by leveraging large-scale genomic knowledge. Additionally, developing more comprehensive in-silico element perturbation analyses will further validate the model’s predictions and provide insights into the functional impact of specific regulatory elements. These advancements, combined with EPInformer’s current flexibility and efficiency, aim to deepen our understanding of regulatory mechanisms and their impact on gene expression and cell type identity. By leveraging CRISPR perturbation datasets and adopting a multi-task learning approach, we expect to refine EPInformer’s predictive capabilities further. Ultimately, these improvements will contribute to a more comprehensive and accurate model of gene regulation, with broad implications for both basic research and potential clinical applications.
Despite these ambitious future directions, the current iteration of EPInformer already represents a significant leap forward in gene expression prediction and enhancer-promoter interaction modeling, providing a powerful and accessible tool for researchers to unravel the complexities of gene regulation.
Methods
Collection and pre-processing of gene expression and epigenomic data
Our study curated three types of datasets for model training and testing: enhancer-related epigenomic data, chromatin contacts, and gene expression (Supplementary Data 1). For the epigenomic data, we obtained DNase and H3K27ac BAM files for all replicates of six cell lines (K562, GM12878, HepG2, NHEK, HUVEC, and H1) from the ENCODE project. Following the ABC model protocol45 (Supplementary Fig. 2), we utilized MACS2 to call peaks from the DNase-seq bam file for each cell line, considering peaks with p < 0.1. We refined these to the top 150,000 regions based on read count, extended from their summits to form 500 bp candidate enhancers, and merging overlapping regions. These extended and merged peaks were defined as candidate elements in our experiments. For promoter elements, we obtained 18,377 protein-coding genes from Xpresso, excluding histone and chromosome Y genes. Following Xpresso’s procedure44, we defined TSSs as the start coordinate of the first exon of the representative transcript; for mRNAs with a dominant CAGE peak, we re-centered the TSS to the coordinate of that peak. For isoform-specific analyses, we aggregated all alternative TSSs for protein-coding genes from GENCODE v41, resulting in 46,616 isoforms with distinct TSSs.
Promoter and putative enhancer sequences were retrieved from the hg38 reference genome. Enhancer sequences exceeding 2 kb were truncated and realigned to center on the DNase-seq peak summit. The 2 kb region surrounding the TSS was designated as the promoter, and candidate enhancers within 100 kb of the TSS, excluding the promoter region, were assigned to the target gene. This process resulted in an average of 338,909 pairs across the six cell lines.
To estimate candidate enhancer activity, we first used the ABC pipeline to compute DNase-seq and H3K27ac ChIP-seq signals from bam files by summing read counts at the candidate enhancer region. Signals from replicate experiments were averaged and quantile-normalized. Based on these normalized signals, the final enhancer activity was then calculated as the geometric mean of DNase and H3K27ac signals.
We compiled a Hi-C dataset to estimate contacts between promoters and candidate enhancers. Cell-type-specific Hi-C contacts were obtained from the 4DN Nucleome database9. Using FANC46, we converted the Hi-C data to bedpe format and applied vanilla coverage normalization at a 5 kb resolution. The ABC pipeline then computed promoter-enhancer contacts by identifying the Hi-C bedpe row containing the gene’s TSS and assigning contact values to enhancer-promoter pairs based on signals at the bin corresponding to the enhancer’s midpoint.
Additionally, we incorporated promoter activity and mRNA half-life features for EPInformer in gene expression modeling. Promoter activity is defined as the geometric mean of H3K27ac and DNase RPM at the 500-bp region around the gene’s TSS; mRNA half-life features, as calculated by Xpresso44 for gene expression modeling, include G/C content, lengths of functional regions (5′ UTRs, ORFs, and 3′ UTRs), intron length, and exon junction density within the open reading frame.
To train and evaluate EPInformer on gene expression prediction, we curated two gene expression datasets, as measured by RNA-seq and CAGE. For CAGE, expression values were determined by aggregating read counts within 384-bp regions centered at each gene’s unique TSS, as per Enformer’s protocol. RNA-Seq expression data were sourced from Xpresso’s training set, quantified by the Roadmap Epigenomics Consortium. To mitigate the right-skewed distribution of gene expression based on raw read count, we applied log10 transformation with a pseudocount of one to handle zero values.
Model architecture
Figure 1a illustrates the model architecture, organized into four key sections: (1) a sequence encoder with 5 residual and 4 dilated convolutional layers plus a linear layer; (2) a fusion layer featuring channel-wise concatenation and 1 × 1 convolution blocks; (3) an interaction encoder with 3 transformer encoders, each having a 4-head self-attention module and a feed-forward layer; (4) a predictor with three dense layers for the gene expression prediction. EPInformer processes input as a one-hot encoded matrix (A = [1, 0, 0, 0], C = [0, 1, 0, 0], G = [0, 0, 1, 0], T = [0, 0, 0, 1], N = [0, 0, 0, 0]), sized (61, 2000, 4), comprising a promoter sequence and 60 candidate enhancer sequences for predicting gene expression. Genes with fewer than 60 candidate enhancers receive padding via zero vectors to ensure uniform dimensions. The sequence encoder first learns sequence embeddings of size (61, 64) for the promoter and its candidate enhancers. The fusion layer concatenates the distances of enhancers to the TSS, enhancer activities, and promoter-enhancer chromatin contacts with sequence embeddings on a channel-wise basis. It then reshapes the concatenated matrix to a size of (61, 64) using a convolution operator. The interaction encoder then captures the interactions between the promoter and the candidate enhancers using self-attention. The attention calculation is based on the matrix operation:
In the attention mechanism, the softmax function generates a probability distribution for promoter-enhancer interactions. A mask vector M is set to a value near negative infinity for padding enhancers, ensuring the interaction encoder disregards these padding embeddings. Interaction encoder learns parameter matrices \({{{\bf{W}}}}^{K}\in {{{\boldsymbol{R}}}}^{{d}_{x}\times {d}_{k}}\), \({{{\bf{W}}}}^{Q}\in {{{\boldsymbol{R}}}}^{{d}_{x}\times {d}_{q}}\) and \({{{\bf{W}}}}^{V}\in {{{\boldsymbol{R}}}}^{{d}_{x}\times {d}_{v}}\) for each head. It transforms promoter-enhancer embedding \({{\bf{X}}}\in {{{\boldsymbol{R}}}}^{61\times {d}_{x}}\) into queries \({{{\bf{Q}}}}_{i}={{{\bf{X}}}}_{i}\times {{{\bf{W}}}}^{Q}\), keys \({{{\bf{K}}}}_{j}={{{\bf{X}}}}_{j}\times {{{\bf{W}}}}^{k}\) and values \({{{\bf{V}}}}_{j}={{{\bf{X}}}}_{j}\times {{{\bf{W}}}}^{V}\). The interaction of promoter \({{{\bf{X}}}}_{P}\) and the \({i}_{{th}}\) enhancer \({{{\bf{X}}}}_{{E}_{i}}\) can be computed as \({a}_{{{\rm{P}}}-{{{\rm{E}}}}_{{{\rm{i}}}}}={{\rm{softmax}}}\left({{{\bf{Q}}}}_{P}{{{\bf{K}}}}_{{E}_{i}}^{T}/\sqrt{{d}_{k}}\right)\), which represents the amount of weight query at promoter puts on the key at the \({i}_{{th}}\) enhancer.
To better capture distal enhancer-gene interactions, we weight each enhancer embedding in the final encoder layer by Hi-C contact (or inverse distance to the TSS if this assay is not available) and enhancer activity. Formally, the interaction of promoter XP and the \({{{\rm{i}}}}_{{{\rm{th}}}}\) enhancer \({{{\rm{X}}}}_{{{{\rm{E}}}}_{{{\rm{I}}}}}\) can be computed as \({{{\rm{a}}}}_{{{\rm{P}}}-{{{\rm{E}}}}_{{{\rm{i}}}}}={{\rm{softmax}}}\left({{{\rm{Q}}}}_{{{\rm{P}}}}{\cdot {{\rm{K}}}}_{{{{\rm{E}}}}_{{{\rm{i}}}}}^{{{\rm{T}}}}/\sqrt{{{{\rm{d}}}}_{{{\rm{k}}}}}{\cdot {{\rm{F}}}}_{{{{\rm{A}}}}_{{{\rm{i}}}}}\cdot {{{\rm{F}}}}_{{{{\rm{C}}}}_{{{\rm{I}}}}}\right)\), where \({{{\rm{F}}}}_{{{{\rm{A}}}}_{{{\rm{I}}}}}\) denotes enhancer activity (geometric mean of H3K27ac and DNase RPM), and \({{{\rm{F}}}}_{{{{\rm{C}}}}_{{{\rm{i}}}}}\) denotes the enhancer-gene Hi-C contacts or 1/distance to gene’s TSS. Each attention head computes a weighted sum over promoter-enhancer pairs: \({{{\rm{a}}}}_{{{\rm{P}}}-{{\rm{E}}}}\times {{\rm{V}}}\). Multiple heads operate with independent parameters; their outputs are concatenated and passed through a linear projection to produce the final layer output. Consequently, in the interaction encoder’s final layer, \({{{\rm{a}}}}_{{{\rm{P}}}-{{\rm{E}}}}\) quantifies the learned enhancer–gene interaction intensity and is used to prioritize enhancers for each gene.
The last transformer encoder outputs the promoter embedding with a size of 64, which embeds all promoter-enhancer pairs. Finally, the predictor concatenates the promoter embedding with 8-bit mRNA half-life features and predicts the gene expression through three dense layers.
In our enhancer activity prediction task, we engineered a model leveraging four pre-trained residual convolutional layers with filter configurations of 128, 64, 64, 128 and kernel sizes 8, 3, 3, 3. Each layer is succeeded by batch normalization, ELU nonlinearity, max pooling (size = 2, stride = 2), and a 1 × 1 convolution step. Beyond the convolutional base, the model employs two fully connected layers, each with 256 neurons, batch normalization, ReLU nonlinearity, and dropout (d = 0.1). The input is a one-hot-encoded 256-bp DNA sequence aimed at predicting enhancer activities. EPInformer-seq takes as input a 256-bp DNA sequence (one-hot encoded) and outputs a single enhancer activity score representing the predicted geometric mean of H3K27ac and DNase signals for that region. For base-resolution analysis, we apply interpretation methods (DeepLIFT or ISM) to derive nucleotide-level attribution scores from the trained model.
Model training and evaluation
As previously proposed by Karbalayghareh et al.29, we implemented a 12-fold cross-chromosome validation strategy. For fold 1 to 10, chromosomes i and i+10 were reserved for validation, while chromosomes \(i+1\) and \(i+11\) were set aside for testing. In fold 11, chromosomes 3 and 21 were used for validation, with chromosomes 22 and X allocated for testing. Fold 12 involved using chromosomes 2 and 22 for validation and chromosomes 1 and Y for testing. The remaining chromosomes were utilized for training in each fold. This evaluation procedure ensures the model is independently assessed across all human chromosomes.
All EPInformer models were implemented in PyTorch (v2.2.0)64 and trained on one A100 GPU with a batch size of 64 using AdamW65 optimizer with a learning rate of \(5\times {10}^{-4}\), a weight decay of \(1\times {10}^{-6}\) and default settings for other hyperparameters: \({\beta }_{1}=0.9\), \({\beta }_{2}=0.99\), \(\varepsilon=1\times {10}^{-8}\). The models were trained using smooth L1 loss66 to align predictive and actual expression levels. To enhance EPInformer’s generalization and mitigate overfitting, we applied early stopping, monitoring the model’s mean square error (MSE) on the validation set and stopping training if there was no MSE improvement for six consecutive epochs. The best-performing model, marked by the lowest MSE on the validation set, was retained for testing on an independent chromosome set, assessing performance through the Pearson Correlation Coefficient. For pre-training and evaluating the EPInformer’s sequence encoder, we adopted the same experimental settings as those used for EPInformer models, with the exception that this model aimed to minimize the loss between predicted and actual enhancer activity, as determined by the geometric mean of H3K27ac ChIP-seq and DNase-seq signals (reads per million (RPM)).
Baseline methods
Five baseline models—Xpresso, Enformer, Borzoi, CREaTor, and Seq-GraphReg—serve as references for gene expression prediction. Enformer, a deep neural network, combines convolutional neural networks (CNNs) with transformer technology, using DNA sequences as input. It processes 196-kbp sequences to predict 5313 genomic tracks for the human genome and 1643 tracks for the mouse genome at 128-bp resolution. However, Enformer and Borzoi’s significant training requirements limit their adaptability across new cell lines, and despite its context spans around 200 kb, it can detect reliably only the impact of proximal enhancers (less than around 10 kb from the TSS)26. Xpresso, a deep learning model, employs CNNs to predict mRNA abundance directly from genomic sequences, focusing on promoter regions and features linked to mRNA stability within a 20 kb range of the TSS. Its reliance on proximal sequences restricts its ability to utilize information from distal enhancers. Seq-GraphReg uses graph attention networks to integrate DNA sequences and HiChIP data, predicting gene expression levels by exploiting chromatin contact signals between distal elements and promoters. CREaTor is a two-stage transformer encoder designed to integrate 200 350 bp DNA sequences and ChIP-seq and DNase-seq signals at CRE regions obtained from SCREEN67, to predict RNA-seq gene expression levels.
To ensure a fair comparison, we aligned the training and testing settings of EPInformer with those of Enformer and Borzoi, respectively. This involved using identical data splits and extracting promoter and potential enhancer sequences from the same regions Enformer/Borzoi was trained on. For EPInformer, gene expression values were determined by summing read counts within a 384-bp window (equivalent to three 128-bp Enformer bins) surrounding each gene’s TSS, using the same data sources (CNhs12333 for GM12878 and CNhs11250 for K562).
For comparison with Xpresso and CREaTor, we retrained and assessed them using the same 12-fold cross-chromosome validation as EPInformer models, focusing on the 18,377 protein-coding genes from the Roadmap Epigenomics Consortium. Seq-GraphReg’s performance was reported from its original study, and we presented EPInformer’s performance using an identical train-test split across all human chromosomes for direct comparison.
Enhancer prioritization
We used the Engreitz Lab combined CRISPR dataset from the CRISPR benchmarking pipeline (https://github.com/EngreitzLab/CRISPR_comparison) and GTEx eQTL variants and RNA-seq data across all tissues included in the eQTL benchmarking pipeline (https://github.com/EngreitzLab/eQTLEnrichment). To prioritize enhancer–gene pairs with EPInformer, we first defined candidate enhancers using ABC-nominated regions and promoters based on each gene’s TSS. Enhancer activity and Hi-C contact were computed using the ABC score pipeline45. For EPInformer, we extracted the attention weights in the last layer of the interaction encoder. Using the promoter token as the query, we aligned keys corresponding to each candidate enhancer, yielding an attention distribution over enhancers for that promoter. We normalized these attention weights per promoter so that the weights across all candidate enhancer pairs summed to one, and used the resulting normalized values as attention scores to quantify the enhancer–gene interaction. The Activity-by-Contact (ABC) score for each enhancer–gene pair was recomputed using the original GitHub implementation (https://github.com/broadinstitute/ABC-Enhancer-Gene-Prediction), using the same enhancer activity and Hi‑C contact inputs that were used to compute attention scores from EPInformer.
Nucleotide contribution and motif discovery
We employed TF-MoDISco-lite36 and Tangermeme37 to analyze TF motifs at putative enhancers based on EPInformer-PE-Activity’s sequence encoder, pre-trained to predict enhancer activity from sequence. TF-MoDISco-lite is a biological motif discovery algorithm that uses attribution scores from a trained deep learning model, in addition to the sequence itself, to guide motif discovery. Tangermeme is a Python package that implements the basic operations necessary to perform sophisticated genomic analyses using machine learning models. It provides a function to perform In-silico Saturation Mutagenesis (ISM) on the model, given a DNA sequence of interest. ISM functions by sequentially substituting each character in a sequence with every other possible character and then assessing the change in the predictive output before and after each substitution. This observed difference is interpreted as a measure of importance or attribution, where a higher magnitude value indicates that the character change has a significant impact on the prediction, thereby suggesting its high importance. Therefore, ISM can be used to uncover blocks of nucleotides corresponding to TF motifs on the putative enhancer sequence.
We utilized Captum (v0.6.0)68 for calculating nucleotide-specific contribution scores in sequences associated with enhancer activity. This process entailed generating 1000 dinucleotide-shuffled variants of each sequence to serve as reference points. Subsequently, the importance scores obtained from DeepLIFT53 for each sequence were combined with their respective one-hot-encoded matrices, yielding the final nucleotide contribution scores.
We utilized TF-MoDISco-lite v2.1.0 (available at https://github.com/jmschrei/tfmodisco-lite) to identify motifs in nucleotide contribution scores across enhancer sequences from the testing set, derived from a 12-fold cross-chromosome validation process. This tool, an efficient version of TF-MoDISco36, was used with its default settings to find seqlet patterns, which were then compared against the JASPAR2024 CORE vertebrates non-redundant database69 using Tomtom54. To analyze nucleotide contributions to enhancer activity predictions accurately, we used In-silico Saturation Mutagenesis (ISM) from Tangermeme v0.2.1, generating attribution scores for each base within targeted regions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genomic datasets analyzed during the current study are available in the ENCODE Project repository (https://www.encodeproject.org/) under the following accession codes: DNase-seq (K562: ENCFF425WDA, ENCFF205FNC; GM12878: ENCFF020WZB, ENCFF729UYK; H1: ENCFF761ZRE; HepG2: ENCFF691HJY; HUVEC: ENCFF091KTX; NHEK: ENCFF117RNM); H3K27ac ChIP-seq (K562: ENCFF600THN, ENCFF232RQF, ENCFF704LGA; GM12878: ENCFF269GKF, ENCFF201OHW; H1: ENCFF693IFG, ENCFF860ABR; HepG2: ENCFF745JCH, ENCFF862NDZ, ENCFF926NHE; HUVEC: ENCFF374DGO, ENCFF609TUB; NHEK: ENCFF051NTC, ENCFF770JWP); and reference Hi-C (ENCFF134PUN [https://www.encodeproject.org/files/ENCFF134PUN]). Additional Hi-C contact matrices are available from the 4D Nucleome Data Portal (https://data.4dnucleome.org/) under accession codes 4DNFITUOMFUQ and 4DNFI1UEG1HD. CAGE data are available from FANTOM5 (https://fantom.gsc.riken.jp/5/sstar/Main_Page) under accession codes CNhs11250 [https://fantom.gsc.riken.jp/5/sstar/FF:10454-106G4] and CNhs12333 [https://fantom.gsc.riken.jp/5/sstar/FF:10823-111C4]. RNA-seq expression profiles are available from the Roadmap Epigenomics Consortium (https://egg2.wustl.edu/roadmap/data/byDataType/rna/expression/57epigenomes.RPKM.pc.gz). The enhancer-gene linkage benchmarking datasets are available in the Engreitz Lab GitHub repositories (https://github.com/EngreitzLab/CRISPR_comparison and https://github.com/EngreitzLab/eQTLEnrichment) and are included in Supplementary Data 2 and 3. The enhancer-gene pair data generated in this study have been deposited in the Zenodo (https://zenodo.org/records/17167181). Source data are provided with this paper.
Code availability
The code used to develop EPInformer, perform the analyses and generate results in this study is publicly available and has been deposited in https://github.com/pinellolab/EPInformer (release version 0.1.1) under the MIT License. The specific version of the code with this publication is archived in Zenodo and is accessible via https://doi.org/10.5281/zenodo.1716718070.
References
Oudelaar, A. M. & Higgs, D. R. The relationship between genome structure and function. Nat. Rev. Genet. 22, 154–168 (2021).
Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020).
Andersson, R. & Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21, 71–87 (2020).
de Boer, C. G. & Taipale, J. Hold out the genome: a roadmap to solving the cis-regulatory code. Nature 625, 41–50 (2024).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012).
Lizio, M. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 22 (2015).
de Hoon, M., Shin, J. W. & Carninci, P. Paradigm shifts in genomics through the FANTOM projects. Mamm. Genome 26, 391–402 (2015).
Reiff, S. B. et al. The 4D nucleome data portal as a resource for searching and visualizing curated nucleomics data. Nat. Commun. 13, 2365 (2022).
Dekker, J. et al. The 4D nucleome project. Nature 549, 219–226 (2017).
Chen, K. M., Wong, A. K., Troyanskaya, O. G. & Zhou, J. A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 54, 940–949 (2022).
Avsec, Ž. et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354–366 (2021).
Gao, Z., Liu, Q., Zeng, W., Jiang, R. & Wong, W. H. EpiGePT: a pretrained transformer-based language model for context-specific human epigenomics. Genome Biol. 25, 310 (2024).
Li, Z. et al. Applications of deep learning in understanding gene regulation. Cell Rep. Methods 3, 100384 (2023).
Zrimec, J. et al. Deep learning suggests that gene expression is encoded in all parts of a co-evolving interacting gene regulatory structure. Nat. Commun. 11, 6141 (2020).
Zhang, Z., Feng, F., Qiu, Y. & Liu, J. A generalizable framework to comprehensively predict epigenome, chromatin organization, and transcriptome. Nucleic Acids Res. 51, 5931–5947 (2023).
Salvatore, M., Horlacher, M., Marsico, A., Winther, O. & Andersson, R. Transfer learning identifies sequence determinants of cell-type specific regulatory element accessibility. NAR Genom. Bioinform. 5, lqad026 (2023).
Seitz, E. E., McCandlish, D. M., Kinney, J. B. & Koo, P. K. Interpreting cis-regulatory mechanisms from genomic deep neural networks using surrogate models. bioRxiv https://doi.org/10.1101/2023.11.14.567120 (2024).
Tan, J. et al. Cell-type-specific prediction of 3D chromatin organization enables high-throughput in silico genetic screening. Nat. Biotechnol. 41, 1140–1150 (2023).
Linder, J., Srivastava, D., Yuan, H., Agarwal, V. & Kelley, D. R. Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation. Nat. Genet. 57, 949–961 (2025).
Zhou, J. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 50, 1171–1179 (2018).
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
Consens, M. E. et al. Transformers and large language models for genomics. Nat. Mach. Intell. 7, 346–362 (2025).
Zhang, S. et al. Applications of transformer-based language models in bioinformatics: a survey. Bioinform. Adv. 3, vbad001 (2023).
Lee, D., Yang, J. & Kim, S. Learning the histone codes with large genomic windows and three-dimensional chromatin interactions using transformer. Nat. Commun. 13, 6678 (2022).
Tang, Z., Toneyan, S. & Koo, P. K. Current approaches to genomic deep learning struggle to fully capture human genetic variation. Nat. Genet. 55, 2021–2022 (2023).
Karollus, A., Mauermeier, T. & Gagneur, J. Current sequence-based models capture gene expression determinants in promoters but mostly ignore distal enhancers. Genome Biol. 24, 56 (2023).
Li, Y. et al. CREaTor: zero-shot cis-regulatory pattern modeling with attention mechanisms. Genome Biol. 24, 266 (2023).
Karbalayghareh, A., Sahin, M. & Leslie, C. S. Chromatin interaction-aware gene regulatory modeling with graph attention networks. Genome Res. 32, 930–944 (2022).
Zhou, Z. et al. DNABERT-2: Efficient foundation model and benchmark for multi-species genome. In Proceedings of the Twelfth International Conference on Learning Representations (ICLR, 2024).
Dalla-Torre, H. et al. Nucleotide transformer: building and evaluating robust foundation models for human genomics. Nat. Methods 1–11, https://doi.org/10.1038/s41592-024-02523-z (2024).
Nguyen, E. et al. HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution. In Advances in Neural Information Processing Systems (NeurIPS, 2023).
Marin, F. I. et al. BEND: Benchmarking DNA language models on biologically meaningful tasks. In Proceedings of the Twelfth International Conference on Learning Representations (ICLR, 2024).
Wang, Y. et al. Genomic touchstone: benchmarking genomic language models in the context of the central dogma. bioRxiv https://doi.org/10.1101/2025.06.25.661622 (2025).
Feng, H. et al. Benchmarking DNA foundation models for genomic and genetic tasks. Nat Commun 16, 10780 (2025).
Shrikumar, A. et al. Technical note on transcription factor motif discovery from importance scores (TF-MoDISco) version 0.5.6.5. Preprint at arXiv https://doi.org/10.48550/arXiv.1811.00416 (2018).
Schreiber, J. tangermeme: A toolkit for understanding cis-regulatory logic using deep learning models. bioRxiv https://doi.org/10.1101/2025.08.08.669296 (2025).
Yuan, H. & Kelley, D. R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022).
Kelley, D. R., Snoek, J. & Rinn, J. L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26, 990–999 (2016).
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015).
Pampari, A. et al. ChromBPNet: Bias Factorized, Base-Resolution Deep Learning Models of Chromatin Accessibility Reveal Cis-Regulatory Sequence Syntax, Transcription Factor Footprints and Regulatory Variants. bioRxiv https://doi.org/10.1101/2024.12.25.630221 (2024).
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (NAACL-HLT, 2019).
Agarwal, V. & Shendure, J. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 31, 107663 (2020).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Kruse, K., Hug, C. B. & Vaquerizas, J. M. FAN-C: a feature-rich framework for the analysis and visualisation of chromosome conformation capture data. Genome Biol. 21, 303 (2020).
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Linder, J., Srivastava, D., Yuan, H., Agarwal, V. & Kelley, D. R. Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation. Nat. Genet. 57, 949–961 (2025).
Koido, M. et al. Prediction of the cell-type-specific transcription of non-coding RNAs from genome sequences via machine learning. Nat. Biomed. Eng. 7, 830–844 (2023).
Nasser, J. et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021).
Gschwind, A. R. et al. An encyclopedia of enhancer-gene regulatory interactions in the human genome. bioRxiv https://doi.org/10.1101/2023.11.09.563812 (2023).
GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Shrikumar, A., Greenside, P. & Kundaje, A. Learning important features through propagating activation differences. In Proceedings of the 34th International Conference on Machine Learning (ICML, 2017).
Gupta, S., Stamatoyannopoulos, J. A., Bailey, T. L. & Noble, W. S. Quantifying similarity between motifs. Genome Biol. 8, R24 (2007).
Schreiber, J. Tomtom-lite: accelerating Tomtom enables large-scale and real-time motif similarity scoring. bioRxiv https://doi.org/10.1101/2025.05.27.656386 (2025).
Doré, L. C. & Crispino, J. D. Transcription factor networks in erythroid cell and megakaryocyte development. Blood 118, 231–239 (2011).
Martin-Rufino, J. D. et al. Transcription factor networks disproportionately enrich for heritability of blood cell phenotypes. Science 388, 52–59 (2025).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Agarwal, V. et al. Massively parallel characterization of transcriptional regulatory elements in three diverse human cell types. Nature, 639, 411–420 (2025)
Fulco, C. P. et al. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354, 769–773 (2016).
De Braekeleer, E. et al. ETV6 fusion genes in hematological malignancies: a review. Leuk. Res. 36, 945–961 (2012).
Bloom, M. et al. ETV6 represses TNF during stress hematopoiesis and regulates HSC self renewal. Blood 140, 2849–2850 (2022).
Kaczynski, J., Cook, T. & Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biol. 4, 206 (2003).
Paszke, A. et al. PyTorch: An imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems (NeurIPS, 2019).
Loshchilov, I. & Hutter, F. Decoupled weight decay regularization. In Proceedings of the Seventh International Conference on Learning Representations (ICLR, 2019).
Girshick, R. Fast R-CNN. In Proceedings of the 2015 IEEE International Conference on Computer Vision (ICCV, 2015).
Moore, J. E. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Miglani, V., Yang, A., Markosyan, A., Garcia-Olano, D. & Kokhlikyan, N. Using Captum to explain generative language models. In Proceedings of the 3rd Workshop for Natural Language Processing Open Source Software (NLP-OSS, 2023).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 52, D174–D182 (2024).
Lin, J. Pinellolab/EPInformer: Release and Storing Also on Zenodo. Zenodo, https://doi.org/10.5281/ZENODO.17167181 (2025).
Acknowledgements
We gratefully acknowledge Simon Senan, Lucas Ferreira DaSilva, and other members of the Pinello Lab for their insightful feedback and discussions. We would also like to thank Maya Sheth and Jesse Engreitz for sharing the data and code for eQTL enrichment analysis. L.P. was partially supported by 1R35HG010717-01 and the Rappaport MGH Research Scholar Award 2024-2029. R.L. was supported by Hong Kong Research Grants Council grants GRF (17113721), TRS (T21-708705/20-N) and the URC fund from HKU.
Author information
Authors and Affiliations
Contributions
L.P. and R.L. conceived the study; L.P. supervised the project. J.L. developed EPInformer and performed computational downstream analysis, including model benchmarking and case studies. J.L., Z.L., Y.Z., and R.L. evaluated the benchmarking results. J.L. and L.P. wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chikashi Terao and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, J., Li, Z., Zhao, Y. et al. EPInformer: scalable and integrative prediction of gene expression from promoter-enhancer sequences with multimodal epigenomic profiles. Nat Commun 17, 3975 (2026). https://doi.org/10.1038/s41467-026-70535-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70535-8
