
Abstract
Despite substantial progress in single-cell screening techniques, antibody (Ab) repertoires still remain enigmatic. Here we show that Ab sequences can be linked to their functionality by using big data obtained from high-throughput sequencing. Using the expansive SARS-CoV-2 pandemic data, we develop an AI-based method to reveal the neutralization potential of Ab repertoires. We employ machine learning to process public 3D structural data of Ab-RBD complexes and create a comprehensive tool, RBD-AIM (https://rbdaim.2a2i.org/), for high-throughput prediction of structural Ab epitopes based on Ab sequence. Using RBD-AIM, we analyze the local big data sources to evaluate the functional biodiversity of native B cell repertoires raised after vaccination and reconstructed in a yeast display system using single-cell microfluidics. This pipeline allows for rapid isolation of neutralizing Abs that promote the survival of transgenic hACE2+ mice in lethal models of SARS-CoV-2 infection. We believe that the AI-guided sequence-functionality link can be successfully employed for further high-throughput discovery of therapeutic Abs and functional analysis of Ab repertoires.
Similar content being viewed by others

Introduction
The SARS-CoV-2 pandemic has raised serious concerns about global biosecurity1,2 and considerably facilitated the development of advanced biotechnological tools3,4,5,6. Meanwhile, as was the case hundreds of years ago, the first as well as the major action to fight against the pandemic was global vaccination7,8,9. The mechanisms underlying the protective effects of vaccination are highly diverse and involve both cell-mediated immunity and antibody-mediated immunity10. However, the human neutralizing antibodies raised against a pathogen per se can ensure the antiviral protection and thus serve the remarkable source for the rapid development of potent and generally safe drug candidates11,12,13. Moreover, the biodiversity of B-cell clones raised against a pathogen provides a functional fingerprint, reflecting its biosafety profile and potential infection severity14. The antigen-Ab interface and the landscape of structural Ab epitopes are critical for developing novel Ab-based therapeutics against infectious diseases15,16, autoimmune disorders17, and cancer18. This makes a comprehensive functional characterization of Ab repertoires a hallmark of modern serology.
Antigen-specific Ab sequences, including the data deposited in public (CoV-AbDab19) or local databases, represent enormous and valuable Big Data sources that lack tools for massive functional characterization20,21. However, since Ab sequences alone provide very limited information on Ab specificity22, uncovering the Ab functionality requires elaborate structural studies23. Most of the existing therapeutic antibodies have complex structural modalities that describe their interactions with antigens24. In-depth functional landscape profiling of Ab epitopes remains challenging. Certain progress has been made using a set of screening technologies and QM/MM applications25,26. In turn, deep mutational scanning (DMS)27,28 is particularly useful as an experimental technique for profiling of antibody epitopes16 and complete mapping of escape mutations29. However, DMS productivity is much less than the amount of Big Data on Ab sequences generated by high-throughput sequencing.
Here, to perform the large-scale structural and functional profiling of Ab repertoires using Ab sequences, we develop a method integrating molecular display and machine learning (Fig. 1). Taking into account the principles behind virus neutralization, we design the AI-guided high-throughput pipeline to predict functional epitopes and create effective Ab-based therapeutics. We choose the receptor-binding domain (RBD) of the SARS-CoV-2 S-protein as a proof-of-concept antigen for epitope prediction since it is one of the main targets of SARS-CoV-2 neutralizing Abs30,31. We create a tool called RBD-AIM (RBD Antibody Interaction Mapper) to bridge the gap between the sequences of RBD-binding Abs and their epitope specificity. The open structural data on RBD-Ab complexes and DMS data are classified using a 12-class model16 and applied to train RBD-AIM for prediction of RBD-Ab complex structures. The AntiBERTy32-based neural network models efficiently predict the class of an epitope, while the AlphaFold33-based refinement further enhances the prediction accuracy and provides the structural model of the RBD-Ab complex. To evaluate the results of AI-guided structural landscape profiling, we employ RBD-AIM to predict the functional properties of the RBD-binding antibodies isolated from the natively paired heavy and light chain antibody repertoires in vaccinated populations. The predictions on Ab epitopes and function are validated by in vitro activity assays, X-ray structural studies, and in vivo studies using a lethal model of SARS-CoV-2 infection. We believe that our approach, as well as AI-based methods in general, will significantly benefit high-throughput sequence-based functional analysis of Ab repertoires and accelerate the development of therapeutic Abs.

Public Big Data sources connecting Ab sequences with their epitopes were used to construct the dataset. A hybrid dataset of 1824 samples, including 450 PDB structures of Ab-RBD complexes supplemented with a functionally annotated DMS dataset16 based on 12-class clustering, was created. The dataset was used to train an AI model, RBD-AIM, for predicting Ab conformational epitopes and providing a genotype-phenotype link between Ab sequence and functionality. To generate local Ab pools, B-cells isolated from vaccinated donors were used for the reconstruction of native B-cell repertoires in a yeast display system. The RBD-binding clones were isolated by double-positive staining with the fluorescently labeled antigen followed by FACS. Local big data were obtained by NGS of the isolated pools. The conformational epitopes of the RBD-specific clones were mapped using RBD-AIM to predict the neutralizing potency and activity spectrum of the RBD-specific Abs based on epitope class and interface Prediction Aligned Error (iPAE) score.
Results
The RBD-AIM model for predicting antibody epitope classes and structural models
To bridge the gap between an Ab sequence and its functional properties, we developed a predictive model called RBD-AIM (Fig. 2). RBD-AIM uses a 12-class model that classifies Ab epitopes by their neutralizing potential and activity spectrum16 (Fig. 2a). Epitope specificity predetermines the basic Ab properties such as ACE2-blocking (Fig. 2b) and neutralization potential (Fig. 2c). RBD-AIM utilizes an ensemble of AI models, including Neural Network classificator (NN-model) and a zero-shot structure prediction model (Fig. 2d). To train and evaluate the models, we applied two datasets: (i) a precise PDB dataset of 450 samples based only on well-characterized Ab-RBD complexes and (ii) a hybrid Ab dataset of 1824 samples having the PDB dataset supplemented with a functionally annotated DMS datset16 based on 12-class clustering. Hence, each epitope class was represented by 73–231 samples in the PDB + DMS dataset (Fig. S1 and Table S11).

a A visual representation of the 12-class model used. All available PDB structures were clustered as following16. The centers of mass belonging to the heavy atoms of RBD in contact with Ab (distance <4 Å) and Ab heavy atoms in contact with RBD, the centers of Cɑ atoms in Ab variable fragments are shown with spheres connected with lines and colored according to their class specificity. ACE2-blocking b and SARS-CoV-2 neutralizing properties c that are available for the PDB + DMS dataset. Data are presented as individual Ab characteristics reported, median values across the class, and interquartile ranges are indicated. d A schematic representation of the RBD-AIM computational pipeline. RBD-AIM includes a trained model based on the AntiBERTy model to predict the epitope class from the input light and heavy chains of an antibody. The model output was used to reconstruct the structural model and adjust the epitope class, particularly in cases when the AlphaFold output metrics indicate a lower iPAE (iPAE<13.5). e The confusion matrix for the predicted antibodies across epitope classes calculated using the RBD-AIM pipeline. f iPAE score predictive accuracy. Scatter plot showing the relationship between Log10(RMSD) and iPAE. Points represent individual observations, the solid line shows the fitted linear regression, and the shaded area indicates the 95% confidence interval, vertical dashed line shows optimal threshold based on ROC AUC plot. ROC curve summarizes iPAE score performance to classify structures with high and low RMSD. g Scatter plot showing the relationship between iPAE and the binding selectivity across RBDs. Points represent individual observations, the solid line shows the fitted linear regression, and the shaded area indicates the 95% confidence interval. h Performance metrics of the RBD-AIM (bold) and homology-based models. The general MCC metrics were calculated both for the full dataset and the PDB dataset; other performance metrics were calculated for the full dataset. Precision and F1 score are weighted by epitope class sizes. Source data are provided as a Source data file.
For the NN-model backbone, we evaluated two comprehensive protein language models: AntiBERTy32, pretrained on a large corpus of antibody sequences, and ESM-234, a masked-language model trained on diverse protein sequences. In each case a linear classification layer was added, using either the concatenated embeddings of the classification (CLS) tokens from the full heavy- and light-chain sequences or the averaged embeddings of the CDR-only regions, and the network was trained to predict the epitope class of the input antibody. The performance metrics of the ESM-2-based models were consistently lower than those of AntiBERTy (Supplementary Note, Table S1), underscoring the effectiveness of antibody-specific pretraining for this task. An ensemble that averages the logits of the AntiBERTy_CLS and AntiBERTy_CDR (AntiBERTy_CDR_CLS) models was therefore selected as the final NN-model. This network outperformed a baseline that assigns classes by Levenshtein-distance similarity to the training set (Table S1 and Fig. S2), demonstrating that it captures predictive features beyond simple sequence homology.
To ensure that the model’s predictive power is not driven by data biases, we conducted several validation experiments. We selected the top 5%, 10%, and 15% most dissimilar samples per class and evaluated performance on these sets. The results provide evidence that RBD-AIM captures predictive features beyond simple sequence homology, even on the most divergent test samples (Table S2). The drop in performance is expected, as the limited size of the training set prevents the model from generalizing well to out-of-distribution samples.
Predicting the structural model of antibody-antigen complexes is a challenging task with low predictive power. However, in case of the RBD domain, there are numerous available 3D RBD-antibody complexes that can be used as templates for predicting the final structures. We selected the most similar antibody within the predicted epitope class by NN-model or the entire dataset as a template reference model, or constructed Ab separately using DeepAb tool35. This structure was further used as an initial template for the AlphaFold2 MSA-free inference36 (see Supplementary Note). The accuracy of the structural prediction was estimated using the confidence metrics of the model, specifically, the average predictive alignment error (PAE) between the interacting subunits (iPAE). The iPAE score had high discriminative power to distinguish between true structures and false predictions (Fig. 2f). The iPAE score allowed for efficient identification of the antibodies with high RMSD (root-mean-square deviation > 2.5 Å) that corresponded to the incorrectly predicted structures, achieving the ROC AUC of 0.91. Moreover, within our settings, the iPAE score reflected variation in the estimation of the Ab binding affinity towards different RBD strains (Fig. 2g). The calculated Pearson correlation R was 0.52, which implies the advantage of this metric for predicting the escape mutations. The best performance metrics with MCC 0.591 are shown by the model where templates are chosen with the predicted NN-class (Table S3). Moreover, our model outperforms other popular structure-prediction models: AlphaFold3, Chai-1, and AlphaFold-Multimer (Table S4 and Fig. 2h).
In practice and functional interpretation, prediction of amino acid contacts and structure quality plays the key role. To address this issue, we calculate Fnat (Fraction of correctly predicted interfacial contacts), RMSD for antibody and interface, and DockQ metrics for successfully predicted complexes for the top two structure models: our structure model and the Chai-1 model, where the class predicted by the NN-model was used as pocket restraint (Chai-1r). These metrics were calculated only for successfully predicted complexes with high quality, according to the model manual: iPAE <13.5 for our model and iPTM > 0.4 for Chai-1r model (Supplementary Note, Table S5). Our model generally outperformed the Chai1r model in terms of both Fnat and DockQ scores across most epitope classes. For example, class A achieved a DockQ of 0.689 and Fnat of 0.594 with our model, compared to 0.575 and 0.355 for Chai-1r. Class B also showed superior performance (DockQ: 0.665, Fnat: 0.509) versus Chai-1r (DockQ: 0.567, Fnat: 0.404). Notable improvements were observed in difficult-to-predict epitope classes such as D1 and D2, with our model achieving DockQ scores of 0.708 and 0.774, respectively, indicating highly accurate interaction modeling. Class E3 and some rare epitopes (e.g., E21) still pose challenges due to limited representation in the training data, leading to lower success rates or lack of confident structure generation in both methods. The accuracy of the epitope-side contact prediction (Fnat_e, fraction of correctly predicted RBD contact residues) was consistently high in our model across most classes, suggesting reliable epitope identification that aligns with experimental structures (Table S5).
Importantly, not all complexes could be successfully predicted using structural modeling, as defined by an iPAE below 13.5 (Fig. 2f). However, for those complexes that met this confidence criterion, the accuracy of epitope class prediction was higher compared to neural network NN–models (Table S6). Thus, as a final model called RBD-AIM, we proposed an ensemble of NN- and structure models, where the final decision is based on the 3D predicted class when the iPAE is below 13.5 and on the NN-model classification in other cases. The ensemble models demonstrated superior predictive accuracy compared to the individual models or the baseline model that relies on the sequence homology-based prediction (Fig. 2h).
The RBD-AIM model successfully assigned the epitope class to a target sequence for most analyzed antibodies during the cross-validation evaluation (Fig. 2e). The best prediction metrics were obtained for A, B, D2, E1, E21, and F3-classes with >75% of correctly assigned epitopes. Lower precision observed for the C (53%) and E3 (52%) classes is likely to arise from the high variability in the binding modes of antibodies for these epitopes. However, most misclassifications were detected in case of adjacent classes, which mitigates their impact on the functional prediction quality. While the 12-class model provides us with a more detailed and interpretable set of results, even higher performance metrics could be achieved with a smaller number of classes (Table S7). However, we preferred to use the 12-class model since it clearly demonstrates the functional difference between all of the classes16.
A web interface (https://rbdaim.2a2i.org/) ensures free access to our pipeline and efficient prediction of antibody-RBD interactions. RBD-AIM represents a significant advancement in linking Ab sequences to their functional properties and structural models, providing a valuable tool for antibody research and development.
Reconstitution of native B cell repertoires by molecular display
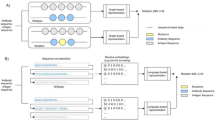
We used B cell stocks as a local big data source to evaluate RBD-AIM efficiency. B cell repertoires were raised by vaccination with different COVID-19 vaccines: mRNA (Pfizer–BioNTech) and adenoviral vector (Sputnik V) based on wild-type (WT) SARS-CoV-2 S-protein. Droplet microfluidics allowed the preservation of native combinations of immunoglobulin variable heavy (VH) and light (VL) chains during single-cell mRNA capture37 and emulsion RT-PCR38 (Fig. 3a). The resulting antibody scFv fragments were anchored to Pichia pastoris yeast cell wall via fusion with α-agglutinin anchor subunit Aga1p from Saccharomyces cerevisiae to provide a self-renewable source of B cell repertoires39. NGS sequencing of scFv libraries demonstrated that microfluidic pairing preserved the native highly degenerated diversity of VH-VL combinations (Fig. 3b). In contrast, combinatorial VH-VL pairing resulted in random VH-VL combinations largely including inactive artifacts, leading to the loss of rare RBD-specific clones. Therefore, employing the natively paired libraries increases the actual diversity of Ab clones by several orders of magnitude compared to conventional combinatorial scFv libraries (Fig. 3c). In line with the previous studies40, we observed a rapid degeneration in diversity of phage display libraries (Fig. S3). Yeast display resulted in a higher diversity of selected RBD-specific clones than phage display. For instance, the clones with complex CDR structures, such as functionally significant cysteine-rich clone T11, were lost during phage display.

a A general outline of the reconstruction of B-cell repertoires using yeast surface display libraries. Droplet microfluidics was used for lysis of individual B-cells and capturing their mRNA with oligo-dT beads in isolated droplets. The resulting mRNA replicas were released from the droplets and processed for emulsion RT-PCR (eRT-PCR). Single beads were further encapsulated into droplets for eRT-PCR amplification of variable fragments of the heavy (VH) and light (VL) immunoglobulin sequences linked by the serine-glycine (G4S)3 linker sequence. The resulting DNA library of natively paired VH-VL sequences was cloned into a vector to produce scFv fragments anchored to the P. pastoris yeast cells or phage display. b A Sankey diagram illustrating representative pairing of the immunoglobulin heavy (H) and light (L) chains in yeast cell libraries (Library) and the pools enriched for RBD binding obtained via microfluidic (Native) and random pairing. RBD+ and RBD+++ indicate the pools selected using 10 nM and 0.3 nM tRBD, respectively. The bar size denotes clone prevalence. The color indicates the percentage of clone heavy (H) and light (L) chains in the library prior to selection. c A theoretical estimate of the library size (Representativity) sufficient to reliably cover a least frequent clone from the given number of the most abundant clones (Diversity), α = 0.05 (See Supplementary Note for details). Frequency distribution of clones was obtained using NGS of the libraries from human donors, n = 11. Square means and ranges are indicated with blue (Native) and red (Random). d The tetrameric complexes of biotinylated RBD with fluorescently labeled streptavidins were used for double-positive staining and FACS of the RBD-binding clones (for more details, see Fig. S21). The first round of RBD-binding clone selection included double-positive staining with 10 nM tRBDs, followed by the isolation of RBD-positive clones with FACS (Fig. S4). During the following selection rounds, staining with 10 nM tRBDs was repeated, and more stringent conditions were applied: tRBD concentration was reduced to 0.3 nM, and 60 nM of soluble ACE2 was added for competition for tRBD binding. Gating strategy: + – 10 nM tRBD, ++ –0.3 nM tRBD, +++ –0.3 nM tRBD top clones; ACE2 –60 nM of soluble ACE2 is added. e The distribution of the most prevalent clones in the libraries before (−) or after (+) vaccination (Vac.), and cell populations selected for RBD-binding according to the plot d. Representative clones from different patients, denoted as T, D, B, L, are separated. The heatmap shows the percentage of individual clones in the samples evaluated by NGS sequencing. Data are represented as means of three technical replicates. Source data are provided as a Source data file.
Isolation of RBD-specific Abs
We used the double-positive staining with RBD-fluorophore tetramers41 to isolate RBD-specific Ab pools from B cell repertoires raised by wild-type (WT) SARS-CoV-2 mRNA and adenoviral vaccines and collected in April 2021, before the delta (B.1.617.2) and omicron (BA.1) waves (Fig. S4). Using more stringent conditions of yeast staining allowed for discriminating the most potent RBD-binding Abs and identifying the Ab clones that compete with ACE2 for the ACE2-binding epitope (Fig. 3d). The B cell repertoires demonstrated high plasticity. The major clones in Ab repertoires before vaccination were replaced with other clones after vaccination and boosting (Fig. 3e). RBD-specific clones were not the most prevalent accounting for 0.02-0.5% of all clones. They were efficiently enriched even after the first round of selection. A panel of particularly potent top-binding RBD-specific clones was selected using subnanomolar antigen concentrations (Fig. S5). A-class Abs (T3 and D1), directly blocking ACE2-RBD interaction, demonstrated decreased enrichment efficacy in the presence of soluble ACE2 (Fig. 3e). Most Abs demonstrated subpicomolar KD with a very slow dissociation rate (kd <10−6s−1) (Fig. S6), while some of them (D16, L1) formed highly stable Ab-RBD complexes (Fig. S7). Although both mRNA-based and adenoviral vaccines raised RBD-specific clones (Supplementary Data 1), mRNA-based vaccination resulted in a higher number of top-binding RBD-specific clones (Fig. S8).
Functional profiling of Ab repertoires using RBD-AIM
Sequences that were repeatedly enriched in RBD-specific pools by two rounds of FACS with 10 nM tRBD staining and one round of FACS with 0.3 nM tRBD staining were suggested as RBD-positive. 30 sequences were considered RBD-positive by these rather strict criteria (Supplementary Note, Supplementary Data 1). Typically, we obtained 5.7 ± 3.2 and 3.2 ± 1.7 (Mean, SD) RBD-positive antibody sequences from each donor vaccinated by mRNA-based (Pfizer–BioNTech) or adenovirus-based (Sputnik V) vaccine. We scrutinized local RBD-specific Ab repertoires using RBD-AIM to identify immunodominant epitopes raised by different vaccines. RBD-AIM showed no significant difference (Fig. S9, P value = 0.62, chi-square test) between the immunodominant epitopes of mRNA and adenoviral vaccines (Fig. 4a). A- and C-class antibodies were the most prevalent among Abs (Fig. 4b). Their function is mediated by a well-known direct mechanism that relies on blocking ACE2 (Fig. 2b), which implies their high neutralization activity towards SARS-CoV-2 (Fig. 2c). Although E22-class antibodies were one of the least presented in the PDB database, we repeatedly observed them in the isolated Ab pools, which indicates their abundance in repertoires raised by vaccination (Fig. 4b). Abs of this class are of particular interest since they have a broader activity spectrum and their neutralizing activity is far from homogeneous (Fig. 2c). Moreover, their neutralization mechanism does not involve direct blockade of ACE2 binding to RBD (Fig. 2b).

a The heatmap illustrates the abundance of the Ab-RBD contacts in Abs raised against adenoviral (Sputnik) and mRNA (Pfizer-BioNTech) vaccines. b The Ab epitopes predicted by RBD-AIM. The centers of mass of Ab heavy atoms interacting with RBD and Cɑ atoms of Ab variable fragments are shown as spheres connected with lines and colored according to their class specificity. The functional properties of Abs from the donors vaccinated with Sputnik (V, L, B) or Pfizer-BioNTech (T, D, TA) were studied for the most abundant clones in the RBD-specific pools. c Strain specificity of Abs predicted by their Ab-RBD structures. The red color (loss of affinity) indicates the number of amino acid substitutions inside the Ab structural epitopes in various SARS-CoV-2 strains, compared to the original (WT) immunogen. Mutations inside the predicted binding epitope were considered to be unfavorable. d The experimental binding spectrum of Abs towards various RBD mutants. Color corresponds to the KD value towards WT RBD, alpha B.1.1.7 (α), gamma P.1 (γ), delta B.1.617.2 (δ), omicron BA.1, and BA.5. e The neutralizing efficiency (IC50, nM) of Abs against pseudoviruses. No Ab exerted any neutralizing activity against BA.5 (IC50 > 50 nM). d, e Data are represented as means of three biological replicates. f The experimental timeline for studying the therapeutic potential of selected neutralizing Abs. Transgenic hACE2+ mice were challenged with lethal doses of WT and omicron BA.1 SARS-CoV-2. On the following day, Abs were i.p. administered. g, h Survival of transgenic hACE2+ mice was monitored for 12–14 days until body weight recovery. g The ACE2-blocking (C-class, D16) and h non-blocking (E221-class, T11) Abs were used. Ab P4A1 was used as a control therapeutic Ab, PBS was used as a negative control without therapy. n = 5–8 mice per group. Two-tailed P values were determined by the Gehan-Breslow-Wilcoxon test. NS not significant. Source data are provided as a Source data file.
Neutralization efficiency of an antibody was estimated by the known characteristic properties of the epitope class (Fig. 2c) it belongs to. Accordingly, classes were subdivided into having high (A, B, C, D1, D2, E1, F2, and F3), medium (E21), mediocre (E22), and low (E3 and F1) neutralizing potential. The epitope class and antigen-binding interface were predicted by RBD-AIM. The number of unfavorable escape mutations in a particular strain regarding the vaccine variant under consideration (WT in our case) was used as a metric of predicted specificity. RBD-AIM estimations proposed high neutralizing efficiency and a narrow activity spectrum for the A- (T3, D1) and C-class (TA9, V1, D16) antibodies (Fig. 4c). On the contrary, it predicted a reduced neutralizing efficiency and broader activity spectrum for the E22-class (L1, B2, T11, T15) and, to a greater extent, for the F1-class (TA4, N1) antibodies (Fig. 4c). The estimated functional features of Abs were validated using a representative panel of RBD-specific clones (Fig. 4d). Abs had an expected cross-competition profile and ACE2-blocking properties (Fig. S10) which is in line with the RBD-AIM prediction. A- and C-class Abs were characterized by a narrow activity spectrum and potent neutralizing activity towards WT SARS-CoV-2 pseudoviruses, whereas they proved inactive against the BA.1 strain (Fig. 4d, e). The E22 antibodies clustered into two subclasses, E221 (L1, B2, T11), and E222 (T15), depending on their activity spectrum and neutralization efficiency (Fig. 4d, e). In contrast to the ACE2-blocking Abs, E221 antibodies exerted a considerable neutralizing activity against BA.1. Despite their high affinity and cross-reactivity towards various RBD variants (Fig. 4d), E222 and F1-class (TA4, N1) Abs exhibited especially low neutralizing activity (Fig. 4e). Reduced neutralizing potential of these Abs was tightly associated with a long distance between their epitopes and ACE2 binding site11,16.
Therapeutic potential of representative ACE2-blocking (D16) and non-ACE2-blocking E221-class (T11) antibodies was evaluated using a lethal model of SARS-CoV-2 infection (Fig. 4f-h). A single administration of a 3 mg/kg dose of Abs cured all animals, in line with the neutralizing profile of selected Abs (Fig. 4g, h). The high therapeutic potential of the selected Abs confirms that the proposed pipeline combining the identification of native potent binders via molecular display followed by AI-based classification provides relevant therapeutic candidates. We could also speculate that the identified Abs have neutralizing potential sufficient to provide protective activity in vaccinated donors.
Structural validation of RBD-AIM predictions
For a clear validation of the RBD-AIM predictions, we obtained the crystal structures of model A (D1), E221 (T11), and F1-class (N1) Abs (Fig. 5a–c), showing no cross competition (Fig. S10) and covering various parts of RBD. RBD-AIM precisely predicted the D1 (RMSD = 2.24 Å) and N1 (RMSD = 3.64 Å) Ab-RBD complex structures (Fig. 5a, c) and Ab-RBD contact interfaces (Fig. S11). Despite the prediction accuracy was moderate (RMSD = 20.8 Å) for the T11-RBD complex, it successfully predicted the center of mass of the T11 epitope and the most important features, such as binding to the “big” RBD T470-P491 loop (BL) (Fig. S11). According to our previous functional studies, D1 binds to the ACE2-binding interface, in contrast to T11 and N1 (Fig. 5d). The Omicron BA.1 mutations were also located inside D1 but not on the T11 or N1 RBD-binding interfaces, which explains a broader cross-reactivity of E221-class Abs. Moreover, E221 and E222-class Abs have different binding modes, which account for their distinct functional properties (Fig. 5e, f). Unlike E222-class, E221-class binds the upper part of the RBD interface, including BL. The alternative chain orientation in T11 and T15 might also serve as a hallmark trait that allows discriminating between E221 and E222 classes.

a–c The crystal structures of the A (D1, PDB ID: 8WFH), E221 (T11, PDB ID: 8WFM), and F1-class (N1, PDB ID: 9JEB) Ab-RBD complexes were aligned with the Ab-RBD complexes predicted by RBD-AIM. RBD is shown in gray, the amino acid residues mutated in the omicron BA.1 variant are shown in green. The Ab fragments in crystal structures are colored with red (D1), cyan (T11), and violet (N1). Predicted Ab structures are shown in white. d The ACE2-RBD complex (PDB ID: 7WBP) with the highlighted ACE2-binding interface. e, f Representative E221 and E222-class binding modes. The crystal structures of the T11-RBD e and T15-RBD models f demonstrating the RBD-binding interfaces and orientation of heavy (H) and light (L) chains, are shown in blue and cyan, respectively.
Mechanistic details of the E221-class neutralizing activity
RBD-AIM allowed for outlining the E221-class Abs from the whole class E22. To further explore the mechanistic details underlying their function, we obtained a comprehensive overview of T11-RBD interactions by X-ray crystallography and molecular modeling (Fig. 6). The T11-binding interface spanned across the vast RBD surface on the opposite side of the ACE2-binding interface (Fig. 6a–c). The structure model of the T11-S protein complex demonstrated that the binding of E221-class Abs occurs only with the open S-protein conformation (Fig. 6d and S12). The neutralizing activity of E221-class Abs is mediated either by transforming the ACE2-binding interface or altering RBD mobility, which is essential for ACE2-RBD complex formation. The binding between T11 and RBD did not prevent the ACE2-RBD complex from being formed (Fig. S13). Moreover, both X-ray crystallography and molecular modeling did not show dramatic differences in RBD conformation after T11 binding (Fig. 6a, e). On the contrary, we observed a drastic decrease in BL mobility both for ACE2-RBD and T11-RBD complexes (Fig. 6f, g). Therefore, we conclude that T11 “freezes” the conformational dynamics of RBD, which is essential for efficient ACE2 binding. The E484 residue, which is frequently mutated in SARS-CoV-2 strains (including the E484A/K mutation in all omicron variants), is the hotspot of RBD plasticity (Fig. 6F). The single E484A mutation in the WT RBD reduced the T11 dissociation constant (kd) by more than two orders (Fig. S5), increased RBD mobility (Fig. S14), and restored the subpicomolar affinity of T11 to RBD, which is typical of the highly mutated omicron BA.1 variant (Fig. S15).

a The crystal structure of the T11-RBD complex and the detailed T11-RBD interface, including heavy b and light c chain interfaces. The amino acid residues mutated in the omicron BA.1 variant are shown in green. d The structure model of the T11-S protein complex. The cryo-EM structure of the SARS-CoV-2 Omicron spike protein with ACE2 (PDB ID: 7WK4) and T11-RBD was aligned with RBD. The S protein monomers are shown in shades of gray, ACE2 is shown in pink, the T11 light chain (L) is cyan, and the T11 heavy chain (H) is blue. e–g Molecular modeling of T11-RBD interaction. e Conformational mobility of the loops in isolated RBD, T11-RBD, and ACE2-RBD complexes. Gray: isolated RBD; blue: T11-RBD; pink: ACE2-RBD. BL: the big loop. For each system, RBD backbone coordinates were averaged over the calculated trajectories. Three replicas were used. RBD residue mobility in the ACE2-RBD f and T11-RBD g complexes. The absolute value of dRMSF is reflected by the thickness of RBD cartoon structure, while residues are colored according to the dRMSF value. The E484 residue is labeled to indicate the position with the greatest decrease in mobility in the ACE2-RBD complex. Source data are provided as a Source data file.
Discussion
Harnessing AI to link genotype to phenotype is an essential step towards the next-level biotechnology. Here, we have integrated AI-based prediction with molecular display for a detailed functional description of native repertoires. RBD-AIM significantly outperformed the existing conventional approaches to predict Ab-RBD complex structures, such as a method based on the AlphaFold modeling. Native B cell repertoires were reconstructed in a yeast display system using the single-cell microfluidics to provide the antigen-specific pools further functionally characterized by RBD-AIM. We developed a streamlined pipeline that provides structural landscapes of Abs raised by vaccination or infection. Molecular display enables the identification of potent antigen binders, and their functionality can be estimated based on the structures of antibody-antigen complexes, facilitating the development of therapeutic Abs. In this case, the primary objective of RBD-AIM is to predict epitope classes, which then serve as templates for structural modeling of antibody-antigen complexes. Moreover, the functionality of Abs from a particular class is similar and well-characterized16, which was used to predict the functionality of Abs identified by molecular display. Structural modeling enables the refinement of antibody-antigen interactions, e.g., prediction of sensitivity to escape mutation, which was also realized in the RBD-AIM model. While similar goals could be achieved through sequence or structure alignment with known antibodies, RBD-AIM enhances the process by incorporating epitope class-specific templates, providing a more targeted approach to structural predictions and outperforming homology-based methods: AlphaFold3, Chai-1, and AlphaFold-Multimer (Table S4 and Fig. 2h).
Although RBD-AIM does not use Ab framework region sequences for training, we observed a strong association of certain germlines with conformational epitopes (Fig. S16), including IGHV 3-53 and IGHV 3-66 (A-class). The probability of generation as well as occurrence frequency of potent ACE2-blocking A-class antibodies have, are significantly higher compared to the other classes (Fig. S17). Moreover, they are efficiently predicted by RBD-AIM, which makes them promising candidates for the rapid development of therapeutic neutralizing Abs. Meanwhile, identifying Abs with a broad activity spectrum (E1 and E21) or an altered mode of action (E221) provides more universal drug candidates or therapeutic options.
RBD-AIM is a powerful AI-based tool for the detailed epitope characterization of vast RBD-binding repertoires. However, it should be stressed that its efficiency entirely relies on the assumption that an Ab sequence under investigation is a validated RBD binder. While AI-based epitope classification can provide insights into an antibody’s anticipated functional profile, it alone does not directly aid in identifying potent antibody sequences. For this issue, high-throughput technologies of selection of potent antigen binders should be applied. Hence, the developed pipeline is based on a synergy between high-throughput identification of potent antigen binders via molecular display and AI-based epitope prediction. In this case, high-affinity Ab candidates selected by yeast display are further curated by AI to prioritize Abs having high neutralizing potential (A, B, C, D1, D2, E1, F2, and F3) or improved cross-reactivity (E1, E21, E221). This methodology we refer to as “AI-guided structural landscape profiling,” may contribute to epitope-specific therapeutic antibody development or rationalize the design of Ab cocktails, having improved virus neutralization potency and cross-reactivity.
In this study, we demonstrated the utility of AI-guided pipeline using the SARS-CoV-2 RBD as a well-characterized benchmark antigen with abundant experimental data. To assess model robustness and data dependency, we evaluated how predictive performance, measured by Matthews correlation coefficient (MCC), varied with different training set sizes, ranging from 90% to 10% of the full dataset (Table S8). For reference, simple homology-based classification methods yielded MCCs of 0.544. In contrast, our deep learning approach outperformed these baselines, reaching an MCC of 0.621 when trained on approximately 1600 samples. We observed a drop in performance when the model was tested on samples most dissimilar from the training set, highlighting a limitation in generalizing to out-of-distribution data. Nevertheless, the model still outperformed homology-based predictions under these challenging conditions.
To explore the applicability of RBD-AIM beyond SARS-CoV-2, we tested the model on antibodies targeting SARS-CoV-1. We analyzed 14 antibody–antigen complexes from the PDB and found that in 9 of these cases, the predicted epitope class was correct, with structural alignment metrics indicating high-quality predictions (LRMS < 1.34 Å, DockQ ranging from 0.450 to 0.560), suggesting that even the existing RBD-AIM model may be relevant to some extent for further SARS-CoV outbreaks. Moreover, to investigate the broader utility of our approach across viral antigens, we conducted a preliminary case study using the H3N2 strain of influenza A. We assembled a dataset comprising 300 antibody–antigen complexes from the PDB and IEDB, and trained the model following the same protocol as RBD-AIM. Antibodies were grouped into six epitope classes based on structural criteria42. The model achieved an overall MCC of 0.204, which improved to 0.302 when considering only epitope classes with more than 20 training samples (Supplementary Note, Table S9). This result suggests that, while initial performance is modest, RBD-AIM can be extended to other pathogens once a critical threshold of training data is met. Our analysis has quantitatively defined this threshold, providing a clear benchmark for future applications. Crucially, generating the necessary data is viably achieved not through low-throughput structural methods, but via high-throughput deep mutational scanning (DMS)—precisely the approach we used for validation—which serves as a scalable source of training data to power the AI-guided pipeline for virtually any target of interest. Similarly to DMS, other high-throughput techniques, including cross-linking mass spectrometry (XL-MS) and cryo-EM epitope mapping, could also be further applied for data supplementation or accelerated generation of training datasets aimed at the creation of even more smart and precise AI-based tools linking sequence with functionality.
Hence, we believe that a combination of molecular display and AI-guided functional analysis represents a next step in the evolution of display technologies, since it provides an example of how molecular display technologies could be further improved by AI to be smart enough to find not only potent but proper binders. With current advances in structural biology and computational science, it could be successfully applied to other antigens and serve as a cutting-edge approach for high-throughput functional mining of Abs.
Methods
The research complies with all relevant ethical regulations and approved by the ethical committee of the Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, State Key Laboratory of Medicinal Chemical Biology and College of Life Sciences, and Scripps Research Institute.
Database for RBD-AIM model training
The Ab database for AI-model training contains the following data: Ab name, amino acid sequences of light and heavy chains, amino acid sequences of CDRs, germline information, and an epitope class. For antibodies with known 3D structure of the Ab-RBD complex, the following data were included: PDB ID, chain ID of RBD, heavy and light chains, coordinates of 3 points for graphical representation of Abs below described, and a list of residues in contact with RBD. The data above was extracted from the Cov-AbDab database19 (version from 8th Feb, 2024) and supplemented with PDB data43 (version dated 1st Mar, 2025) for all available entries of human antibodies against SARS-CoV-2 RBD. Positions of CDRs and germlines were calculated using ANARCI utilities44 with the IMGT Chothia scheme. Only Abs with complete CDR sequences were used. If a PDB file contained a multimer structure of the Ab-RBD complex, the monomer with the maximal number of contacts between Ab and RBD was selected. To assess the similarity of Abs binding modes, we first superimposed the reference RBD structure with every Ab-RBD complex using the Combinatorial Extension algorithm45. Then, for each complex, we calculated the Cartesian coordinates of three points describing the Ab position relative to RBD: the center of RBD heavy atoms in contact (i.e., within 4 Å) with Ab heavy atoms; the center of Ab heavy atoms in contact with RBD heavy atoms; the center of Cɑ-atoms of variable fragments of both light and heavy chains (Cɑ-center). Finally, the classification data from a Supplementary Table 2 from16, which contained amino acid sequences and epitope classes of 1640 Abs, was used to classify epitopes in our database and expand the dataset. Entries with identical CDRs or having one substitution in CDRs were considered duplicates and removed. Then, the geometric centers of all three points, describing the Ab position relative to the RBD, were calculated. For each class, the mean of coordinates was calculated after discarding outliers. For Abs from the PDB database, classes were assigned based on the nearest class. The resulting classes of Abs were corrected according to structural data if latter conflicted with DMS data (supplementary table 2 from16). For more detailed information, please refer to Supplementary Note and Table S11.
AI model for the conformational epitope prediction (RBD-AIM)
The epitope prediction based on antibody sequences was considered as a classification task that involved two sequences: the heavy and light chains. For this purpose, several models were developed.
The first set of models utilized a large neural network transformer-based model pre-trained on a large dataset of unlabeled sequences of heavy and light antibody sequences. For this purpose, AntiBERTy32 and ESM-234 models were used as baseline models. Both models combine sequence representations of heavy and light chains into a single feature used for prediction and were created in two versions: utilized concatenated CLS token representations from the heavy and light Fab chains, concatenate average representations from the CDR regions of antibody light and heavy chains (See Supplementary Note for details). The second set of models is complex structure prediction models. We improved standard AlphaFold2 pipeline for our purpose. The model takes RBD, heavy, and light antibody sequences as input. Then choose the template for the Ab structure and combine it with the RBD template to construct the final Ab-RBD complex. We considered three variants of Ab template selection: (i) comparison based on Levenshtein distance, where the nearest complex was selected as the template; (ii) comparison based on Levenshtein distance, where the nearest complex within the class predicted by NN-model was selected as the template; (iii) construction of Ab templates by DeepAb tool35, which specialized on antibody structure predictions (See Supplementary Note for details).
The final RBD-AIM model is an ensemble of NN- and structure models, called RBD-AIM. The final decision of RBD-AIM is based on the 3D predicted class when the iPAE is below 13.5 and on the NN-model classification in other cases (See Supplementary Note for details).
To ensure robust evaluation and mitigate data leakage or class underrepresentation, we employed 10-fold stratified cross-validation, which involved splitting the dataset into 10 non-intersecting subsets (Supplementary Table S12). Each subset was further used as a test set, while the rest were used as a train set. This approach preserves class proportions across all folds, ensuring each class is proportionally represented in both training and test sets. Performance metrics were computed on aggregated predictions.
We considered five performance metrics: Accuracy, weighted Precision, weighted Recall, weighted F1-score, and Matthews Correlation Coefficient (MCC). Accuracy was calculated as
where TP, TN, FP, and FN denote true positives, true negatives, false positives, and false negatives, respectively. To account for class imbalance, weighted forms of Precision, Recall, and F1-score were used. Weighted Precision was defined as
where wi represents the proportion of instances in epitope class i, and C is the total number of classes. Weighted Recall was computed as
and the weighted F1-score as
Finally, MCC, which provides a balanced measure even in the presence of class imbalance, was calculated as
The final RBD-AIM model was trained on the full dataset. Training was conducted utilizing an NVIDIA A100 GPU, which required approximately 12 h. The trained model is hosted on a publicly accessible web server designed to provide users with an interface for performing model inference.
Vaccinated donors
All donors were confirmed to be COVID-19 negative according to the quantitative reverse transcription PCR (RT-qPCR). Prior to vaccination, no nucleocapsid (N)- and receptor-binding domain (RBD)-specific IgGs were detected in the sera of naïve individuals without COVID-19 symptoms. Vaccinated donors received two doses of adenoviral Gam-COVID-Vac (Sputnik V) or mRNA Pfizer-BioNTech BNT162b2 vaccine with a 21-day interval. None of the volunteers had experienced serious adverse effects after vaccination. The study protocol was approved by the independent Ethic Committee of Central Clinical Hospital of the Russian Academy of Sciences (protocol #143). The study was conducted according to the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki 1964, along with its later amendments. All participants provided written informed consent. Plasma samples were centrifuged for 10 min at 1000 × g to clarify the supernatant before use. Venous blood for B cell isolation was collected into EDTA Vacutainers (BD, USA). PBMC were isolated with Ficoll gradient centrifugation, and B cells were enriched with Dynabeads Untouched Human B Cell kit (negative selection, Invitrogen, Thermo Fisher) as described previously46.
Generation of natively paired VH-VL antibody repertoires
Ig repertoires with natively paired variable fragments of heavy and light chains were obtained using a microfluidic platform similar to the previous workflow47. Briefly, the OB1-Mk3 piezoelectric pressure controller (Elveflow, France) was utilized to generate W/O emulsion with three independent streams: (i) B cell suspension, (ii) the suspension of oligo d(T)25 magnetic beads in lysis buffer (100 mM Tris pH 7.5, 500 mM LiCl, 10 mM EDTA, 1% lithium dodecyl sulfate, 5 mM dithiothreitol), and (iii) Pico-Surf 2% (w/w) in Novec™ 7500 (Sphere Fluidics, UK). Custom hydrophobized microfluidic chips with flow-focusing geometry and square 60 µm orifice39 were used with 6.5:6.5:7 µL/min low rates for (i):(ii):(iii) streams. A single-cell regime, with the average droplet occupancy (λ) = 0.1, was used, which corresponded to a concentration of 106 B cells/ml. For each donor, at least 200,000 B cells were used for capturing single-cell mRNA. After 30 min incubation on ice, perfluoro-1-octanol (Sigma-Aldrich, USA) was added to break the emulsion. The magnetic beads were thoroughly washed48, resuspended in a one-step RT-PCR solution (QIAGEN OneStep RT-PCR Kit, Cat. No./ID: 210212) with the overlap extension VH and VL primer set described previously47. Overlap-extension emulsion eRT-PCR was performed under the following conditions: 30 min at 55 °C followed by 15 min at 95 °C; 4 cycles of 94 °C for 40 s, 50 °C for 30 s, 72 °C for 1 min 40 s; 4 cycles of 94 °C for 40 s, 55 °C for 30 s, 72 °C for 1 min 40 s; 32 cycles of 94 °C for 40 s, 60 °C for 30 s, 72 °C for 1 min 40 s; 72 °C for 10 min; hold at 4 °C according to the previously described protocol38. Amplicons were extracted from the emulsions, further amplified using a nested PCR47, and either sequenced using 2×300 paired-end Illumina MiSeq or ligated into the modified expression vector pPic9k-α-SfiI-FLAG-anchor based on pPIC9k (Invitrogen, USA) using SfiI sites39. The representativity of the resulting libraries was determined by plating serial cell dilutions on 2YT agar plates supplemented with 100 μg/mL ampicillin. Bacterial transformants were scraped off the plate into 2YT media, and the library DNA was purified using the Midiprep kit (QIAgen) according to the manufacturer’s instructions. For more detailed information, please refer to Supplementary Note.
Yeast display of natively paired VH-VL antibodies
The plasmid libraries were linearized with PmeI and transformed into the P. pastoris GS115 electrocompetent cells as previously described49. Up to 20 transformations with 1 µg of DNA were pooled for each library. Transformants were selected by incubation at 30 °C for 48 h on histidine-deficient RDB plates (1 M sorbitol, 1% dextrose, 4 × 10−5% biotin, 5 × 10−3% amino acids, 1.34% yeast nitrogen base, and 2% agar). The representativity of the resulting yeast libraries was determined by plating serial cell dilutions. Yeast transformants were scraped off the plate into YPD media. 5 × 108 cells were used for inoculation of 100 mL (OD600 = 0.1) of YPD (2% peptone, 1% yeast extract, 2% dextrose) in 250 mL. Yeast libraries were cultivated overnight at 30 °C under 200 rpm. Expression of the anchored scFvs was induced with methanol by diluting the culture to OD600 = 2 with BMMY media (1% yeast extract, 2% peptone, 1.34% YNB, 100 mM KH2PO4, 4 × 10−5% biotin, 0.5% (v/v) methanol). The culture was incubated at 30 °C, 200 rpm for 48 h with 0.5% (v/v) methanol addition every 24 h. Cells were washed twice with the conjugate buffer (PBS, 0.5% BSA, 2 mM EDTA) and incubated for 30 min with the tetrameric complexes41 of enzymatically biotinylated RBD with a streptavidin-R-phycoerythrin conjugate (SAPE, Invitrogen) and streptavidin-Cy5 conjugate (Abcam)—tRBDs. Gating was defined to remove debris and doublet cells using FSC and SSC. Positive cells were defined as cells having more than one order of magnitude higher fluorescence both in tRBD-PE and tRBD-Cy5 channels (Fig. S4). The first round of RBD-binding clone selection included double-positive staining with 10 nM tRBDs, followed by the isolation of RBD-positive clones with FACS (Fig. S4). During the following selection rounds, staining conditions were more stringent: tRBD concentration was reduced to 0.3 nM, and 60 nM of soluble ACE2 was added for competition for tRBD binding (Fig. 3d). The isolated clones were regenerated by agar plating and massively analyzed by deep sequencing or picked for verification of the most abundant top-binding clones by Sanger sequencing.
Deep sequencing and in silico reconstruction of VH-VL combinations
The DNA of isolated RBD-binding yeast clones was extracted according to50. To overcome degeneration of biodiversity, ePCR was used to prepare VH-VL amplicons38. The detailed protocol of Illumina library preparation and sequencing was described previously51. The list of primers used for this study is available in Supplementary Table S10. Briefly, VH-VL amplicons were prepared using a-short For (1) and FLAG Rev (2) primers. The obtained VH-VL amplicon was further utilized for two separate PCR reactions to generate overlapping amplicons covering the heavy and light IG chains. The overlap contains the IG heavy chain CDR3 used for in silico chain pairing after sequencing. Amplicon I was synthetized with CVD-Amp1 For (3)/ CVD-Amp1 Rev (4) primers. Amplicon II was prepared using the IGH multiplex primer mix (MiLaboratories, USA)/CVD-Amp2L Rev (5) and CVD-Amp2K Rev (6) mix. The obtained PCR product was subjected to another PCR reaction with the same amplification profile as the first PCR and underwent 6 cycles of amplification. The PCR mix contained 200 µM of each dNTP, Unique Dual Indexing primer mix (Illumina, USA), and Tersus polymerase in Tersus Buffer. The obtained libraries were pooled and sequenced on Illumina MiSeq (paired-end 300 + 300). The paired-end libraries of both sequenced VH and VL amplicons were filtered by the corresponding primer sequences using cutadapt v4.152 (--discard-untrimmed, --action=retain). All steps of the Ig repertoire extraction from sequencing data were performed using MiXCR v4.5.053 (mixcr analyze module, generic-amplicon, --dna --floating-left-alignment-boundary --floating-right-alignment-boundary J --assemble-contigs-by CDR3 --add-step assembleContigs). Data from the VH amplicon was used for the full chain sequence and clarification of the V gene. Before the extraction of an Ig repertoire from the VL amplicones, we split paired-end samples into separate forward and reverse sequencing datasets containing the fragments of light and heavy Ig sequences, respectively. To filter out the clonotypes with a high error rate, we used only sequences covered by at least 3 reads. The information about VH-VL combinations within a repertoire was collected based on the simultaneous presence of these chains in the VL amplicon. Since the read IDs in split forward and reverse reads are identical for an initial paired sequence, the statistics of the clonotypes corresponding to the reads can also be applied to the heavy-light clonotype pairs. To detect the heavy Ig sequence from a VH amplicon associated with a VH-VL pair, we overlapped VH and VL datasets based on the identical D/J genes and the CDR3 heavy chain nucleotide sequence. To evaluate the specificity of a given VH-VL pair towards the corresponding VH and VL clonotypes, we introduced the IGH_sym/IGKL_sym (symmetry) measure. To obtain it, we first calculated the match fraction (IGH_matchFrac and IGKL_matchFrac) of the VH-VL pair as the number of clonotype reads involved in pair formation divided by the total number of reads covering this clonotype in the repertoire. The following formulas were used to calculate the symmetry measures:
IGKL_sym = IGH_freq/(IGKL_freq * IGKL_matchFrac)
IGH_sym = (IGH_freq * IGH_matchFrac)/IGKL_freq
To normalize the symmetry measures to the same scale, the obtained values were used as a power of 100 (100^sym if sym ≤ 1; or 100^(1/x) otherwise). To use both symmetry values for VH and VL as filtering parameters for pair selection, we calculated their harmonic mean. For more detailed information, please refer to Supplementary Note.
Human IgG construction, expression, and purification
Selected antibodies were expressed as human monoclonal IgG1 as described previously51. Briefly, heavy and light chains were cloned in the pFUSE-CHIg-hG1 and pFUSE2-CLIg-hK or pFUSE2-CLIg-hlambda vectors (InvivoGen, USA), respectively. Full-sized human IgGs were expressed in HEK 293-F cells in serum-free medium FreeStyle (Thermo Fisher Scientific, USA) for 4–6 days. The PEI transfection agent (Thermo Fisher Scientific, USA) was used for transfection. Immunoglobulins were purified from the cultivation medium on HiTrap Protein G HP columns (Merck, Germany), followed by size exclusion chromatography using a Superdex 200 column (Cytiva, USA). The purified immunoglobulins were quantified using the home-made Human IgG ELISA Quantitation Set and Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, USA), 12% denaturing SDS-PAGE under reducing conditions.
Verification of IgG specificity by ELISA
To test monoclonal recombinant IgG binding to RBD protein, ELISA assays were performed as previously described51. Briefly, 96 well MaxiSorp plates (Nunc, Denmark) were coated with 50 µL of recombinant protein at a concentration of 2 µg/mL in carbonate buffer, and the protein was allowed to coat the wells overnight at 4 °C. Plates were then washed with 250 µL of the wash buffer (phosphate buffered saline (PBS) with 0.01% Tween-20 [PBST]) and blocked with 250 µL of 2% nonfat dry milk in PBS for 1 h at 37 °C. The purified antibodies were diluted in the conjugate buffer (PBST with 0.5% nonfat dry milk) to the concentrations in the range from 2000 to 0.2 ng/mL. 50 µL of the purified antibodies was added to each well and incubated for 1 h at 37 °C. Next, the plates were washed 3 times with the wash buffer. Goat anti-human anti-IgG-Fc horseradish peroxidase (HRP)-conjugated antibodies (Millipore Cat# AP113P, RRID:AB_11214132) were diluted 1:5000 in the conjugate buffer, and 50 µL was added to each well. After 1 h at 37 °C, the plates were washed 5 times with wash buffer, and 50 µL of TMB substrate was added. After 10 min at room temperature, the reaction was stopped with 50 µL of 10% phosphate acid, and the OD450 was read using the VarioScan plate reader. All isolated antibodies demonstrated no cross-reactive binding to non-relevant proteins.
Protein expression and purification
To isolate of RBD-binding clones, a codon-optimized DNA fragment encoding the S-protein RBD of the SARS-CoV-2 virus (amino acid residues 330–528, Wuhan Hu-1 strain) and an extracellular fragment of ACE2 (amino acid residues 18–740) were chemically synthesized and cloned in frame with the interleukin-2 signal sequence, 6xHis tag, and C-terminal AviTag into the pcDNA3.1/Hygro vector using NheI and XhoI sites. RBD was expressed by transient transfection in HEK 293-F cells using FreeStyle medium (Gibco, USA) for seven days at 37 °C with 8% CO2 and 135 rpm. To provide structural studies, the SARS-CoV-2 spike RBDs (WT, Omicron BA.1, Omicron BA.4/5) were constructed, expressed, and purified following the previously described protocols54,55. The genes encoding single-chain variable fragments (scFv) of Abs were synthesized and inserted into pAcGP67 vector. The resulting recombinant expression plasmid was then transformed into DH10Bac component cells, and Sf9 cells were transfected with the extracted bacmid using Cellfectin II Reagent (Invitrogen, USA). Low-titer viruses were harvested and subsequently amplified to generate high-titer viral stocks. The viral particles were used to infect Hi5 cells. After 48 h of infection, the cell supernatant was collected. The target protein was first purified using a Ni-NTA affinity column (GE Healthcare, USA) and eluted with a buffer containing 500 mM imidazole. The eluted protein was then concentrated and further purified using a Superdex 75 Increase 10/300 size-exclusion column (GE Healthcare, USA). The target protein was collected, concentrated, and stored at −80 °C. Avi-tagged RBD was enzymatically labeled with biotin according to the manufacturer’s instructions (Avidity LLC, USA). The biotinylation of RBD was estimated by Western blot using HRP-conjugated streptavidin (Thermo Fisher Scientific).
SARS-CoV-2 pseudovirus neutralization assay
The pseudovirus neutralization assay was performed as previously described56. The 96-well plates were seeded at a density of 2 × 104 cells/well with HEK 293T-ACE2 cells. After 24 h of incubation, the culture supernatant was aspirated gently to leave 45 μL in each well. Then, 5 µL of antibody serial dilutions were added to each well, followed by the infection of HEK 293T-ACE2 with pseudoviruses (50 μL per well). 3-fold serial dilutions of antibody samples were made in triplicate with the starting dilution of 100 nM in a final volume of 100 μL. 3 wells without antibodies and 3 wells without pseudoviral particles served as the virus controls and cell controls, respectively. After 48 h, the culture supernatant was aspirated. Cells were lysed with 100 µL lysis buffer (25 mM Tris-PO4, pH 7.8, 1% Triton X-100, 10% glycerol, 2 mM DTT, 2 mM EDTA) for 5 min at room temperature; 80 µL aliquots of cell lysate were transferred to 96-well black-walled plates, followed by addition of 20 µL of luciferase substrate (Bright-Glo™ Reagent, Promega, USA). Luciferase activity was measured using the Thermo Scientific Varioskan Flash. The data were processed with Origin Software. The half-maximal neutralizing concentration (IC50) of antibodies was defined as an antibody concentration required for reducing the relative light units (RLUs) by 50% compared with the virus control wells after subtracting the background RLUs in the control groups with cell controls.
Surface plasmon resonance (SPR)
SPR experiments were performed using the Biacore T200 system (GE Healthcare, USA). In brief, experiments were performed at 25 °C in HBS-EP+ buffer (Cytiva, USA). RBD was immobilized onto a CM5 chip (GE Healthcare, USA) in the 0.01 M sodium acetate buffer, pH 5.0, using the standard EDC/NHS protocol (Cytiva, USA). For competitive binding experiments, RBD-modified chips were saturated with 100 nM of the first antibody, followed by an injection of 100 nM of the second antibody or 2 mM of soluble ACE2 at a flow rate of 20 μL/min.
In vivo efficacy of antibodies in the lethal model of SARS-CoV-2 infection
CAG-hACE2-IRES-Luc-Tg transgenic mice (NM-TG-200002, C57BL/6Smoc-Tgtn(CAG-human ACE2-IRES-Luciferase-WPREpolyA)Smoc, C57BL/6 background), received from Shanghai Model Organisms Center, PRC, expressing high levels of hACE2 (Tg hACE2+), were used as a lethal SARS-CoV-2 infection model. Transgenic mice (8-10-week-old, male) were housed in the Animal Breeding Facility of BIBCh, RAS (the Unique Research Unit Bio-Model of the IBCh, RAS; the Bioresource Collection – Collection of SPF-Laboratory Rodents for Fundamental, Biomedical and Pharmacological Studies, contract #075-15-2025-486). The genotype of all mice was confirmed for the presence of the hACE2 gene by PCR. During the experiment, all mice were maintained in a specific pathogen-free (SPF) state under a strict light cycle (lights on at 07:00 h and off at 19:00 h), at 22 ± 2 °C and 50 ± 10% relative humidity. Animals were provided ad libitum access to a standard laboratory autoclavable rodent diet. All procedures with animals were performed according to the primary standards of AAALACi. All experiments and manipulations were approved by the institutional animal care and use committee of the Branch of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Science, Pushchino, Moscow region, Russia (IACUC #874/23 from 14.03.2023). Live virus protection experiments were conducted under the conditions of a specialized BSL-3 laboratory with local legislation and institutional requirements. Tg hACE2+ mice were inoculated intranasally under anesthesia with lethal doses (LD100) of SARS-CoV-2 corresponding to 103 and 104 plaque-forming units (PFU) for Wuhan strain (WT) and omicron BA.1, respectively. After 24 h, the mice were administered with 100 µL of antibody solution in PBS (0.3, 3, and 10 mg/kg dose) or vehicle intraperitoneally. The therapeutic neutralizing antibody P4A137 was used as a control. The survival and body weight were monitored until body weight recovery. n = 5–8 mice per group (for details, see Source data file). Euthanasia was performed by cervical dislocation. Two-tailed P values were determined by the Gehan-Breslow-Wilcoxon test using Prism 9.1.1 (GraphPad, USA).
Crystallization
The scFv of Ab and the SARS-CoV-2 RBD protein were mixed at a molar ratio of 1:1.2 and incubated on ice for 2 h. The complex was purified using a Superdex 75 Increase 10/300 size-exclusion column (GE Healthcare, USA). The purified complex was concentrated to achieve the concentration of 5 or 10 mg/mL for crystallization screening using the vapor-diffusion sitting-drop method at 16 °C. Screening kits used included PEG/Ion, Crystal Screen, Index, SaltRX from Hampton Research, and Wizard I–IV from Emerald BioSystems. The diffraction crystals of BA.4/5-D1 complex were formed in the mother liquor containing 25% w/v PEG 1500, SPG Buffer/NaOH pH 8.5. Crystals were cryo-protected in 4 M sodium formate at 100 K for X-ray data collection. The diffraction crystals of Omicron BA.1-T11 complex were formed in the mother liquor containing 0.05 M Citric acid, 0.05 M BIS-TRIS propane, pH 5.0, 16% w/v Polyethylene glycol 3350. Crystals were cryo-protected in 4 M Sodium formate at 100 K for X-ray data collection. The diffraction crystals of SARS-CoV-2 RBD-N1 complex were formed in the mother liquor containing 0.2 M Magnesium sulfate heptahydrate, 20% w/v Polyethylene glycol 3350, pH 6.0. Crystals were cryo-protected in 20% glycerol, 0.16 M Ammonium tartrate dibasic pH 7.0, 16 % w/v Polyethylene glycol 3350 at 100 K for X-ray data collection.
X-ray data collection, processing, and structure determination
Diffraction data were collected at the Shanghai Synchrotron Radiation Facility BL18U1 and BL10U and processed using the HKL2000 software package57. The structures were determined by molecular replacement using Phenix58, with the crystal structure of the SARS-CoV-2-TH003 antibody (PDB code: 8GPY) serving as the molecular replacement model. The initial model was built into the modified experimental electron density using Coot59 and further refined in Phenix. Model geometry was validated using MolProbity, and figures were made by PyMOL (http://www.pymol.org).
MD simulations
MD simulations of all the studied proteins and their complexes in explicit aqueous solution were performed using the GROMACS 2020.4 package60 and the CHARMM36 force field61,62,63,64,65). An integration time step of 2 fs was used, and 3D periodic boundary conditions were imposed. The spherical cut-off function (12 Å) was used to truncate van der Waals interactions. Electrostatic interactions were treated using the particle mesh Ewald (PME) method66 (real space cutoff 12 and 1.2 Å grid with fourth-order spline interpolation). The TIP3P water model was used67, and Na+ and Cl− ion parameters were implemented. Apart from counterions, 150 mM NaCl was added to each system. Simulations were performed at 310 K temperature and 1 bar isotropic pressure maintained using the V-rescale68 and the Parrinello–Rahman69 algorithms with 0.5 and 5.0 ps relaxation parameters, respectively, and a compressibility of 4.5 × 10−5 bar−1 for the barostat. The protein and solvent molecules were coupled separately. Before the production MD runs, all systems were minimized over 5000 steps using a conjugate gradients algorithm, followed by heating from 5 K to 310 K over 50,000 steps, during which internal coordinates of the protein heavy atoms were restrained. Production runs were simulated for 1.0 μs. Bonds with an H atom were constrained via implementing LINCS70. All MD trajectories were processed using the trjconv utility from the GROMACS 2020.4 package to get the protein centered in the box, 3D periodic boundary conditions removed, and to obtain an output frequency of 100 ps per frame. The values of root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) were calculated using the rms and rmsf utilities, respectively, both from the GROMACS package. RMSF values were evaluated for portions of the trajectories proven to be equilibrated (from 600 ns to 1000 ns). When three replicas were calculated, RMSF values were calculated for 100 ns intervals of the equilibrated segments, followed by the calculation of the mean value and standard deviation for each residue. Molecular editing and graphics rendering were performed in PyMOL v. 2.4.0 (The PyMOL Molecular Graphics System, Schrödinger, LLC) and UCSF Chimera71.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RBD-AIM is free available with a web interface (https://rbdaim.2a2i.org/). The dataset and RBD-AIM data generated in this study have been deposited in GitHub under accession code https://github.com/tanishAI/rbd-aim and Zenodo under accession code https://zenodo.org/records/15330778, https://doi.org/10.5281/zenodo.15330778. The RBD-specific Ab sequences are provided in the Supplementary Information (Supplementary Data 1). The database of Abs used for RBD-AIM model training is available in Supplementary Information (Supplementary Data 2). The sequence of expression vector pPic9k-α-SfiI-FLAG-anchor that was applied for the yeast display of native B cell repertoires is provided in the Supplementary Information (Supplementary Data 3). The atomic models generated from X-ray crystallographic studies of the Ab-RBD complexes have been deposited at the Protein Data Bank (PDB, http://www.rcsb.org/) under accession codes PDB ID: 8WFH (Omicron BA.4/5-D1 complex) PDB https://doi.org/10.2210/pdb8WFH/pdb, PDB ID: 8WFM (Omicron BA.1-T11 complex) PDB https://doi.org/10.2210/pdb8WFM/pdb, PDB ID: 9JEB (SARS-CoV-2 RBD-N1 complex) PDB https://doi.org/10.2210/pdb9JEB/pdb. The omit maps are provided in the Supplementary Information (Supplementary Data 4). The table summarizing structural and refinement statistics is provided in the Supplementary Information (Table S13). Source data are provided with this paper. The authors declare that all data are available in the article, its Supplementary Information files, and the Source Data file and deposited in Zenodo, GitHub, and PDB or from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
The source code of RBD-AIM is available at https://github.com/tanishAI/rbd-aim and https://zenodo.org/records/15330778, https://doi.org/10.5281/zenodo.15330778. The software to prepare antibody database is available at https://github.com/d-nolde/sars-cov2-ab, https://doi.org/10.5281/zenodo.18392067. The bioinformatic pipeline for the identification of natively paired VH-VL antibodies is available at https://github.com/smiranast/VH-VL-merger/.
References
Chakraborty, I. & Maity, P. COVID-19 outbreak: migration, effects on society, global environment and prevention. Sci. Total Environ. 728, 138882 (2020).
Hu, B., Guo, H., Zhou, P. & Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154 (2021).
Zost, S. J. et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. 26, 1422–1427 (2020).
Kevadiya, B. D. et al. Diagnostics for SARS-CoV-2 infections. Nat. Mater. 20, 593–605 (2021).
Zhou, Y. et al. A comprehensive SARS-CoV-2–human protein–protein interactome reveals COVID-19 pathobiology and potential host therapeutic targets. Nat. Biotechnol. 41, 128–139 (2023).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Logunov, D. Y. et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 397, 671–681 (2021).
Jackson, L. A. et al. An mRNA vaccine against SARS-CoV-2 — preliminary report. N. Engl. J. Med. 383, 1920–1931 (2020).
Leung, N. H. L. et al. Comparative antibody and cell-mediated immune responses, reactogenicity, and efficacy of homologous and heterologous boosting with CoronaVac and BNT162b2 (Cobovax): an open-label, randomised trial. Lancet Microbe 4, e670–e682 (2023).
Chen, Y. et al. Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses. Nat. Rev. Immunol. 23, 189–199 (2023).
Follmann, D. et al. Examining protective effects of SARS-CoV-2 neutralizing antibodies after vaccination or monoclonal antibody administration. Nat. Commun. 14, 3605 (2023).
Copin, R. et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell 184, 3949–3961.e3911 (2021).
Voss, W. N. et al. Hybrid immunity to SARS-CoV-2 arises from serological recall of IgG antibodies distinctly imprinted by infection or vaccination. Cell Rep. Med. 5, 101668 (2024).
Barnes, C. O. et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 588, 682–687 (2020).
Cao, Y. et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602 (2022).
Heggelund, J. E., Das, S., Stamnaes, J., Iversen, R. & Sollid, L. M. Autoantibody binding and unique enzyme-substrate intermediate conformation of human transglutaminase 3. Nat. Commun. 14, 6216 (2023).
Mandal, K. et al. Structural surfaceomics reveals an AML-specific conformation of integrin β2 as a CAR T cellular therapy target. Nat. Cancer 4, 1592–1609 (2023).
Raybould, M. I. J., Kovaltsuk, A., Marks, C. & Deane, C. M. CoV-AbDab: the coronavirus antibody database. Bioinformatics 37, 734–735 (2020).
Li, X., Van Deventer, J. A. & Hassoun, S. ASAP-SML: an antibody sequence analysis pipeline using statistical testing and machine learning. PLOS Computat. Biol. 16, e1007779 (2020).
Ruffolo, J. A., Chu, L.-S., Mahajan, S. P. & Gray, J. J. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat. Commun. 14, 2389 (2023).
Robert, P. A. et al. Unconstrained generation of synthetic antibody–antigen structures to guide machine learning methodology for antibody specificity prediction. Nat. Comput. Sci. 2, 845–865 (2022).
Antanasijevic, A. et al. From structure to sequence: antibody discovery using cryoEM. Sci. Adv. 8, eabk2039 (2022).
Rougé, L. et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science 367, 1224–1230 (2020).
Blanchard, J. W. et al. Replacing reprogramming factors with antibodies selected from combinatorial antibody libraries. Nat. Biotechnol. 35, 960–968 (2017).
Smirnov, I. V. et al. Robotic QM/MM-driven maturation of antibody combining sites. Sci. Adv. 2, e1501695 (2016).
Fowler, D. M. et al. High-resolution mapping of protein sequence-function relationships. Nat. Methods 7, 741–746 (2010).
Fowler, D. M. & Fields, S. Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 (2014).
Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29, 44–57.e49 (2021).
Zost, S. J. et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 584, 443–449 (2020).
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020).
Ruffolo, J. A., Gray, J. J. & Sulam, J. Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv preprint https://doi.org/10.48550/arXiv.2112.07782 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Ruffolo, J. A., Sulam, J. & Gray, J. J. Antibody structure prediction using interpretable deep learning. Patterns 3, 100406 (2022).
Ahdritz, G. et al. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nat. Methods 21, 1514–1524 (2024).
Guo, Y. et al. A SARS-CoV-2 neutralizing antibody with extensive Spike binding coverage and modified for optimal therapeutic outcomes. Nat. Commun. 12, 2623 (2021).
Terekhov, S. S. et al. Liquid drop of DNA libraries reveals total genome information. Proc. Natl. Acad. Sci. USA 117, 27300–27306 (2020).
Terekhov, S. S. et al. Microfluidic droplet platform for ultrahigh-throughput single-cell screening of biodiversity. Proc. Natl. Acad. Sci. USA 114, 2550–2555 (2017).
Zhang, H. et al. Phenotype-information-phenotype cycle for deconvolution of combinatorial antibody libraries selected against complex systems. Proc. Natl. Acad. Sci. 108, 13456–13461 (2011).
Franz, B., May, K. F. Jr, Dranoff, G. & Wucherpfennig, K. Ex vivo characterization and isolation of rare memory B cells with antigen tetramers. Blood 118, 348–357 (2011).
Wiley, D. C., Wilson, I. A. & Skehel, J. J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378 (1981).
Burley, S. K. et al. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 51, D488–D508 (2022).
Dunbar, J. & Deane, C. M. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics 32, 298–300 (2015).
Shindyalov, I. N. & Bourne, P. E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. Des. Select. 11, 739–747 (1998).
Lomakin, Y. A. et al. Deconvolution of B cell receptor repertoire in multiple sclerosis patients revealed a delay in tBreg maturation. Front. Immunol. 13, 803229 (2022).
Zhou, X. et al. Molecular deconvolution of the neutralizing antibodies induced by an inactivated SARS-CoV-2 virus vaccine. Protein Cell 12, 818–823 (2021).
McDaniel, J. R., DeKosky, B. J., Tanno, H., Ellington, A. D. & Georgiou, G. Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat. Protoc. 11, 429–442 (2016).
Wu, S. & Letchworth, G. J. High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. BioTechniques 36, 152–154 (2004).
Lõoke, M., Kristjuhan, K. & Kristjuhan, A. Extraction of genomic DNA from yeasts for PCR-based applications. BioTechniques 50, 325–328 (2011).
Lomakin, Y. A. et al. Two-dimensional high-throughput on-cell screening of immunoglobulins against broad antigen repertoires. Commun. Biol. 7, 842 (2024).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Bolotin, D. A. et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381 (2015).
Fu, D. et al. Structural basis for SARS-CoV-2 neutralizing antibodies with novel binding epitopes. PLoS Biol. 19, e3001209 (2021).
Guo, Y. et al. Discovery and characterization of potent pan-variant SARS-CoV-2 neutralizing antibodies from individuals with Omicron breakthrough infection. Nat. Commun. 14, 3537 (2023).
Crawford, K. H. D. et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses 12, 513 (2020).
Otwinowski, Z. & Minor, W. in Methods in Enzymology 307-326 (Academic Press, 1997).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. 66, 486–501 (2010).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
MacKerell, A. D. Jr et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
Klauda, J. B., Monje, V., Kim, T. & Im, W. Improving the CHARMM force field for polyunsaturated fatty acid chains. J. Phys. Chem. B 116, 9424–9431 (2012).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Pettersen, E. F. et al. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
This work was supported by a Ministry of Science and Higher Education of the Russian Federation grant no. 075-15-2025-488; H.Z. was supported by the National Natural Science Foundation of China, grant no. 82261138553, and the National Key Research and Development plan of China, grant no. 2018YFE0200400. Y.G. was supported by the National Natural Science Foundation of China, grant no. 32271256.
Author information
Authors and Affiliations
Contributions
S.S.T., N.V.I., N.Z., Y.A.M., D.E.N., Y.A.L., A.Z., L.A.O., D.M.M., M.N.B., T.S., E.A., M.Z., K.G., A.O.S., I.M., T.V.B., N.N.K., A.S.C., I.E.E., and I.Y., performed experiments, analyzed and interpreted data; S.S.T., I.V.S conceptualized application of microfluidic approach to isolate single B cells, S.S.T., Y.A.M., Y.A.L., L.A.O., D.M.M., M.N.B., and T.V.B. performed Ab identification and functional analyses; N.V.I., D.E.N., A.Z., T.S., and O.K. build RBD-AIM model; N.Z., M.Z., K.G., and Y.G. performed structural studies; E.A. performed MD modelling; T.S., S.D., N.A., and S.M., built a web interface; A.O.S., I.M., and I.Y. performed NGS sequencing and data analysis; T.V.B. and N.N.K. performed protein expression and purification; A.S.C. performed in vivo studies; I.E.E. and V.M.B. contributed to SPR studies; A.V.S. and D.Z., analyzed and interpreted data; R.L., A.G.G., R.D.K. conceptualized the study, designed the research; S.S.T., N.V.I., Y.A.M., T.S., R.G.E., I.V.S., O.K., H.Z., Y.G., A.V.S., A.G.G., and R.D.K. designed the research, analyzed and interpreted data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Terekhov, S.S., Ivanisenko, N.V., Zhang, N. et al. Mining antibody functionality via AI-guided structural landscape profiling. Nat Commun 17, 4009 (2026). https://doi.org/10.1038/s41467-026-70553-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70553-6
