
Abstract
Covalent organic frameworks-based solid-state electrolytes have attracted significant attention in recent years due to their design flexibility, intrinsic porosity, and environmentally friendly characteristics. However, their practical application in batteries remains limited by inadequate ionic conductivity and Li+ transference number, primarily arising from the absence of effective strategies to modulate the pore chemical environment for ion transport. In this study, we introduce a pore-engineering approach by incorporating alternating oxyethylene and perfluoroalkyl chains into the covalent organic frameworks. This sequence-controlled modification simultaneously suppresses anion migration and mitigates lithium-ion aggregation, thereby constructing a continuous and efficient site-to-site Li+ transport pathway. Benefiting from this design, the resulting covalent organic framework exhibits a high Li+ conductivity of 1.06 mS·cm−1 at 25 °C and an Li+ transference number of 0.9. A symmetric Li | |Li cell delivers Li plating/stripping stability over 7500 hours with minimal voltage polarization at 0.2 mA·cm−2 and areal capacity of 0.2 mAh·cm−2. Furthermore, solid-state Li | |LiNi0.8Mn0.1Co0.1O2 battery demonstrates a specific capacity of 180 mAh·g−1 at 1 C (1 C = 200 mA·g−1) and long-term stability at 5 C, retaining 80% capacity after 700 cycles. Here we report pore design strategy and open avenues for the development of high-performance, fast-charging solid-state lithium batteries.
Similar content being viewed by others

Introduction
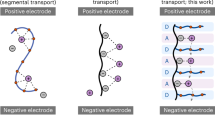
Owing to the serious safety concerns of organic-based liquid electrolytes, the research on solid-state electrolytes (SSEs) is considered a crucial direction for lithium-ion batteries (LIBs). Covalent organic frameworks (COFs)-based SSEs are classified as polymer-based SSEs with structural diversity, crystallinity and high design flexibility1,2,3,4,5. Although polymer-based SSEs are pivotal for high interfacial stability and safety, their application is hindered by intrinsic limitations6,7,8, such as low ionic conductivity due to poor Li-salt dissociation and inefficient Li+ migration pathways9,10,11,12. For instance, polyethylene oxide (PEO) possesses abundant Li+ hopping sites, Li-metal compatibility, and reductive stability, yet the incompatibility with high-voltage cathodes restricts its utility (Fig. 1a)13,14,15. Especially, fluoroalkyl chains exhibit high dielectric constants to enhance Li-salt dissociation and oxidative stability for high-voltage operation (Fig. 1b)16, which suggests combining PEO with fluoroalkyl moieties could synergistically optimize electronic distribution, stability, and chemical environment of Li+ migration (Fig. 1c). Nevertheless, there is competing requirements for ionic conduction, electrochemical stability, and controlled Li+ migration environments to achieve such combination. Further complications arise from Li+/ion-pair aggregation and uneven charge distribution in traditional polymer matrices, which stem from disordered chain arrangements and exacerbate strong Li+-anion interactions, reducing conductivity and transference numbers17,18,19,20,21.

Electrostatic potential, structure and features of PEO (a), fluoroalkyl (b), and compositions (c); structural scheme of no pore modification COF (d) and pore modification COF (e); the structural advantages of PF–COF (f); electrostatic potential distribution of TA–COF and PF–COF (g); schematic illustration of high-selectivity Li+ migration in PF–COF solid-state electrolyte for dual-stable-interface (h).
Designing periodically sequenced polymer electrolytes could balance the competing requirements for the combination and gain fundamental insights into enhancing Li-salt dissociation and migration pathways, as well as resolving oxidative/reductive stability trade-offs. COF-based SSEs offer great opportunities in designing sequenced polymers with ordered functional-group distributions to regulate charge/ion transport. However, synthesizing such COF-based SSEs is significantly challenging, as existing COF-based solid electrolytes rely exclusively on rudimentary post-synthetic modifications or passive pore infiltration, demonstrating limited efficacy in modulating Li+ migration pathways and enhancing ionic conductivity22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39. Especially, the periodic incorporation of donor-acceptor moieties within COF pore architectures presents substantial synthetic challenges in the precise spatial control of functional group alignment and maintenance of structural integrity during framework assembly. Single functionalized fragment-regulated COF-based SSEs often demonstrate an intrinsic limitation in concurrently achieving high ionic conductivity, rapidly migrated kinetics of Li+ and optimal interfacial compatibility with electrodes. Therefore, designing a strategy of integrating alternating sequencing polymer chains during COF synthesis directly incorporates various functional monomers into the COF backbone, enabling precise control over the arrangement of donor/acceptor moieties and pore distribution. Critically, alternating sequencing polymer-modified COFs exhibit advantages over existing COF-based electrolytes. The ordered insertion of polymer chains optimizes charge distribution and enhances Li-salt dissociation, which could address the key limitation of low ionic conductivity in polymer electrolytes. The donor-acceptor feature and nanoconfined pore arrangement regulate the chemical environment for Li+ migration and improve ion transport efficiency. The crystallinity of COFs ensures structural stability while the sequenced polymers introduce dynamic functionality.
Here, we present a pore modification strategy through the alternating insertion of oxyethylene chains and perfluoroalkyl chains (denoted as PF) in COF material. Compared with bare pore COF (denoted as TA–COF, Fig. 1d), this sequencing pore modification COF-based solid-state electrolyte (denoted as PF–COF) creates a favorable environment for Li+ migration, prevents Li+ aggregation and provides abundant site-to-site Li+ migration pathways. The PF–COF electrolyte also exhibits interfacial stability, with a robust LiF-rich solid electrolyte interphase (SEI). Compared with the electrostatic potential of TA–COF (Fig. 1g), the PF–COF (Fig. 1g) shows a reallocated charge distribution. Due to the strong electron-withdrawing nature of C-F2, the surface charge of PF–COF is positive, which effectively limits TFSI− migration and promotes highly selective Li+ transference (Fig. 1h). As a result, a Li+ conductivity of 1.06 × 10−3 S/cm and transference number of 0.9 are achieved in this sequencing pore modified COF-based solid-state electrolyte, which is highly competitive among leading SSE technologies and surpass most oxides and polymers. Moreover, the assembled solid-state Li||LiNi0.8Mn0.1Co0.1O2 battery delivers a specific capacity (180 mAh g−1 at 1 C) and demonstrates capacity retention at 5 C (80% retention after 700 cycles). The interaction mechanism between COF materials and ions (Li+ and TFSI−) is thoroughly investigated by molecular dynamics (MD) simulations and spectroscopic characterization techniques. Moreover, PF–COF offers distinct practical advantages, including facile solution processability at room temperature, intrinsic flexibility, and low cost compared to sulfide or rare-earth-containing oxide SSEs. This design overcomes the challenges of traditional polymer and reported-COFs electrolytes (e.g., random chain arrangement, poor ion mobility), and opens avenues for practical SSEs with programmable properties for solid-state batteries.
Results and discussions
Designs and characterization of Li–TA–COF and Li–PF–COF electrolytes
Designing a functional unit of polymers containing PEO and C–F2 functional groups is challenging; thus, we adopted a practically synthetic route of PF after thorough consideration (Fig. 2a). The detailed synthesis process is presented in the Supporting Information. Related nuclear magnetic resonance (1H NMR and 13C NMR) and mass spectrometry (Supplementary Figs. 1–12) indicate that the PF monomer is successfully synthesized with high purity. Subsequently, the synthesis routes of target materials TA–COF and PF–COF are optimized to yield high crystallinity. The COFs are obtained by the solvothermal method, and the preparation procedure is provided in the Supporting Information. Chemical structures of PF–COF and TA–COF are shown (Supplementary Figs. 13 and 14), where the PEO and fluoroalkyl chains are alternatively distributed in the pores. Crystallinity of PF–COF is studied by powder X-ray diffraction (PXRD). Two distinct peaks at 2.5 and 5 ° are observed in the PF–COF PXRD pattern, corresponding to the (100) and (200) planes, respectively. The PXRD pattern of PF–COF and starting materials (PF and TFPB) is demonstrated (Supplementary Fig. 15), suggesting a complete chemical conversion into PF–COF. In addition, the PXRD result is highly consistent with simulation of AA stacking (Supplementary Fig. 16). Pawley-refinement is applied to assess the structural integrality and the Pawley-refined result for AA stacking is consistent with experimental profile (Rwp = 5.23%, Rp = 4.13%, Fig. 2b). The simulated structural model for AA stacking is displayed in Fig. 2d, e, indicating PEO and fluoroalkyl chains are following an alternating sequence. Therefore, the analyses suggest the layer structures and high crystallinity of PF–COF. Similar results can be found in PXRD and Pawley-refined profiles of TA–COF (Fig. 2c, Supplementary Figs. 17 and 18). The most obvious difference between PF–COF and TA–COF is that no chains are distributed inside the pore of TA–COF. Characteristic chemical bond of C=N in TA–COF and PF–COF is proved by Fourier transform infrared (FT-IR) spectrum (Supplementary Fig. 19) with a wavenumber of 1670 cm−1 and solid-state 13C nuclear magnetic resonance (NMR) (Supplementary Fig. 20) with chemical shift of 150 ppm40. The FT-IR spectra of PF–COF and TA–COF with respective starting units (Supplementary Figs. 21 and 22) demonstrate a complete formation of crystalline COFs without the presence of building blocks.

Synthetic route of PF (a); PXRD and Pawley-refined patterns of PF–COF (b) and simulated AA stacking mode (d); PXRD and Pawley-refined patterns of TA–COF (c) and simulated AA stacking mode (e).
Transmission electron microscopy (TEM) (Supplementary Fig. 23) and scanning electron microscopy (SEM) (Supplementary Fig. 24) results demonstrate that the PF–COF is presented in the form of nanosheets, and the C, N, O, and F elements are uniformly distributed from the EDS mapping. SEM (Supplementary Fig. 25) and TEM (Supplementary Fig. 26) images of TA–COF indicate a rod-shaped morphology. Surface area of PF–COF and TA–COF is measured by N2 adsorption experiments at 77 K. As shown (Supplementary Fig. 27), PF–COF has a low BET surface area with 142 m2 g−1, and the pore distribution is calculated to be 2 nm, which is smaller than TA–COF (Supplementary Fig. 28). The significant decline of pore size is ascribed to the insertion of flexible PEO and fluoroalkyl chains. Compared with TA–COF, PF–COF demonstrates thermal stability and negligible weight loss up to 300 °C (Supplementary Fig. 29) in the thermogravimetric test. Therefore, thermal stability is significantly improved by introducing PEO and fluoroalkyl groups, which is beneficial for the safety of solid-state batteries at high temperature40. Structural stability of PF–COF is further investigated in various chemical environments. Supplementary Figs. 30 and 31 show the PXRD and FT-IR patterns of PF–COF after immersing in acid, base and other organic solvents, implying the structural stability of PF–COF. Therefore, it is evident that flexible PEO and fluoroalkyl chains dramatically enhance the stability of the PF–COF solid-state electrolyte. Finally, X-ray photoelectron spectroscopy (XPS) spectra (Supplementary Fig. 32) of TA–COF and PF–COF suggest the consistency of chemical structure and functional groups.
Li salt dissociation and interaction
The solid-state electrolytes (namely Li–TA–COF and Li–PF–COF) are prepared by mixing LiTFSI and COF materials (TA–COF and PF–COF). The degree of Li+ dissociation and relative interaction in COF-based solid-state electrolyte are studied by MD simulation (the raw file for calculation is provided in Supplementary Data 1, 2, 4, and 5. The results of MD simulations for Li–TA–COF and Li–PF–COF are shown in Fig. 3a, b, Supplementary Data 3 and Supplementary Data 6. LiTFSI is uniformly distributed in the PF–COF framework, whereas aggregated LiTFSI is found in the TA–COF. Their different distribution suggests that sequencing polyethylene oxide and perfluoroalkyl chains in Li–PF–COF enhances Li+ dissociation. Therefore, the spacing distance (L) between TFSI− units on the Li–PF–COF backbone is larger and much more ordered than that in the Li–TA–COF backbone41. According to the Manning counterion condensation theory42, the ion (Li+) dissociation will be increased with enhanced L. In addition, reduced density of Li+ in Li–PF–COF matrix promotes higher Li+ dissociation than Li–TA–COF (Fig. 3c). Radical distribution function is applied to investigate the interaction sites with Li+ in COF materials. Possible coordination sites between Li+ with O in PEO (termed as O2PF–COF) and skeleton (termed as O1PF–COF) are provided (Supplementary Fig. 33a). The coordination sites between Li+ with O in TA–COF skeleton (termed as OTA–COF) and N in skeleton (termed as NTA–COF) are provided (Supplementary Fig. 33b). As shown in Fig. 3d, the average coordination number of Li-O2PF–COF, Li-O1PF–COF, Li-NTA–COF, and Li-OTA–COF is calculated to be 0.82, 0.64, 0.71, and 0.53, respectively. Therefore, the interaction between Li+ and PEO in Li–PF–COF is the strongest among all the sites, implying high Li salt dissociation and Li+ conductivity in Li–PF–COF solid state electrolyte43. In addition, the radical distribution function of Li+–Li+ in Li–PF–COF shows a strong peak at 0.36 Å, corresponding to the distance between adjacent Li+–Li+. Nevertheless, additional subtle peaks ranging from 0.65 to 1.1 Å are observed for Li–PF–COF (Fig. 3e). The MD analyses indicate that Li+ distribution in PEO and fluoroalkyl-linked material is more homogeneous and less aggregated than in Li–TA–COF. The source files for the MD simulations have been deposited in the Supplementary Data and referenced in the manuscript.

Snapshot of MD simulation for Li–TA–COF (a) and Li–PF–COF (b); Li+ density distribution, Z represents the distance from the positive electrode plate (c), radical distribution functions and coordination number calculated from MD simulation of Li–PF–COF (d); radical distribution functions of Li pairs in Li–TA–COF and Li–PF–COF (e); solid-state 7Li NMR of LiTFSI, Li–TA–COF and Li–PF–COF (f); FT-IR spectra of LiTFSI, PF–COF and Li–PF–COF (g); zeta potential of TA–COF and PF–COF (h).
Strong interaction between Li+ with PF–COF is further verified by solid-state 7Li-NMR. A higher chemical shift of 0.037 ppm of Li in Li–PF–COF is observed in Fig. 3f, while a lower chemical shift of −0.53 ppm for Li–TA–COF and −0.56 ppm for LiTFSI is observed. This high chemical shift indicates greater Li dissociation and conductivity for Li–PF–COF44. Wavenumber changes from 1132 cm−1 to 1138 cm−1 of the C–O–C bond in the FT-IR spectrum of PF–COF and Li–PF–COF confirm the strong interaction between Li+ with PF–COF (Fig. 3g). The measured zeta potential disparity between TA–COF (−2.7 mV) and PF–COF (+5.2 mV) (Fig. 3h) establishes a critical electrostatic gradient that governs anion immobilization. This net positive surface charge in PF–COF generates a Coulombic attraction force with TFSI- anion, effectively anchoring them within the framework’s nanochannels. Such electrostatic confinement creates anion-depleted regions that enhance Li+ transference number (0.9) and boost ionic conductivity (1.06 mS cm−1) in PF–COF solid-state electrolyte. As a result, the solid-state electrolyte Li–PF–COF has high Li+ conductivity and transfer number.
The anti-oxidation ability of TA–COF, PF–COF and some conventional electrolyte molecules (ethylene carbonate: EC, fluoroethylene carbonate: FEC, propylene carbonate: PC, diethyl carbonate: DEC, 1,3-dioxolane: DOL, 1,2-dimethoxyethane: DME, LiTFSI) is assessed by theoretical calculation. Figure 4a displays the HOMO energy level distribution of these electrolytes, in which PF–COF has the lowest HOMO energy level, indicating anti-oxidation ability. The electron location function (ELF) demonstrates greater localization of PF–COF in the polyethylene oxide and perfluoroalkyl chains, indicating electron migration by donor-acceptor interaction (Fig. 4b). Additionally, the electron localization function of PF–COF shows more nucleophilic sites than TA–COF, resulting in strong interaction with Li+ (Fig. 4c). It is noteworthy that the alternative sequencing polymer chains altered the electron distribution, rendering the C=O and O in PEO of PF–COF with more negative electrostatic potential predicted by Fukui function (Fig. 4d), promoting this region a prime candidate for Li+ adsorption and migration. Moreover, the interaction affinity between PF–COF and Li+ (−6.11 eV) is stronger than TA–COF and Li+ (−5.13 eV), as shown in Fig. 4e. The attractive ability between PF–COF and Li+ breaks the strong interaction in TFSI– and Li+ and contributes to free Li+, thereby achieving highly-selective Li+ migration and increasing the Li+ dissociation, transference number and ionic conductivity.

HOMO energy level of PF–COF, TA–COF, FEC, LiTFSI, EC, PC, DEC, DME, DOL electrolytes (a); electron localization function of TA–COF and PF–COF (b), average local ionization energy of TA–COF and PF–COF (c), and orbit weight Fukui function (d) of TA–COF and PF–COF; adsorption energy TA–COF and PF–COF towards Li+ (e).
Alternative sequencing-assisted Li+ transport and conduction
Ionic conductivity of Li–TA–COF and Li–PF–COF is measured by electrochemical impedance spectroscopy (EIS) and calculated to be 1.06 × 10−3 S/cm for Li–PF–COF and 3.67 × 10−4 S/cm for Li–TA–COF (Fig. 5a) at room temperature, respectively. Conductivity at various temperatures is tested and displayed in Supplementary Figs. 34 and 35 and Supplementary Table 1. Therefore, according to the Arrhenius plot, the activation energy (Ea) is 0.17 eV for Li–PF–COF, and 0.18 eV for Li–TA–COF (Fig. 5b). Moreover, the Li+ transference number of Li–PF–COF reaches a high value of 0.9, which is more than doubled from that of Li–TA–COF (Fig. 5c, Supplementary Figs. 36 and 37). High Li+ transference number of Li–PF–COF demonstrates that the TFSI− is constrained in the framework of PF–COF. This is because PEO and fluoroalkyl chains change the charge state and regulate the electron density of Li–PF–COF45. This result also highly agrees with Zeta potential analyses (Fig. 3h). Therefore, the negative TFSI− is captured by positive Li–PF–COF by electrostatic interaction, resulting in highly selective Li+ migration and conductivity. The high ionic conductivity and transference number have rarely been reported for COF-based and other polymer-based solid-state electrolytes (Fig. 5d, Supplementary Table 2). Especially, the obtained transference number of Li–PF–COF can only be achieved in a single-ion conductor, which is useful for fast-charging solid-state batteries. Enhanced ionic conductivity and decreased Ea from Li–PF–COF to Li–TA–COF suggest that alternative sequencing of PEO and fluoroalkyl chains remarkably improves the electrochemical performances of COF-based solid-state electrolyte41.

EIS of Li–TA–COF and Li–PF–COF electrolytes (a); Ea of Li–TA–COF and Li–PF–COF electrolytes (b); Li+ transfer number of Li–TA–COF and Li–PF–COF electrolytes (c); comparison of Li+ transference number and Li+ conductivity of COF-based solid-state electrolyte (d), the source of the literature data shown in this Figure can be found in Supplementary Table 2; electrochemical stable window of Li–TA–COF and Li–PF–COF (e); galvanostatic voltage profiles of Li||Li symmetric batteries using Li–TA–COF and Li–PF–COF electrolytes at 0.2 mA cm−2 with 0.2 mAh cm−2 (f); EIS of Li||Li symmetric batteries using Li–PF–COF electrolytes at various cycles (g); Li+ migrated pathway in Li–PF–COF electrolyte by MD simulation (h).
The electrochemical window of Li–PF–COF is extended to 5.2 V compared to Li–TA–COF (Fig. 5e), which ensures Li–PF–COF is compatible with high-nickel cathode materials (such as NCM811). The compatibility between designed solid-state electrolytes and Li anode is evaluated by Li||Li symmetrical batteries. The Li|Li–PF–COF|Li battery can be operated for 7500 h at 0.2 mA cm−2 without evident voltage fluctuation (Fig. 5f). By contrast, the Li|Li–TA–COF|Li battery exhibited a shorter cycling for 1600 h with increasing overpotential. From the EIS spectra at various cyclic numbers in Li||Li symmetrical batteries (Fig. 5g), it is indicative that the Li–PF–COF has interfacial stability with Li metal than Li–TA–COF (Supplementary Fig. 38). The compatibility with Li metal anode is rarely reported in other COF-based solid-state electrolytes. In addition, after pore modification by alternative sequencing of PEO and fluoroalkyl chains, the critical current density of Li–PF–COF solid-state electrolyte is 1 mA cm−2, which is higher than that of Li–TA–COF (0.8 mA cm−2, Supplementary Fig. 39).
To understand the Li+ migrated procedures, the Li+ movement and pathway in alternative sequencing of PEO and fluoroalkyl chains modified Li–PF–COF are collected by MD simulations (Fig. 5h). Initially, Li+ is interacted with N atom in TFSI− (state Ⅰ). Subsequently, Li+ dissociates from TFSI− under strong interaction with the CF2 chain in PF–COF, where it undergoes as an intermediate state (Ⅱ) and moves to the adjacent PEO chain. Thirdly, Li+ hops along the alternating structure and forms the Li–O coordination (Ⅲ)41. Subsequently, the Li+ repeatably migrates back to another PEO and CF2 centers and returns to state Ⅰ, indicating the PF–COF electrolyte has continuous channels for Li+ transport. Therefore, this pore modification by alternative sequencing of PEO and fluoroalkyl chains in the COF electrolyte can create a site-to-site continuous pathway for Li+ migration.
Interfacial stability of Li–TA–COF and Li–PF–COF with Li metal
Smooth and uniform surface of Li anode in Li|Li–PF–COF|Li battery after cycling is observed in SEM image (Fig. 6a), revealing Li–PF–COF electrolyte can suppress the formation of Li dendrites and promote uniform Li deposition. However, obvious Li dendrites and dead Li are observed on the surface of the cycled Li anode by SEM in Li|Li–TA–COF|Li (Fig. 6c). The SEI components of the cyclic Li anode are analyzed by HRTEM. Compared with Li–TA–COF (Fig. 6d), the deposits in Li–PF–COF are much more abundant (Fig. 6b), which is attributed to LiF and Li2O nanoparticles. Furthermore, the lattice of LiF and Li2O nanoparticles can be measured to be 0.23 (Fig. 6e) nm and 0.26 nm (Fig. 6f), corresponding to the (111) planes46. These inorganic species are important and beneficial components of SEI. By contrast, the lattice is indistinguishable in Li–TA–COF (Fig. 6g, h), suggesting its weak capability of restraining dendrites. In addition, the SEI structure is further revealed by time-of-flight secondary ion mass spectrometry (ToF-SIMS). Some typical fragments of LiF−, Li2O−, and Li2–CO2− are analyzed. As demonstrated in Fig. 6i, j, the spatial distributions of LiF−, Li2O−, and Li2–CO2− are revealed by the depth profile of ToF-SIMS47. It is evident that the contents of LiF−, Li2O−, Li2–CO2−, and corresponding depth profiles in Li–PF–COF-based battery are higher than Li–TA–COF-based battery. The robust SEI plays a crucial role in stabilizing the interface between the solid-state electrolyte and the Li anode. Surface species of cycled Li metal anode are further evaluated by XPS. From Fig. 6k–n, there is a higher amount of LiF, Li2O, and Li2–CO2 in Li–PF–COF than in Li–TA–COF48, which is in high accordance with ToF-SIMS analyses. Therefore, benefiting from the structure of alternative sequencing of PEO and fluoroalkyl chains, the Li–PF–COF solid-state electrolyte can generate a robust SEI. Consequently, Li dendrite is efficiently suppressed, and cycling stability is obtained in a Li–PF–COF solid-state battery.

SEM (a) and TEM (b) image of Li anode in Li–PF–COF assembled battery; SEM (c) and TEM (d) image of Li anode in Li–TA–COF assembled battery; HRTEM image of LiF (e) and Li2O (f) in Li–PF–COF assembled battery; HRTEM image of LiF (g) and Li2O (h) in Li–TA–COF assembled battery; three-dimensional and spatial structures of Li2–CO2−, Li2O−, LiF− (i) and corresponding depth profiles (j) by ToF–SIMS in Li–PF–COF and Li–TA–COF assembled batteries; HRXPS spectra of C1s (k), Li 1s (l), O 1s (m), and F 1s (n) in Li–PF–COF and Li–TA–COF assembled batteries.
Performances of Li||NCM811 solid-state battery
Li–PF–COF solid-state electrolyte film is prepared, and flexibility is demonstrated (Fig. 7a). The flexible property may reduce the interfacial resistance due to good contact between electrolyte and electrodes. Moreover, the nanoconfined PC solvents within the COF nanochannels create continuous pathways that enable high Li+ conductivity, while the inherent flexibility of the COF membrane, combined with applied stack pressure, ensures conformal physical contact with the electrodes. This synergy between rapid bulk ion transport and intimate interfacial contact achieves effective electrochemical wetting, leading to low interfacial resistance and stable cycling in solid-state batteries. The thickness of Li–PF–COF solid-state electrolyte film is measured to be ~118 μm (Supplementary Fig. 40). In addition, Li–PF–COF solid-state electrolyte film shows stronger tensile strengths than Li–TA–COF (Supplementary Fig. 41). The infinitesimal PC molecule in Li–PF–COF solid-state electrolyte film is determined to be 6 wt% by TGA (Supplementary Fig. 42). Li|Li–PF–COF | NCM811 shows specific capacity with 204 mAh g−1 at 0.2 C, 192 mAh g−1 at 0.5 C, 180 mAh·g−1 at 1 C, 160 mAh g−1 at 2 C, 134 mAh g−1 at 3 C, 119 mAh g−1 at 5 C (C = 200 mA g−1), respectively. Especially, the specific capacity at high current density of 5 C is dramatically higher than Li|Li–TA–COF | NCM811 (100 mAh g−1) and Li|liquid|NCM811 (92 mAh g−1), as shown in Fig. 7b. Rate capability of Li|Li–PF–COF|NCM811 (Fig. 7c) is ascribed to a higher Li+ transfer number than Li|Li–TA–COF|NCM811 (Supplementary Fig. 43) and Li|liquid|NCM811 (Supplementary Fig. 44).

Digital photograph of Li–PF–COF solid-state electrolyte film (a); rate performances of Li||NCM811 batteries by various electrolytes (b); charge/discharge curves at various current density of Li|Li–PF–COF|NCM811 (c), (C = 200 mA g−1); cycling stability of Li||NCM811 batteries by various electrolytes at 1 C (d); charge/discharge curves at various cycles of Li|Li–PF–COF|NCM811 (e); SEM images of cycling NCM811 material in liquid battery (f), Li–TA–COF (g) and Li–PF–COF (h); cycling stability of Li||NCM811 battery based on Li–PF–COF electrolyte at 5 C (i); comparison of Li||NCM811 performance with reported works from refs. 49,50,51,52,53,54,55,56 (j); the Li||NCM811 pouch cell (monolayer, mass loading: 9.5–10 mg cm2, N/P:1.15–1.2, C = 200 mA g−1) with Li–PF–COF solid-state electrolyte (k).
The cycling performance of assembled batteries is assessed by a charging/discharging test at 1 C. Li|Li–PF–COF|NCM811 shows a capacity retention of 93% after 100 cycles and 82% after 200 cycles at room temperature (Fig. 7d, e). By contrast, Li|Li–TA–COF|NCM811 exhibited a poor capacity retention of 72% after 130 cycles, and Li|liquid|NCM811 has low-capacity retention of 40% after 100 cycles (Fig. 7d). The favorable cycling stability and high discharge capacity of Li|Li–PF–COF|NCM811 are ascribed to its intrinsic structure, which effectively improves the ionic conductivity, transfer number, and interfacial stability.
For Ni-rich materials, the capacity fading is mainly caused by the microcracks during the H2–H3 phase transition. Microcracks create pathways for electrolyte molecules to attack and accelerate the NCM811 cathode material degradation, resulting in fast capacity fading. Therefore, the Li–PF–COF polymer electrolyte can effectively slow down the microcrack formation and improve the cycling stability in the solid-state batteries. In order to study the microcracks in the battery systems of Li|Li–PF–COF|NCM811, Li|Li–TA–COF|NCM811 and Li|liquid|NCM811, the microstructure of NCM811 materials after cycling is measured by SEM. Compared with Li|liquid|NCM811 (Fig. 7f), the microcracks are rarely observed in Li|Li–TA–COF|NCM811 (Fig. 7g), implying the microcrack formation is reduced in solid-state batteries. More importantly, it is obvious that microcrack is not generated in Li|Li–PF–COF|NCM811 battery (Fig. 6h), which is presented in Li|Li–TA–COF|NCM811 (Fig. 7g). Li–PF–COF solid-state electrolyte creates a favorable environment which limits anionic migration, avoids Li+ aggregation, provides site-to-site Li+ migrated pathways, and promotes uniform deposition. Especially, the ordered Li+ migration and deposition can timely replenish the Li+ in the Li layer of NCM811, which stabilizes the layered crystal structure, resulting in cycling performance. For a solid-state NCM811 battery, the fast-charging performance will play a crucial role in electric vehicles and hold great promise for commercialization.
Fast-charging capability is a critical requirement for solid-state batteries targeting electric vehicle applications and represents a major focus of current research. Significant progress has been made in enhancing the rate capability of solid-state systems utilizing various electrolyte classes (e.g., sulfides, oxides, polymers). However, achieving stable, high-capacity cycling under high current densities (≥5 C) with high-nickel cathodes like NCM811 remains particularly challenging, primarily due to exacerbated interfacial degradation, lithium plating risks, and insufficient ionic kinetics under polarization. Reports demonstrating long-term cyclability (>500 cycles) with capacity retention at these rates are comparatively scarce. The Li|Li–PF–COF|NCM811 delivers high discharge capacity at 5 C. Moreover, the capacity retention can be maintained up to 80.5% after 700 cycles (Fig. 7i), suggesting fast-charging capacity of Li|Li–PF–COF|NCM811 solid-state battery. More importantly, as the fast-charging behavior is rarely studied and reported, Li|Li–PF–COF|NCM811 demonstrates promising potential for practical application. It is worth noting that the capacity of the Li|NCM811 battery assembled by Li–PF–COF solid-state electrolyte is markedly higher than reported polymer electrolyte49,50,51,52,53,54,55,56 (Fig. 7j, Supplementary Table 3). Therefore, the above results indicate that alternative sequencing of PEO and fluoroalkyl chains in COF electrolytes has great potential in solid-state batteries. Finally, we evaluate Li–PF–COF as a solid-state electrolyte in practical Li||NCM811 pouch cells, which delivered initial capacity and cycling stability—94% capacity retention after 120 cycles at 0.5 C and room temperature (Supplementary Fig. 45). The cell also withstood severe mechanical abuse such as folding and twisting without failure, indicating flexibility and robust interfacial contact. The structural integrity of the pore-functionalized PF–COF—evidenced directly by the retention of its crystalline framework and chemical bonding post-cycling and indirectly by long-term performance—is maintained despite a favorable morphological reorganization during membrane processing, which collectively underpins its electrochemical durability (Supplementary Fig. 46). These results demonstrate that Li–PF–COF combines high electrochemical performance with mechanical resilience, making it a highly promising candidate for real-world solid-state batteries, including in flexible form factors.
To sum up, we propose a strategy of pore modification by alternative sequencing of PEO and fluoroalkyl chains in the COF electrolyte to boost the performance of a solid-state Li||NCM811 battery at high current density with interfacial stability for the Li metal anode. Due to the strong electron-withdrawing of C–F2, the Li–PF–COF delivers a positive charge state and effectively adsorbs TFSI−, leading to highly-selective Li transference (0.9) and high ionic conductivity of 1.06 × 10−3 S/cm. Relevant interaction mechanisms between COF materials and ions (Li+ and TFSI−) are thoroughly studied by molecular dynamics simulations, FT-IR, and 7Li NMR. The interfacial regulation capability of Li–PF–COF electrolyte ensures the assembled solid-state lithium metal symmetrical battery with cycling stability (7500 h at 0.2 mA cm−2) with low polarization voltage. Moreover, the assembled solid-state Li||NCM811 battery demonstrates high specific capacity (180 mAh g−1 at 1 C) and fast-charge performance (700 cycles with 80% at 5 C). We provide an encouraging method for fast-charging solid-state batteries realized via functional COF materials.
Methods
Synthesis of TA–COF
The synthesis of TA–COF was conducted following the protocol illustrated in Supplementary Fig. 13. Precise amounts of TFPB (39 mg, 0.10 mmol) and TA (29.1 mg, 0.15 mmol) were transferred to a Pyrex tube. The mixed precursors were dissolved in a 1:1 (v/v) solvent mixture of 1,4-dioxane and mesitylene (3 mL total volume), followed by ultrasonic homogenization for 10 min. Subsequently, glacial acetic acid (0.2 mL, 6.0 M) was added, and a second ultrasonic treatment (10 min) was carried out to ensure complete dissolution. The homogeneous solution was rapidly frozen in liquid nitrogen under vacuum (0 mbar) before flame-sealing the Pyrex tube. Thermal polymerization was conducted at 120 °C under static conditions for 72 h following gradual thermal equilibration to 25 °C. Post-synthesis processing involved mechanical opening of the sealed tube followed by centrifugal isolation of the crude product. Sequential purification included three wash cycles (50 mL × 3) with acetone and prolonged immersion in anhydrous acetone (12 h) with solvent exchange at 3 h intervals. Final activation was achieved through vacuum desiccation at 80 °C for 12 h, yielding TA–COF as a crystalline yellow powder.
Synthesis of PF–COF
The synthesis of PF–COF was conducted following the protocol illustrated in Supplementary Fig. 14. Precise amounts of TFPB (39 mg, 0.10 mmol) and PF (92.7 mg, 0.15 mmol) were transferred to a Pyrex tube. The mixed precursors were dissolved in mesitylene (3 mL), followed by ultrasonic homogenization for 10 min. Subsequently, glacial acetic acid (0.4 mL, 6.0 M) was added, and a second ultrasonic treatment (10 min) was carried out to ensure complete dissolution. The homogeneous solution was rapidly frozen in liquid nitrogen under vacuum (0 mbar) before flame-sealing the Pyrex tube. Thermal polymerization was conducted at 120 °C under static conditions for 72 h following gradual thermal equilibration to 25 °C. Post-synthesis processing involved mechanical opening of the sealed tube followed by centrifugal isolation of the crude product. Sequential purification included three wash cycles (50 mL × 3) with acetone and prolonged immersion in anhydrous acetone (12 h) with solvent exchange at 3 h intervals. Final activation was achieved through vacuum desiccation at 80 °C for 12 h, yielding PF–COF as a crystalline yellow powder.
Preparation of COFs solid-state electrolyte film
The TA–COF/PF–COF samples (100 mg) were immersed in 20 mL of a 1 M LiTFSI solution in THF and kept at 25 °C for 12 h. After filtration, the collected solids were vacuum-dried at 80 °C for 12 h. The resulting Li+-incorporated materials were designated as Li–TA–COF and Li–PF–COF–Li, respectively. To prepare the COF-based solid-state electrolyte membrane, the COF material was homogeneously blended with a 1 wt% polytetrafluoroethylene (PTFE) solution (60 wt% dispersion in H2O). The mixture was ground for 20 min until a dough-like consistency was achieved, followed by rolling into a freestanding membrane. The membrane was punched into discs (16 mm in diameter, mass loading:17.5 mg cm–2) and activated by soaking in a 1 M LiTFSI solution (dissolved in PC) for 12 h. Subsequently, the discs were dried at 80 °C for 12 h to eliminate residual moisture. The final SSE membranes (thickness: ~118 μm, PC residue of 6 wt% measured by TGA) were stored in an argon-filled glove box for subsequent use.
Preparation of NCM811 solid-state battery
NCM811 cathodes were fabricated by blending NCM811 powder, Super P, and poly(vinylidene fluoride) at a mass ratio of 90:5:5 in N-methyl-2-pyrrolidone to obtain a uniform slurry. This slurry was cast onto aluminum foil and dried under vacuum at 80 °C for 10 h. The dried electrodes were cut into discs with a diameter of 14.0 mm, yielding an active material loading of approximately 6.0 mg cm−2. Solid-state cells with a Li|COF|NCM811 configuration were assembled using the prepared NCM811 cathode, COF-based solid-state electrolyte separator, and Li anode (thickness: 450 μm). Galvanostatic charge/discharge cycling was performed at 30 °C on a Neware CT-4008-5V50mA-164 test system within a voltage window of 2.7–4.3 V under different current densities. For reference, the C-rate in this work is defined relative to a specific current of 200 mA g−1 (1 C = 200 mA g−1). All reported electrochemical data and related discussions are based on this specification.
Data availability
Source data are provided with this paper.
References
Yang, J. et al. Oxygen- and proton-transporting open framework ionomer for medium-temperature fuel cells. Science 385, 1115–1120 (2024).
Li, X. et al. Interface preassembly oriented growth strategy towards flexible crystalline covalent organic framework films for OLEDs. Nat. Commun. 16, 3321 (2025).
Wu, J., Zhang, S., Gu, Q. & Zhang, Q. Recent progress in covalent organic frameworks for flexible electronic devices. FlexMat 1, 160 (2024).
Liu, X. et al. Polyimide covalent organic frameworks as efficient solid-state Li+ electrolytes. Sci. China Chem. 67, 1647–1652 (2024).
Zhang, X. et al. Topology fortified anodes powered high-energy all-solid-state lithium batteries. Adv. Mater. 37, 2506298 (2025).
Zhang, W. et al. Single-phase local-high-concentration solid polymer electrolytes for lithium-metal batteries. Nat. Energy 9, 386–400 (2024).
Song, Z. et al. A reflection on polymer electrolytes for solid-state lithium metal batteries. Nat. Commun. 14, 4884 (2023).
Liu, Y. et al. Achieving a high loading of cathode in PVDF-based solid-state battery. Energy Environ. Sci. 17, 344–353 (2024).
Zhang, S. et al. Cubic iodide LixYI3+x superionic conductors through defect manipulation for all-solid-state Li batteries. Angew. Chem. Int. Ed. 63, e202316360 (2024).
Kmiec, S., Ruoff, E. & Manthiram, A. A new class of oxyhalide solid electrolytes NaNbCl6-2xOx for solid-state sodium batteries. Angew. Chem. Int. Ed. 64, e202416979 (2024).
Wang, G. et al. Oxychloride polyanion clustered solid-state electrolytes via hydrate-assisted synthesis for all-solid-state batteries. Adv. Mater. 37, 2410402 (2024).
Li, H. et al. A cost-effective sulfide solid electrolyte Li7P3S7.5O3.5 with low density and excellent anode compatibility. Angew. Chem. Int. Ed. 63, e20240789 (2024).
Li, R. et al. The deconstruction of a polymeric solvation cage: a critical promotion strategy for PEO-based all-solid polymer electrolytes. Energy Environ. Sci. 17, 5601–5612 (2024).
Zheng, G. et al. Anion-mediated interphase construction enabling high-voltage solid-state lithium metal batteries. Nano Energy 125, 109617 (2024).
Su, X. et al. Polyethylene oxide-based composite solid electrolytes for lithium batteries: current progress, low-temperature and high-voltage limitations, and prospects. Electrochem. Energy Rev. 7, 2 (2024).
Jia, Z. et al. Configuration design toward sustainably-released polymer electrolytes for enhancing ionic transport and cycle stability of solid lithium batteries. Energy Storage Mater 68, 103325 (2024).
Wang, S. et al. Organic cationic-coordinated perfluoropolymer electrolytes with strong Li+-solvent interaction for solid state Li-metal batteries. Angew. Chem. Int. Ed. 63, e202412434 (2023).
Chen, Y. et al. Monodispersed sub-1 nm inorganic cluster chains in polymers for solid electrolytes with enhanced Li-ion transport. Adv. Mater. 35, 2303226 (2023).
Song, Z. et al. Designer anions for better rechargeable lithium batteries and beyond. Adv. Mater. 36, 2310245 (2024).
Wang, S. et al. Highly conjugated three-dimensional covalent organic frameworks with enhanced Li-ion conductivity as solid-state electrolytes for high performance lithium metal batteries. J. Mater. Chem. A 10, 8761 (2022).
Antonopoulou, M. et al. Concurrent control over sequence and dispersity in multiblock copolymers. Nat. Chem. 14, 304–312 (2022).
Wang, S. et al. Highly elastic energy storage device based on intrinsically super-stretchable polymer lithium-ion conductor with high conductivity. Fundam. Res. 4, 140–146 (2024).
Shi, P. et al. Symmetric variation of monomers for constructing 1D/2D imine-based covalent organic frameworks. CCS Chem. 6, 941–952 (2024).
Niu, C., Zhao, S. & Xu, Y. In situ gelled covalent organic frameworks electrolyte with long-range interconnected skeletons for superior ionic conductivity. J. Am. Chem. Soc. 146, 3114 (2024).
Li, Z. et al. Olefin-linked covalent organic frameworks with electronegative channels as cationic highways for sustainable lithium metal battery anodes. Angew. Chem. Int. Ed. 62, e202307459 (2023).
Gong, W. et al. Covalent organic framework with multi-cationic molecular chains for gate mechanism controlled superionic conduction in all-solid-state batteries. Angew. Chem. Int. Ed. 62, e202302505 (2023).
Jiang, G. Lithium-ion accelerated regulators by locally-zwitterionic covalent organic framework nanosheets. Adv. Energy Mater. 14, 2303672 (2024).
Guo, Z. et al. Fast ion transport pathway provided by polyethylene glycol confined in covalent organic frameworks. J. Am. Chem. Soc. 141, 1923 (2019).
Du, Y. et al. Ionic covalent organic frameworks with spiroborate linkage. Angew. Chem. Int. Ed. 55, 1737 (2016).
Chen, H. et al. Cationic covalent organic framework nanosheets for fast Li-ion conduction. J. Am. Chem. Soc. 140, 896 (2018).
Jeong, K. et al. Solvent-free, single lithium-ion conducting covalent organic frameworks. J. Am. Chem. Soc. 141, 5880 (2019).
Li, X. et al. Solution-processable covalent organic framework electrolytes for all-solid-state Li−organic batteries. ACS Energy Lett. 5, 3498 (2020).
Li, Z. et al. Cationic covalent organic framework based all-solid-state electrolytes. Mater. Chem. Front. 4, 1164 (2020).
Niu, C., Luo, W., Dai, C., Yu, C. & Xu, Y. High-voltage-tolerant covalent organic framework electrolyte with holistically oriented channels for solid-state lithium metal batteries with nickel-rich cathodes. Angew. Chem. Int. Ed. 60, 24915 (2021).
Liu, J. et al. Cationic covalent organic framework with ultralow HOMO energy used as scaffolds for 5.2 V solid polycarbonate electrolytes. Adv. Sci. 9, 2200390 (2022).
Guo, D. et al. Foldable solid-state batteries enabled by electrolyte mediation in covalent organic frameworks. Adv. Mater. 34, 2201410 (2022).
Woog Kang, T. et al. An ion-channel-restructured zwitterionic covalent organic framework solid electrolyte for all-solid-state lithium-metal batteries. Adv. Mater. 35, 2301308 (2023).
Lee, J.-H. et al. Multicomponent covalent organic framework solid electrolyte allowing effective Li-ion dissociation and diffusion for all-solid-state batteries. ACS Nano 17, 17372 (2023).
Li, W. et al. High-voltage single-ion covalent organic framework electrolytes enabled by nitrile migration ladders for lithium metal batteries. Angew. Chem. Int. Ed. 63, e202410392 (2024).
Wu, X. et al. Perfluoroalkyl-functionalized covalent organic frameworks with superhydrophobicity for anhydrous proton conduction. J. Am. Chem. Soc. 142, 14357–14364 (2020).
Han, S. et al. Sequencing polymers to enable solid-state lithium batteries. Nat. Mater. 22, 1515–1522 (2023).
Zhang, W. X. et al. Molecularly tunable polyanions for single-ion conductors and poly(solvate ionic liquids). Chem. Mater. 33, 524–534 (2021).
Shi, J. et al. An amphiphilic molecule-regulated core-shell-solvation electrolyte for Li-metal batteries at ultra-low temperature. Angew. Chem. Int. Ed. 62, e202218151 (2023).
Hu, C. et al. Superionic conductors via bulk interfacial conduction. J. Am. Chem. Soc. 142, 18035–18041 (2020).
Zhao, G. et al. Constructing donor–acceptor‑linked COFs electrolytes to regulate electron density and accelerate the Li+ migration in quasi‑solid‑state battery. Nano-Micro Lett 17, 21 (2025).
Liu, Y. et al. Self-assembled monolayers direct a LiF-rich interphase toward long-life lithium metal batteries. Science 375, 739 (2022).
Zhang, Q.-K. et al. Homogeneous and mechanically stable solid–electrolyte interphase enabled by trioxane-modulated electrolytes for lithium metal batteries. Nat. Energy 8, 725–735 (2023).
Ma, M. et al. Reactive solid polymer layer: from a single fluoropolymer to divergent fluorinated interface. Angew. Chem. Int. Ed. 63, e202407304 (2024).
Huang, Y. et al. A relaxor ferroelectric polymer with an ultrahigh dielectric constant largely promotes the dissociation of lithium salts to achieve high ionic conductivity. Energy Environ. Sci. 14, 6021–6029 (2021).
Ma, Y. et al. Competitive Li-ion coordination for constructing a three-dimensional transport network to achieve ultra-high ionic conductivity of a composite solid-state electrolyte. Energy Environ. Sci. 17, 8274 (2024).
Xia, S. et al. Ultrathin layered double hydroxide nanosheets enabling composite polymer electrolyte for all-solid-state lithium batteries at room temperature. Adv. Funct. Mater. 31, 2101168 (2021).
Li, M. et al. Ion−dipole-interaction-induced encapsulation of free residual solvent for long-cycle solid-state lithium metal batteries. J. Am. Chem. Soc. 145, 25632–25642 (2023).
Pan, J. et al. A quasi-double-layer solid electrolyte with adjustable interphases enabling high-voltage solid-state batteries. Adv. Mater. 34, 2107183 (2022).
Yang, K. et al. Stable interface chemistry and multiple ion transport of composite electrolyte contribute to ultra-long cycling solid-state LiNi0.8Co0.1Mn0.1O2/lithium metal batteries. Angew. Chem. Int. Ed. 60, 24668–24675 (2021).
Shi, P. et al. A dielectric electrolyte composite with high lithium-ion conductivity for high-voltage solid-state lithium metal batteries. Nat. Nanotechnol. 18, 602–610 (2023).
Yang, W. et al. Solvation-tailored PVDF-based solid-state electrolyte for high-voltage lithium metal batteries. Angew. Chem. Int. Ed. 63, e202401428 (2024).
Acknowledgements
The authors would like to acknowledge financial support provided by National Natural Science Foundation of China (52303283, 52372232), the Major Science and Technology Projects of Yunnan Province (202403AA080019, 202302AB080019-3), Natural Science Foundation of Yunnan Province (202301AS070040, 202401AU070201), the Analysis and Measurements Center of Yunnan University for the sample testing service, the Electron Microscope Center of Yunnan University for the support of this work. The authors would like to thank the Shiyanjia Lab (www.shiyanjia.com) for the XPS test. Prof. C. Wang would like to acknowledge the financial support from the National Natural Science Foundation of China (Grant Nos. W2441017 and 22409103), the “Innovation Yongjiang 2035” Key R&D Program (Grant Nos. 2024Z040 and 2025Z063).
Author information
Authors and Affiliations
Contributions
G.F. contributed to all experiments, data curation, original draft—writing, and conceptualization; M.Y. synthesized the COF. Z.H., S.Y., H.Y., and Y.J. performed the morphology characterization and analysis; C.H. and Y.P. reviewed and revised the paper; H.G. contributed to conceptualization, review and supervision. All authors commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41467_2026_70591_MOESM3_ESM.zip (download ZIP )
Supplementary Data 1, Supplementary Data 2, Supplementary Data 3, Supplementary Data 4, Supplementary Data 5, Supplementary Data 6
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, G., Yang, M., Zhang, Z. et al. Alternating-sequence polymer chain facilitating Li+ transport in covalent organic frameworks. Nat Commun 17, 2442 (2026). https://doi.org/10.1038/s41467-026-70591-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70591-0
