
Abstract
Coherent driving of optical transitions in matter using laser fields has been established as an important paradigm in quantum optics, quantum information processing and Floquet engineering. Recent studies extended coherent driving to solution-processed materials such as lead halide perovskites and colloidal semiconductor nanocrystals. Such systems, however, often feature broad and continuous absorption profiles that limit the studies mostly to below-bandgap driving, while a comprehensive understanding of coherent light-matter interaction requires both nonresonant and resonant driving. Here we utilize magic-size clusters of Cd3P2 in the limit of extreme quantum confinement to solve these issues. Their sharp, well-isolated and linearly-polarized band-edge exciton transition allows to coherently drive it at resonance, producing a Mollow-like lineshape using cross-polarized transient absorption measurements, which is also captured by our quantum simulation using the density matrix method. Analyzing the spectral responses under both nonresonant and resonant driving consistently yields a transition dipole moment exceeding 20 Debye.
Similar content being viewed by others

Introduction
A long-lasting theme in the field of light-matter interaction is coherent driving of the matter states by laser fields. Such a topic has experienced different phases of research interest and motivation in the past decades, from early efforts of understanding the fundamental principles of light-matter interaction1,2 to recent interest in quantum information processing and quantum light sources3,4, and to the emerging field of Floquet engineering of nonequilibrium matter states5,6. The subject of research has also been greatly expanded in this journey. Early studies mostly focused on atoms and molecules in gas phase7,8, which possess sharp optical transitions that are particularly suitable for optical driving and detection. The experiments and principles were then translated to studies of artificial systems with discrete transitions, such as epitaxially grown III-V group quantum dots (QDs; also called “artificial atoms”) and quantum wells (QWs) under cryogenic temperatures9,10,11,12. More lately, monolayer or few-layer van der Waals materials have also been established as a versatile platform to explore various types of coherent driving phenomena, including but not limited to, valley-selective optical Stark effect (OSE)13,14, biexcitonic OSE15,16, Autler-Towns splitting (ATS) under intraexciton driving17, quantum interference18, and Floquet engineering of band structures19,20,21,22.
A highly interdisciplinary field emerging in the past few years is coherent driving of solution-processed semiconductor materials, including lead halide perovskites and colloidal semiconductor nanocrystals23,24,25,26,27,28,29,30,31. These materials allow for conducting coherent driving experiments in ambient conditions at room temperature, which is of direct relevance to applications such as optoelectronic devices and photochemical reactions29. However, due to the broad and continuous optical transitions in these systems, these experiments were mostly restricted to observing OSE under non-resonant driving by negatively detuned laser pulses such as to avoid direct excitation of continuous transitions23,24,25,26,27. A typical observation is that the lowest energy transition is transiently blue shifted by the driving pulse, whereas interesting physics beyond this phenomenon remains to be explored. Although colloidal QDs have relatively narrow and isolated transitions, their room temperature absorption profiles are often still dominated by strongly overlapped manifolds of transitions24,25,26. In a recent study, colloidal nanoplatelets with three-level-like energy structure resembling that of QWs were used to demonstrate the ATS line shapes, which entails resonant/nonresonant driving of the “empty” intersubband transition using near-infrared pulses and probing of the interband absorption changes using visible pulses28.
Much richer physics will emerge if a two-level system can be driven both nonresonantly and resonantly. Beyond a laser-induced spectral shift, exotic phenomena such as Mollow triplet emission profile32, quantum interference33 and gain without inversion34,35 can be observed. This has been nicely illustrated in a classical study on epitaxial QDs at 5 K12; while strong driving of an exciton transition and probing of another coupled exciton transition resulted in expected ATS lineshapes, driving and probing the same exciton transition gave complex Mollow-like absorption profiles that include “negative” absorption region (i.e., optical gain). This optical gain originates from coherent exchange of energy in the pump and probe fields through the commonly coupled QD exciton transition, and it should be regarded as gain without inversion, as there is no population inversion in this system12. Inspired by these studies, a solution-processed material system that could be examined for comprehensive driving experiments at room temperature should satisfy the following criteria: (i) it should have narrow and well-isolated band-edge exciton transitions almost free from inhomogeneous broadening; (ii) it should have a large transition dipole moment that facilitates the realization of strong driving; and (iii) ideally the transition is highly polarized that enables rejection of unwanted signals (e.g., scattering) through manipulation of laser polarizations.
A potential candidate that satisfies the above requirements is the family of magic-size clusters (MSCs)36,37,38,39,40,41,42,43. Their atomically precise structures essentially eliminate inhomogeneous broadening encountered in typical colloidal nanocrystals39,43,44, and their sizes (typically <2 nm) are situated in a stronger confinement regime than typical QDs38,41,42. These features lead to molecular-like discrete electronic levels with sharp and well-isolated optical transitions at the ensemble level. Moreover, in this extreme confinement regime, even slight symmetric breaking will cause highly anisotropic, linearly polarized transitions, which has already been exploited to demonstrate aggregation-induced macroscopic chirality45,46,47. Here we choose extremely-confined Cd3P2 MSCs, with strong optical transition in the visible region, to investigate their transient optical responses under resonant and nonresonant driving conditions using femtosecond pump-probe transient absorption (TA) spectroscopy at room temperature. We observe not only the typical blue-shifted OSE lineshape under negatively detuned driving, but also peak splitting near time zero under resonant driving. A Mollow-like lineshape is extracted by a global fitting procedure, which is also successfully captured by our quantum simulation using the density matrix approach. Importantly, in the resonant case, the splitting of the Rabi sidebands scales linearly with the electric field strength, which is distinct from nonresonant OSE with energy shift scaling linearly with the light intensity. Resonant and nonresonant driving conditions consistently give a large transition dipole moment exceeding 20 Debye which underlies the observed strong optical responses.
Results
Principles and system design
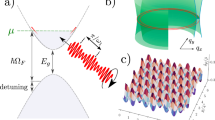
Figure 1 summarizes the principles of coherent interaction between a two-level system (comprising the excited state |e〉 and the ground state |g〉 separated by ћωeg) and a driving light field with photon energy of ћωp. The coherent absorption and emission of photons yield a series of photon-dressed states (also called Floquet replica states), and those with strongest interaction with the equilibrium matter states are denoted as |e - ћωp〉 and |g + ћωp〉, as indicated in the figure. With a negatively detuned driving photon (i.e., Δ = ћωp - ћωeg < 0), the photon-dressed states hybridize with the matter states to produce a blue shifted transition that carries the major oscillator strength of the original matter states (Fig. 1a), thereby generating a derivative-like TA spectrum (Fig. 1b). When ћωp becomes in resonance with ћωeg, the photon-dressed states are in degeneracy with the matter states, resulting in strongest hybridization. The results are four levels that are half-light and half-matter in nature (Fig. 1c). The absorption profile should consist of three lines similar to the Mollow triplet in emission, with the strength of the center band equaling the sum of the two Rabi sidebands (Fig. 1d). A hallmark feature of this resonant driving scheme is that the splitting of these two Rabi sidebands is proportional to the electric field strength12, whereas for large-detuned nonresonant driving, the spectral shift is approximately proportional to the light intensity23,24,25,27. Finally, when ћωp exceeds ћωeg, the light-matter interaction becomes similar to the negatively detuned driving condition, except that the transition that carries the major oscillator strength is now a redshifted one (Fig. 1e). The TA spectrum is also derivative-like (Fig. 1f). A realistic complexity associated with resonant and positively detuned driving is excitation of real populations that gives large ground-state bleach and excited-state absorption signals overwhelming the coherent driving signals. Due to this issue, such experiments have never been conducted for solution-processed semiconductor materials.

a Negatively detuned driving photon (ћωp <ћωeg) induces a blue shifted energy arising from the hybridization of the photon-dressed states (dashed line) and the matter state (solid line). e Positively detuned driving photon (ћωp > ћωeg) generates a redshifted one. c Zero-detuned driving photon produces Rabi splitting resulting in three types of transitions profiles. The Gaussian line shapes (upper panels) are used to represent the absorption profile of band-gap exciton and then the differential spectra before and after energy shifted can be derived (lower panels), corresponding to blue-shifted (b), Rabi splitting (d), and red-shifted (f) cases, respectively.
We note that the spectral lineshape illustrated for resonant driving in Fig. 1d is a simplified one without considering the interference effect. In this scheme, the strong driving pulse couples to the transition and modifies the energy levels, whilst the weak probe pulse serves as a spectator to measure the modified transitions. In a complete picture, however, the electric field (ε) that interacts with the transition dipole (μ) is a sum of the driving and probe fields (ε = εd + εp)12. This is the fundamental reason that the pump and probe fields can coherently exchange energy through interacting with the common transition, resulting in complex interference spectral lineshapes12.
Cd3P2 MSCs were chosen for the present study as the small bandgap of bulk Cd3P2 (~0.55 eV)48,49 allows the extremely-confined MSCs to still absorb in the visible spectral range that can be facilely accessed by pump-probe TA spectroscopy, whereas CdS and CdSe MSCs commonly studied in the synthesis literature typically absorb in the ultraviolet spectral range38,39,44. Colloidal Cd3P2 MSCs were synthesized following the method reported in the literature50, as detailed in the Methods section. Figure 2a presents the steady-state absorption spectrum of Cd3P2 MSCs dispersed in hexane, which is dominated by a sharp exciton peak at ~2.75 eV. This peak is almost completely isolated from higher-energy absorption bands, which is distinct from the spectra of typical colloidal QDs. The photoluminescence (PL) spectrum of Cd3P2 MSCs also shows a sharp peak centered at ~2.70 eV (Fig. 2b).

a Steady-state absorption spectrum, with the band-edge exciton peak at ~2.75 eV. The spectra of the driving pulses (shaded pulses) are included for comparison. b Steady-state photoluminescence (PL) spectrum; inset shows polarization angle-dependent PL intensity at 2.69 eV with linearly polarized pump energy of 2.88 eV.
Previous transmission electron microscope (TEM) characterization of Cd3P2 MSCs gave a diameter <2 nm, which is complicated by the issue that electron beams can induce cluster aggregation51. Even 2 nm is already 20-fold smaller than the Bohr exciton diameter of ~40 nm for bulk Cd3P249. Reliable determination of the exact structure and morphology of these Cd3P2 MSCs remains challenging. Here we carried out pair distribution function (PDF) analysis of Cd3P2 MSCs using synchrotron X-ray total scattering (see Methods). As shown in Supplementary Fig. 1, the PDF in general resembles that of α-Cd37S20 MSCs reported in the literature44, which suggests that the size and structure of these two types of MSCs are similar. The α-Cd37S20 MSCs were reported to take an anisotropic shape that leads to linearly polarized transition dipoles52. Such a property enables the formation of chiral assemblies from these achiral MSCs45. In principle, such a linearly polarized transition should also benefit our coherent driving experiments, which leads us to first investigate the polarization characteristics of our Cd3P2 MSCs.
Identification of linearly-polarized exciton transition
We first measured the PL anisotropy of Cd3P2 MSCs by rotating a polarizer placed in front of the detector. As shown in the inset of Fig. 2b, when these MSCs are excited by linearly polarized light (at 2.88 eV), the PL intensity measured at 2.69 eV is strongly dependent on the polarization angle, consistent with a linear polarization of the band edge exciton. Considering that the emissive state is Stokes-shifted than the absorptive state, absorption anisotropy should be more relevant to our coherent driving experiments than emission anisotropy. However, unlike PL measurements in which we can select a subset of MSCs with their transition dipoles aligned along the electric field direction of the excitation light, direct measurements of absorption anisotropy would not work due to the random orientation of the MSCs in the ensemble. Instead, we can conduct absorption anisotropy measurements of Cd3P2 MSCs using polarization-controlled femtosecond pump-probe TA spectroscopy in which the linearly polarized pump beam can also select a subset of MSCs with their transition dipoles aligned along the electric field direction of the pump beam53,54,55; see Methods for details. The linearly polarized laser pulses are labeled as V (vertical) and H (horizontal), and VV and VH stand for experiments with mutually parallel and perpendicular pump-probe polarizations, respectively. The pump photon energy is at 2.82 eV and the pump pulse energy is controlled low (~7 μJ cm-2). Figures 3a, b present the VV and VH TA spectra, respectively, at indicated pump-probe delays from 1 ps to 1 ns. The spectra can be understood as a bleach of the lowest energy exciton peak superimposed on a broad absorption feature56. The latter suggests that the biexciton states in these small-size MSCs are not bound states; otherwise, one would expect a red-shifted absorptive feature indicative of biexciton binding, as commonly observed on the TA spectra of colloidal QDs57.

a, b TA spectra of Cd3P2 MSCs at indicated delays obtained with (a) VV and (b) VH pump-probe configurations, with ћωp = 2.82 eV. c Bottom: comparison of the averaged TA kinetics within 2.73–2.79 eV under VV (red circles) and VH (yellow circles) configurations (lower panel); Top: calculated time-dependent anisotropy (blue dots). d TA anisotropy value averaged within 100 ps (blue dots) as a function of pump photon energy. The dashed line with shading is the steady-state absorption spectrum.
Importantly, although the spectral shapes of VV and VH spectra have no difference, the signal amplitude in the former configuration (Fig. 3a) is about three times larger than that in the latter (Fig. 3b). Figure 3c plots the bleach recovery dynamics averaged in the range of 2.73–2.79 eV (lower panel), from which the time-dependent anisotropy r(t) can be calculated (upper panel) according to: \(r(t)=\left({S}_{{{\rm{VV}}}}(t)-{S}_{{{\rm{VH}}}}(t)\right)/\left({S}_{{{\rm{VV}}}}\left(t\right)+2{S}_{{{\rm{VH}}}}(t)\right)\), where SVV(t) and SVH(t) stand for VV and VH signal sizes, respectively. The obtained anisotropy is close to 0.4 at the early time, which indicates a near fully linearly polarized absorption dipole58,59. Interestingly, r(t) shows only slight decay within 100 ps and then negligible decay in a few ns, which likely correspond to aggregation-free individual clusters and cluster aggregates, respectively. Using the anisotropy decay time of 100 ps, the individual cluster size is estimated to be ~1 nm; see Supplementary Note 1 for details.
Pump photon energy (ћωp) dependent TA anisotropy was further conducted. In this experiment, the pump photon energy was tuned from 2.8 eV to 4 eV, and the anisotropy of the TA signals near the band edge was calculated. The early time r(t) is plotted in Fig. 3d along with the absorption spectrum of the MSCs. The anisotropic value drops substantially when the pump photon energy exceeds 3.0 eV, which is stabilized at ~0.2 for photon energy larger than 3.5 eV. There are two possible reasons accounting for the reduced TA anisotropy: (1) The higher energy transitions are linearly polarized but there is an angle of ~35° between their transition dipoles and the band-edge transition dipole; or (2) the higher energy transitions are not linearly polarized. Nevertheless, the anisotropic value close to 0.4 for pump photon energy below 3 eV establishes a highly linearly polarized band-edge exciton transition in these MSCs. By contrast, TA spectra of these Cd3P2 MSCs collected with co- and counter-circularly polarized pump-probe pulses have similar signal amplitudes (Supplementary Fig. 2), indicating that the exciton transition is not circularly polarized.
In general, linearly polarized excitons in nanostructures can result from intrinsic electronic structure and/or extrinsic dielectric confinement effect60. The latter typically requires highly anisotropic geometric shapes such as 1D nanorods61,62 or nanowires63 and 2D nanosheets64, which unlikely applies to our small clusters with only slight shape anisotropy. For the intrinsic electronic structure, short-range and long-range exchange interaction of the electron-hole pairs in QDs leads to a series of fine structure levels with well-defined quantum numbers and therefore well-defined linear or circular polarizations65. In particular, the anisotropic long-range exchange interaction arising from morphological and/or lattice asymmetry often creates linearly polarized excitons66. However, the fine structure splitting in typical QDs ranges from μeV to meV65, resulting in negligible polarization at room temperature due to thermal mixing of different states. In contrast, MSCs represent an extremely strong confinement region, which should give giant fine structure splitting. Qualitatively, one can also understand the highly linearly-polarized transition dipole of the MSCs by treating them as small molecules. Most molecules have anisotropic molecular shape and their lowest energy transition is strongly linearly polarized, giving rise to transient absorption anisotropy also close to 0.452. This similarity between MSCs and molecules further corroborate the atomically precise structure of the MSCs, that is, the structural anisotropy is nearly identical for each MSC. As a comparison, we also conducted linearly polarized TA measurements for ~2.6 nm Cd3P2 QDs (Supplementary Fig. 3), which gives a very small anisotropy value of ~0.05 (close to zero) standing in stark contrast to the Cd3P2 MSCs. Microscopically, each QD in the sample has a different size and morphology, and therefore each QD should also have a different degree of polarization for the band edge exciton transition, resulting in negligible anisotropy at the ensemble level.
Non-resonant and resonant coherent driving
We then conducted coherent driving experiments for Cd3P2 MSCs using two different photons energies, ћωp = 2.12 and 2.75 eV, corresponding to the negative detuning (Δ = −630 meV) and resonant (Δ ≈ 0) conditions, respectively. The spectra of these driving pulses are plotted in Fig. 2a for comparison with the absorption spectrum of Cd3P2 MSCs. All the experiments were performed at room temperature and, unless otherwise specified, the VH pump-probe configuration was adopted in order to suppress the leakage of the pump pulse into the TA spectra, which is especially important for resonant driving.
2D pseudocolor TA spectra acquired with 2.12 and 2.75 eV driving are presented in Figs. 4a, c, respectively, with the driving power density indicated in each panel. In these spectra, we focus on the data within ~200 fs around time zero, which are the spectral features arising from coherent driving. In Fig. 4a, negatively detuned driving leads to a typical derivative-like (i.e., with negative and positive alternating amplitudes) TA signal due to normal OSE blueshift, and this signal fades away after the duration of the driving pulse. Importantly, for the resonant driving case in Fig. 4c, we observe a narrowed negative feature sandwiched between two narrowed positive wings near time zero, which resembles the schematic spectrum presented in Fig. 1d. This Mollow-splitting like spectrum implies we might have indeed achieved resonant coherent driving of the band-edge exciton in Cd3P2 MSCs. After the driving pulse duration, the spectrum evolves into a different one that is consistent with the real excitation spectra presented in Fig. 3. This transformation could arise from dephasing of the coherent driving signals and/or direct real excitation of the band-edge exciton29. Strictly speaking, real excitation also originates from coherent driving, but represents a case of very rapid dephasing (driving while dephasing)29. Considering that we are dealing with an ensemble sample at room temperature, distinct dephasing times should exist in the sample.

a, c 2D pseudocolor TA spectra of Cd3P2 MSCs measured with VH pump-probe configuration using ћωp of (a) 2.12 eV (Δ = −630 meV) and (c) 2.75 eV (Δ ≈ 0). The incident pump intensities are indicated in each panel. Note the maximum bleach signal (c) is beyond the color scales in the insets; the color scales are chosen for better visualization of the coherent signals near time zero. b, d Their corresponding TA dynamics probed at indicated energy. The blue shaded area near time zero represents the driving pulse duration. e, f Simulated 2D TA spectra under resonant driving with (e) or without (f) the coherent species Scoh. included; see main text for details.
The kinetic trace of the bleach probed at 2.72 eV of Fig. 4a is plotted in Fig. 4b, and the response of this signal is comparable to the driving pulse duration (~170 fs). For resonant driving, the kinetic traces probed at 2.74 and 2.81 eV (i.e., the bleach and the blue-side positive wing, respectively, in Fig. 4c) are shown in Fig. 4d. The rapid decay of the signal within the pulse duration can be clearly observed, and the rest signals due to real population shows oscillations arising from electron–phonon coupling (see Supplementary Fig. 4 for details).
In order to isolate the coherent driving signals from real excitation signals for resonant driving, we performed global fitting using a sequential kinetic model; see Supplementary Note 2. Details for the fits are provided in Figs. S5-S7. In this global fitting, we can isolate three spectral species, a coherent one (Scoh.), S1 and S2 (Supplementary Fig. 5). Scoh. is not symmetric due to the asymmetric lineshape of the band-edge exciton (Fig. 2a), but its temporal profile is consistent with its coherent nature (Supplementary Fig. 5). S1 and S2 can be assigned to multiparticle and single-exciton species, respectively, which are consistent with our previous study56. The ~1.35 ps time for the decay of S1 and growth of S2 is the characteristic time for multiparticle Auger-like annihilation (Supplementary Fig. 5)56. Note that the multiparticle species S1 actually contains contributions from both single-exciton and biexciton states. We cannot differentiate the spectra of clean single-exciton and biexciton species due to the complicated distribution of absorbed photons per MSC at time zero across the ensemble sample. In the case of excitation well above the band edge in QDs, this distribution is often assumed to be Poissonian, but here we are not in this excitation regime, and therefore the distribution remains unknown. In this case, our global fitting gives an “average” result that as time goes on the initial spectrum that contains both single-excitons and biexcitons (S1) gradually evolves into a spectrum that is dominated by single-excitons (S2). We have conducted the global fitting for several different driving intensities (Supplementary Figs. 6 and 7), and obtained the spectra of S1 and S2 as shown in Supplementary Fig. 8. The ratio between the spectral amplitudes of S1 and S2 indeed first increases with pump intensity and then gradually approaches saturation (Supplementary Fig. 8), consistent with the increase of biexciton populations with increasing pump intensity.
Importantly, in order to substantiate the existence of Scoh. and its assignment, we have compared the results of global fitting with or without the inclusion of Scoh., whereas the rest components as well as their time evolution are kept identical. As shown in Fig. 4e, f, the simulated spectrum without Scoh. shows a substantial discrepancy compared to the experimental spectrum, especially within ~200 fs around time zero. Additionally, the kinetic traces probed at 2.82 and 2.75 eV, for example, are compared in Figures S5e and S5f, from which the inclusion of Scoh. clearly reproduces the experimental traces much better than the one without Scoh.. This analysis further confirms the observation of a Mollow-splitting spectrum due to quantum coherent driving.
The driving power dependent coherent spectra isolated from direct observation and global fitting are plotted in Fig. 5a, b for negatively detuned and resonant driving, respectively. In both cases, the signal amplitudes increase with increasing driving powers. Importantly, these spectra can be well simulated by spectral shifting (for nonresonant driving) or splitting (for resonant driving) of the experimental band-edge exciton absorption according to the principles illustrated in Fig. 1; see Supplementary Note 3. The simulated spectra are plotted in Figs. 5c, d, with the shifting or splitting energy indicated in each panel. As noted above, the asymmetric spectra of resonant driving can be well reproduced by using the asymmetric absorption profile of Cd3P2 MSCs.

a, b Coherent spectra obtained with (a) ћωp = 2.12 eV and (b) ћωp = 2.75 eV using varying driving power intensities. a is directly taken from experimental spectra and (b) is obtained from global fitting of the experimental spectra. c, d Simulated coherent spectra by spectral shifting/splitting of the ground state band-edge exciton absorption, with the shifting/splitting energies indicated. The simulated spectra are slightly different from the experimental spectra in terms of transition energies, because the steady-state absorption and TA spectrometers are not perfectly calibrated in absolute wavelengths. e Driving power intensity dependent SWT (red circles) and Stark shift (δE; blue circles) and their linear fits (solid lines), for ћωp = 2.12 eV. The calculated transition dipole under this VH configuration (μVH) is indicated. (f) Driving field amplitude (square root of power intensity) dependent Rabi splitting (δERabi; green circles) and its linear fit (solid line), for ћωp = 2.75 eV. Inset shows the plot of δERabi over power intensity, which is clearly nonlinear.
The driving power dependent coherent spectra can be used to quantify the spectral shifting or splitting, and therefore, the magnitude of transition dipole moment (μ) of Cd3P2 MSCs. For the case of negatively detuned driving, we adopt a well-established spectral-weight-transfer (SWT) method13; see Supplementary Note 4. The obtained power-dependent SWT and OSE shift (δE) are both plotted in Fig. 5e. The derived energy shift scales linearly with the driving power, which is in good agreement with Eq. 123:
where \({\hslash \varOmega }_{{{\rm{R}}}}\) is the Rabi energy equaling με, ε is the amplitude of the electric field and I0 is the driving power. The value of μ quantified from Fig. 5e using Eq. 1 is ~13.2 D; see Supplementary Note 5 for calculation details. Notably, however, this experiment is conducted with the VH pump-probe configuration, indicating that the calculated dipole moment is an underestimated one. For negatively detuned driving, it is also convenient to measure the OSE shift using the VV pump-probe configuration, and the results are provided in Supplementary Fig. 9. Under an identical driving power, the signal amplitude ratio of VV and VH configurations is close to 3:1, which is consistent with our TA anisotropy measurements discussed above. Using the VV signals, we estimate μVV to be ~22.8 D, satisfying the theoretical prediction of μVV2/μVH2 = 3/1. This value (22.8 D) is comparable with those derived for much larger size colloidal QDs of various compositions (e.g., 24 D for 3.6 nm CdSe QDs25; 34 D for 5 nm CsPbI3 QDs24). The extreme confinement in these Cd3P2 MSCs concentrates the oscillator strength to the band-edge exciton, which underlies the strong optical responses we observed in these coherent driving experiments.
As for the resonant driving condition, the splitting of the two Rabi sidebands with respect to the original absorption peak should satisfy the following equation12:
Experimentally, however, it is not straightforward to directly readout the Rabi splitting from the TA spectra, due to the spectral overlap between the two Rabi sidebands and the center band bleach. We therefore use the simulated TA spectra in Fig. 5d to match the experimental spectra extracted from global fitting in Fig. 5b. From the best match, we can estimate the splitting energy, which is found to scale linearly with the square root of the driving power intensity (i.e., the electric field strength) instead of the power itself (Fig. 5f), consistent with Eq. 2. The calculated μVH is ~11.8 D. Considering that the calculation of the transition dipole moment relies critically on the pump intensity (I0) which in turn depends sensitively on beam-size and pulse-duration measurements, the consistency among the values of μVH calculated from resonant and nonresonant driving conditions is remarkable. Moreover, although the signal amplitude at each probe energy increases with the driving power, the total integrated TA signal remains close to zero (Supplementary Fig. 10). This behavior reflects the conservation of total oscillator strength, i.e., the original transition strength is redistributed to dressed states under strong optical driving, as schematically illustrated in Fig. 1d. We have also conducted the positively detuned driving experiment in order to reproduce the schemes in Fig. 1e, f, but the results are much more complex, as discussed in Supplementary Note 6 and Supplementary Figs. 11 and 12.
Discussion
As we noted in the discussion of the theoretical lineshape in Fig. 1d, besides a simple Rabi splitting, quantum interference between the pump and probe beams and gain without inversion should also be expected under very strong resonant driving, which has been clearly illustrated for a single InAs QD at 5 K12. In that experiment, the Rabi splitting in the range of 1 to 2 GHz substantially exceeded the absorption linewidth of ca. 190 MHz, thereby enabling the strongly-driven phenomena. In our present case, although the large transition dipole and the intense femtosecond driving pulse has led to Rabi splitting reaching 20 meV, it is still much smaller than the room-temperature ensemble absorption linewidth of ~100 meV. Therefore, it would be interesting if these MSCs can be driven under cryogenic temperatures which should help reduce the homogeneous linewidth. At present this is prohibited by the weak stability of these MSCs, restricting our experiments to room temperature measurements of MSCs in circulated solvent. Future efforts will be invested to stabilize these MSCs by, e.g., choosing surface ligands and polymer matrix67,68,69 that can improve the stability of these small-size clusters under strong laser pulse driving.
Nevertheless, a major advancement of the present study is the observation of a Mollow-splitting like TA spectrum of the Cd3P2 MSCs under resonant driving at room temperature, which is validated by comparing the experimental TA spectra to the global fitting results with or without including the Mollow-splitting species (Fig. 4), and by a linear scaling relationship between the splitting energy and the driving electric field strength instead of power intensity (Fig. 5f). In order to gain further physical insight into the coherent driving case, we also conducted a quantum simulation using the Lindblad master equation based a density matrix approach. While it is unrealistic to perform full quantum optical simulation for a system as complex as the Cd3P2 MSCs here at room temperature, this Lindblad equation simulation can capture the physical essence of a two-level system under laser driving and in the presence of dephasing, as established in our previous study29. Details of the simulation is provided in Supplementary Note 7. As shown in Supplementary Fig. 13, this simulation predicts fine structures exhibited as multiple fringes in both time and frequency domains, consistent with previous models70. These features are interpreted as the interference between fields emitted at different times during the driving pulse, but they are typically obscured in experiments by the phase fluctuations of the pump and probe pulses and by many other competing processes29,71. The overall Mollow-like TA spectral profile remains robust (Supplementary Fig. 13) and is successfully observed in experiments.
In summary, we have systematically investigated resonant and non-resonant coherent driving of linearly-polarized excitons in colloidal Cd3P2 MSCs at room temperature by applying polarization-controlled TA spectroscopy. Their sharp and well-isolated band-edge exciton transition enabled us to go beyond previous below-gap driving experiments for solution-processed semiconductor materials and to conduct resonant driving experiments. Although several other systems such as colloidal nanoplatelets (NPLs) or monolayer transition metal dichalcogenides (TMDs) also exhibit narrow linewidths at room temperature, they each have their own complexity. NPLs have linearly-polarized in-plane exciton transitions, but there is a strong overlap between the heavy- and light-hole exciton transitions with the continuous band. Moreover, their intersubband transitions can also be driven28, which strongly complicates the analysis of band-edge exciton Rabi splitting. For TMDs, the exciton-to-biexciton transition is exactly of opposite circular polarization compared to the ground-to-exciton transition15, which prohibits the use of cross-polarized TA to conduct resonant driving of the ground-to-exciton transition. The unique features of the MSCs allowed us to observe a Mollow-like lineshape under resonant driving, with the splitting of the Rabi sidebands scaling linearly with the electric field strength, which is distinct from nonresonant OSE with energy shift being proportional to the light intensity. The transition dipole moment derived from these coherent deriving experiments exceeds 20 Debye, which arises from extreme confinement that concentrates the oscillator strength to the band-edge exciton. Our results may pave the way for the exploration of the linearly-polarized excitons of colloidal MSCs for exotic phenomena such as quantum interference and gain without inversion, as well as for new platforms of optical manipulation of quantum states and quantum information processing.
Methods
Synthesis of Cd3P2 MSCs
The colloidal Cd3P2 MSCs were synthesized following the procedure reported by Yu et al.50. Briefly, 0.533 g (0.2 mmol) of Cadmium acetate dihydrate, 0.2 mmol oleic acid and 5 mL 1-octadecene were loaded in a 100-mL three-neck flask and then degassed at 100 °C for 2 hours. The phosphorus precursor prepared by mixing 0.05 mmol of Tris(trimethylsilyl)phosphine and 1 g of 1-octadecene, was quickly injected into the flask. The reaction was kept at 100 °C for 20 seconds under nitrogen flow, after which it was stopped by removing the heating mantle and was cooled down to room temperature. The obtained yellow solution was washed with excess ethanol to remove solvent, excessive ligands and unreacted precursors. The resulting precipitate was collected and dissolved in n-hexane or heptane for spectroscopic measurements.
Synthesis of Cd3P2 QDs
Cd3P2 QDs were synthesized according to the procedure developed by Peng et al.72. Briefly, a mixture of 77 mg of Cadmium Oxide, 0.3 mL of oleic acid and 9 mL of 1-octadecene was heated at 230 °C under nitrogen flow until it turned clear. 1 mL of 0.2 M Tris(trimethylsilyl)phosphine in 1-octadecene (with a small amount of hexane) was swiftly injected into the flask and the reaction was kept for 5 or 6 seconds at 230 °C. The crude solution was washed for several times by adding ethanol.
X-ray total scattering measurements
PDF data were collected at the PD beamline at the Australian Synchrotron using a Mythen II detector over a 2θ range of 120°. Samples were packed into 0.7 mm borosilicate capillaries and rotated during data collection. The wavelength, zero error and instrument contribution to the peak shape was refined to 0.59 Å using NIST LaB6 660b. Energy was calibrated with NIST LaB6 standard. We further convert the experimental integrated scattering data into the scattering function G(r) by using available software package PDFgetx3. The function [G(r)] of Cd3P2 MSCs is plotted against the interatomic distance, as demonstrated in Supplementary Fig. 1. The coordination numbers of anions and cations in both cubic and tetragonal phases of Cd3P2 are inconsistent with those of the well-known InP or CdSe MSCs. The amount of calculation is beyond the scope of this study. Nevertheless, we find some similarities between PDFs of Cd3P2 MSCs and α-Cd37S20 MSCs44. The major peak positions at 5-10 Å of Cd3P2 MSCs match reasonably well with those of α-Cd37S20 MSCs, and the strongest peak position is also close to that of α-Cd37S20 MSCs, indicating their similar overall sizes.
PL anisotropy measurements
Steady-state PL spectra are collected by a setup that consists of a monochromator (SpectraPro-2300i, Acton Research Co., USA) and a charge-coupled device camera (PIXIS 100, Princeton Instruments, USA), and the same microscope objective was used for excitation and PL collection. Polarization-dependent PL measurements were conducted by rotating a linear polarizer placed after the sample, while another linear polarizer for the excitation beam remained unchanged.
Femtosecond pump-probe spectroscopy
Femtosecond transient absorption measurements were conducted using a Pharos femtosecond laser system (Light Conversion; 1030 nm, 10 kHz repetition rate). The output laser was split in two beams with a beam splitter, one of which was directed to an optical parametric amplifier (Orpheus-HP; Light Conversion) to generate wavelength-tunable pump pulses, and the other being delayed by a motorized delay stage and then focused onto a BBO frequency-doubling crystal to generate 515 nm beam and then focused into a 2 mm thick sapphire window to generate a white light continuum (WLC) used for probe beam. The probe beam was focused with an Al parabolic reflector onto the sample. The probe beam was then collimated and focused into a fiber-coupled spectrometer with a line scan camera and detected at a frequency of 10 kHz. Circular and linear polarized pump and probe beams were obtained by separately inserting two sets of linear polarizers (Thorlabs) and quarter-wave or half-wave plates (Thorlabs) right before the sample. Samples were placed in a flow cell and were vigorously circulated during the measurements to avoid photodamaging, which was further confirmed by the same absorption spectra of samples before and after TA measurements. All measurements were carried out at room temperature. For above-bandgap and resonant cases, the optical density at excitonic absorption peak (2.75 eV) is around 0.31; and it was changed to 0.57 for below-bandgap case.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Information. They are also available at figshare, https://doi.org/10.6084/m9.figshare.31321438. Source data are provided with this paper.
Code availability
All codes are available from J.Z. or K.W. upon request.
References
Shirley, J. H. Solution of the Schrödinger equation with a Hamiltonian periodic in time. Phys. Rev. 138, B979 (1965).
Autler, S. H. & Townes, C. H. Stark effect in rapidly varying fields. Phys. Rev. 100, 703 (1955).
Monroe, C. Quantum information processing with atoms and photons. Nature 416, 238–246 (2002).
Shields, A. J. Semiconductor quantum light sources. Nat. Photonics 1, 215–223 (2007).
Oka, T. & Kitamura, S. Floquet engineering of quantum materials. Annu. Rev. Condens. Matter Phys. 10, 387–408 (2019).
Rudner, M. S. & Lindner, N. H. Band structure engineering and non-equilibrium dynamics in Floquet topological insulators. Nat. Rev. Phys. 2, 229–244 (2020).
Tamarat, P. et al. Pump-probe experiments with a single molecule: ac-Stark effect and nonlinear optical response. Phys. Rev. Lett. 75, 1514 (1995).
Sautenkov, V. A., Rostovtsev, Y. V. & Eliel, E. R. Observation of narrow Autler-Townes components in the resonant response of a dense atomic gas. Phys. Rev. A—At., Mol., Optical Phys. 78, 013802 (2008).
Mysyrowicz, A. et al. “Dressed excitons” in a multiple-quantum-well structure: evidence for an optical Stark effect with femtosecond response time. Phys. Rev. Lett. 56, 2748 (1986).
Von Lehmen, A., Chemla, D. S., Zucker, J. & Heritage, J. P. Optical Stark effect on excitons in GaAs quantum wells. Opt. Lett. 11, 609–611 (1986).
Muller, A., Fang, W., Lawall, J. & Solomon, G. S. Creating polarization-entangled photon pairs from a semiconductor quantum dot using the optical Stark effect. Phys. Rev. Lett. 103, 217402 (2009).
Xu, X. et al. Coherent optical spectroscopy of a strongly driven quantum dot. Science 317, 929–932 (2007).
Sie, E. J. et al. Valley-selective optical Stark effect in monolayer WS2. Nat. Mater. 14, 290–294 (2014).
Kim, J. et al. Ultrafast generation of pseudo-magnetic field for valley excitons in WSe2 monolayers. Science 346, 1205–1208 (2014).
Yong, C.-K. et al. Biexcitonic optical Stark effects in monolayer molybdenum diselenide. Nat. Phys. 14, 1092–1096 (2018).
Sie, E. J., Lui, C. H., Lee, Y.-H., Kong, J. & Gedik, N. Observation of intervalley biexcitonic optical Stark effect in monolayer WS2. Nano Lett. 16, 7421–7426 (2016).
Yong, C.-K. et al. Valley-dependent exciton fine structure and Autler–Townes doublets from Berry phases in monolayer MoSe2. Nat. Mater. 18, 1065–1070 (2019).
Lin, K.-Q., Bange, S. & Lupton, J. M. Quantum interference in second-harmonic generation from monolayer WSe2. Nat. Phys. 15, 242–246 (2019).
Zhou, S. et al. Pseudospin-selective Floquet band engineering in black phosphorus. Nature 614, 75–80 (2023).
Kobayashi, Y. et al. Floquet engineering of strongly driven excitons in monolayer tungsten disulfide. Nat. Phys. 19, 171–176 (2023).
Merboldt, M. et al. Observation of Floquet states in graphene. Nat. Phys. 21, 1093–1099 (2025).
Choi, D. et al. Observation of Floquet–Bloch states in monolayer graphene. Nat. Phys. 21, 1100–1105 (2025).
Giovanni, D. et al. Tunable room-temperature spin-selective optical Stark effect in solution-processed layered halide perovskites. Sci. Adv. 2, e1600477 (2016).
Li, Y., He, S., Luo, X., Lu, X. & Wu, K. Strong spin-selective optical Stark effect in lead halide perovskite quantum dots. J. Phys. Chem. Lett. 11, 3594–3600 (2020).
Tang, Y., Saniepay, M., Mi, C., Beaulac, R. & McGuire, J. A. Optical Stark metrology of CdSe quantum dots: Reconciling the size-dependent oscillator strength with theory. arXiv preprint arXiv:1703.00530 (2017).
Diroll, B. T. Circularly polarized optical Stark effect in CdSe colloidal quantum wells. Nano Lett. 20, 7889–7895 (2020).
Xiang, D., Li, Y., Wang, L., Zhao, Y. & Wu, K. Coupled double optical Stark effect in CdSe colloidal nanoplatelets. ACS Photonics 8, 745–751 (2020).
Li, Y., Wang, L., Wang, J., Zhu, J. & Wu, K. Autler-Townes splitting and linear dichroism in colloidal CdSe nanoplatelets driven by near-infrared pulses. Sci. Adv. 10, eabq2729 (2024).
Li, Y., Yang, Y., Liu, Y., Zhu, J. & Wu, K. Observation of Floquet states and their dephasing in colloidal nanoplatelets driven by visible pulses. Nat. Photonics 18, 1044–1051 (2024).
Yang, Y. et al. Large polarization-dependent exciton optical Stark effect in lead iodide perovskites. Nat. Commun. 7, 12613 (2016).
Zhang, J., Tang, Y., Lee, K. & Ouyang, M. Tailoring light–matter–spin interactions in colloidal hetero-nanostructures. Nature 466, 91–95 (2010).
Mollow, B. Power spectrum of light scattered by two-level systems. Phys. Rev. 188, 1969–1975 (1969).
Dynes, J., Frogley, M., Beck, M., Faist, J. & Phillips, C. ac Stark splitting and quantum interference with intersubband transitions in quantum wells. Phys. Rev. Lett. 94, 157403 (2005).
Wu, F. Y., Ezekiel, S., Ducloy, M. & Mollow, B. Observation of amplification in a strongly driven two-level atomic system at optical frequencies. Phys. Rev. Lett. 38, 1077–1080 (1977).
Kauranen, M., Maki, J. J., Gaeta, A. L. & Boyd, R. W. Two-beam-excited conical emission. Opt. Lett. 16, 943–945 (1991).
Herron, N., Calabrese, J., Farneth, W. & Wang, Y. Crystal structure and optical properties of Cd32S14 (SC6H5)36. DMF4, a cluster with a 15 Angstrom CdS core. Science 259, 1426–1428 (1993).
Zhang, B. et al. Thermally-induced reversible structural isomerization in colloidal semiconductor CdS magic-size clusters. Nat. Commun. 9, 2499 (2018).
Bootharaju, M. S. et al. Magic-sized stoichiometric II–VI nanoclusters. Small 17, 2002067 (2021).
Ripberger, H. H., Sandeno, S. F., Eagle, F. W., Nguyen, H. A. & Cossairt, B. M. Structure and reactivity of II–VI and III–V magic-sized clusters: understanding and expanding the scope of accessible form and function. Acc. Mater. Res. 5, 726–738 (2024).
Mule, A. S. et al. Unraveling the growth mechanism of magic-sized semiconductor nanocrystals. J. Am. Chem. Soc. 143, 2037–2048 (2021).
Cossairt, B. M. & Owen, J. S. CdSe clusters: at the interface of small molecules and quantum dots. Chem. Mater. 23, 3114–3119 (2011).
Chen, Z., Manian, A., Widmer-Cooper, A., Russo, S. P. & Mulvaney, P. Semiconductor quantum dots in the cluster regime: focus review. Chem. Rev. 9, 4359–4396 (2025).
Zhang, B. et al. Stable CsPbBr3 nanoclusters feature a disk-like shape and a distorted orthorhombic structure. J. Am. Chem. Soc. 144, 5059–5066 (2022).
Williamson, C. B. et al. Chemically reversible isomerization of inorganic clusters. Science 363, 731–735 (2019).
Ugras, T. J. et al. Transforming achiral semiconductors into chiral domains with exceptional circular dichroism. Science 387, eado7201 (2025).
Zhang, B. et al. CsPbBr3 superstructures with circularly polarized photoluminescence obtained by the self-assembly and annealing of nanoclusters. Angewandte Chemie, 64, e202423272 (2025).
Yao, Y. et al. Extracting pure circular dichroism from hierarchically structured CdS magic cluster films. ACS Nano 16, 20457–20469 (2022).
Zdanowicz, W. & Zdanowicz, L. Semiconducting compounds of the AII BV group. Annu. Rev. Mater. Sci. 5, 301–328 (1975).
Andrzejewski, J. & Misiewicz, J. Energy band structure of Zn3P2-type semiconductors: analysis of the crystal structure simplifications and energy band calculations. Phys. Status Solidi (b) 227, 515–540 (2001).
Wang, R. et al. Magic-sized Cd3P2 II− V nanoparticles exhibiting bandgap photoemission. J. Phys. Chem. C. 113, 17979–17982 (2009).
Palencia, C., Yu, K. & Boldt, K. The future of colloidal semiconductor magic-size clusters. ACS Nano 14, 1227–1235 (2020).
Nevers, D. R. et al. Mesophase formation stabilizes high-purity magic-sized clusters. J. Am. Chem. Soc. 140, 3652–3662 (2018).
Jonas, D. M., Lang, M. J., Nagasawa, Y., Joo, T. & Fleming, G. R. Pump− probe polarization anisotropy study of femtosecond energy transfer within the photosynthetic reaction center of Rhodobacter sphaeroides R26. J. Phys. Chem. 100, 12660–12673 (1996).
Yeh, A. T., Shank, C. V. & McCusker, J. K. Ultrafast electron localization dynamics following photo-induced charge transfer. Science 289, 935–938 (2000).
Zhu, J. et al. Photoselective ultrafast investigation of xanthorhodopsin and its carotenoid antenna salinixanthin. J. Phys. Chem. B 114, 3038–3045 (2010).
Liu, Y., Li, Y., Yang, Y., Zhu, J. & Wu, K. Transient spectral and dynamic properties of magic-size Cd3P2 nanoclusters in the limit of strong confinement. Sci. China Chem. 66, 3628–3635 (2023).
Klimov, V. I. Spectral and dynamical properties of multiexcitons in semiconductor nanocrystals. Annu. Rev. Phys. Chem. 58, 635–673 (2007).
Jameson, D. M. & Ross, J. A. Fluorescence polarization/anisotropy in diagnostics and imaging. Chem. Rev. 110, 2685–2708 (2010).
Diroll, B. T., Dadosh, T., Koschitzky, A., Goldman, Y. E. & Murray, C. B. Interpreting the energy-dependent anisotropy of colloidal nanorods using ensemble and single-particle spectroscopy. J. Phys. Chem. C. 117, 23928–23937 (2013).
Rodina, A. V. & Efros, A. L. Effect of dielectric confinement on optical properties of colloidal nanostructures. J. Exp. Theor. Phys. 122, 554–566 (2016).
Hu, J. et al. Linearly polarized emission from colloidal semiconductor quantum rods. Science 292, 2060–2063 (2001).
Hadar, I., Hitin, G. B., Sitt, A., Faust, A. & Banin, U. Polarization properties of semiconductor nanorod heterostructures: from single particles to the ensemble. J. Phys. Chem. Lett. 4, 502–507 (2013).
McDonald, M. P., Vietmeyer, F. & Kuno, M. Direct measurement of single CdSe nanowire extinction polarization anisotropies. J. Phys. Chem. Lett. 3, 2215–2220 (2012).
Ma, X. et al. Anisotropic photoluminescence from isotropic optical transition dipoles in semiconductor nanoplatelets. Nano Lett. 18, 4647–4652 (2018).
Tamarat, P. et al. The ground exciton state of formamidinium lead bromide perovskite nanocrystals is a singlet dark state. Nat. Mater. 18, 717–724 (2019).
Wang, X. et al. Polarization-sensitive halide perovskites for polarized luminescence and detection: recent advances and perspectives. Adv. Mater. 33, 2003615 (2021).
Sandeno, S., Harvey, S., Lin, V. & Cossairt, B. Narrow blue emission from cadmium phosphide clusters enhanced through phosphinate ligation. Nano Lett. 25, 9410–9416 (2025).
Sandeno, S. F. et al. Ligand steric profile tunes the reactivity of indium phosphide clusters. J. Am. Chem. Soc. 146, 3102–3113 (2024).
Kim, K. et al. Vapor infiltration synthesis of indium sulfide magic size cluster. ACS Nano 18, 31372–31380 (2024).
Moelbjerg, A., Kaer, P., Lorke, M. & Mørk, J. Resonance fluorescence from semiconductor quantum dots: beyond the Mollow triplet. Phys. Rev. Lett. 108, 017401 (2012).
Uchida, K., Kusaba, S., Nagai, K., Ikeda, T. N. & Tanaka, K. Diabatic and adiabatic transitions between Floquet states imprinted in coherent exciton emission in monolayer WSe2. Sci. Adv. 8, eabq7281 (2022).
Xie, R., Zhang, J., Zhao, F., Yang, W. & Peng, X. Synthesis of monodisperse, highly emissive, and size-tunable Cd3P2 nanocrystals. Chem. Mater. 22, 3820–3822 (2010).
Acknowledgements
We acknowledge financial support from the Chinese Academy of Sciences (XDB0970303), the National Natural Science Foundation of China (22573105), the Fundamental Research Funds for the Central Universities (20720220009) and the New Cornerstone Science Foundation.
Author information
Authors and Affiliations
Contributions
K.W. supervised the project. Y.Liu synthesized the samples, characterized them and measured the spectroscopy under the supervision of J.Z. J.Z. conducted global fitting analysis. Y.Li and Y.Y. helped with spectroscopy experiments and contributed to discussion. K.W. and Y.Liu wrote the manuscript with inputs from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gabriel Nagamine, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Li, Y., Yang, Y. et al. Resonant and non-resonant driving of linearly-polarized excitons in Cd3P2 magic-size clusters. Nat Commun 17, 4022 (2026). https://doi.org/10.1038/s41467-026-70674-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70674-y
