
Abstract
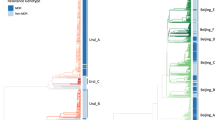
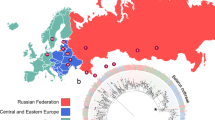
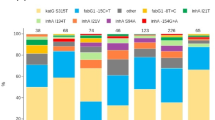
Multidrug resistant tuberculosis (MDR-TB) epidemics are sustained by transmission of reproductively fit MDR M. tuberculosis (Mtb) strains. We search a large publicly available dataset of ~200,000 Mtb whole genome sequences to identify strains related to a highly successful MDR clade circulating in Moldova belonging to lineage 4.2.1/Ural. We characterize a clade of 1604 drug-resistant Mtb sequences harboring conserved resistance-conferring mutations. We identify the Russian Federation as the most likely country of origin for this clade and infer several independent migration events from Russia and Moldova to other European and Asian countries. We estimate that this clade is expanding more rapidly than comparable clades of lineage 4.2.1/Ural. The broad dispersal of this highly successful clade is an urgent global health threat. Genomic surveillance is essential to track the evolution and spread of this and other strains of concern.
Similar content being viewed by others
Data availability
All data were accessed from the European Nucleotide Archive. Run accession numbers, country of origin, and inferred sample dates are available in Supplementary Data 1.
References
Kendall, E. A., Fofana, M. O. & Dowdy, D. W. Burden of transmitted multidrug resistance in epidemics of tuberculosis: a transmission modelling analysis. Lancet Respir. Med. 3, 963–972 (2015).
Yang, C. et al. Phylogeography and transmission of M. tuberculosis in Moldova: a prospective genomic analysis. PLoS Med. 19, e1003933 (2022).
Merker, M. et al. Transcontinental spread and evolution of Mycobacterium tuberculosis W148 European/Russian clade toward extensively drug resistant tuberculosis. Nat. Commun. 13, 5105 (2022).
Loiseau, C. et al. The relative transmission fitness of multidrug-resistant Mycobacterium tuberculosis in a drug resistance hotspot. Nat. Commun. 14, 1988 (2023).
Eldholm, V. et al. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 113, 13881–13886 (2016).
World Health Organization. Global Tuberculosis Report 2024. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2024 (WHO, 2024).
Goig G. A. et al. Ecology, global diversity and evolutionary mechanisms in the Mycobacterium tuberculosis complex. Nat. Rev. Microbiol. 23, 602–614 (2025).
Stucki, D. et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 48, 1535–1543 (2016).
Mokrousov, I. The quiet and controversial: Ural family of Mycobacterium tuberculosis. Infect. Genet. Evol. 12, 619–629 (2012).
Sinkov, V. et al. New epidemic cluster of pre-extensively drug resistant isolates of Mycobacterium tuberculosis Ural family emerging in Eastern Europe. BMC Genom. 19, 762 (2018).
Brown T. S. et al. Evolution and emergence of multidrug-resistant Mycobacterium tuberculosis in Chisinau, Moldova. Microb Genom. 7, 000620 (2021).
Lan Y. et al. Identifying local foci of tuberculosis transmission in Moldova using a spatial multinomial logistic regression model. eBioMedicine. 102, 105085 (2024).
Chitwood, M. H. et al. The recent rapid expansion of multidrug resistant Ural lineage Mycobacterium tuberculosis in Moldova. Nat. Commun. 15, 2962 (2024).
Brynildsrud, O. B. et al. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci. Adv. 4, eaat5869 (2018).
Napier, G., Campino, S., Phelan, J. E. & Clark, T. G. Large-scale genomic analysis of Mycobacterium tuberculosis reveals extent of target and compensatory mutations linked to multi-drug resistant tuberculosis. Sci. Rep. 13, 623 (2023).
Song, Y., Gill, I., MacPherson, A. & Colijn, C. Sampling aware ancestral state inference. bioRxiv. 2025:2025.05.20.655151.
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2018).
Kidenya, B. R., Mshana, S. E., Fitzgerald, D. W. & Ocheretina, O. Genotypic drug resistance using whole-genome sequencing of Mycobacterium tuberculosis clinical isolates from North-western Tanzania. Tuberculosis 109, 97–101 (2018).
Napier, G. et al. Characterisation of drug-resistant Mycobacterium tuberculosis mutations and transmission in Pakistan. Sci. Rep. 12, 7703 (2022).
Roberts, L. W. et al. MmpR5 protein truncation and bedaquiline resistance in Mycobacterium tuberculosis isolates from South Africa: a genomic analysis. Lancet Microbe 5, 100847 (2024).
Neher, R. A., Russell, C. A. & Shraiman, B. I. Predicting evolution from the shape of genealogical trees. eLife 3, e03568 (2014).
Zwyer, M. et al. Back-to-Africa introductions of Mycobacterium tuberculosis as the main cause of tuberculosis in Dar es Salaam, Tanzania. PLoS Pathog. 19, e1010893 (2023).
Gygli, S. M. et al. Prisons as ecological drivers of fitness-compensated multidrug-resistant Mycobacterium tuberculosis. Nat. Med. 27, 1171–1177 (2021).
Arandjelović, I. et al. Longitudinal Outbreak of Multidrug-Resistant Tuberculosis in a Hospital Setting, Serbia. Emerg. Infect. Dis. 25, 555–558 (2019).
Phelan, J. E. et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 11, 41 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Sobkowiak, B. et al. A new method for detecting mixed Mycobacterium tuberculosis infection and reconstructing constituent strains provides insights into transmission. Genome Med. 17, 8 (2025).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Didelot, X., Croucher, N. J., Bentley, S. D., Harris, S. R. & Wilson, D. J. Bayesian inference of ancestral dates on bacterial phylogenetic trees. Nucleic Acids Res. 46, e134-e (2018).
Walker, T. M. et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 13, 137–146 (2013).
Volz, E. M. et al. Identification of hidden population structure in time-scaled phylogenies. Syst. Biol. 69, 884–896 (2020).
Maddison, W. P., Midford, P. E. & Otto, S. P. Estimating a binary character’s effect on speciation and extinction. Syst. Biol. 56, 701–710 (2007).
Stadler, T. et al. Estimating the basic reproductive number from viral sequence data. Mol. Biol. Evol. 29, 347–357 (2011).
Schliep, K. P. phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593 (2010).
World Health Organization. Catalogue of Mutations in Mycobacterium tuberculosis Complex and their Association with Drug Resistance (World Health Organization, 2023).
Field, N. et al. Strengthening the reporting of molecular epidemiology for infectious diseases (STROME-ID): an extension of the STROBE statement. Lancet Infect. Dis. 14, 341–352 (2014).
Acknowledgements
The authors report funding from the National Institutes of Health (R01AI180209: M.H.C., B.P., T.C., B.S., and P01AI159402: M.H.C., T.C., and B.S.), and the Medical Research Council (UKRI1414: B.S.).
Author information
Authors and Affiliations
Contributions
C.C. and B.S. conceived the study. IR and YTC assembled the data. M.H.C., Y.S., and B.S. analyzed the data. M.H.C., Y.S., B.I.P., and B.S. visualized results. T.C. secured funding. M.H.C., I.R., Y.S., B.I.P., Y.T.C., N.C., V.C., C.C., T.C., and B.S. reviewed results, contributed to manuscript drafting, and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks David Couvin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chitwood, M.H., Rancu, I., Song, Y. et al. The global phylogeography of rapidly expanding multidrug resistant Ural lineage 4.2 Mycobacterium tuberculosis. Nat Commun (2026). https://doi.org/10.1038/s41467-026-71193-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-026-71193-6
