
Abstract
Neuronal intranuclear inclusion disease is caused by abnormal GGC repeat expansion in the NOTCH2NLC gene, though its pathogenic mechanism remains incompletely understood. This study shows that the abnormally expanded polyG-uN2C protein, encoded by the repeat sequence, contains intrinsically disordered regions and forms aggregates, leading to mislocalization of nucleophosmin and downregulation of fibrillarin. PolyG aggregates interact with nucleophosmin and rRNA, disrupting ribosomal homeostasis. Furthermore, polyG facilitates the downregulation of chromatin structural proteins CTCF and RAD21, thereby impairing chromatin organization. This pathological manifestation can be mitigated by restoring CTCF/RAD21 expression. Furthermore, in brain organoids derived from neuronal intranuclear inclusion disease patients, we observe nucleolar stress accompanied by genome-wide chromatin structural alterations. These changes correlate with increased DNA damage and cellular senescence phenotypes. Notably, antisense oligonucleotides targeting GGC effectively reduce polyG aggregation and ameliorate related molecular defects, ultimately alleviating senescence-associated phenotypes. These findings establish key mechanisms underlying neuronal intranuclear inclusion disease pathogenesis and provide proof-of-concept for targeted therapy.
Similar content being viewed by others


Introduction
Neuronal intranuclear inclusion disease (NIID) is a progressive neurodegenerative disorder characterized by a wide spectrum of clinical manifestations, including cognitive impairment, dystonia, and motor dysfunction1,2,3. Research has established that abnormal GGC repeat expansions (repeat number >60) in the 5′ untranslated region (UTR) of the NOTCH2NLC gene are causative for NIID4,5. These expanded GGC repeats can induce the translation of an upstream open reading frame (uORF), producing a toxic polyglycine (polyG) protein, which is considered a key driver of NIID pathogenesis6,7,8,9.
Our previous studies demonstrated that polyG predominantly forms intranuclear inclusions and triggers nucleolar stress by inducing the nucleocytoplasmic translocation of nucleophosmin (NPM1). NPM1 plays critical roles in DNA damage repair10 and the maintenance of three-dimensional (3D) genome architecture11. Under physiological conditions, NPM1 is recruited to DNA damage sites to facilitate repair processes10; its dysfunction may lead to impaired DNA repair and increased genomic instability, ultimately accelerating cellular senescence. Furthermore, as a nucleolar protein, NPM1 interacts with the chromatin structural protein CTCF12, anchoring chromatin around the nucleolus to regulate gene expression. CTCF, in cooperation with RAD21, maintains 3D chromatin organization by binding specific loci to facilitate the formation of chromatin loops and topologically associating domains (TADs)13. These findings suggest that in NIID, polyG-induced NPM1 dysfunction may contribute to DNA damage and aberrant 3D chromatin structure, though the precise mechanisms remain unclear.
In recent years, antisense oligonucleotides (ASOs) have emerged as promising therapeutic agents for rare neurological disorders, including spinal muscular atrophy14, myotonic dystrophy15,16,17, and amyotrophic lateral sclerosis (ALS)18,19,20,21. Notably, in fragile X-associated tremor/ataxia syndrome (FXTAS), another disorder caused by GGC repeat expansions, steric-blocking ASOs targeting the repeat tract effectively reduced the accumulation of toxic FMRpolyG and demonstrated significant therapeutic efficacy. This raises the intriguing possibility that GGC-targeting ASOs may also hold therapeutic potential for NIID.
In this study, we further elucidate how NPM1 dysfunction in NIID models leads to increased genomic instability and disrupted 3D chromatin architecture. Moreover, we demonstrate that ASO-mediated targeting of GGC repeat expansions rescues these pathological phenotypes, highlighting its potential as a therapeutic strategy for NIID. Our findings provide insights into the molecular mechanisms underlying NIID and establish a theoretical foundation for the future development of gene-targeted therapies.
Results
PolyG disrupts nucleolar structure and function
PONDR22 (predictor of natural disordered regions) is a bioinformatics tool specifically designed for predicting intrinsically disordered regions (IDRs) in protein sequences. By analyzing amino acid sequence features, such as composition and hydrophobicity, it assesses the propensity of protein regions to lack stable three-dimensional structures. Analysis using this tool revealed that polyG contains segments with scores exceeding the threshold (>0.5), indicating potential intrinsic disorder. This characteristic suggests its possible involvement in biological functions, such as phase separation or multivalent interactions (Fig. 1a). The IDR feature of polyG resembles that of pathogenic repeat polypeptides generated by trinucleotide expansions in FMR1 and C9orf72, as previously reported23,24. Although our prior studies demonstrated that pathogenic polyG induces nucleolar stress marked by NPM1 nucleocytoplasmic mislocalization8, its specific impact on nucleolar structure and function remained unclear. The nucleolus consists of fibrillar centers (FCs) enriched with NPM1 and granular components (GCs) enriched with fibrillarin (FBL), where the IDR of NPM1 plays a critical role in maintaining liquid-liquid phase separation (LLPS) homeostasis and structural integrity. We thus hypothesized that pathological polyG, harboring IDR, may disrupt nucleolar structure and function.

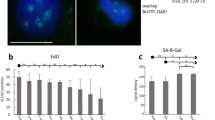
a PONDR prediction of potential intrinsically disordered regions (IDRs) in 90✕-polyG sequences. Scores above 0.5 indicate disorder. b Cell viability curves of HEK-293T cells transfected with 20✕-polyG or 90✕-polyG constructs over a 0–96 h time course, measured by CCK-8 assay (OD450). Data are presented as mean ± SD (n = 3 independent experiments). Statistical significance at each time point was determined by unpaired two-tailed Student’s t test. c Immunofluorescence images showing 90✕-polyG aggregates and the subcellular distribution of NPM1 in HEK-293T cells. The negative control cells (only with transfection reagent) exhibit normal nucleolar NPM1 localization. In contrast, the positive control cells show nucleoplasmic mislocalization of NPM1. n = 3 independent experiments. Scale bar: 10 μm. d Violin plots quantifying the frequency of NPM1 nucleoplasmic mislocalization. Analysis was performed specifically on successfully transfected cells from the conditions indicated in (c) (blank, 20✕-polyG, and 90✕-polyG group). Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test. n = 3 independent experiments. e Capillary electrophoresis immunoblotting analysis of NPM1 and FBL protein levels in blank (untransfected), 20✕-polyG-, and 90✕-polyG-transfected HEK-293T cells. n = 3 independent experiments. f, h Violin plots showing relative protein expression levels of NPM1 (e) and FBL (f) in blank, 20✕-polyG-, and 90✕-polyG-transfected HEK-293T cells, normalized to GAPDH. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test (n = 3 independent experiments). g Immunofluorescence images showing 90✕-polyG aggregates and the distribution of FBL and NPM1 in HEK-293T cells. n = 3 independent experiments. Scale bar: 5 μm. i Capillary electrophoresis immunoblotting analysis of NPM1 and FBL protein levels in control (GGCnor) and polyG-expressing (GGCexp) neurons at 30 days in vitro (DIV). n = 3 biological replicates. j, k Violin plots showing relative protein expression levels of NPM1 (h) and FBL (i) in GGCnor and GGCexp neurons at 30 DIV, normalized to GAPDH. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Data are presented as mean ± SD.
To test this hypothesis, we transfected HEK-293T cells with our previously constructed 20✕- and 90✕-polyG overexpression plasmids8. Specifically, we generated expression vectors containing the 5′-UTR of NOTCH2NLC with 20 (normal repeat) or 90 (pathogenic repeat) GGC repeats, fused to mCherry (polyG-mCherry). We observed that 90✕-polyG formed more abundant aggregates in HEK-293T cells compared to the control groups (Supplementary Fig. 1a), while demonstrating significant spatial colocalization with ubiquitin and p62 (Supplementary Fig. 1b)—a characteristic pathological hallmark of NIID4,6,7,9,25. An mCherry-only plasmid served as a negative control. CCK8 assays revealed that 90✕-polyG significantly reduced cellular metabolic activity compared to 20✕-polyG (Fig. 1b), suggesting its cytotoxic potential.
NSC348884 can specifically induce nucleocytoplasmic translocation of NPM1. Our experiments revealed that the 90✕-polyG group induced nucleocytoplasmic translocation of NPM1, a phenotype consistent with that observed in the positive control group treated with the specific inducer NSC348884 (Fig. 1c). This effect was not observed in the negative control (transfection reagent only), mCherry, or 20✕-polyG groups (Fig. 1c). This conclusion was further supported by statistical analysis of the occurrence frequency (Fig. 1d). Moreover, we found that 90✕-polyG upregulated the expression of NPM1 (Fig. 1e, f). These results suggest that 90✕-polyG induces nucleolar stress. Concurrently, 90✕-polyG disrupted the GC layer (Fig. 1g) and downregulated FBL expression (Fig. 1e, h) in our cellular model. Furthermore, in HEK-293T cells, the 90✕-polyG-induced alterations in NPM1 and FBL were consistent with changes observed at the mRNA level (Supplementary Fig. 1c, d), suggesting that polyG exerts its effects through transcriptional regulation. To validate these findings, we confirmed the presence of aggregates co-formed by 90✕-polyG, ubiquitin, and p62 in the neurogenic mouse hippocampal cell line HT-22 (Supplementary Fig. 1e). Moreover, overexpression of 90✕-polyG in HT-22 cells similarly resulted in increased NPM1 and decreased FBL at both transcriptional and translational levels (Supplementary Fig. 1f–j), accompanied by loss of normal structural localization of both NPM1 (Supplementary Fig. 1k) and FBL (Supplementary Fig. 1l). In summary, these results demonstrate that 90✕-polyG disrupts nucleolar structure and function.
In our earlier work, we successfully established three iPSC lines from NIID patients carrying NOTCH2NLC GGC repeat expansions (130, 109, and 139 repeats) and differentiated them into 3DCOs (Supplementary Fig. 2a–c) and neurons (Supplementary Fig. 2d)8. Age- and sex-matched normal control-derived neurons and 3DCOs were used as normal controls. To evaluate the developmental stability of the 3DCOs, we analyzed the structural integrity and cellular composition of three normal control-derived 3DCOs. The results demonstrated consistent thickness of SOX2+ pseudoventricles and stable TBR2+/SOX2+ area ratios across samples (Supplementary Fig. 2e–g). Furthermore, immunofluorescence analysis revealed maintained GFAP distribution area, as well as fluorescence intensity of NeuN (MNs) and DCX (immature neurons) (Supplementary Figs. 2h–j, and 3a, b). Additionally, comparative analysis of three independent batches from one normal control confirmed consistent SOX2+ thickness, TBR2+/SOX2+ area ratio, GFAP distribution area, and NeuN fluorescence intensity across batches (Supplementary Fig. 3c–h). Likewise, we observed typical cortical pseudoventricular structures in three patient-derived cerebral organoids, with no significant differences in the thickness of SOX2+ or the TBR2+/SOX2+ area ratio (Supplementary Fig. 3i–k).
We previously demonstrated that these patient-derived 3DCOs retained NOTCH2NLC GGC repeat mutations and exhibited characteristic NIID pathological features, including p62- and ubiquitin-positive intranuclear inclusions (Supplementary Fig. 4a–d) 8. Furthermore, we observed that these inclusions were present across multiple cell types in 3DCOs, specifically within DCX+ immature neurons, MAP2+ MNs and GFAP+ astrocytes (Supplementary Fig. 4e). Comparative analysis revealed significantly elevated NPM1 expression (Fig. 1i, j, and Supplementary Fig. 4f, g) and reduced FBL levels (Fig. 1i, k, and Supplementary Fig. 4f, h) in NIID patient-derived neurons and 3DCOs. These alterations at the protein level were consistent with corresponding changes in their transcriptional levels (Supplementary Fig. 4i–l). Collectively, these results demonstrate that pathogenic polyG disrupts nucleolar structure and function.
Further observation revealed that a subset of inclusions exhibited nuclear localization across these models. We quantified the nuclear localization rates of inclusions in different models. Based on data from a published genetically engineered mouse model (32-day-old)9, the nuclear localization rates varied across brain regions: 14% in the hippocampus versus 24% in the cortex. The nuclear localization rates in our patient-derived neurons (30-day-old, 25%) and cortical organoids (90-day-old, 23%) closely matched that observed in the cortical region of the mouse model (24%) (Supplementary Fig. 4l). Similarly, the nuclear localization rate in HT-22 cells (14%) aligned with that in the hippocampal tissue of the mouse model (14%) (Supplementary Fig. 4l). These data indicate that while overexpression models may exhibit supraphysiological expression levels, both patient-derived models and HT-22 cells show remarkable consistency with the genetically engineered animal model in key pathological features-particularly in nuclear localization rates of inclusions-reflecting the dynamic progression of polyG-mediated pathogenesis across different spatiotemporal contexts.
Pathogenic polyG activates the NPM1-centric protein quality control system
Notably, we observed colocalization between 90✕-polyG and NPM1 in HEK-293T, HT-22 and neurons cellular models (Supplementary Fig. 5a–c), suggesting a potential interaction. To investigate this, we controlled plasmid transfection duration in HEK-293T cells and found that 90✕-polyG (mCherry-tagged) upregulated NPM1 expression in a time-dependent manner (Supplementary Fig. 5d–f). At 12 h post-transfection, 90✕-polyG aggregates became detectable (Fig. 2a) and progressively enlarged (Supplementary Fig. 5g). Intriguingly, 90✕-polyG initially colocalized with NPM1 at 12 h (Fig. 2a). We observed colocalization of 90✕-polyG with NPM1 in patient-derived neurons (Fig. 2b and Supplementary Fig. 5c). Given NPM1’s role in the protein quality control system26, pathological polyG may activate this pathway. Specifically, in the nervous system, misfolded proteins enter the nucleolus and bind NPM1 to mitigate toxicity, with HSP70 partially restoring refolding capacity26. To test whether polyG engages this mechanism, immunofluorescence confirmed robust colocalization between 90✕-polyG and HSP70 in HEK-293T and HT-22 cells (Fig. 2c and Supplementary Fig. 5h), alongside significantly elevated HSP70 levels (Fig. 2d and Supplementary Fig. 5i, j). Consistent with observations in cellular models, we found that inclusions in NIID patient-derived neurons (Fig. 2e) and 3DCOs (Supplementary Fig. 5k) also exhibited spatial colocalization with HSP70, along with a significant upregulation of HSP70 expression (Fig. 2f and Supplementary Fig. 5l, m), when compared to normal controls. These findings suggest that 90✕-polyG activates an NPM1-centric protein quality control response.

a Temporal immunofluorescence analysis showing the distribution of 90✕-polyG and NPM1 in HEK-293T cells over time. n = 3 independent experiments. Scale bar: 5 μm. b Immunofluorescence images showing specific colocalization of ubiquitin with NPM1 in GGCexp neurons, but not in GGCnor neurons, at 30 DIV. Arrows indicate inclusions. n = 3 biological replicates. Scale bar: 10 μm. c Immunofluorescence images showing colocalization of 90✕-polyG with HSP70 in HEK-293T cells (not observed in 20✕-polyG-transfected or blank control cells). n = 3 independent experiments. Scale bar: 5 μm. d Immunoblotting and quantitative analysis showing a significant increase in HSP70 protein levels in 90✕-polyG-transfected HEK-293T cells compared to blank and 20✕-polyG-transfected controls, normalized to GAPDH. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test (n = 3 independent experiments). e Immunofluorescence images showing colocalization of ubiquitin with HSP70 in GGCexp neurons (not observed in GGCnor neurons) at 30 DIV. Arrows indicate inclusions. n = 3 biological replicates. Scale bar: 10 μm. f Quantitative analysis showing significantly higher HSP70 protein levels in GGCexp neurons compared to GGCnor neurons at 30 DIV, normalized to GAPDH. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Data are presented as mean ± SD.
Since NPM1 binds rRNA through its IDR to regulate protein biosynthesis, we discovered that rRNA also participates in inclusion body formation (Supplementary Fig. 5n). However, whether 90✕-polyG colocalizes with rRNA in an IDR-dependent manner remains unclear. To test this hypothesis, siRNA-mediated NPM1 knockdown (>90% efficiency, Supplementary Fig. 5o) abolished 90✕-polyG-NPM1 colocalization but preserved polyG-rRNA binding (Supplementary Fig. 5p), indicating that polyG independently associates with rRNA, yet preferentially interacts with NPM1. This behavior contrasts with C9orf72-derived repeat dipeptides, which preferentially bind rRNA27. Together, our results demonstrate that 90✕-polyG independently engages both NPM1 and rRNA, with binding priority for NPM1.
PolyG-induced nucleolar dysfunction disrupts 3D chromatin architecture
Nucleolar homeostasis plays a critical role in maintaining 3D genome organization. Previous studies have shown that NPM1 interacts with the chromatin structural protein CTCF to regulate the formation of 3D chromatin architecture12. Additionally, RAD21 cooperates with CTCF to participate in the formation of TADs and chromatin loops13. We therefore hypothesized that 90✕-polyG-mediated perturbation of NPM1 might induce alterations in 3D genome organization. To test this hypothesis, we found that compared to 20✕-polyG and empty vector controls, 90✕-polyG significantly reduced both the fluorescence intensity of CTCF and RAD21 (Fig. 3a, b), as well as their protein expression levels (Fig. 3c–e) in HEK-293T models. We consistently observed these effects in 90✕-polyG-expressing HT-22 models (Supplementary Fig. 6a–e). Furthermore, 90✕-polyG reduce CTCF and RAD21 expression in HEK-293T cells in a time-dependent manner (Supplementary Fig. 6f–h), suggesting sustained effects on 3D chromatin structure proteins. Consistent with these findings, NIID patient-derived neurons (Fig. 3f–h) and 3DCOs (Supplementary Fig. 6i–k) also showed downregulation of CTCF and RAD21. Collectively, these results providing a potential mechanistic basis for the subsequent alterations in 3D chromatin structure.

Immunofluorescence images showing reduced fluorescence intensity of CTCF (a) and RAD21 (b) in 90✕-polyG-transfected HEK-293T cells compared to controls. n = 3 independent experiments. Scale bar: 5 μm. c Capillary electrophoresis immunoblotting analysis of CTCF and RAD21 protein levels in blank, 20✕-polyG-, and 90✕-polyG-transfected HEK-293T cells. n = 3 independent experiments. Violin plots showing relative protein expression levels of CTCF (d) and RAD21 (e) in blank, 20✕-polyG-, and 90✕-polyG-transfected HEK-293T cells, normalized to GAPDH. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test (n = 3 independent experiments). f Capillary electrophoresis immunoblotting analysis of CTCF and RAD21 protein levels in GGCnor and GGCexp neurons at 30 DIV. n = 3 biological replicates. Violin plots showing relative protein expression levels of CTCF (g) and RAD21 (h) in GGCnor and GGCexp neurons at 30 DIV, normalized to GAPDH. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Data are presented as mean ± SD.
Furthermore, patient-derived 3DCOs exhibited a more diffuse nuclear distribution pattern of CTCF compared to controls (Supplementary Fig. 7a), suggesting potential functional impairment. Notably, control 3DCOs demonstrated significant co-expression of RAD21 with the neural progenitor marker PAX6 (Supplementary Fig. 7b), consistent with RAD21’s role in maintaining stem cell identity. In contrast, this co-expression pattern was altered in patient-derived 3DCOs (Supplementary Fig. 7c) and associated with reduced signal intensity of the pan-neuronal marker TUJ1 (Supplementary Fig. 7d). These findings indicate that pathogenic polyG-induced downregulation and mislocalization of CTCF/RAD21 underlie the abnormalities in neural progenitor identity by dysregulating the expression of genes involved in neural differentiation.
To further characterize polyG-mediated 3D chromatin structural changes, we performed Hi-C sequencing on 90-day-old 3DCOs derived from three NIID patients and three normal controls. Using Hi-C 2.0 technology, we obtained approximately 3 billion paired-end reads per sample, generating about 1 billion uniquely mapped valid interactions in total. Data quality was verified by cis/trans interaction ratios and distance-dependent interaction frequency decay analysis (Supplementary Fig. 8a). The interaction matrices showed high correlation between groups (Pearson correlation coefficient R = 0.96) (Supplementary Fig. 8b) at 5-kb resolution (Supplementary Fig. 8c), enabling multiscale analysis of 3D chromatin structure, including compartments, TADs, and loops.
Chromatin is spatially segregated into generally active A compartments and repressive B compartments, with A compartments located in the nuclear interior and B compartments anchored at the nuclear periphery via Lamin B1. Compared to normal controls, disease 3DCOs showed significant compartment changes (Fig. 4a). Detailed analysis revealed that 512 compartments (8.9%) underwent switching, including 295 (5.1%) A-to-B and 217 (3.8%) B-to-A conversions, while 5231 (91.1%) compartments remained stable (Fig. 4b). Previous finding that 90✕-polyG induces Lamin B1 disruption7 may explain these compartment conversions.

a Log2 ratio of contact frequencies (GGCexp/GGCnor). b Analysis of compartment switching in GGCexp versus GGCnor 3DCOs. c Hi-C contact maps illustrating chromatin compartments, topologically associating domains (TADs), and chromatin loops (indicated by blue arrows). d Insulation scores for the genomic bins corresponding to the region shown in the middle panel of (c). Red and green arrows mark TAD boundaries and interiors, respectively. e Fluorescence images confirming successful lentivirus-mediated overexpression (OE) of CTCF and RAD21 in GGCexp-3DCOs. 60-day-old 3DCOs were infected with lentivirus at an MOI of 40 and analyzed one week later. n = 3 biological replicates. Scale bar: 100 μm. f Log2 ratio of contact frequencies (OE/GGCnor). g Hi-C contact maps illustrating chromatin compartments, TADs, and chromatin loops (indicated by blue arrows) in the OE condition. h Insulation scores for the genomic bins corresponding to the region shown in the middle panel of (g). Yellow and green arrows mark TAD boundaries and interiors, respectively.
Given that CTCF and RAD21 cooperatively regulate TAD and loop formation, we further investigated the effects of their dysregulation. We identified 5592 and 5568 TADs in disease and control groups, respectively. Compared to normal controls, the disease group showed loss of 2026 TADs and gain of 2050 TADs. Control TADs exhibited sharp boundaries, whereas disease TAD boundaries appeared blurred (Fig. 4c, middle), indicating more frequent intra-TAD interactions in controls. Quantification of boundary insulation strength revealed higher insulation scores at TAD boundaries in NIID 3DCOs (Fig. 4d), indicating reduced boundary strength. Moreover, intra-TAD insulation scores were lower in NIID 3DCOs (Fig. 4d), suggesting decreased interaction frequencies within TADs.
Loop analysis identified 4990 and 5456 loops in the disease and control groups, respectively. The disease group showed loss of 2308 loops and gain of 1845 loops compared to controls. Among 3153 conserved loops, 191 exhibited enhanced interaction strength (fold change ≥2) while 441 showed weakened interactions (fold change ≤0.5). As shown in Fig. 4c (right), loop interactions were generally reduced in NIID 3DCOs. Loop anchors are enriched with enhancer-promoter interactions. To further clarify the potential functional impact of the differential loops between the two groups, we analyzed genes located near loop anchors and those within loops. The results showed that there were 5419 genes within 50 kb of loop anchors and 8549 genes located inside loops. These gene sets overlapped with the DEGs from scRNA-seq results by 587 and 506 genes, respectively (Supplementary Data 1). Enrichment analysis revealed that the DEGs near anchors were associated with functions, such as ribosome, translation, DNA mismatch repair, DNA replication, and the ubiquitin-proteasomal system. In contrast, the DEGs within loops were linked to 5q35 copy number variation and apoptosis (Supplementary Data 1). In summary, these results indicate that the dysregulation of loop structures in patient-derived cerebral organoids is consistent with pathological features, and genes near loop anchors appear to play a more significant role.
To further elucidate the role of CTCF and RAD21 in maintaining 3D chromatin architecture in NIID patient-derived 3DCOs, we performed lentivirus-mediated rescue of CTCF/RAD21 expression. We transduced 60-day-old 3DCOs from three patients with lentiviral vectors. One week post-transduction, robust expression of CTCF (EGFP-tagged) and RAD21 (mCherry-tagged) was confirmed (Fig. 4e) and further validated by protein quantification (Supplementary Fig. 8d–f). Hi-C sequencing was subsequently performed 30 days post-transduction (OE group).
Quality control assessments using cis/trans interaction ratios and distance-dependent interaction decay confirmed high-quality Hi-C data (Supplementary Fig. 8g). The OE group exhibited higher correlation with normal controls (NC group) (Pearson R = 0.97) than with the disease group (Supplementary Fig. 8h), with achieved resolution of 5 kb (Supplementary Fig. 8i). Notably, the OE group partially reversed disease-associated compartment switching, as visualized by interaction frequency heatmaps (Fig. 4f). Among previously switched compartments, 245 of 512 (47.9%) were restored (135 B-to-A and 110 A-to-B reversions), while 5499 compartments remained conserved. We identified 5577 TADs and 5519 chromatin loops in the OE group. Specifically, 707 previously lost TADs were reestablished, and 889 aberrantly gained TADs were eliminated (Fig. 4g). Insulation score analysis demonstrated enhanced TAD boundary strength and increased intra-TAD contact frequency in the OE group compared to the NIID group (Fig. 4h). Furthermore, 2129 (51.3%) altered loops were reversed, with 717 loops regained and 1412 loops eliminated. Using the aforementioned method, we identified 319 differentially DEGs located near the rescued loop anchors in the OE group (Supplementary Data 1), which remain associated with functions, such as ribosome and mRNA processing (Supplementary Data 1). In summary, CTCF/RAD21 overexpression partially rescues the 3D chromatin structural abnormalities induced by NOTCH2NLC GGC repeat expansions. In summary, our results demonstrate that CTCF and RAD21 significantly contribute to the abnormalities in 3D chromatin architecture observed in NIID 3DCOs models.
NPM1 dysfunction induces DNA damage and cellular senescence in NIID pathogenesis
As NPM1 is recruited to DNA damage sites and participates in DNA repair processes28, we hypothesized that 90✕-polyG-induced nucleocytoplasmic mislocalization of NPM1 might lead to genomic instability. Phosphorylated γH2AX (p-γH2AX), a well-established marker of DNA double-strand breaks29, demonstrated significantly elevated fluorescence intensity and a higher percentage of positive cells in both HEK-293Tand HT-22 cells transfected with 90✕-polyG, compared to both 20✕-polyG and empty vector controls (Fig. 5a, b, and Supplementary Fig. 9a–c). Among HEK-293T cells successfully expressing 90✕-polyG, approximately 86.0% exhibited DNA damage, while this proportion reached 92.3% in HT-22 cells. In contrast, only 36.7% of HEK-293T cells expressing mCherry alone or 20✕-polyG showed DNA damage, decreasing further to 14.0% in HT-22 cells. Protein quantification further confirmed that 90✕-polyG significantly increased p-γH2AX levels in both HEK-293T and HT-22 cells (Fig. 5c, d, and Supplementary Fig. 9d, e). Notably, p-γH2AX induction appeared independent of 90✕-polyG aggregation, as enhanced p-γH2AX fluorescence was still observed in cells without visible aggregates (Supplementary Fig. 9a, b).

a Immunofluorescence images showing p-γH2AX in HEK-293T cells transfected with 90✕-polyG, 20✕-polyG, or blank control. n = 3 independent experiments. Scale bar: 5 μm. b Quantification of p-γH2AX-positive cells from (a). One-way ANOVA with Tukey’s test (n = 3). c Immunoblotting of p-γH2AX in HEK-293T cells (90✕-polyG vs controls) and in GGCnor vs GGCexp neurons at 30 DIV. HEK-293T: n = 3 independent experiments; neurons: n = 3 biological replicates. d Quantification of p-γH2AX levels in HEK-293T cells from (c), normalized to GAPDH. One-way ANOVA with Tukey’s test (n = 3). e Immunofluorescence images showing ubiquitin inclusions and p-γH2AX in GGCnor and GGCexp neurons at 30 DIV. n = 3 biological replicates. Arrows indicate cells positive for both ubiquitin inclusions and γH2AX foci. Scale bar: 50 μm. f Quantification of p-γH2AX-positive neurons from (e). Two-tailed paired Student’s t test (n = 3). g Quantification of p-γH2AX protein levels in neurons from (c), normalized to GAPDH. Two-tailed paired Student’s t test (n = 3). GSVA enrichment scores for DNA damage response (h) and TP53 pathway (i) at day 90. Each point represents the pseudobulk score of one donor (n = 3/group). Two-sided Wilcoxon test. Box plots show median and quartiles. j, k Enrichment plots of senescence pathways significantly enriched in DEGs from 90×-polyG-expressing HEK-293T cells (red boxes). DEGs were derived from three independent RNA-seq experiments. Enrichment was assessed using Enrichr (Fisher's exact test, Benjamini–Hochberg, adjusted p < 0.05). An asterisk (*) denotes a significant adjusted p-value. See Supplementary Data 1 for exact adjusted p-values. β-galactosidase staining (l) and quantification (m) in HEK-293T cells. One-way ANOVA with Tukey’s test (n = 3). Scale bar: 50 μm. β-galactosidase staining (n) and quantification (o) in GGCnor vs GGCexp neurons at 30 DIV. Two-tailed paired Student’s t test (n = 3). Scale bar: 20 μm. p GSVA enrichment scores for DNA damage, telomere stress-induced senescence pathway at day 90. Each point represents the pseudobulk score of one donor (n = 3/group). Two-sided Wilcoxon test. Box plots show median and quartiles. Data are mean ± SD.
We next conducted related experiments in neurons and 3DCOs derived from three NIID patients. Consistent with our earlier findings, DEGs located near differential loop anchors between the two groups in cerebral organoids were significantly enriched in pathways related to DNA damage, such as DNA mismatch repair (Supplementary Data 1). Consistent with previous findings, NIID patient-derived neurons showed a significantly higher percentage of p-γH2AX-positive events (approximately 18.0%) along with elevated p-γH2AX expression compared to normal controls (Fig. 5c, e, f). Notably, we observed that over 80% of neurons containing inclusions exhibited enhanced p-γH2AX fluorescence. Similarly, patient-derived 3DCOs demonstrated a substantially increased proportion of p-γH2AX-positive events (approximately 14%) (Supplementary Fig. 9f, g) accompanied by upregulated p-γH2AX expression (Supplementary Fig. 9h, i). Furthermore, to comprehensively evaluate DNA damage levels in patient-derived 3DCOs, we performed GSVA on our previously published scRNA-seq data8. The results demonstrated significantly enhanced DNA damage responses across all major cell types, including NPCs, neuroblasts (NBs), neurons, and glutamatergic neurons, radial glial cells (Fig. 5h and Supplementary Fig. 9j). Consistent with these findings, we observed significant activation of the TP53 pathway—a key downstream signaling cascade of DNA damage—in patient-derived 3DCOs (Fig. 5i). These findings collectively indicate widespread genomic instability in NIID disease models.
Since accumulated DNA damage is a known driver of cellular senescence30, we next investigated whether NIID models exhibit accelerated aging phenotypes. Bulk RNA sequencing of HEK-293T cells transfected with 20✕-polyG, 90✕-polyG, or empty vector revealed distinct transcriptional profiles. Principal component analysis showed clear separation between groups while maintaining high intra-group consistency (Supplementary Fig. 10a). Hierarchical clustering analysis further demonstrated that 20✕-polyG-transfected cells clustered with empty vector controls, whereas 90✕-polyG-expressing cells formed a distinct group (Supplementary Fig. 10b), suggesting that 90✕-polyG induces unique transcriptional alterations. Venn diagram analysis illustrated the numbers of differentially expressed genes among groups (Supplementary Fig. 10c), and heatmap visualization confirmed the specific gene expression pattern in 90✕-polyG cells (Supplementary Fig. 10d). Notably, pathway enrichment analysis of upregulated genes in 90✕-polyG cells revealed significant enrichment in senescence-associated pathways (the SHAP pathway; Fig. 5j, k), while 20✕-polyG cells showed no such enrichment compared to controls (Supplementary Fig. 10e), indicating that senescence phenotypes primarily occur in 90✕-polyG cells.
Consistent with cellular senescence characteristics, we observed significantly increased β-galactosidase activity in both HEK-293T (Fig. 5l, m) and HT-22 cells (Supplementary Fig. 10f, g) expressing 90✕-polyG. Similarly, patient-derived neurons (Fig. 5n, o) and 3DCOs (Supplementary Fig. 10h, i) exhibited markedly elevated β-galactosidase activity compared to controls. Additionally, GSVA analysis of scRNA-seq data from patient-derived 3DCOs confirmed elevated senescence signatures associated with DNA damage responses (Fig. 5p). Interestingly, neurons in the scRNA-seq showed lower senescence scores than controls (Supplementary Fig. 10j), potentially due to their heightened vulnerability to damage and subsequent loss.
In conclusion, our results demonstrate that accumulation of DNA damage events in NIID disease models promotes cellular senescence. These findings reveal that polyG disrupts nucleolar function and NPM1 homeostasis, thereby inducing DNA damage and cellular senescence.
ASO reduces polyG biosynthesis in NIID models
ASOs have demonstrated therapeutic efficacy in treating FMR1 repeat expansion-associated disorders29,31. Given the similar repeat mutations and pathogenic mechanisms between FMR1 and NOTCH2NLC, we investigated whether ASOs could be applied to treat NIID caused by NOTCH2NLC GGC repeat expansions. Based on previous studies, we designed ASO-GGC, ASO-GGC-cy3, and control ASO-ctr, which block the translation of repeat RNAs through a steric hindrance mechanism32. Our experiments showed that ASO-GGC-cy3 (200 nM) could efficiently enter HEK-293T cells via lipofection (Supplementary Fig. 11a). To evaluate the cytotoxicity of ASO-ctr and ASO-GGC, we measured cell viability and found no significant effects compared to the transfection reagent-only control group (Supplementary Fig. 11b). Additionally, apoptosis and necrosis assays revealed that neither ASO-ctr nor ASO-GGC significantly increased cell death (Supplementary Fig. 11c, d). These results confirm the minimal cytotoxicity of ASO-ctr and ASO-GGC, prompting us to further assess their therapeutic potential in NIID models.
We co-transfected HEK-293T cells with 90✕-polyG plasmids and 200 nM ASO-GGC or ASO-ctr, followed by analysis after 48 h. ASO-GGC significantly reduced 90✕-polyG (mCherry) expression (Fig. 6a, b), accompanied by a marked decrease in 90✕polyG inclusions (Fig. 6c, d). To determine whether ASO-GGC suppresses translation by degrading 90✕-polyG-mCherry mRNA, we quantified mRNA levels and observed a significant increase in mCherry mRNA (Fig. 6c, d). This suggests that the steric hindrance imposed by ASO-GGC may stabilize the mRNA structure, reducing its degradation while simultaneously preventing ribosomal binding and subsequent translation. These results indicate that ASO-GGC reduces 90✕-polyG production primarily by inhibiting translation rather than degrading mRNA. Further evaluation of endogenous NOTCH2NLC demonstrated that ASO-GGC did not significantly affect its protein expression (Supplementary Fig. 11e, f) while notably increasing NOTCH2NLC transcript levels (Supplementary Fig. 11g).

Immunoblotting (a) and quantitative analysis (b) showing that ASO-GGC treatment reduces mCherry-polyG fusion protein levels in HEK-293T cells compared to the control ASO (ASO-ctr), normalized to tubulin. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 independent experiments). Immunofluorescence images (c) and quantitative analysis (d) showing that ASO-GGC treatment decreases the number of 90✕-polyG inclusion bodies in HEK-293T cells compared to ASO-ctr. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 independent experiments). Scale bar: 20 μm. e qPCR analysis showing that ASO-GGC treatment enhances mCherry transcription in HEK-293T cells. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 independent experiments). Immunofluorescence images (f) and quantitative analysis (g) showing that ASO-GGC (200 nM) reduces the number of p62/ubiquitin-positive inclusions in patient-derived GGCexp neurons compared to ASO-ctr. Neurons were treated at 30 DIV for 2 weeks prior to analysis. Arrows indicate cells showing colocalization of ubiquitin and p62. n = 3 biological replicates; statistical analysis was performed using a two-tailed paired Student’s t test. Scale bar: 20 μm. Immunofluorescence images (h) and quantitative analysis (i) showing that ASO-GGC (200 nM) reduces the number of p62/ubiquitin-positive inclusions in patient-derived GGCexp-3DCOs at 90 days compared to ASO-ctr. Organoids were treated at 76 DIV for 2 weeks prior to analysis; three organoids were selected per sample for analysis. n = 3 biological replicates; statistical analysis was performed using a two-tailed paired Student’s t test. Scale bar: 10 μm. Data are presented as mean ± SD.
These findings suggest that ASO-GGC specifically binds to the GGC repeat expansion in the 5′UTR of NOTCH2NLC mature mRNA, thereby impeding ribosomal progression after translation initiation. This steric hindrance prevents efficient readthrough of the GGC repeat region, leading to substantially reduced polyG production. Meanwhile, the ASO binding stabilizes the mRNA structure, slowing its degradation and resulting in elevated mRNA levels32. Although ribosomes are blocked at the ASO-bound repeat region, they can bypass this 5′ UTR segment to translate full-length, normal NOTCH2NLC protein downstream. To validate ASO efficacy in patient-derived 3DCOs, we first treated 3DCOs with 200 nM ASO-GGC-cy3 for 2 weeks, confirming successful ASO delivery (Supplementary Fig. 11a). We subsequently treated patient-derived neurons and 3DCOs from three NIID cases with 200 nM ASO-GGC to evaluate therapeutic effects, with ASO-ctrl-treated patient-derived models serving as controls. During the treatment period, ASO-GGC or ASO-ctrl was dissolved in culture medium and replenished with each medium change for 2 consecutive weeks. Immunofluorescence analysis demonstrated that ASO-GGC significantly reduced the number of p62- and ubiquitin-positive intranuclear inclusions in these models (Fig. 6f–i), indicating decreased polyG production. Consistent with observations in cellular models, ASO-GGC increased endogenous NOTCH2NLC mRNA levels in both patient-derived neurons and 3DCOs without affecting its protein expression (Supplementary Fig. 11h–k).
In conclusion, these findings demonstrate that ASO-GGC effectively reduces polyG biosynthesis and aggregation in NIID models by inhibiting translation, while preserving endogenous NOTCH2NLC protein expression.
ASO rescues 3D genomic disorganization and senescence in NIID models
Given that ASO-GGC significantly reduces polyG expression and aggregation, we investigated its potential to ameliorate genomic instability and senescence in NIID models. In HEK-293T cells co-transfected with 90✕-polyG/ASO-GGC and 90✕-polyG/ASO-ctr, ASO-GGC significantly decreased NPM1 expression levels (Supplementary Fig. 11l, m), indicating alleviated nucleolar stress. Concurrently, ASO-GGC substantially reduced DNA damage and senescence phenotypes in 90✕-polyG-expressing HEK-293T cells, as demonstrated by decreased p-γH2AX levels (Supplementary Fig. 11l, n) and β-galactosidase activity (Supplementary Fig. 11o, p). Similarly, in NIID patient-derived neurons and 3DCOs, ASO-GGC significantly improved pathological conditions compared to the ASO-ctrl group. Specifically, ASO-GGC ameliorated nucleolar stress by reducing NPM1 expression (Fig. 7a, b, d, e). Furthermore, ASO-GGC attenuated DNA damage in patient-derived models, as evidenced by reduced p-γH2AX levels (Fig. 7a, c, d, f) and fewer DNA damage foci (Fig. 7g–j), concomitant with elevated TUJ1 expression in 3DCOs (Supplementary Fig. 11q). Finally, ASO-GGC significantly reduced senescence in both patient-derived neurons (Fig. 7k, l) and 3DCOs (Fig. 7m, n).

Immunoblotting analysis (a) and quantitative analysis of NPM1 (b) and p-γH2AX (c) protein levels demonstrating that ASO-GGC treatment significantly reduces their levels in patient-derived GGCexp neurons compared to ASO-ctr, normalized to GAPDH. Neurons were treated at 30 DIV for 2 weeks prior to analysis. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Immunoblotting analysis (d) and quantitative analysis of NPM1 (e) and p-γH2AX (f) protein levels demonstrating that ASO-GGC treatment significantly reduces their levels in patient-derived GGCexp-3DCOs compared to ASO-ctr, normalized to GAPDH. Organoids were treated at 76 DIV for 2 weeks prior to analysis; three organoids were selected per sample for analysis. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Immunofluorescence images (g) and quantitative analysis (h) showing that ASO-GGC treatment reduces the percentage of p-γH2AX-positive cells in patient-derived GGCexp neurons compared to ASO-ctr. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Arrows indicate cells positive for both ubiquitin inclusions and γH2AX foci. Scale bar: 50 μm. Immunofluorescence images (i) and quantitative analysis (j) showing that ASO-GGC treatment reduces the percentage of p-γH2AX-positive cells in patient-derived GGCexp-3DCOs compared to ASO-ctr. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Scale bar: 20 μm. Images of β-galactosidase staining (k) and quantitative analysis (l) showing a significant decrease in the area of β-galactosidase-positive blue precipitates in ASO-GGC-treated patient-derived GGCexp neurons compared to ASO-ctr. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Scare bar: 20 μm. Images of β-galactosidase staining (m) and quantitative analysis (n) showing a significant decrease in the area of β-galactosidase-positive blue precipitates in ASO-GGC-treated patient-derived GGCexp-3DCOs compared to ASO-ctr. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Scare bar: 50 μm. Data are presented as mean ± SD.
To further elucidate the therapeutic effects of ASO-GGC on NIID, we performed scRNA-seq on ASO-GGC- and ASO-ctr-treated NIID patient-derived 3DCOs. Specifically, 3DCOs from three patients were randomly divided into two groups and treated with either ASO-GGC or ASO-ctr for 2 weeks. After dissociation into single-cell suspensions and equal mixing, we obtained 26,423 and 14,284 high-quality cells from ASO-GGC and ASO-ctr groups, respectively. ScType analysis identified six cell types: NPCs, NBs, MNs, GABAergic neurons (GNs), radial glial cells (RGCs), and oligodendrocyte precursor cells (OPCs) (Fig. 8a and Supplementary Fig. 12a). Bubble plots (Supplementary Fig. 12b) and heatmaps (Supplementary Fig. 12c) of marker genes are provided. Cell proportion analysis revealed that following ASO-GGC treatment, the proportions of OPCs, GNs, and NPCs increased, whereas the proportions of RGCs, MNs, and NBs decreased (Supplementary Fig. 12d). We identified 325 differentially expressed genes between groups (Fig. 8b), which were significantly enriched in pathways related to ribosome biogenesis and polypeptide elongation (Fig. 8c), closely associated with translation and macromolecular biosynthesis (Fig. 8d). Subsequently, we identified DEGs within each major cell subtype between ASO-treated and control organoids, including NPCs (883 DEGs), NBs (476 DEGs), MNs (468 DEGs), GNs (605 DEGs), RGCs (763 DEGs), and OPCs (218 DEGs) (Supplementary Data 2). Furthermore, we performed functional enrichment analysis on DEGs, the results revealed significant enrichment in pathways related to rRNA processing and translation (Supplementary Fig. 13a–j). Notably, our previous study showed impaired ribosomal and translational functions in NIID models8, suggesting ASO-GGC may exert therapeutic effects by restoring these functions. The proportion of neuronal populations showed no significant change before and after ASO-GGC treatment (ASO-ctrl group: 44.3% vs. ASO-GGC group: 41.2%). GSVA further confirmed that ASO-GGC significantly attenuated DNA damage (Fig. 8e), suppressed TP53 pathway activation (Fig. 8f), and reduced senescence-associated signatures (Fig. 8g). These improvements were primarily observed in neural progenitor cells, glutamatergic neurons, and oligodendrocyte precursor cells (Supplementary Fig. 14a, b). Interestingly, we noted a slight but statistically significant increase in DNA damage scores in neuroblasts, indicating that the efficacy of polyG-clearing therapy may exhibit cell-type-specific heterogeneity.

a uMAP projection identifying six major neural cell types in GGCexp-3DCOs treated with either ASO-ctr or ASO-GGC (200 nM). Patient-derived GGCexp-3DCOs were treated for 2 weeks prior to analysis at 90 days; three organoids per sample were pooled for cell counting and sequencing after treatment. b Volcano plot displaying differentially expressed genes (DEGs) between ASO-ctr and ASO-GGC treated groups. DEGs were identified using the two-sided Wilcoxon rank-sum test implemented in Seurat. For visualization clarity, genes with |log₂(fold change)| <3.5 or −log₁₀ (p-value) <60 are filtered out. c, d Functional enrichment analysis of DEGs, performed via Enrichr (Fisher's exact test, Benjamini–Hochberg correction), reveals significant associations with pathways related to ribosome subunits, translation, and macromolecule biosynthesis. Adjusted p < 0.05 was considered significant. An asterisk (*) denotes a significant adjusted p-value. GSVA enrichment scores for the DNA damage response pathway (e), TP53 pathway (f), and DNA damage telomere stress-induced senescence pathway (g) in GGCnor-3DCOs and ASO-GGC-treated GGCexp-3DCOs at day 90. Each data point represents the pseudobulk GSVA score of an individual donor (n = 3 per group, derived from the single-cell data by aggregating cells from each donor). Statistical significance was assessed using the two-sided Wilcoxon rank-sum test (***: p < 0.001). Box plots show the median (center line) and the first and third quartiles (box bounds). h hdWGCNA identifies nine co-expression gene modules from the single-cell RNA sequencing data. i Four gene modules show the most significant changes in expression following ASO-GGC treatment. j, k Module enrichment analysis reveals significant involvement in pathways related to mRNA metabolism, ribosome biogenesis, and translation. Enrichment was assessed using the Fisher exact test (Enrichr) with Benjamini–Hochberg correction. An asterisk (*) indicates a significant adjusted p-value (<0.05).
Additionally, hdWGCNA identified nine gene modules (Fig. 8h and Supplementary Data 3), among which the pink (90 genes), blue (929 genes), red (231 genes), and turquoise (1913 genes) modules were most significantly affected by ASO-GGC (Fig. 8i). Functional enrichment analysis of all genes within these modules revealed strong associations with pathways involved in mRNA metabolic processes, ribosome biogenesis, and translational regulation (Fig. 8j, k), further supporting ASO-mediated restoration of translational function. Detailed enrichment results for these four individual modules are provided in Supplementary Fig. 15 and Supplementary Data 3.
In summary, our results demonstrate that ASO-GGC can significantly improve DNA damage and senescence in NIID models by restoring nucleolar function, reducing genomic instability, and rescuing the dysregulated expression patterns of genes associated with neuronal development.
To determine whether ASO-GGC could rescue 3D chromatin structural abnormalities in NIID models, we first evaluated changes in chromatin structural proteins. Results showed that compared to ASO-ctr, ASO-GGC significantly upregulated CTCF and RAD21 expression in HEK-293T cell models (Supplementary Fig. 16a–c). Similarly, in patient-derived models, ASO-GGC increased CTCF and RAD21 expression (Fig. 8a–f). Concurrently, we observed that the intranuclear distribution of CTCF in patient-derived 3DCOs reverted from a diffuse pattern to more distinct focal puncta following treatment (Supplementary Fig. 16d), indicating restoration of normal CTCF localization. Collectively, these findings demonstrate the therapeutic potential of ASO-GGC in ameliorating polyG-induced chromatin architectural abnormalities.
To further assess the structural restoration by ASO-GGC, we performed Hi-C sequencing on treated patient-derived 3DCOs, generating approximately 3 billion paired-end reads and ~1 billion uniquely mapped valid interactions. Data quality was verified by cis/trans interaction ratios and distance-dependent interaction frequency decay (Supplementary Fig. 16e). Compared to pre-treatment Hi-C results, ASO-GGC-treated samples showed higher correlation with normal controls (Pearson’s R = 0.97) (Supplementary Fig. 16f) at 5-kb resolution (Supplementary Fig. 16g). Heatmaps displayed compartment changes (Fig. 9g). Detailed analysis revealed that ASO-GGC restored 407/512 (79.5%) aberrant compartments (244 B to A and 163 A to B reversions) (Fig. 9h). We identified 5973 TADs and 5673 loops in treated samples, with ASO-GGC restoring TAD boundaries and loop structures on chromosome 8 (Fig. 9i). Specifically, 1182 lost TADs were regained while 427 aberrant TADs disappeared. Insulation score analysis showed ASO-GGC enhanced TAD boundary insulation and partially restored intra-TAD contact frequencies (Fig. 9j). Moreover, ASO-GGC reversed 2082/4159 (50.1%) altered loops (977 regained, 1105 lost). Using the aforementioned method, we identified 329 DEGs located near the restored loop anchors after ASO-GGC treatment (Supplementary Data 1), which remained associated with functions, such as ribosome and mRNA processing (Supplementary Data 1). To compare whether OE and ASO-GGC exert similar therapeutic effects, we found that among the DEGs near the rescued loops, there were 227 overlapping genes, which were also enriched in pathways related to ribosome and mRNA processing (Supplementary Data 1). These findings demonstrate that ASO-GGC can partially rescue NOTCH2NLC GGC repeat expansion-induced 3D chromatin abnormalities, further supporting its therapeutic potential for NIID and providing mechanistic insights.

Immunoblotting analysis (a) and quantitative analysis confirming that ASO-GGC upregulates CTCF (b) and RAD21 (c) protein expression in patient-derived GGCexp neurons relative to ASO-ctr, normalized to GAPDH. Neurons were treated at 30 DIV for 2 weeks prior to analysis. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). Immunoblotting analysis (d) and quantitative analysis confirming that ASO-GGC upregulates CTCF (e) and RAD21 (f) protein expression in patient-derived GGCexp-3DCOs relative to ASO-ctr, normalized to GAPDH. Organoids were treated at 76 DIV for 2 weeks prior to analysis; three organoids were selected per sample for analysis. Statistical analysis was performed using a two-tailed paired Student’s t test (n = 3 biological replicates). g Log2 ratio of contact frequencies (ASO-GGC-treated GGCexp/untreated GGCexp). h Analysis of compartment switching patterns in ASO-GGC-treated versus untreated GGCexp 3DCOs. i Hi-C contact maps illustrating chromatin compartments, TADs, and chromatin loops (indicated by blue arrows) in ASO-GGC-treated 3DCOs. j Insulation scores for the genomic bins corresponding to the region shown in the middle panel of (i). Yellow and green arrows mark TAD boundaries and interiors, respectively. Data are presented as mean ± SD.
Comparative analysis with healthy control-derived 3DCOs revealed that ASO-GGC prompted switching in 212 compartments (3.7%), comprising 90 A-to-B and 122 B-to-A transitions. At the TAD level, ASO-GGC triggered the formation of 1321 and the disappearance of 1293 TADs. Regarding chromatin loops, the treatment led to the emergence of 1989 loops and the loss of 2210 existing ones. We propose three potential explanations for these observations: (1) partial restoration, where treatment incompletely reverts the genome to its precise healthy baseline; (2) potential off-target effects; or (3) dynamic genomic recalibration representing a transitional state during recovery from pathological conditions.
Discussion
Similar to the repeat-associated polypeptides derived from C9ORF7227, the polyG protein encoded by NOTCH2NLC GGC repeats also contains IDRs. These IDRs can interact with rRNA and other biomolecules, modulating diverse physiological processes. Although prior studies have demonstrated the co-localization of polyG with rRNA8, whether NPM1 mediates this interaction remains unclear. Here, we confirmed that polyG can independently bind both NPM1 and rRNA but exhibits a preferential interaction with NPM1. In contrast, C9ORF72-polyRP primarily associates with rRNA before engaging with NPM127. This difference may stem from their distinct amino acid compositions: polyG is glycine-rich, whereas polyRP is arginine-rich. Arginine, the largest amino acid, possesses extended side chains and charged groups that facilitate interactions with other molecules. In contrast, glycine—the smallest amino acid-lacks such side chains or charges, likely contributing to the observed binding preference.
PolyG undergoes liquid-liquid phase separation in cells and forms aggregates in a concentration-dependent manner, as visually confirmed by prior live-cell imaging7. Aggregated polyG induces nucleocytoplasmic mislocalization of NPM1 and demonstrates direct colocalization with it. This observation suggests that NPM1 mitigates polyG toxicity by sequestering it, while HSP70 may partially restore its refolding capacity26. However, persistent polyG accumulation may overwhelm NPM1 and HSP70 functionality, leading to decompensation26. Notably, both HSPB1 and HSP70 function as molecular chaperones that co-regulate the liquid-phase properties of TDP-43 in ALS33. Moreover, HSPB1 has been demonstrated to mitigate cellular damage induced by GIPC1-polyG34, suggesting potential shared pathological mechanisms among polyG-related disorders. Simultaneously, the ensuing nucleolar dysfunction disrupts not only the FC and GC architecture but also triggers broader cellular dysregulation, including impaired rRNA biogenesis and defective DNA damage repair. Beyond genomic instability, polyG disrupts nucleolar LLPS, altering 3D chromatin organization. Using Hi-C sequencing in NIID cerebral organoids, we demonstrated significant 3D chromatin structural abnormalities, which, alongside increased genomic instability, likely accelerate cellular senescence.
NPM1 is recruited to DNA damage sites and participates in DNA damage repair30. Consequently, polyG-induced dysfunction of NPM1 significantly increases DNA damage events. Previous studies have found that C9ORF72-related repeat polypeptides similarly exacerbate DNA damage35,36, suggesting common pathological mechanisms may exist among these disorders. Furthermore, inclusion bodies in both NIID and FXTAS contain numerous RNA-binding proteins23,37, some of which are also involved in DNA damage repair pathways. Therefore, dysfunction of these RBPs may further aggravate polyG-induced genomic damage.
Currently, pathogenic mechanisms of repeat expansion mutations involve loss-of-function, repeat RNA toxicity, and toxic poly-dipeptide production. ASOs exert therapeutic effects primarily via RNase H1-dependent RNA degradation or steric blockade of translation. While no effective treatment exists for NIID, ASOs have demonstrated promise in disorders like C9ORF72-ALS/FTD and FMR1-associated FXTAS by targeting repeat expansions. For instance, ASOs ameliorate FMR1 GGC repeat pathology by disrupting repeat RNA-RBP interactions32,38, thereby restoring RBP function, and by suppressing translation of toxic poly-dipeptides.
In this study, GGC-targeting ASOs suppressed polyG translation in NIID models, markedly reducing inclusion body formation and NPM1 expression—a potential indicator of alleviated nucleolar stress. Moreover, ASO-GGC significantly diminished DNA damage and senescence phenotypes in vitro. Similarly, in patient-derived cerebral organoids, ASO-GGC reduced senescence levels induced by genomic instability, and Hi-C analysis confirmed rescue of 3D chromatin architecture. However, potential off-target effects of ASOs must be considered in therapeutic development.
Notably, NOTCH2NLC is a human-specific gene, and patient-derived cerebral organoids may better recapitulate NIID pathology. The therapeutic efficacy of ASOs in this model provides compelling evidence for gene-targeted therapy in NIID and establishes a paradigm for accelerating the design and preclinical evaluation of genetic therapies for related disorders.
Although the pathogenic mechanisms of NIID have been extensively studied, research focusing on specific cell types remains limited. Furthermore, due to current technical constraints, we were unable to achieve precise quantification of polyG aggregation frequency across different neural cell types. Therefore, elucidating the precise impact of polyG on neural lineage differentiation and the underlying cell type-specific susceptibility mechanisms represents a critical direction for future investigation, requiring more refined models and dynamic analytical approaches.
In summary, leveraging patient-derived organoids, our study elucidates how polyG drives NIID pathogenesis by disrupting nucleolar homeostasis, inducing DNA damage, and perturbing 3D genome organization. These findings not only advance the mechanistic understanding of NIID but also provide experimental support for ASO-based therapeutic strategies, laying a foundation for future clinical translation.
Methods
Ethics statement
The induced pluripotent stem cell (iPSC) lines used in this study were derived from skin biopsies obtained in our previous work8, which was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. All participants provided written informed consent for the collection and use of samples for research purposes. All experiments conducted in this study were performed in accordance with relevant guidelines and regulations. Since this study involved only the use of previously established iPSC lines and did not recruit new human subjects or collect new human tissues/data, additional ethical approval was not required.
Generation of iPSC lines
Briefly, skin fibroblasts were obtained from three NIID patients harboring pathogenic NOTCH2NLC GGC repeat expansions and three healthy controls with normal NOTCH2NLC alleles. Fibroblasts were cultured in fibroblast medium containing 10% fetal bovine serum (FBS; Gibco). Reprogramming was performed using the CytoTune™-iPS 2.0 Sendai Reprogramming Kit (Invitrogen) according to the manufacturer’s protocol. After infection, cells were plated onto Matrigel-coated plates (Corning) and cultured in fibroblast medium for 7 days, followed by replacement with essential 8™ complete medium (Gibco), changed daily. iPSC colonies emerged after 2–3 weeks and were manually picked and expanded. Colonies were passaged using EDTA (Invitrogen) and treated with 10 μM Y-27632 (Rock inhibitor, Sigma-Aldrich) for the first 24 h after passaging. All iPSC lines used in this study (passages 20–30) were confirmed to have normal karyotypes prior to differentiation.
Cerebral three-dimensional cerebral organoids (3DCOs) differentiation
3DCOs were generated using the STEMdiff™ Cerebral Organoid Kit (Stemcell, 08,570)39. Briefly, iPSCs with optimal growth morphology were maintained on Matrigel-coated 6-well plates in mTeSR Plus medium (Stemcell, 100-0276). At ~80% confluency, cells were dissociated into single-cell suspensions using gentle cell dissociation reagent (Stemcell, 07,174) and seeded in low-attachment 96-well U-bottom plates with EB seeding medium to form embryoid bodies (EBs). On day 5, EBs were transferred to low-attachment 24-well plates containing induction medium. At day 7, EBs were embedded in Matrigel (Corning, 354,277) and cultured in 6-well plates with expansion medium to initiate neuroepithelium formation. From day 10 onward, 3DCOs were maintained in maturation medium on an orbital shaker, with medium changes every 3 days until full maturation. The disease and control groups consisted of 3DCOs derived from three NIID patients and three healthy controls, respectively. Both cohorts were sex-matched (each comprising two males and one female) and were exclusively of Han Chinese ethnicity.
Culture of iPSC-derived neurons
The differentiation of iPSCs into neurons was performed as previously described with modifications40. Briefly, iPSCs at 80–90% confluence were dissociated into single cells using StemPro Accutase and seeded onto Matrigel (Corning, 354,277)-coated plates at a density of 2.5 − 3 × 10⁵ cells per well in mTeSR Plus medium (Stemcell, 100-0276). On the following day (day 0 of neural induction), the medium was replaced with complete PSC neural induction medium (Neurobasal medium supplemented with 10% GIBCO neural induction supplement). A 50% medium change was performed on day 2, followed by complete medium changes every other day until day 7. The resulting neural progenitor cells (NPCs) at passage 0 (P0) were harvested using Accutase and passaged at a ratio between 1:3 and 1:6 onto new Matrigel-coated plates for subsequent neuronal differentiation. For neuronal differentiation, NPCs were seeded onto Matrigel-coated plates and maintained in complete BrainPhys neuronal medium (Stemcell, 05,795). The culture was fed by replacing 50% of the medium every 2–3 days. Mature neurons (MNs) were obtained after 30 days of differentiation and used for subsequent experiments. Neurons were derived from three patients (two males and one female) as the disease group and three normal controls (two males and one female) as the control group.
Genetic constructs
GGC repeat-containing plasmids were constructed as previously described6,8. Briefly, the human NOTCH2NLC exon 1 sequence (including 5′-UTR GGC repeats) was cloned into pcDNA3.1 fused to an ATG-deleted mCherry reporter. The construct initiates translation from the uN2C ORF start codon and terminates at the first stop codon. We generated two variants: 20 × GGC-mCherry (control) and 90 × GGC-mCherry (pathogenic expansion), along with an mCherry-only empty vector. All plasmids were transformed into DH5α competent cells to ensure repeat stability and validated by Sanger sequencing.
Synthesis of ASOs
ASOs were designed based on prior research32. The 11-nt ASO-GGC (5′-CCGCCGCCGCC-3′) contained 10 locked nucleic acid (LNA) modifications and a 3′ 2′-O-methyl group, with phosphorothioate linkages at all LNA positions. Control ASO-ctrl (5′-GCGUATTATAGCCGATTAAC-3′) and Cy3-labeled ASO-GGC-cy3 (5′-CCGCCGCCG-3′) were synthesized by Genema Gene with HPLC purification.
Cell culture and transfection
The HEK-293T human embryonic kidney cell line used in this study was purchased from the American Type Culture Collection (ATCC, catalog number: CRL-3216). The HT22 mouse hippocampal neuronal cell line was obtained from Ubigene Biosciences (Guangzhou, China; catalog number: YC-A004). HEK-293T and HT-22 cells were cultured in high-glucose DMEM (Gibco) supplemented with 10% FBS, 1% L-glutamine (Lonza), and 1% penicillin/streptomycin at 37 °C with 5% CO₂. For transfection, cells were seeded in culture plates and transfected at ~80% confluency using Lipofectamine ® 3000 transfection kit (Thermo Fisher Scientific) per manufacturer’s protocol. ASOs were denatured at 95 °C for 30 s and chilled on ice before transfection. NSC348,884 (7 μM) was administered for 24 h to specifically induce nucleocytoplasmic translocation of NPM1.
Cell viability assay
HEK-293T cells (5 × 10³/well in 96-well plates) were treated with CCK-8 reagent (10 μL/well) (Dojindo, CK04) for 2 h at 37 °C. Absorbance was measured at 450 nm (reference: 650 nm). Viability was calculated as: (ODtreatment − ODblank)/(ODcontrol − ODblank) × 100.
Apoptosis and necrosis detection
Cells were stained with YO-PRO-1 (491 nm excitation) and propidium iodide (PI; 618 nm). YO-PRO-1+ cells indicated apoptosis; PI+ cells indicated necrosis.
Tissue immunofluorescence
Well-developed 3DCOs were fixed in 4% paraformaldehyde (PFA) for 30 min at room temperature, washed three times with PBS, then dehydrated in 30% sucrose solution at 4 °C until sedimentation. The dehydrated 3DCOs were embedded in OCT compound and sectioned at 14 μm thickness using a Leica cryostat (Germany), with sections stored at −80 °C until use. For immunofluorescence, thawed sections were permeabilized with 0.3% Triton X-100 in PBS, blocked with 5% BSA for 30 min at room temperature, then incubated with primary antibodies overnight at 4 °C. After PBS washes, sections were incubated with fluorophore-conjugated secondary antibodies (1:100) for 1 h at room temperature, followed by nuclear counterstaining with DAPI (Southern Biotech, 0100-20). Fluorescence images were acquired using a Zeiss confocal microscope. Primary antibodies included: TUJ1 (Proteintech, 66,375-1-Ig, 1:100), CTIP2 (Proteintech, 55,414-1-AP, 1:100), PAX6 (Abcam, ab78,545, 1:100; ab5790, 1:50), TBR2 (Abcam, ab216,870, 1:400), GFAP (Proteintech, 16,825-1-AP, 1:100), NeuN (Proteintech, 66,836-1-Ig, 1:100), DCX (Proteintech,13,925-1-AP, 1:100), NPM1 (Proteintech, 60,096-1-Ig/10,306-1-AP, 1:200), FBL (Proteintech, 16,021-1-AP, 1:400), CTCF (Proteintech, 10,915-1-AP, 1:200), RAD21 (Proteintech, 27,071-1-AP, 1:50), p-γH2AX (Proteintech, 29,380-1-AP, 1:100), HSP70 (Proteintech, 10,995-1-AP, 1:200), rRNA (Santa Cruz, sc-33,678, 1:50), p62 (Proteintech, 66,184-1-Ig, 1:200), and ubiquitin (Proteintech, 10,201-2-AP, 1:200). Secondary antibodies were: FITC-conjugated goat anti-mouse/rat IgG (Proteintech, SA00003-1/11, 1:100), Cy3-conjugated goat anti-mouse/rabbit IgG (Proteintech, SA00009-1/2, 1:100), and CoraLite Plus 647-conjugated goat anti-mouse IgG (Proteintech, RGAM005, 1:100).
Cell immunofluorescence
Cells were fixed (4% PFA, 15 min), permeabilized (0.3% Triton X-100, 30 min), and stained as above.
High-throughput/resolution chromosome conformation capture (Hi-C) and analysis
Hi-C libraries were constructed from pooled samples of each group (5 × 10⁴ cells per sample) using an in situ Hi-C protocol. Briefly, cells were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched with 0.125 M glycine. After lysis, chromatin was digested with 100 U of MboI (NEB), and the resulting fragments were labeled with biotin-14-dCTP (Invitrogen). Ligation was performed with 50 U of T4 DNA ligase (NEB), followed by reversal of cross-linking and DNA purification using the QIAamp DNA Mini Kit. The purified DNA was sheared to 300–500 bp, and a sequencing library was prepared through end repair, A-tailing, adapter ligation, and streptavidin-based pull-down of biotinylated fragments, followed by PCR amplification. The final libraries were quantified and sequenced on the MGISEQ platform (BGI, China) to a depth of approximately 300 million reads per library to enable robust detection of 3D chromatin structures.
We processed high-resolution Hi-C data using the HiC-Pro pipeline (v3.1.0)41. Briefly, raw Hi-C reads were aligned to the human reference genome (hg38) using Bowtie2 (v2.4.4), followed by filtering of duplicates and low-quality reads to extract valid interaction pairs. The contact matrices were normalized using iterative correction and eigenvector decomposition (ICE) to remove technical biases. Hi-C map resolution was defined as described previously42, calculated as the smallest bin size where ≥80% of bins contained ≥1000 contact reads.
Compartments (A/B) were assigned following established methods43. Expected contact frequencies were calculated based on genomic distance between loci, and Pearson correlation matrices were generated from observed/expected matrices. Principal component analysis (PCA) was performed on the correlation matrices, with the first principal component (PC1) used for compartment calling. Genomic regions with positive PC1 values (higher gene density) were designated as compartment A, while negative values indicated compartment B.
TADs were identified using the insulation score method44 with 20 kb bins and 1 Mb sliding windows. Insulation scores were normalized by the mean score across all bins per chromosome, with TAD boundaries called at local minima (FAN-C boundaries). Chromatin loops were detected using the HiCCUPS algorithm in Juicer Tools (v1.9.9) at 5-kb resolution with a significance threshold of FDR < 0.2. Differential chromatin loops were defined as those with interaction strength fold changes >2 or <0.5. Hi-C interaction matrices were visualized using Juicebox (v1.9.8).
Analysis of spatial association between chromatin loops and genes
To ensure the biological relevance of subsequent analyses, we constructed a stringently filtered gene set based on the hg38 genome annotation. Through systematic exclusion based on naming conventions, we removed all non‑coding RNAs (e.g., LINC‑, MIR‑, etc.), pseudogenes, and low‑confidence predicted genes (e.g., LOC‑, AC‑ prefixes). Simultaneously, gene‑length filtering (500 bp–5 Mb) was applied to remove annotation noise, while all ribosomal protein genes (RPL‑/RPS‑) were intentionally retained. Ultimately, we obtained a set of 20,190 high‑confidence protein‑coding genes, which served as a reliable basis for subsequent chromatin loop association analysis. To identify genes potentially regulated by differential chromatin loops, we performed spatial proximity analysis between all differential loops and the above gene set. Specifically, for each loop, we calculated the centroid coordinates of its two anchors. A gene was considered spatially associated with the loop if its transcription start site fell within a 50‑kb window upstream or downstream of the centroid of either anchor. To ensure specificity of associations, only the three closest genes per anchor were retained, and each gene was uniquely assigned to its nearest loop to avoid multiple counting. The resulting candidate gene set was subsequently used for downstream functional enrichment analysis.
Co-expression of CTCF and RAD21 via lentiviral transduction
To co-express CTCF and RAD21 in 3DCOs, we transduced three patient-derived 3DCOs (60-day-old) using lentiviral vectors. Equal titers of CTCF-GFP and RAD21-mCherry lentiviral preparations were pooled and directly added to the organoid culture medium at a total MOI of 40. To enhance transduction efficiency, a transduction enhancer was supplemented during the procedure. After 48-h incubation in virus-containing medium, the organoids were transferred to fresh maintenance medium. Fluorescence detection and protein quantification were performed 7 days post-transduction, followed by Hi-C sequencing at 30 days after infection.
Bulk RNA sequencing and bioinformatics
Bulk RNA sequencing was performed by BGI Genomics using the following workflow: total RNA was extracted from plasmid-transfected HEK-293T cells using TRIzol reagent, with concentration and purity assessed by Nanodrop 2000 and RNA integrity verified using Agilent 2100 Bioanalyzer. Potential genomic DNA contamination was excluded through agarose gel electrophoresis. For library preparation, 2 μg of total RNA per sample underwent poly(A) enrichment using oligo magnetic beads, followed by strand-specific library construction. Sequencing was performed on the MGI platform with 150 bp paired-end reads.
For data analysis, clean reads were aligned to the hg38 reference genome using HISAT2 (v2.2.1)45. Gene expression levels were quantified as FPKM values and subjected to PCA and hierarchical clustering. Differential expression analysis was performed using DESeq2 (v3.19)46, with significantly differentially expressed genes defined as those with adjusted p < 0.05 and absolute fold change >2. Functional enrichment analysis of these genes was subsequently conducted using Enrichr47.
β-galactosidase staining
Cells/tissue sections were fixed, permeabilized, and incubated with β-gal (1 mg/mL, 37 °C overnight). Blue precipitate area was quantified using ImageJ (v1.53).
Capillary electrophoresis immunoassay (Jess)
Cells were lysed in ice-cold lysis buffer containing protease inhibitors and 1 mM PMSF using ultrasonication, followed by centrifugation at 12,000 × g for 15 min at 4 °C to collect the protein supernatant, with concentrations determined by BCA assay. For automated capillary Western blot analysis using the ProteinSimple Jess system, protein samples were mixed with master mix and sample buffer, denatured at 95 °C for 5 min, then processed according to manufacturer’s protocol (https://www.bio-techne.com/resources/protocols/jess-abby-wes-protocols) with sequential loading of antibody diluent, primary antibodies [TUJ1 (Proteintech, 66,375-1-Ig, 1:100), NPM1 (Proteintech, 60,096-1-Ig/10,306-1-AP, 1:200), FBL (Proteintech, 16,021-1-AP, 1:50), CTCF (Proteintech, 10,915-1-AP, 1:100), RAD21 (Proteintech, 27,071-1-AP, 1:100), p-γH2AX (Proteintech, 29,380-1-AP, 1:30), HSP70 (Proteintech, 10,995-1-AP, 1:200), rRNA (Santa Cruz, sc-33,678, 1:50), NOTCH2NL (Santa Cruz, sc-100,307), GAPDH (Proteintech, 10,494-1-AP, 1:400)], HRP-conjugated secondary antibodies (1:100), chemiluminescent substrate and wash buffer, followed by automated electrophoresis, immunodetection and image acquisition using default settings, with data analysis performed using Compass software and signal intensity quantified by ImageJ (v1.53). Uncropped and unprocessed images of all immunoblots are provided in Supplementary Fig. 17.
RT-PCR and qPCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen), followed by cDNA synthesis with the HiScript IV all-in-one ultra RT SuperMix kit (Vazyme, R433-01) according to the manufacturer’s protocol. Quantitative PCR was performed using SupRealQ Ultra Hunter SYBR qPCR Master Mix (Vazyme, Q713-02) on a CFX96 Deep Well Real-Time System (Bio-Rad) under the following cycling conditions: initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. GAPDH served as the internal reference gene, and relative expression levels were calculated using the 2−ΔΔCt method. The following primer pairs were used: NPM1 (forward: 5′-GCTGTGGAGGAAGATGCAGAGTC-3′, reverse: 5′-GGGCAGACCGTCTTCCAGATATAC-3′), FBL (forward: 5′-GCCACACCTTCCTGCGTAATG-3′, reverse: 5′-TGCGGCTTCATGTTCTCCTGTTG-3′), mCherry (forward: 5′-ACGACGCCGAGGTGAAGAC-3′, reverse: 5′-CTCGTTGTGGCTGGTGATGTC-3′), GAPDH (forward: 5′-GAGTCAACGGATTTGGTCGT-3′, reverse: 5′-TTGATTTTGGAGGGATCTCG-3′), and NOTCH2NLC (forward: 5′-CTACCAGTGCCAGTGCCTTCAG-3′, reverse: 5′-CCGACAGGTGCCTCCATTGAC-3′).
Single-cell RNA sequencing (scRNA-seq) analysis
For scRNA-seq, patient-derived 3DCOs were cultured for 90 days and then treated with either 200 nM control ASO (ASO-ctr) or target ASO (ASO-GGC) for 2 weeks. 3DCOs from each treatment group were enzymatically dissociated into single-cell suspensions, counted, and pooled in equal numbers for sequencing on the Novogene platform, generating approximately 100 GB of raw sequencing data per 10,000 cells. The raw FASTQ data were processed using Celescope (v1.5.2) for quality filtering and gene alignment, generating standardized output files compatible with Seurat (v5.1.0)48. Quality control metrics were applied to retain cells expressing 200–4500 genes while excluding those with >20% mitochondrial RNA content. Following normalization, we identified 2000 highly variable genes for downstream analysis. Principal component analysis was performed using Seurat’s “ScaleData” and “RunPCA” functions, with Harmony (v1.2.1) 49 employed for batch effect correction. Unsupervised clustering was conducted at a resolution of 0.3 using the “FindClusters” and “FindNeighbors” functions, with results visualized via UMAP projection. Cell type annotation was performed using ScType50.
In the scRNA-seq, differentially expressed genes (DEGs) were identified using the FindMarkers function from the Seurat package (v5.1.0), based on a nonparametric Wilcoxon rank-sum test. The significance thresholds were set at an adjusted p < 0.05 and an absolute fold change > 0.25. For network analysis, we implemented single-cell weighted gene co-expression network analysis (hdWGCNA v0.4.00) 51 by first creating a metacell expression matrix through k-nearest neighbors aggregation (“MetacellsByGroups”) of genes detected in ≥5% of cells. The optimal soft threshold power was determined via the “TestSoftPowers” function, followed by Pearson correlation-based network construction (“ConstructNetwork”). Module eigengenes were derived through linear regression (“ModuleEigengenes”), with differential module analysis performed using Wilcoxon tests (“FindDMEs”). Full methodological details are available at: https://smorabit.github.io/hdWGCNA/articles/basic_tutorial.html. Functional enrichment analysis was conducted using Enrichr47 (significance threshold: p < 0.05), while pathway activity was assessed via gene set variation analysis (GSVA v1.52.3)52 using gene sets obtained from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/human/search.jsp). GSVA scores were calculated using single-sample gene set enrichment analysis (ssGSEA) based on gene sets associated with specific pathways. The following gene sets were utilized in this study: DNA damage response (WP707, https://www.wikipathways.org/pathways/WP707), DNA damage response-TP5353, and DNA damage/telomere stress-induced senescence (R-HSA-2,559,586, https://www.reactome.org/content/detail/R-HSA-2559586).
Statistical and reproducibility
To minimize bias, all image analyses were performed using either fully automated pipelines or blinded manual assessment. Statistical analyses were conducted using GraphPad Prism (v9.0.0), with data presented as mean ± standard deviation (SD). Data normality was tested. No covariates were included in the statistical models. Data are shown as violin plots showing the distribution of all values. In violin plots, the upper, middle, and lower horizontal lines represent the 75th percentile (Q3), median (Q2), and 25th percentile (Q1), respectively. All experiments were independently repeated three times. For each replicate in 2D culture models, quantitative data were obtained by averaging measurements from 3 randomly selected fields of view. For 3DCOs, five random, nonoverlapping fields of view were imaged per stained section to account for organoid heterogeneity. Measurements from these five fields were averaged to generate a single data point per organoid. Data from three organoids per condition were then used for statistical analysis. For statistical comparisons between two groups, two-tailed paired Student’s t test was employed; for comparisons among three or more groups, one-way ANOVA with Tukey’s or Dunnett’s post-hoc test was applied. For all experiments, p < 0.05 were considered statistically significant.
All newly generated materials are available from the corresponding author upon reasonable request.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequence data generated in this study have been deposited in the National Genomics Data Center (https://ngdc.cncb.ac.cn/gsa) under BioProject accessions PRJCA055018, PRJCA054996 [https://ngdc.cncb.ac.cn/bioproject/browse/054996], PRJCA058665, and PRJCA054993. The processed data generated in this study are provided in the Supplementary Information/Source data file. Source data are provided with this paper.
References
Sone, J. et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 139, 3170–3186 (2016).
Tian, Y. et al. Clinical features of NOTCH2NLC-related neuronal intranuclear inclusion disease. J. Neurol. Neurosurg. Psychiatry 93, 1289–1298 (2022).
Fan, Y., Xu, Y. & Shi, C. NOTCH2NLC-related disorders: the widening spectrum and genotype-phenotype correlation. J. Med. Genet. 59, 1–9 (2022).
Sone, J. et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 51, 1215–1221 (2019).
Deng, J. et al. Long-read sequencing identified repeat expansions in the 5’UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J. Med. Genet. 56, 758–764 (2019).
Boivin, M. et al. Translation of GGC repeat expansions into a toxic polyglycine protein in NIID defines a novel class of human genetic disorders: the polyG diseases. Neuron 109, 1825–1835 (2021).
Zhong, S. et al. Upstream open reading frame with NOTCH2NLC GGC expansion generates polyglycine aggregates and disrupts nucleocytoplasmic transport: implications for polyglycine diseases. Acta Neuropathol. 142, 1003–1023 (2021).
Fan, Y. et al. GGC repeat expansion in NOTCH2NLC induces dysfunction in ribosome biogenesis and translation. Brain 146, 3373–3391 (2023).
Liu, Q. et al. Expression of expanded GGC repeats within NOTCH2NLC causes behavioral deficits and neurodegeneration in a mouse model of neuronal intranuclear inclusion disease. Sci. Adv. 8, eadd6391 (2022).
He, L. et al. C9orf72 functions in the nucleus to regulate DNA damage repair. Cell Death Differ. 30, 716–730 (2023).
Escobar, T. M. et al. Inheritance of repressed chromatin domains during S phase requires the histone chaperone NPM1. Sci. Adv. 8, eabm3945 (2022).
Wang, A. J., Han, Y., Jia, N., Chen, P. & Minden, M. D. NPM1c impedes CTCF functions through cytoplasmic mislocalization in acute myeloid leukemia. Leukemia 34, 1278–1290 (2020).
Davidson, I. F. et al. CTCF is a DNA-tension-dependent barrier to cohesin-mediated loop extrusion. Nature 616, 822–827 (2023).
Finkel, R. S. et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: final report of a phase 2, open-label, multicentre, dose-escalation study. Lancet Child Adolesc. Health 5, 491–500 (2021).
Mulders, S. A. et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 106, 13915–13920 (2009).
Pandey, S. K. et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmcol. Exp. Ther. 355, 329–340 (2015).
Wheeler, T. M. et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115 (2012).
Smith, R. A. et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 116, 2290–2296 (2006).
Donnelly, C. J. et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013).
Tran, H. et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 28, 117–124 (2022).
Becker, L. A. et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371 (2017).
Romero, P. et al. Sequence complexity of disordered protein. Proteins 42, 38–48 (2001).
Asamitsu, S. et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci. Adv. 7, eabd9440 (2021).
Lee, K. H. et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788 (2016).
Yang, J. et al. Polyglycine-mediated aggregation of FAM98B disrupts tRNA processing in GGC repeat disorders. Science 389, eado2403 (2025).
Frottin, F. et al. The nucleolus functions as a phase-separated protein quality control compartment. Science 365, 342–347 (2019).
White, M. R. et al. C9orf72 poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol. Cell 74, 713–728 (2019).
Box, J. K. et al. Nucleophosmin: from structure and function to disease development. BMC Mol. Biol. 17, 19 (2016).
Derbis, M., Konieczny, P., Walczak, A., Sekrecki, M. & Sobczak, K. Quantitative evaluation of toxic polyglycine biosynthesis and aggregation in cell models expressing expanded CGG repeats. Front. Genet. 9, 216 (2018).
Dileep, V. et al. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell 186, 4404–4421 (2023).
Shah, S. et al. Antisense oligonucleotide rescue of CGG expansion-dependent FMR1 mis-splicing in fragile X syndrome restores FMRP. Proc. Natl. Acad. Sci. USA 120, e2302534120 (2023).
Derbis, M. et al. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 12, 1265 (2021).
Lu, S. et al. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol. 24, 1378–1393 (2022).
Ding, N. et al. Heat-shock chaperone HSPB1 mitigates poly-glycine-induced neurodegeneration via restoration of autophagic flux. Autophagy 21, 1298–1315 (2025).
Andrade, N. S. et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 15, 13 (2020).
Nihei, Y. et al. Poly-glycine-alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathol. 139, 99–118 (2020).
Tchurikov, N. A. et al. DNA double-strand breaks coupled with PARP1 and HNRNPA2B1 binding sites flank coordinately expressed domains in human chromosomes. PLoS Genet. 9, e1003429 (2013).
Iwahashi, C. K. et al. Protein composition of the intranuclear inclusions of FXTAS. Brain 129, 256–271 (2006).
Lancaster, M. A. & Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340 (2014).
Bardy, C. et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc. Natl. Acad. Sci. USA 112, E2725–2734 (2015).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Crane, E. et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244 (2015).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Xie, Z. et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 1, e90 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 16, 1289–1296 (2019).
Ianevski, A., Giri, A. K. & Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 13, 1246 (2022).
Morabito, S., Reese, F., Rahimzadeh, N., Miyoshi, E. & Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 3, 100498 (2023).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Amundson, S. A. et al. Stress-specific signatures: expression profiling of p53 wild-type and -null human cells. Oncogene 24, 4572–4579 (2005).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 82171247 and 82371433 to C.S.), the Funding for Scientific Research and Innovation Team of the First Affiliated Hospital of Zhengzhou University (grant no. ZYCXTD2023011 to C.S.), the Henan Provincial Leading Talent Program (grant no. 254000510049 to C.S.), and the Henan Clinical Medical Scientist Program (grant no. HNCMS202425 to C.S.).
Author information
Authors and Affiliations
Contributions
C.H.S., A.R.L.S. designed the project, M.J.L., M.B.T., H.Y.L., and Y.L.Y. performed the experiments, X.Y.H., Z.Y.W., and R.W.Y. analyzed the data. Z.W.H., Y.M.F., Y.M.S., Z.Y.Z., and P.L.J. performed the analysis of Hi-C sequencing, D.R.M., S.J.L., Y.Y.L., and C.W.H. performed the RNA sequencing, and scRNA-seq experiments. Z.Q.Z., C.Y.M., D.Y., and H.C. supervised the data analysis. S.S.Q., C.W., and C.Y.Z. performed experiments and analyzed qPCR data related to shRNA knockdown. M.J.L., M.B.T., C.Y.M., and S.J.L wrote the manuscript with the critical suggestions from S.T., A.R.L.S., Z.Q.Z., Y.M.X., S.S., Q.W., and H.C. C.H.S., A.R.L.S. conceived the whole study and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: a Chinese patent application related to the ASOs and their use in treating NIID has been filed by the First Affiliated Hospital of Zhengzhou University. The inventors are Changhe Shi and Mengjie Li. The application number is 202511263079.1, and its status is pending. This patent covers the ASO sequences and their therapeutic applications described in this manuscript. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Oliver Davis and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, M., Tang, M., Hao, X. et al. ASO therapy rescues NOTCH2NLC GGC repeat expansion-induced genomic damage, 3D chromatin structural abnormalities, and senescence. Nat Commun 17, 4925 (2026). https://doi.org/10.1038/s41467-026-71516-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-71516-7
