
Abstract
While simple spin-boson models have been realized on quantum hardware, simulating extended electronic networks with local vibrational environments remains a fundamental challenge in the presence of non-equilibrium, long-lived electronic-vibrational (vibronic) coherence. We present a framework for the digital-analog simulation of open quantum systems governed by Hamiltonians with linear-vibronic coupling (LVC) and structured vibrational environments. Our approach exploits the intrinsic dissipation of qubits in near-term quantum hardware as a resource to emulate vibrational relaxation, combined with a model-specific error mitigation scheme to filter out noise sources incompatible with the target open system. We validate our strategy by resolving the vibronic transfer spectra of a one-dimensional donor-acceptor chain on IBM superconducting processors, reproducing non-Markovian dynamics and scaling the chain length up to 10 electronic sites, an unprecedented scale for chemical dynamics on quantum computers. Our model of vibronic electron transfer offers a portable, application-oriented benchmark for simulating long-lived entangled states on NISQ computers.
Similar content being viewed by others

Introduction
The transformation of the energy and mobility sectors demands the development of novel and powerful simulation tools such as quantum computers, supporting the design of new materials operating across a wide range of environmental conditions. Kinetic theories at thermal equilibrium fail to accurately capture transfer rates in relevant scenarios, such as the inverted region in the Marcus theory of electron transfer1,2. This regime is for example relevant for batteries with large overpotentials, indicative of high reorganization energies and complex solvation shells3,4 that are linked to dissipative processes. A proper understanding of heat loss at the microscopic scale is crucial in the engineering of next-generation devices for energy storage and production.
Nonequilibrium processes on the picosecond scale (10−12 s) are coming into focus thanks to the increasing time resolution of spectroscopic techniques5,6. Nonequilibrium quantum effects are a promising route to increase the efficiency of energy materials thanks to their unusual thermodynamic properties7, such as long-lived electronic-vibrational (vibronic) coherence8,9 or the slow cooling of hot-carriers10,11. These processes are found to strongly affect the efficiency and directionality of electron transfer (ET) between reactant molecules and electrode surfaces12,13,14, as well as the ultrafast charge separation in organic photovoltaics15 or photosynthetic systems16, and are also believed to strongly influence the transport of charges in battery electrodes17,18 and perovskites19.
Quantum computers bring the potential to simulate the nonequilibrium dynamics of materials in operando conditions to reveal new insights into the underlying mechanisms that govern their performance20. As such models are described by open quantum systems, noise processes in the quantum hardware that are compatible with the target open quantum system can be exploited21,22,23,24,25, while unwanted sources of noise are mitigated insofar as possible26,27. There is a growing interest in using dissipation and structured noise as a resource in quantum computing, from the acceleration of variational optimization workflows28 to the engineering of initial states29,30,31,32. In particular, novel quantum platforms that naturally realize quantized oscillators via bosonic elements in the hardware have the potential to become powerful toolboxes for the simulation of open quantum systems33,34,35,36, avoiding resource-intensive boson-to-qubit encodings23. On trapped ions, vibronic electron transfer with tunable dissipation has been demonstrated for dimer systems37,38, as well as the engineering of structured environments for spin-boson models39,40,41. Although quantum algorithms for the simulation of extended networks like the Holstein model have been put forth42,43,44, experimental demonstrations with large problem sizes are still notoriously challenging.
We focus on the non-equilibrium dynamics of vibronic networks, where ultrafast ET ( ≲ 100 fs) can be observed in the non-Markovian dynamics, whose duration is determined by the finite lifetime of vibrational motion45. While ultrafast ET can happen via purely electronic resonance between donor and acceptor states46, it can remarkably also occur via vibronic resonance mediated by the coherent interaction of electrons and vibrations like C=C stretch modes in organic photovoltaics47, whose frequencies are one order of magnitude above room temperature. Such effects are supported by pump-probe experiments showing clear evidence of long-lived ( ≲ 0.5 ps) vibronic coherence in the charge separation of electronic excitations15,48,49,50.
Our approach uses the pseudomode formalism to simulate the dynamics of the open vibronic system51,52,53,54,55,56,57, as depicted in Fig. 1a. Using auxiliary harmonic oscillators (pseudomodes) that are locally damped by their respective Markovian baths, one is able to engineer arbitrary spectral densities to simulate the non-Markovian dynamics of the reduced system of interest (the electronic network). One can substantially lower the memory requirements in numerical simulations when combining pseudomodes with tensor networks techniques5,58,59, because local damping naturally destroys correlations between oscillators. This logic can be extended to quantum algorithms that exploit local dissipation as a quantum resource. By using damped oscillators in the simulation of open quantum systems on NISQ computers, entanglement requirements may be lowered and adapted to the lifetime of system-environment correlations60. While classical methods such as HEOM or T-TEDOPA are capable of simulating small systems with highly structured environments61, they face severe limitations when scaling to larger networks, illustrating that the primary challenge in simulating open quantum systems lies in the scaling of the system size, showing the potential utility for quantum algorithms like the one presented here.

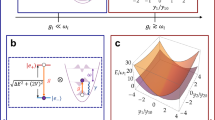
a The left panel shows a schematic of the one-dimensional LVC Hamiltonian with nearest-neighbors electronic interactions of coupling strength J, where each site of energy Ωn is locally coupled to a single quantum harmonic oscillator of frequency ω0 with vibronic coupling strength g. These oscillators may be damped by independent vibrational environments with a rate γ that are characterised by the width of the peak of the same spectral density \({{{\mathcal{J}}}}(\omega )\) (Eq. (5)), shown in the center panel. The lifetime of vibrational excitations is given by 1/γ and determines the duration of system-environment correlations (non-Markovian dynamics), which can be observed in the right panel with the bath correlation function \({{{\mathcal{C}}}}(t)\) (Eq. (13)). b On the quantum computer, each electronic site is mapped to a single qubit while oscillators are mapped to qubits using a boson-to-spin encoding (left panel). Trotterized circuits corresponding to the closed model (γ = 0) simulate the dynamics of the open model (γ > 0) with a time step Δt due to the intrinsic damping of oscillator qubits. The center panel illustrates a distribution of coherence times of qubits involved in the circuit, together with the threshold required to simulate weakly damped C=C stretch modes. Note that the threshold depends on the choice of Δt and the circuit execution time on the hardware (Eq. (18)). We found that the shortest T1 or T2 time determines the effective damping rate ΓQC, that lies for the simulations presented here between (50 fs)−1 and (150 fs)−1 (right panel). c Experimental results (gray markers) for vibronic electron transfer in a chain of N = 7 sites (14 qubits) are compared to an emulation of the open quantum system (magenta), where all oscillator qubits experience the same damping rate γ = (100 fs)−1 (see Fig. 5d, e).
When simulating the Trotterized dynamics of the closed model on the quantum computer with a time step Δt as shown in Fig. 1, the intrinsic noise of the quantum processor imprints an effective damping rate ΓQC, which is determined by the T1, T2 times of the involved qubits (see Eq. (18)). Rather than targeting a specific damping rate, this work focuses on maximizing simulation time to identify the longest vibrational relaxation lifetime ΓQC achievable on the quantum processor. The effective damping rate ΓQC can be tuned by adjusting the Trotter step Δt: larger steps reduce dissipation imprinted on the resulting dynamics, while smaller steps increase it, as demonstrated previously23. However, this tuning is constrained by two key limits. First, Δt must be sufficiently small to fully resolve the spectral content of the Hamiltonian, determined by its largest possible frequency. Second, excessively small Δt leads to deep quantum circuits, increasing the number of time steps required to reach a given evolution time. In general, target models with stronger damping rates (γ ≫ ΓQC) can also be implemented either via delay instructions on environment qubits32 or alternatively, by coupling the latter to an ancillary qubit with periodic reset operations62. However, the simulation of target models with longer vibrational lifetimes (γ ≪ ΓQC) requires hardware with larger qubit coherence times beyond what is available today. For a faithful simulation only qubits that are used to encode oscillators should be subject to dissipation, while noise on other qubits requires mitigation.
In this work, we present a protocol to simulate non-equilibrium dynamics of vibronic networks, and validate the approach by simulating a microscopic model of electron transfer on the superconducting quantum computer ibm_aachen. The model consists of an electronic chain of donor and acceptor sites, each of which are coupled to a local vibrational mode. Our experimental results reproduce ET dynamics on up to 10 electronic sites (20 qubits) and lead to an effective lifetime 1/ΓQC that lies between 50 and 150 fs (Fig. 1b), approaching the long-lived motion of intramolecular vibrational modes in organic molecules (1/γ ≳ 500 fs), a fundamental challenge in classical simulations of open quantum systems where low damping rates render perturbative methods highly inaccurate5.
Results
Electron transfer model
We consider a microscopic model of electron transfer from a single donor to N − 1 acceptor sites, coupled with nearest-neighbor electronic interactions (see Fig. 1a). The excitation initially sits at the donor site (n = 0), and the first acceptor site (n = 1) acts as an energetic trap that competes with successful charge separation. In the thermodynamic limit (N → ∞), this constitutes the archetypal problem in the theory of electron transfer where a single donor is coupled to a continuous band of acceptor states63.
The Hamiltonian parameters are chosen to model the ultrafast charge separation in a prototypical donor-acceptor blend from organic photovoltaics, consisting of a donor polymer like P3HT (Poly(3-hexylthiophene)) and functionalized fullerenes like PCBM ([6,6]-phenyl-C61-butyric acid methyl ester) as acceptor molecules47. As represented by colored circles in Fig. 1a, each molecule is treated as a two-level system that corresponds to its highest occupied (HOMO) and lowest unnocupied (LUMO) molecular orbitals. The coupling between LUMO levels leads to electronic hopping between neighboring sites.
The initial state is given by a single electron-hole pair at the donor site (shown in green in Fig. 2a), after which the electron may transfer to the acceptor molecules, while the hole is assumed to be frozen at the donor because of its lower mobility. This allows us to focus on the dynamics of the electron in the Coulomb potential that is induced by the hole, reflecting the bound nature of electron-hole pairs (excitons) in organic molecules. Charge separation is thought to take place faster than the creation of excitons via the absorption of photons in the donor. It is therefore common to assume that the dynamics remain in the single-excitation manifold at all times. The Hamiltonian incorporates both electronic and vibrational degrees of freedom, and is given by
The electronic part of the Hamiltonian is modeled as a tight-binding chain \({\widehat{H}}_{{{{\rm{el}}}}}\)
where J = 500 cm−1 is the coupling strength between sites, Ω0 is the energy of the LUMO level of the single donor site, Ωn = − V/n (n≥1) is the LUMO energy of the n-th acceptor site, V = 2420 cm−1 is the Coulomb binding energy (V = ∣Ω1 − Ωn∣ for n → ∞) and \({\widehat{a}}_{n}^{{{\dagger}} }\) (\({\widehat{a}}_{n}\)) are the creation (annihilation) operators for an electron on site n. The local energy levels Ωn are visualized in Fig. 2a, where the first acceptor site (n = 1) clearly constitutes an energetic trap. Note that despite the assumption of a single-excitation, the occupied Hilbert space of the vibronic Hamiltonian in Eq. (1) generally scales exponentially with the system size N because of the vibronic terms.

a Schematic of the one-dimensional donor-acceptor interface with vibronic coupling to high-frequency, intramolecular C=C stretch modes. We vary the driving force Δ of the donor site (n = 0) to study the probability of electron transfer Ptransfer(Δ, Time) to the acceptor sites (n > 1). b Donor energy and eigenenergies of the acceptor subsystem \({\widehat{H}}_{{{{\rm{el}}}}}^{({{{\rm{Acc.}}}})}\) (see Eq. (8)) for typical electronic and vibronic resonances. Both mechanisms induce ultrafast ET by creating superpositions of states with mixed donor/acceptor character. For the vibronic case (right-hand side), the superposition is created by coherent electron-oscillator (vibronic) coupling.
The vibrational part of the Hamiltonian \({\widehat{H}}_{{{{\rm{vib}}}}}\) couples each electronic site n locally to a set of Mn quantum harmonic oscillators
where \({\widehat{b}}_{n,m}^{{{\dagger}} }\) and \({\widehat{b}}_{n,m}\) are the creation and annihilation operators for the m-th vibrational oscillator at site n, and frequency ωn,m. The linear vibronic coupling (LVC) term \({\widehat{H}}_{{{{\rm{el}}}}-{{{\rm{vib}}}}}\) describes the interaction between the electronic states and oscillators
where gn,m is the vibronic coupling strength between the n-th site and the m-th vibrational oscillator.
The interaction between site n and its local vibrational environment is captured in the spectral density function
that becomes a continuous function in the thermodynamic limit Mn → ∞ of the bath. We focus on modeling high-frequency, dispersionless, intramolecular vibrations like C=C stretch modes that are intrinsic to organic molecules. Therefore, we assume that every site is coupled to identical vibrational environments that are independent from each other
and are characterized by a single Lorentzian peak around ωn,0 ≡ ω0 with ω0 = 1500 cm−1. Therefore, identifying Mn ≡ M, \({\widehat{b}}_{n,0}\equiv {\widehat{b}}_{n}\) and gn,0 ≡ g0, we model \({{{\mathcal{J}}}}(\omega )\) with a single pseudomode (see Methods), i.e. a single damped oscillator (M = 1) of frequency ω0 and vibronic coupling strength \({g}_{0}={\omega }_{0}\,\sqrt{s}\), where s = 0.05 is the dimensionless Huang-Rhys factor. The damping rate γ is determined by the width of the Lorentzian peak in the spectral density \({{{\mathcal{J}}}}(\omega )\) (see Fig. 1a).
The pseudomode formalism leads to the following master equation for the density matrix
where \(\widehat{H}\) is the full vibronic Hamiltonian in Eq. (1) and \({{{{\mathcal{L}}}}}_{{{{\rm{vib}}}}}^{(n)}(\widehat{\rho })\) are the Lindblad superoperators describing the local damping of each oscillator. Although Eq. (7) takes the form of a Lindblad master equation, the pseudomode approach in principle allows to reproduce arbitrary non-Markovian dynamics in the the electronic degrees of freedom (\({\widehat{\rho }}_{{{{\rm{el}}}}}={{{{\rm{Tr}}}}}_{{{{\rm{vib}}}}}\widehat{\rho }\)), where the pseudomodes are traced out51,52,53.
We probe two different mechanisms for ET: depending on the driving force Δ = Ω0 − Ω1, ET occurs via electronic or vibronic resonances, as depicted in Fig. 2b. For purely electronic resonances, the donor energy Ω0 is resonant with an eigenenergy Δe of the acceptor subsystem
defined in the same manner as \({\widehat{H}}_{{{{\rm{el}}}}}\), but restricted in the sums to n > 0 to exclude the donor site at n = 0. Vibronic coupling is not required for electronic resonances to occur. Vibronic resonances require vibronic coupling and occur for driving forces Δv ≈ Δe + ω0, when the energy difference between the donor and one of the acceptor eigenstates of \({\widehat{H}}_{{{{\rm{el}}}}}^{({{{\rm{Acc.}}}})}\) is matched by the energy ω0 of a vibrational excitation. The mechanism of vibronic ET is thus driven by a non-separable (entangled) superposition of electronic and vibrational excitations (right panel in Fig. 2b). Further details are explained in Supplementary Notes.
Quantum simulations, emulations and classical simulations
This work aims at quantum simulations of the presented ET-Model with a single pseudomode per electronic site, for which Nb = 2 energy levels per oscillator are sufficient, as validated in the Supplementary Notes. We describe a more general qubit encoding, suitable for generic vibronic Hamiltonians with various oscillators and multiple energy levels, in the Supplementary Methods. We show the scalability of the approach in Discussion.
For a model with N sites, N qubits are used to simulate the electronic subspace and N further qubits for the damped oscillators (pseudomodes). We perform quantum simulations of this model as depicted in Fig. 1a by using quantum circuits corresponding to the full Hamiltonian in Eq. (1), employing Trotterized time evolution on the quantum computer with 104 shots per time step. Our model-specific error-mitigation scheme, which is based on discarding shots from the raw measurement results, filters out sources of noise like depolarization that are incompatible with the pseudomode formalism while retaining damping in the hardware qubits (see Methods). The resulting ET dynamics on the quantum computer exhibits an effective damping rate ΓQC > 0 that is strongly influenced by the qubits with the worst properties in the circuit, such as short coherence time and large gate errors (see Fig. 1b). Note that we attempt to reach the longest possible lifetime by choosing the qubits with the best properties, instead of targeting a specific damping rate.
To validate the performance of our quantum simulations, we compare against two different types of numerical simulations. We refer to numerical simulations of the quantum circuits (Δt = 4 fs, Nb = 2) as emulations. They involve no noise besides amplitude damping on qubits encoding oscillators and are obtained with the QuEST toolkit64. Simulations of the target model using a smaller time step Δt = 0.5 fs and Nb = 5 levels per oscillators are referred to as classical simulations and obtained using the Dissipation-Assisted Matrix Product Factorization (DAMPF) method, which efficiently applies the pseudomode approach in tensor networks53,59. Comparing our quantum simulations against emulations allows us to assess the effects of noise in the quantum computer, while classical simulations can be considered an exact benchmark with negligible Trotter error and negligible oscillator truncation error (see Supplementary Notes).
Probing electronic and vibronic resonances for N = 5 sites
We evolve an initial excitation in the donor site and aim for a qualitative reproduction of the time-averaged probability of electron transfer Ptransfer(Δ, T),
as a function of the driving force Δ and integration time T, where Pn(t) is the population of site n. The transfer probability describes the probability of an electron moving from the donor to the acceptor without getting stuck in the trap, see Fig. 2a. We expect two types of peaks in Ptransfer(Δ, T), associated with the electronic and vibronic mechanism of ET illustrated in Fig. 2b.
In Fig. 3a we show error-mitigated experimental results for the site population dynamics Pn(t) on ibm_aachen at an electronic resonance with driving force Δe = 1435 cm−1 for the case without vibronic coupling (g = 0). For comparison, the lower panel shows the classical simulation (using Δt = 0.5 fs). We observe both slow and fast modulations of the population in time. The slow modulations stem from the superposition of a pair of eigenstates of the electronic Hamiltonian \({\widehat{H}}_{{{{\rm{el}}}}}\) that arise from the mixing of donor-acceptor states when the donor is resonant with an eigenstate of the acceptor subsystem, as schematically shown in Fig. 2b. The fast modulations correspond to an energy gap around Δe, indicating a contribution of the ground state near − V to the superposition. We refer to Supplementary Notes for a detailed analysis of the states participating in ET.

a The top panel shows a quantum simulation of the population dynamics Pn(t) for an electronic resonance at Δe = 1435 cm−1 without vibronic coupling (g = 0) on 5 qubits. The bottom panel shows a classical simulation (Δt = 0.5 fs) for reference. To enhance visibility for the population dynamics over few sites and many time steps, the contour plot shows lines of equal population, using a linear interpolation of the position coordinate. b Transfer probability Ptransfer(Δ, T) for T = 75 fs (top panel) and T = 200 fs (bottom panel) for the cases with (g = g0 in orange, 10 qubits) and without (g = 0 in blue, 5 qubits) vibronic coupling. The points show expectation values obtained from the experiment, with error bars accounting for shot noise. The curves display classical simulations with a damping rate of γ = (500 fs)−1. The simulations shown in a and c are marked in red. c The top panels shows a quantum simulation of the population dynamics Pn(t) at the vibronic resonance (Δv = 3010 cm−1) without (g = 0, blue) and with (g = g0, orange) vibronic coupling. The bottom panels show classical simulations (Δt = 0.5 fs, Nb = 5) of the target model with a damping rate of (112.5 fs)−1 as reference. d. Population dynamics Pn(t) of the run with Δv and g = g0 (also shown in c) with and without error mitigation, and three classical simulations with increasing damping rates of (500 fs)−1 (solid lines), (112.5 fs)−1 (dashed lines) and (50 fs)−1 (dashed-dotted lines). Each time point corresponds to a unique quantum circuit sampled with 104 shots.
In Fig. 3b we show Ptransfer(Δ, T) resulting from simulations with different Δ after T = 75 fs and T = 200 fs (18 and 50 time steps). We compare quantum simulations without vibronic coupling (g = 0 in Eq. (4)) using 5 qubits for the sites and simulations incorporating vibronic coupling (g = g0) to local oscillators using 5+5 qubits for sites and oscillators. The structure of Ptransfer is well reproduced and the peaks are revealed at the expected driving forces. Notably, the quantum hardware is able to resolve the vibronic resonance at Δv = 3010 cm−1 for the case of vibronic coupling (g = g0), which only starts to build up after a few vibrational cycles of ≈ 22 fs and continues to rise within the lifetime of vibrational excitations. This is evident when comparing Ptransfer(75 fs) with Ptransfer(200 fs). Depolarising noise tends to equilibrate the populations of all qubits and thus artificially raises Ptransfer. This effect is most prominent in regions where neither electronic nor vibronic resonances are present65, leading to an overestimation of Ptransfer, as seen for example around Δ = 2000 cm−1 in Ptransfer(200 fs). The existing noise in the quantum hardware therefore sets a limit to the maximum time that one can integrate the probability of transfer while revealing structure, rather than adding a global shift in Ptransfer. The electronic resonance shown in Fig. 3a is marked with a red cross at Δe, while the vibronic resonance case shown in Fig. 3c is indicated in red markers at Δv.
In Fig. 3c we show the population dynamics Pn(t) of the quantum simulations with driving force Δv and compare them in the bottom panel to classical simulations (Δt = 0.5 fs, Nb = 5) with a damping rate of (112.5 fs)−1, which we identified as the effective damping rate ΓQC (see Supplementary Fig. 4). The dynamics showcases the slow build-up of populations at sites n = 2 and n = 4 in the case of vibronic coupling (right), while in the case of no coupling (left) the excitation stays localized at the donor. This confirms that vibronic coupling is behind this mechanism by entangling electronic eigenstates with oscillator excitations. Maintaining the contrast in Ptransfer between g = 0 and g = g0 at vibronic resonances is the basis for our proposal to use microscopic ET as an application-based benchmark.
In Fig. 3d we show the evolution of populations Pn(t) at the vibronic resonance Δv = 3010 cm−1 in the presence of vibronic coupling (g = g0). We show the raw experimental data (black crosses) and after error mitigation (orange circles), demonstrating the impact of our model-specific mitigation protocol (see Methods). While the raw data exhibits a fast equalization of the populations of all qubits, error-mitigated data is well matched to classical simulations. We show three classical simulations with increasing damping rates, where the highly damped simulation shows a stronger increase of the trap population, while the weakly damped simulation has a more effective electron transfer to the acceptor sites. The periodic pumping of population from the donor to acceptor sites gives rise to a ratchet-like growth of the populations P2(t), P3(t), P4(t), along with the quenching of population on the acceptor trap P1(t). These are signatures of a quasi-stationary, nonequilibrium state that provides ultrafast vibronic ET. Interestingly, the populations Pn(t) of the sites with the largest contribution to this vibronic resonance (n = 2 and n = 4) are better reproduced than the population of spectator sites like n = 3, whose population stays relatively low and it is overestimated on the quantum computer (see Supplementary Notes for the involved eigenstates).
Scaling Up the Number of Sites
In this section we probe the capacity of the quantum computer to scale up the problem size N by simulating vibronic resonances from N = 3 sites up to N = 10 on ibm_aachen. These simulations of ET pose an application-based benchmark, because the vibronic mechanism of ET relies on non-separable states where electronic sites and oscillators are entangled (see Supplementary Notes), and test the ability of the quantum processor to maintain entangled bipartitions of sites and oscillators over a considerable number of time steps. We simulate an ET-model with vibronic coupling (g = g0) on 10 different days using N + N qubits and compare against an ET-model without vibronic coupling (g = 0) at the same driving force, where ET is suppressed, on 3 different days using N qubits. The constant depth of the Trotter circuits with respect to the number of sites N thereby ensures comparability (see Discussion).
In Fig. 4 we show experimental results for the transfer probability Ptransfer(Δv(N), T) after T = 200 fs (50 time steps) for system sizes N = 3 − 10 using up to 20 qubits. For the ET-model with g = g0 (g = 0), the orange circles (blue crosses) give the median value for Ptransfer of the 10 (3) simulations while the error bars indicate the lowest and highest result. As shown in Fig. 4a, the energy landscape and the structure of vibronic resonances change with the problem size N, requiring us to carefully select for each N a driving force that corresponds to a genuine vibronic resonance. The specific driving forces Δv(N) for each N are given in Supplementary Notes.

For each system size N we show experimental results for Ptransfer(T = 200 fs) obtained from 10 different quantum simulations on ibm_aachen for an ET-model with vibronic coupling (g = g0) on up to 20 qubits, and 3 different quantum simulations without vibronic coupling (g = 0) on up to 10 qubits. The orange circles (blue crosses) show the median transfer probability for g = g0 (g = 0), while the error bars indicate the smallest and largest value. a. We show the experimental results for N = 3, 6 and 8 over the driving force Δ, together with classical simulations (Δt = 0.5 fs, Nb = 5). The vertical lines in magenta highlight the vibronic peaks which we chose for our quantum simulations. b. We show experimental results over the number of sites N, compared against noiseless emulations (Δt = 4 fs, Nb = 2) of the undamped model. Note that the height varies between vibronic peaks due to the changing energy landscape.
In Fig. 4b we compare the experimental results for different system sizes N against noiseless emulations. For every case, we obtained at least one run with very strong agreement to the emulation when using N + N qubits in the presence of vibronic coupling (g = g0). In the absence of vibronic coupling (g = 0), the probability of transfer at Δv(N) gets suppressed, and the experimental values using N qubits lie even closer to the emulated values. The variance of experimental results across independent runs is significantly larger for g = g0 (N + N qubits) than for g = 0 (N qubits). We attribute this to the availability of a sufficiently large set of connected qubits with low error rates, which is fluctuating between days for the used device. This is also illustrated by the outlier for g = 0 and N = 10, which compared to the simulations for N≤9 from the same day had to use one additional qubit with considerably worse error rates.
Furthermore, our simulations show good separation between the cases g = 0 and g = g0 for all system sizes N, verifying that the increase in Ptransfer was indeed mediated by vibronic coupling and entangled site-oscillator states in the quantum processor, instead of being a product of depolarising noise.
To investigate the accuracy of our quantum simulations and to attribute an effective damping rate ΓQC, we define the time-averaged deviation
with respect to the populations of the electronic system sites \({P}_{n}^{({{{\rm{ref.}}}})}(t)\) calculated with a reference method, either emulations (Δt = 4 fs, Nb = 2) or classical simulations (Δt = 0.5 fs, Nb = 5). The deviation function ϵ(T) strongly depends on the value γ used in reference calculations, and this dependence becomes more prominent for times T ≳ γ−1. A high ϵ(T) relative to γ = 0, for example, does not necessarily indicate poor performance, as this deviation can be significantly reduced when comparing against reference calculations with finite damping (γ > 0). First, we compare our experimental results on the quantum processor to undamped (γ = 0, Fig. 5a–c) reference simulations, providing a clear benchmark that quantifies the hardware’s capability to simulate long-lived, weakly damped dynamics (1/γ ≳ 500 fs), a regime that remains challenging for classical simulations5,47. Furthermore, we compare experimental results against damped (γ > 0, Fig. 5d–e) reference simulations, which allows to determine the effective damping rate ΓQC imprinted onto the pseudomode ET-model.

a Quantum simulations at different levels of error mitigation (see Methods) compared against undamped emulations (γ = 0, Δt = 4 fs, Nb = 2 levels per oscillator) at T = 200 fs. The minimum, maximum and median of 10 simulations across different days is shown for each N. b Comparison against undamped classical simulations (γ = 0, Δt = 0.5 fs, Nb = 5) for T = 200 fs. c Heatmap showing the time-averaged deviation ϵ(T) compared against the undamped emulations, as a function of the total evolution time T of the fully mitigated results, whereby for each N and T the run with the lowest deviation is shown. The time corresponding to the errors shown in a and b is indicated with a dashed line. d, e Extraction of ΓQC by comparing two quantum simulation for N = 7 sites from different days against classical simulations with γ ranging between (12.5 fs)−1 and (500 fs)−1. The dot markers indicate the minimum of each curve.
In Fig. 5a we show the spread of ϵ(T) values at T = 200 fs (50 Trotter steps), for each N across 10 independent runs, when compared to emulations (Δt = 4 fs, Nb = 2) of the vibronic ET-model (g = g0) without damping (γ = 0). While the deviation clearly increases as a function of the system size before error-mitigation (black crosses), discarding shots from the raw measurement results that violate the conservation of site excitations (\({\widehat{H}}_{{{{\rm{el}}}}}\), blue markers) allows us to reduce the median deviation below 10% to a value that is approximately constant for N = 3 − 7 sites. In addition, when we discard shots with more than a single vibrational excitation (\({\widehat{H}}_{{{{\rm{el}}}}}\), \({\widehat{H}}_{{{{\rm{vib}}}}}\), orange markers), the time-averaged deviation ϵ(T) stays now below 10% for all cases N = 3 − 10. Remarkably, the lowest deviation ϵ(T) achieved for any of these simulations is for N = 7 (7+7 qubits). This finding is in accordance to the hardware calibration data, as the qubit properties over multiple days are found to be best on average for N = 7. Besides, the experimental run for N = 7 with the lowest deviation corresponds to the experiment with the best qubit properties, in particular the highest minimum T1, T2 times and lowest gate errors (see Supplementary Fig. 6).
In Fig. 5b we show the same analysis when comparing against classical simulations (Δt = 0.5 fs, Nb = 5) with a shorter time step and up to 5 levels per oscillator. This comparison allows us to assess the impact of the Trotter error and the truncation of oscillators in our experiments with respect to a classical simulation, where both errors are negligible and can be considered numerically exact. While the Trotter error introduces an overall shift to ϵ(T) that is approximately constant for all N, truncation errors from oscillators are more pronounced for small system sizes (N≤6). Note that the error mitigation, which discards shots with more than a single vibrational excitation (orange markers), leads to a higher deviation for the smallest system size N = 3, as processes with two vibrational excitations have a non-negligible contribution to vibronic ET (see Supplementary Notes for a separate analysis of Trotter and truncation errors).
In Fig. 5c we plot the time evolution of the time-averaged deviation ϵ(T) with respect to emulations, showing for each system size N and time point T the fully mitigated experimental data with the lowest deviation. Surprisingly, the deviation does not increase with the system size N. This is consistent with the fact that we obtained for every N at least one very accurate simulation, and with our observation that the accuracy of quantum simulations only depends on the availability of a sufficiently large connected set of good qubits. We observe slowly increasing deviation from the undamped dynamics as the simulation time increases, reaching a time-averaged deviation of 10% after around 300 fs, indicating the current limit in simulation time.
In Fig. 5d we assess the effective damping rate ΓQC of the quantum simulation with N = 7 sites from May 28 which is the run with the overall lowest deviation shown in Fig. 5b. To do so, we compare that quantum simulation to classical simulations (Δt = 0.5 fs, Nb = 5) of the target model with different fixed damping rates γ. We observe a minimum of ϵ(T) at \({\Gamma }_{{{{\rm{QC}}}}}^{-1}=62.5\,{{{\rm{fs}}}}\), for which the value of the time-averaged deviation does not increase further with T, i.e. ϵ(T) ≈ ϵ(T + Δt), indicating a successful reproduction of pseudomode damping. In Fig. 5e we show a quantum simulation with N = 7 from a different day (May 07). Compared to the May 28 run, the deviation ϵ(T) is generally higher and keeps increasing with no clear minimum at any damping rates γ. Overall, this indicates a larger amount of noise that is incompatible with pseudomode damping and overshadows the effect of an effective damping rate ΓQC. The two simulations shown here are among those with most and least favorable average two-qubit gate fidelities and minimum T1 times on the quantum circuit (see Supplementary Fig. 6). In summary, we find that the effective damping rates ΓQC vary substantially between different runs of the same experiment, and are mostly determined by the quality of entangling gates and the lowest T1, T2 times of qubits.
Amount of shots required for error mitigation
Since we apply error mitigation as a post-processing method by discarding shots from the raw measurements (see Methods), the number of shots left for the computation of expectation values is an important metric. We discard shots that do not comply with particle conservation in the electronic subsystem (\({\sum }_{n=0}^{N-1}{\widehat{a}}_{n}^{{{\dagger}} }{\widehat{a}}_{n}=1\) in \({\widehat{H}}_{{{{\rm{el}}}}}\)). Furthermore, we set a limit to the number of vibrational excitations (\({\sum }_{n=0}^{N-1}{\widehat{b}}_{n}^{{{\dagger}} }{\widehat{b}}_{n}\le {N}_{\max }\) in \({\widehat{H}}_{{{{\rm{vib}}}}}\)). While we chose \({N}_{\max }=1\) for all results presented above, the analysis of remaining shots in this subsection here chooses \({N}_{\max }=2\), motivated by the fact that states with two vibrational excitations participate to a small extent in the charge transfer process for the smallest system sizes (N = 3 or N = 4), ensuring that all discarded shots are truly stemming from errors. Nonetheless, it is worth noting that the choice of \({N}_{\max }\) depends on the structure of the vibrational environment and the energy of the initial state.
To estimate every population Pn(t) with the same accuracy ϵshot-noise, the number of shots scales logarithmically with the size of the system \({N}_{{{{\rm{shots}}}}} \sim \log (N)/{\epsilon }_{{{{\rm{shot-noise}}}}}^{2}\)66. This scaling behavior stems from the statistical nature of quantum measurements (shot-noise) and is already present for ideal quantum computers without noise. In the presence of noise, more shots are demanded to compensate for the loss of incompatible shots after error-mitigation. To evaluate the rate at which shots are lost due to errors in the hardware, every circuit was sampled with the same number of shots (Nshots = 104), regardless of its depth and number of qubits.
The number of remaining shots SN(t) as a function of the number of time steps is shown in Fig. 6a. We also plot in dashed lines the number of shots \({S}_{N,{{{\rm{mixed}}}}}=N(1+N+\left(\begin{array}{c}N\\ 2\end{array}\right))/{4}^{N}\) that would remain if the quantum processor was in the completely mixed state, i.e. after loosing all quantum information due to noise. Expectedly, the number of remaining shots SN(t) decays toward this bound for every system size N. It is worth noting that after 100 time steps, SN(t) remains above the completely mixed state SN,mixed in all cases.

a Number of remaining shots as a function of the number of Trotter steps (1 step ≡ Δt=4 fs), after discarding measurements corresponding to unphysical states with more than two vibrational excitations. For each system size N = 3 − 10, the shaded area indicates the highest and lowest number of shots among the ten independent simulations with vibronic coupling (g = g0). The dashed horizontal lines refer to the expected number of shots if the processor is in the completely mixed state, marked with a dagger for N = 3 (6 qubits). The solid lines correspond to a numerical fit (Eq. (11)), interpolating between a power law and an exponential decay towards the completely mixed state, with this transition indicated with star markers. b Remaining number of shots as a function of the number of qubits evaluated at increasing number of Trotter steps (0 − 300 fs). The solid lines correspond to a numerical fit with an exponential decay (Eq. (12)). The green area indicates the largest Trotter step for which the total execution time lies within the lowest T1, T2 times of qubits in the circuit ( ~ 30 μs for Nqubits≤16 and ~ 10 μs for Nqubits = 20).
A possible explanation for the loss of shots with increasing number of time steps are the different types of noise occurring on the hardware. Over the first time steps, we find a power-law behavior that we attribute to gate infidelities. This power-law decay is stronger for more qubits, corresponding to the increasing average gate errors as shown in Supplementary Table 1. After enough time steps, we find that the loss of shots transitions from power-law to exponential. This happens when the execution time of the circuit on the hardware approaches the regime of the smallest T1 and T2 times involved in the circuit, indicating the effect of continuous noise. To test this claim, we fitted for each N the simple model
to the quantum simulation with the largest number of remaining shots SN(t) at the end of the simulation (t = 400 fs). Notably, the parameter τ (indicated with star markers in Fig. 6a) marks the transition from the power-law behavior of gate error propagation to the exponential scaling of damping and dephasing noise, and is indeed in good agreement with the lowest T1, T2 times when considering the circuit execution times in Supplementary Table 1. In particular, the transition occurs twice as late for N = 3, where no SWAP gates are required, reflecting that the corresponding circuit execution time is half as long as for N > 3.
In Fig. 6b we show the number of remaining shots as a function of the number of qubits at specific time steps. The solid lines at each Trotter step were obtained with a numerical fit
with prefactor C and rate r. While the number of remaining shots has an exponential dependence with the total number of qubits Nqubits, the rate r is highly dependent on the number of time steps. For steps 0, 5, 10, 25, 50 and 75 we find 1/r values of 278, 31, 18, 8, 5 and 4 respectively, indicating at each Trotter step, the number of qubits for which the number of remaining shots is around 1/e of its initial value. The values of the prefactor C in Eq. (12) stay close to the initial number of shots in our experiments (104) until 25 Trotter steps, while they almost double for 50 steps, indicating the scaling becomes worse than exponential after 25 steps (T = 100 fs). The execution time of circuits for 25 steps coincides with the lowest T1, T2 values in the circuit, illustrating again the importance of the minimum qubit coherence time.
To summarize, in the ideal scenario of a noiseless quantum computer, increasing the system size has only a logarithmic impact on the number of shots that are required to estimate individual qubit populations with the same level of accuracy. In the presence of noise, additional shots are needed to compensate for the loss of incompatible shots after error-mitigation. Our simulations reveal that the number of remaining shots has an exponential dependence on the system size N with a small exponent, that increases for simulation times that exceed the coherence time of qubits in the circuit. Regarding the number of time steps, the loss of compatible shots scales polynomially within this coherence time, and becomes exponential for longer circuits.
Discussion
This section investigates how far our quantum simulations can be extended to compete with classical simulations. First, we consider the quantum circuit corresponding to a single Trotter step, as outlined in Fig. 7a. The gates can be organized in four parallel layers, such that the circuit depth does not grow when increasing the number of sites N, enabling the simulation of arbitrarily large systems with near-constant quantum runtime. For system sizes with more than N = 3 sites (3 + 3 qubits), an additional layer of parallelized SWAP gates has to be introduced when mapping the model to the heavy-hex layout processor of ibm_aachen (see Fig. 1b for N = 7). While the mapping onto the heavy-hex topology is already very efficient, even shorter circuits with no SWAP gates and therefore longer time evolution are possible for quantum computers with a square topology.

a Quantum circuit corresponding to a single Trotter step, consisting of four parallelisable layers. The vibronic coupling term \({\widehat{H}}_{{{{\rm{el}}}}-{{{\rm{vib}}}}}\) in magenta consists of parallelized two-qubit entangling gates between site qubits and their corresponding oscillator qubit. b Vibronic models with a single oscillator per site can be scaled up without increasing circuit depth. The example shows a model with N = 50 sites (100 qubits) on a 10 × 10 square layout processor. c When scaling up the vibrational structure by adding multiple oscillators per site, the local vibrational environment of site n can be encoded in the upper/lower rows at the n-th column.
To assess how the quantum approach presented here performs relative to state-of-the-art classical techniques, we compare to the tensor-network DAMPF algorithm59, where computation time scales as \({{{\mathcal{O}}}}({N}^{3}M{N}_{b}^{2}{\chi }^{2})\) for a parallelized HPC-implementation of the 1D chain model as presented here with N sites and M oscillators per site, Nb energy levels per oscillator and χ as the bond dimension of the matrix product operators, underlining that the main challenge for classical simulations lies in the scaling of the system size N. We note that while in general the complexity of DAMPF scales with \({(\dim {\widehat{H}}_{{{{\rm{el}}}}})}^{3}\), the single-excitation-manifold yields \(\dim {\widehat{H}}_{{{{\rm{el}}}}}=N\).
In comparison, the number of qubits in our quantum algorithm scales linearly with the system size N, as illustrated in Fig. 7b with an efficient routing strategy for a square topology to scale up the number of sites to N = 50 using 100 qubits with M = 1, Nb = 2.
While the circuit execution time is constant with N, the current bottleneck lies in the amount of shots discarded in the post-processing of the error mitigation scheme. Extrapolating from our analysis in Fig. 6b, we expect simulations with 100 qubits over 100 timesteps will become feasible when gate fidelities and qubit coherence times have improved by another order of magnitude: Assuming hardware improvements lead to 10 times longer simulations with the same error and shot loss as today, we estimate using Eq. (12) that millions of shots will be required to simulate 100 time steps at a target accuracy of ϵshot-noise = 0.01, which takes tens of minutes to sample at the high shot rates of superconducting processors, which sampled 104 shots in our simulations within few seconds.
Although models with more complex vibrational structures are outside the scope of this work, it is also possible to include more oscillators and energy levels in our approach, as shown in Fig. 7c. Such multi-pseudomode models can be used to fit arbitrary spectral densities \({{{\mathcal{J}}}}(\omega )\), including features like thermal environments. If every site is coupled to independent vibrational environments, the number of qubits scales linearly with the number of oscillators M. When using a straight-forward encoding where each additional phonon level is mapped to a single qubit (see Supplementary Methods), the number of required qubits scales as \({{{\mathcal{O}}}}(NM{N}_{b})\). More efficient encodings of oscillators could in principle lead to a logarithmic scaling in Nb. The quantum circuits can be parallelized with respect to N, similar to the Trotter circuit in Fig. 7a. Each oscillator qubit that couples to an electronic site adds an additional gate layer, causing the Trotter circuit depth to scale as \({{{\mathcal{O}}}}(M{N}_{b})\). Note that this scaling holds for All-To-All, Square, and Heavy-Hex topologies. However, the latter require several layers of SWAP gates to move the system sites along the oscillator chains.
Significantly, classical simulations quickly become intractable in non-perturbative regimes where vibronic coupling is comparable to the strength of electronic interactions (g0 ~ J) and oscillators are weakly damped (γ ≪ g0, J)39,59, as exemplified in DAMPF, where the bond dimension that is required to stay below a certain error threshold is determined by the balance between the spread of quantum correlations (entanglement) and local dissipation60. In contrast, the quantum algorithm is independent of bond dimensions χ as the quantum processor naturally encodes the full vibronic state in the qubit-mapped Hamiltonian.
Beyond the model studied here, extensions of our approach to include arbitrary numbers of electronic excitations, or to include electron-hole dynamics with a second register of site qubits, promise an even larger speedup over classical methods like DAMPF, that scales with \({(\dim {\widehat{H}}_{{{{\rm{el}}}}})}^{3}\). Furthermore, our approach can be generalized to 2D lattice geometries, where the entanglement area law leads to even larger bond dimensions that challenge classical tensor network methods, while our quantum simulation does not suffer from this dimensional bottleneck, in particular when employing platforms with flexible coupling topologies: trapped ions with long-range connectivity and neutral atoms with reconfigurable geometries can accommodate 2D layouts. Similarly, shared bath models, where multiple electronic sites couple to a common environment, can be implemented efficiently on such platforms by routing several sites to the same oscillator, capturing environment-mediated correlations that are particularly costly to treat classically. More broadly, new quantum hardware designs make use of native bosonic elements with parametrizable damping rates, like the motion of trapped ions and superconducting resonators34,35. In particular, ion traps excel at capturing more complex vibrational environments36,38,39.
In conclusion, the pseudomode approach opens an unconventional avenue in quantum computing. While here we have compared our approach to classical methods such as DAMPF, which can target arbitrary decay rates for each oscillator, the damping rates in superconducting processors are not freely tunable: they are determined by the intrinsic qubit dissipation, governed by hardware coherence times T1, T2 and the Trotter time step. For the target model considered here with equal damping rates on each oscillator, our qubit selection strategy addresses this constraint by prioritizing oscillator qubits with similar and maximally long coherence times. The residual inhomogeneities in dissipation rates remain inconsequential for simulation times T ≲ 1/ΓQC, when the precise distribution of hardware damping rates is not yet resolved by the dynamics. Importantly, our quantum simulations were able to accurately resolve the peak structure of the time-averaged probability Ptransfer, because the accuracy of individual populations are less relevant for spectral-dependent figures of merit like Ptransfer, similar to the success of shadow spectroscopic techniques67. Long-lived correlations on the picosecond scale are industrially relevant for the design of new energy materials. While the quantum simulations presented here exhibit effective damping rates ΓQC in the order of (100 fs)−1, we find the main bottleneck for large-scale simulations to be the limited number of qubits that are connected by high-fidelity gates and with sufficiently large coherence times. Simulating models with arbitrarily high damping rates is technologically feasible by artificially amplifying a particular type of noise32,62, however the fundamental challenge still lies in the simulation of target models with lower damping rates. The simulation of vibronic ET for increasingly large problem sizes constitutes an application-based benchmark that, since the vibronic mechanism is driven by the entanglement between electronic sites and oscillators, measures the capacity of the quantum computer to sustain and scale up long-lived entangled states between well-defined bipartitions of the processor.
Methods
Pseudomode description of the vibrational environment
The width of a peak in the spectral density \({{{\mathcal{J}}}}(\omega )\) is inversely proportional to the lifetime of vibrational excitations (see Fig. 1a). This width also determines the duration of system-environment correlations as illustrated by the decay of the correlation function of the environment68
where β is the inverse temperature (see Fig. 1a). Here we fix the temperature to zero, where the bath correlation function \({{{\mathcal{C}}}}(t)\) is the one-sided Fourier transform of the spectral density \({{{\mathcal{J}}}}(\omega )\). As shown in47, thermal effects only become apparent after 150 fs, suppressing electronic resonances at high driving forces which require longer evolution to be resolved. In contrast, vibronic resonances are very robust against thermal effects, because they are driven by long-lived, intramolecular vibrations whose frequencies (ω0 = 1500 cm−1) are much higher than room temperature (kBTroom ≈ 200 cm−1). Incorporating finite temperatures in simulations is possible by introducing a low-frequency background that requires a higher number of pseudomodes.
A single broadened peak in the spectral density \({{{\mathcal{J}}}}(\omega )\), corresponding to a finite lifetime of vibrational excitations, can be captured in unitary dynamics using an infinite amount of oscillators (M → ∞) around the frequency ω0 of the associated peak. An equivalent and simpler approach to reproduce such a broadened peak is achieved by coupling a single oscillator (M = 1, bn ≡ bn,1) with frequency ω0 to a secondary bath. Such a damped oscillator is called a pseudomode53,54,55,56,57.
The resulting dynamics are governed by the following master equation for the density matrix
where \(\widehat{H}\) is the Hamiltonian of the total vibronic system in Eq. (1) and \({{{{\mathcal{L}}}}}_{{{{\rm{vib}}}}}(\widehat{\rho })\) represents the Lindblad superoperator
that describes the damping of the n-th oscillator due to its coupling to an auxiliary bath with a rate γn (see Fig. 1b). As thermal effects are negligible, only amplitude damping towards the ground state is considered in Eq. (15).
After tracing out the vibrational degrees of freedom, this approach allows to engineer non-Markovian dynamics in the reduced state of the electronic system (\({\widehat{\rho }}_{{{{\rm{el}}}}}={{{{\rm{Tr}}}}}_{{{{\rm{vib}}}}}\widehat{\rho }\)) in a non-perturbative manner. For a target spectral density \({{{\mathcal{J}}}}(\omega )\) of the physical model that one wants to simulate, the equivalence between the unitary dynamics of \(\widehat{H}\) with a continuous distribution of harmonic oscillators and the non-unitary dynamics of the pseudomode model with a single damped oscillator relies on matching the correlation function \({{{\mathcal{C}}}}(t)\) of the environment in Eq. (13)53,54. We note that an anti-symmetrized Lorentzian spectral function is required to satisfy the property of vanishing coupling at zero energy \({{{\mathcal{J}}}}(0)=0\), which stems from thermodynamic constrains68. This would require using more than a single pseudomode to properly fit the anti-symmetrized Lorentzian. Nonetheless, when a single Lorentzian function is located at high-frequency, this only leads to negligible artifacts and electronic and vibronic resonances are still well resolved. We refer to Ref. 47 for a full analysis with numerically exact simulations.
Quantum algorithm
We perform a Trotter expansion of the time-evolution operator of the full Hamiltonian in Eq. (1) over time tm = m Δt,
For all our simulations on the quantum computer, we evolve an initial excitation in the donor site with a fixed time step Δt = 4 fs. This value represents a good compromise between total circuit depth and Trotter error and is motivated by the fact that the highest frequency in our Hamiltonian (Eq. (1)) is around (11 fs)−1, stemming from the large Coulomb binding energy V = 2420 cm−1 of bound electron-hole pairs in organic molecules. To validate the Trotter approximation, we compare to classical simulations with a smaller time step of Δt = 0.5 fs in Supplementary Notes.
We encode the Hamiltonian by mapping the N nearest-neighbor coupled electronic sites on a chain of N qubits, where measuring a qubit in state \(\left|1\right\rangle\) means localization of an electron at the corresponding site. The N quantum oscillators are truncated after the first excited state (involving Nb = 2 levels) and each oscillator is mapped onto a single qubit by identifying the number operator with the Pauli \(\widehat{Z}\) operator and the displacement operator with Pauli \(\widehat{X}\). The truncation is motivated by the fact that high-frequency modes cannot be thermally activated, and validated with simulations with Nb = 5 levels in Supplementary Notes. We refer to Supplementary Methods for the explicit qubit mapping, the involved gates and hardware execution times of the circuits. Note that the mapping presented there also allows for more levels per oscillator or the addition of more pseudomodes. Furthermore, we refer to Supplementary Fig. 5 for a comparison between simulations compiled with the variable-angle RX gate and the fixed-angle SqrtX gate as single-qubit gates.
Exploiting hardware noise as computational resource
We assume that noisy processes in the hardware stem from two major sources. One noise channel contains amplitude damping and pure dephasing, which are continuously affecting all qubits, including idle qubits. Another noise channel is depolarisation from gate noise. On the other hand, imperfections in the hardware implementation of single and two-qubit gates introduce additional detrimental effects such as depolarising noise that progressively destroys quantum coherence among qubits. Altough we acknowledge there are other sources of noise, it is sufficient for our purposes to consider noise channels that can be described by Markovian theory. After mapping the full Hamiltonian Eq. (1) to operators acting on qubits in the processor, we model hardware noise with a master equation within the Lindbladian formalism
where the operators \({\widehat{L}}_{n,\alpha }\) refer to noise processes acting on the n-th qubit, and the α index distinguishes between different noise processes. The core idea is to match the Lindbladian \({{{{\mathcal{L}}}}}_{{{{\rm{vib}}}}}\) of the target open quantum system in Eq. (7) to the noise operators \({\widehat{L}}_{n,\alpha }\). While this approach works generally on any type of quantum hardware, target models with pseudomodes are well suited to superconducting hardware due to the large availability of amplitude damping. As summarised in Table 1, we exploit the intrinsic amplitude damping of the qubits that are used to realize damped oscillators (pseudomodes) in Eq. (15) on the quantum processor. Because amplitude damping also introduces dephasing, pure dephasing acting on oscillator qubits can also be properly accommodated by considering an effective damping rate that depends on the T1, T2 times of each particular qubit. Furthermore, pure dephasing on qubits assigned to electronic sites can be identified with a constant offset in the spectral density23, although this already constitutes a deviation from our target model. We refer to the Supplementary Methods for more details. Note that other noise processes are detrimental and need to be mitigated (see next subsection).
For any experiment of Trotterized dynamics on the quantum computer, the correspondence between hardware error rates and the effective damping rate ΓQC that one is able to simulate depends on the hardware execution time \({T}_{{{{\rm{exec}}}}}^{{{{\rm{Trot.}}}}}\) of one Trotter circuit and the particular choice of simulation time step, which we fixed to Δt=4 fs for all our experiments on the quantum computer. For our simulations of the ET-model, a reasonable lower bound for the effective damping rate ΓQC is given by
where \({\min }_{q}({T}_{1}^{(q)},{T}_{2}^{(q)})\) refers to the smallest T1 or T2 time among the qubits in the circuit. In the general case, the inequality reflects the fact that ΓQC is limited by the qubit in the chain with the strongest damping. This mirrors kinetic competition in our electron transfer model (depicted in Fig. 1a): productive vibronic separation and lossy trapping constitute parallel pathways, where the faster channel dominates the quantum yield by outcompeting alternatives. Similarly, among independent decoherence channels acting on different qubits, the fastest decay governs ΓQC, establishing a hardware-imposed ceiling on simulable vibrational lifetimes. Note that Eq. (18) becomes equal for the case of equal damping rates on each qubit, while the exact value of ΓQC depends on the full distribution of T1,T2 times. This bound represents a qualitative statement, as it disregards other sources of noise in the quantum computer.
For selecting suitable hardware qubits in the processor we use Mapomatic69, where most emphasis is given on finding configurations of qubits with the lowest amount of gate errors (single and two-qubit entangling gates) and low readout error. Among such configurations, we choose T1, T2 times that are as large as possible. We reserve the best qubits for encoding electronic sites, since only noisy oscillator qubits are compatible with our model (see Table 1). Nonetheless, we find that gate fidelities and the minimum of the T1, T2 times are the main limiting factor for accurate simulations when encoding either sites or oscillators in the quantum processor. We refer to the Supplementary Methods for further details about our criteria to select suitable qubit layouts.
Error mitigation
To enable accurate simulations over many time steps while retaining a meaningful damping rate, we need to filter out other sources of noise that are incompatible with the target model insofar as possible. Entangling gates in superconducting processors can introduce errors such as strong depolarising noise that tends to equilibrate the populations of all qubits. We employ two levels of a model-specific error mitigation scheme, that is hardware- and implementation-agnostic and can partially correct for depolarizing errors and amplitude damping on electronic sites (see Table 1). It is complemented with readout error mitigation70 for the results presented in this work.
First, we discard shots that do not comply with particle conservation in the electronic subsystem (\({\sum }_{n=0}^{N-1}{\widehat{a}}_{n}^{{{\dagger}} }{\widehat{a}}_{n}=1\) in \({\widehat{H}}_{{{{\rm{el}}}}}\)), since the creation or annihilation of electronic excitations is not possible in the Hamiltonian model (Eq. (1)). This particle conservation constitutes a powerful error mitigation strategy at the post-processing stage that is crucial for long-time evolution62.
Secondly, we set a limit to the maximum number of vibrational excitations (\({\sum }_{n=0}^{N-1}{\widehat{b}}_{n}^{{{\dagger}} }{\widehat{b}}_{n}\,\le 1\) in \({\widehat{H}}_{{{{\rm{vib}}}}}\)), as the total energy in the system is usually around at most 2ω0 and spreads over all electronic sites and all oscillators. As schematically shown in the right panel of Fig. 2b, vibronic resonances are driven by the entanglement between electronic states and quantized vibrations, forming non-separable superpositions of zero and one-phonon configurations. For this reason, we only keep shots with a small number of vibrational quanta. Note that the number of vibrational quanta should not be limited for models with oscillators that can be thermally activated, nevertheless, extensions to the pseudomode formalism with coupled oscillators can accurately reproduce nonzero temperatures while initialising all oscillators to their ground state, regardless of their frequencies53.
When combined with noise characterization and error mitigation techniques like randomized compiling and probabilistic error cancellation, partial control over error rates in the qubits could be achieved26. However, we note that since one goal of this work is to maximize vibrational lifetimes and maintain system-bath coherence as long as possible, we did not apply such mitigation techniques that would address coherent gate errors.
Data availability
Source data underlying all figures and tables are provided with the paper. The raw data are available under restricted access for institutional property regulations. They can be requested from the corresponding authors for verification and reproducibility purposes, subject to institutional approval. Source data are provided with this paper.
Code availability
The code used to generate the data for this study is partly based on proprietary software developed at HQS Quantum Simulations GmbH and the German Aerospace Center (DLR). Due to institutional intellectual property restrictions, this part of the code cannot be made publicly available. Non-proprietary components and example scripts are available from the corresponding authors along with the raw data.
References
Fraggedakis, D. et al. Theory of coupled ion-electron transfer kinetics. Electrochim. Acta 367, 137432 (2021).
Parada, G. A. et al. Concerted proton-electron transfer reactions in the Marcus inverted region. Science 364, 471 (2019).
Williams, N. J. et al. Proton-coupled electron transfer at SOFC electrodes. J. Chem. Phys. 158, 244107 (2023).
Cheng, H. et al. Emerging Era of Electrolyte Solvation Structure and Interfacial Model in Batteries. ACS Energy Lett. 7, 490 (2022).
Lorenzoni, N. et al. Full microscopic simulations uncover persistent quantum effects in primary photosynthesis. Sci. Adv. 11, eady6751 (2025).
Giannetti, C. et al. Ultrafast optical spectroscopy of strongly correlated materials and high-temperature superconductors: a non-equilibrium approach. Adv. Phys. 65, 58 (2016).
Cheng, C.-H. & Lai, P.-Y. Theory of nonequilibrium asymptotic state thermodynamics: Interacting Ehrenfest urn ring as an example. Phys. Rev. E 108, 064114 (2023).
Mikhailova, V. A. & Ivanov, A. I. Effect of Relaxation of Intramolecular High-Frequency Vibrational Mode on Nonthermal Electron Transfer Probability. Stochastic Point-Transition Approach. J. Phys. Chem. C. 111, 4445 (2007).
Zheng, J.-C. et al. Non-Markovian dynamics with λ-type atomic systems in a single end photonic waveguide https://doi.org/10.48550/arXiv.2503.15216 (2025).
Guo, Z. et al. Long-range hot-carrier transport in hybrid perovskites visualized by ultrafast microscopy. Science 356, 59 (2017).
Kahmann, S. & A. Loi, M. Hot carrier solar cells and the potential of perovskites for breaking the Shockley-Queisser limit. J. Mater. Chem. C. 7, 2471 (2019).
Preston, R. J. et al. Nonadiabatic Quantum Dynamics of Molecules Scattering from Metal Surfaces. J. Chem. Theory Comput. 21, 1054 (2025).
Ghan, S., Diesen, E., Kunkel, C., Reuter, K. & Oberhofer, H. Interpreting ultrafast electron transfer on surfaces with a converged first-principles Newns-Anderson chemisorption function. J. Chem. Phys. 158, 234103 (2023).
Utz, A. L. Vibrations that live long and prosper. Nat. Chem. 10, 577 (2018).
Bian, Q. et al. Vibronic coherence contributes to photocurrent generation in organic semiconductor heterojunction diodes. Nat. Commun. 11, 617 (2020).
Ma, F., Romero, E., Jones, M. R., Novoderezhkin, V. I. & van Grondelle, R. Both electronic and vibrational coherences are involved in primary electron transfer in bacterial reaction center. Nat. Commun. 10, 933 (2019).
Schleker, P. P. M. et al. Electrolyte contact changes nano-Li4Ti5O12 bulk properties via surface polarons. Commun. Chem. 6, 1 (2023).
Kick, M., Scheurer, C. & Oberhofer, H. Polaron-Assisted Charge Transport in Li-Ion Battery Anode Materials. ACS Appl. Energy Mater. 4, 8583 (2021).
Windsor, D. & Xu, H. Polarons: Energetics and their structural and electronic effects in ATiO3 perovskite systems. Phys. Rev. Mater. 7, 055004 (2023).
Kemper, A., Abdurazakov, O. & Freericks, J. General Principles for the Nonequilibrium Relaxation of Populations in Quantum Materials. Phys. Rev. X 8, 041009 (2018).
Fratus, K. R. et al. Describing Trotterized Time Evolutions on Noisy Quantum Computers via Static Effective Lindbladians. Quantum 9, 1854 https://doi.org/10.22331/q-2025-09-11-1854 (2025).
Fratus, K. R., Leppäkangas, J., Marthaler, M. and Reiner, J.-M. The Discrete Noise Approximation in Quantum Circuits https://doi.org/10.48550/arXiv.2311.00135 (2023).
Leppäkangas, J. et al. Quantum algorithm for solving open-system dynamics on quantum computers using noise. Phys. Rev. A 108, 062424 (2023).
Chen, I.-C., Pollock, K., Yao, Y.-X., Orth, P. P. & Iadecola, T. Problem-tailored Simulation of Energy Transport on Noisy Quantum Computers. Quantum 8, 1545 (2024).
Rose, M., Kais, S. & Mazziotti, D. A. Open-quantum-system simulation through exploiting noise on quantum computers. Phys. Rev. A 111, 052612 (2025).
Guimarães, J. D., Lim, J., Vasilevskiy, M. I., Huelga, S. F. & Plenio, M. B. Noise-Assisted Digital Quantum Simulation of Open Systems Using Partial Probabilistic Error Cancellation. PRX Quantum 4, 040329 (2023).
Guimarães, J. D. et al. Optimized noise-assisted simulation of the Lindblad equation with time-dependent coefficients on a noisy quantum processor. Phys. Rev. A 109, 052224 (2024).
Wang, T., Zhang, Z., Lu, B.-N., Cirio, M. and Li, Y. Super-bath Quantum Eigensolver https://doi.org/10.48550/arXiv.2412.19599 (2024).
Verstraete, F., Wolf, M. and Ignacio Cirac, J. Quantum computation and quantum-state engineering driven by dissipation, Nature Physics, 633 https://doi.org/10.1038/nphys1342 (2009).
Micheli, A., Kraus, B. & Büchler, H. Quantum states and phases in driven open quantum systems with cold atoms. Nat. Phys. 4, 878 (2008).
Horstmann, B., Cirac, J. I. & Giedke, G. Noise-driven dynamics and phase transitions in fermionic systems. Phys. Rev. A 87, 012108 (2013).
Stadler, P. et al. Demonstration of system-bath physics on a gate-based quantum computer. Phys. Rev. A 111, 022614 (2025).
von Lüpke, U., Rodrigues, I. C., Yang, Y., Fadel, M. & Chu, Y. Engineering multimode interactions in circuit quantum acoustodynamics. Nat. Phys. 20, 564 (2024).
Bozkurt, A. B., Golami, O., Yu, Y., Tian, H. and Mirhosseini, M. A mechanical quantum memory for microwave photons, Nature Physics, 1 https://doi.org/10.1038/s41567-025-02975-w (2025).
Leppäkangas, J. et al. Quantum algorithms for simulating systems coupled to bosonic modes using a hybrid resonator-qubit quantum computer https://doi.org/10.48550/arXiv.2503.11507 (2025).
Navickas, T. et al. Experimental Quantum Simulation of Chemical Dynamics. J. Am. Chem. Soc. 147, 23566 (2025).
Whitlow, J. et al. Quantum simulation of conical intersections using trapped ions. Nat. Chem. 15, 1509 (2023).
So, V. et al. Trapped-ion quantum simulation of electron transfer models with tunable dissipation. Sci. Adv. 10, eads8011 (2024).
Sun, K. et al. Quantum simulation of spin-boson models with structured bath. Nat. Commun. 16, 4042 (2025).
So, V. et al. Quantum simulations of charge and exciton transfer in multi-mode models using engineered reservoirs. Nat Commun 17, 438 https://doi.org/10.1038/s41467-025-67116-6 (2026).
Olaya-Agudelo, V. C. et al. Simulating open-system molecular dynamics on analog quantum computers. Phys. Rev. Res. 7, 023215 (2025).
Knörzer, J., Shi, T., Demler, E. & Cirac, J. Spin-Holstein Models in Trapped-Ion Systems. Phys. Rev. Lett. 128, 120404 (2022).
Motlagh, D. et al. Quantum Algorithm for Vibronic Dynamics: Case Study on Singlet Fission Solar Cell Design. Quantum Sci. Technol. 10 045048 https://doi.org/10.1088/2058-9565/ae0828 (2025).
Kumar, S. et al. Digital-analog quantum computing of fermion-boson models in superconducting circuits. npj Quantum Inf. 11, 43 (2025).
Dan, X., Geva, E. & Batista, V. S. Simulating Non-Markovian Quantum Dynamics on NISQ Computers Using the Hierarchical Equations of Motion. J. Chem. Theory Comput. 21, 1530 (2025).
Xue, W. et al. Reducing the Excitonic Loss at Donor/Acceptor Heterojunction with Negligible Exciton Dissociation Driving Force. ACS Appl. Energy Mater. 5, 9929 (2022).
Somoza, A. D., Lorenzoni, N., Lim, J., Huelga, S. F. & Plenio, M. B. Driving force and nonequilibrium vibronic dynamics in charge separation of strongly bound electron-hole pairs. Commun. Phys. 6, 65 (2023).
Bakulin, A. A. et al. The role of driving energy and delocalized states for charge separation in organic semiconductors. Science 335, 1340 (2012).
Song, Y., Clafton, S. N., Pensack, R. D., Kee, T. W. & Scholes, G. D. Vibrational coherence probes the mechanism of ultrafast electron transfer in polymer-fullerene blends. Nat. Commun. 5, 4933 (2014).
Xu, Z. et al. Coherent real-space charge transport across a donor-acceptor interface mediated by vibronic couplings. Nano Lett. 19, 8630 (2019).
Imamoglu, A. Stochastic wave-function approach to non-Markovian systems. Phys. Rev. A 50, 3650 (1994).
Garraway, B. M. Nonperturbative decay of an atomic system in a cavity. Phys. Rev. A 55, 2290 (1997).
Mascherpa, F. et al. Optimized auxiliary oscillators for the simulation of general open quantum systems. Phys. Rev. A 101, 052108 (2020).
Tamascelli, D., Smirne, A., Huelga, S. & Plenio, M. Nonperturbative Treatment of non-Markovian Dynamics of Open Quantum Systems. Phys. Rev. Lett. 120, 030402 (2018).
Cirio, M. et al. Pseudofermion method for the exact description of fermionic environments: From single-molecule electronics to the Kondo resonance. Phys. Rev. Res. 5, 033011 (2023).
Cirio, M., Luo, S., Liang, P., Nori, F. & Lambert, N. Modeling the unphysical pseudomode model with physical ensembles: Simulation, mitigation, and restructuring of non-Markovian quantum noise. Phys. Rev. Res. 6, 033083 (2024).
Menczel, P., Funo, K., Cirio, M., Lambert, N. & Nori, F. Non-hermitian pseudomodes for strongly coupled open quantum systems: Unravelings, correlations, and thermodynamics. Phys. Rev. Res. 6, 033237 (2024).
Lorenzoni, N. et al. Systematic coarse graining of environments for the nonperturbative simulation of open quantum systems. Phys. Rev. Lett. 132, 100403 (2024).
Somoza, A. D., Marty, O., Lim, J., Huelga, S. F. & Plenio, M. B. Dissipation-Assisted Matrix Product Factorization. Phys. Rev. Lett. 123, 100502 (2019).
Kashyap, V., Styliaris, G., Mouradian, S., Cirac, J. I. & Trivedi, R. Accuracy guarantees and quantum advantage in analog open quantum simulation with and without noise. Phys. Rev. X 15, 021017 (2025).
Xu, M., Vadimov, V., Stockburger, J. T. & Ankerhold, J. Colloquium: Simulating non-markovian dynamics in open quantum systems. Rev. Mod. Phys. 98, 021002 https://doi.org/10.1103/w3nw-hbjc (2026).
Rost, B. et al. Long-time error-mitigating simulation of open quantum systems on near term quantum computers. npj Quantum Inf. 11, 1 (2025).
May, V. and Kühn, O.Charge and Energy Transfer Dynamics in Molecular Systems, 3rd ed. (WILEY-VCH Verlag GmbH & Co. KGaA, 2011)
Jones, T., Brown, A., Bush, I. & Benjamin, S. C. Quest and high performance simulation of quantum computers. Sci. Rep. 9, 10736 (2019).
Zhao, Q., Zhou, Y. & Childs, A. M. Entanglement accelerates quantum simulation. Nat. Phys. 21, 1338 (2025).
Pashayan, H., Bartlett, S. D. & Gross, D. From estimation of quantum probabilities to simulation of quantum circuits. Quantum 4, 223 (2020).
Chan, H. H. S., Meister, R., Goh, M. L. & Koczor, B. Algorithmic shadow spectroscopy. PRX Quantum 6, 010352 (2025).
Breuer, H.-P. and Petruccione, F., The Theory of Open Quantum Systems https://doi.org/10.1093/acprof:oso/9780199213900.001.0001 (Oxford University Press, 2007).
Nation, P. D. & Treinish, M. Suppressing quantum circuit errors due to system variability. PRX Quantum 4, 010327 (2023).
Nation, P. D., Kang, H., Sundaresan, N. & Gambetta, J. M. Scalable mitigation of measurement errors on quantum computers. PRX Quantum 2, 040326 (2021).
Acknowledgements
We thank Elias Walter and Arta Schellhorn for helpful discussions.
Funding
This project was made possible by the DLR Quantum Computing Initiative and the Federal Ministry for Economic Affairs and Climate Action; M.G., A.D.S., P.S., M.M. and B.H. were supported by BASIQ (qci.dlr.de/projects/basiq), G.S. by ALQU (qci.dlr.de/projects/alqu), and M.G., A.D.S. and G.S. by the project ELEVATE (Enhanced Problem Solving with Quantum Computers). M.G., A.D.S. and B.H. further acknowledge support for the following points: From the state of Baden-Württemberg (KQCBW) for the use of the ibm_aachen quantum computer. For classical simulations, the usage of the JUSTUS 2 cluster, supported by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no. INST 40/575-1 FUGG. For emulations, scientific support and HPC resources were provided by the German Aerospace Center (DLR). The HPC system CARA is partially funded by “Saxon State Ministry for Economic Affairs, Labour and Transport” and “Federal Ministry for Economic Affairs and Climate Action”. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.D.S. and M.G. conceived the idea and developed the model. M.G., A.D.S., P.S., and M.M. build the methodology for simulations on the quantum computer. M.G. performed the experiments and emulations. A.D.S. performed classical simulations. M.G., A.D.S., and G.S. analyzed the results and wrote the original draft. M.M. and B.H. supervised the project and revised the manuscript. B.H. was responsible for the acquisition of funding. All authors discussed the results and reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gajewski, M., Somoza, A.D., Schmiedinghoff, G. et al. Simulating electron transfer on noisy quantum computers. Nat Commun 17, 4779 (2026). https://doi.org/10.1038/s41467-026-73700-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-73700-1
