
Abstract
The utilization of artificial intelligence in studying the dysregulation of gene expression in Alzheimer’s disease (AD) affected brain tissues remains underexplored, particularly in delineating common and specific transcriptomic signatures across different brain regions implicated in AD-related cellular and molecular processes, which could help illuminate novel disease biology for biomarker and target discovery. Herein we developed a deep learning framework, which consisted of multi-layer perceptron (MLP) models to classify neuropathologically confirmed AD versus controls, using bulk tissue RNA-seq data from the RNAseq Harmonization Study of the Accelerating Medicines Project for Alzheimer’s Disease (AMP-AD) consortium. The models were trained based on data from three distinct brain regions, including dorsolateral prefrontal cortex (DLPFC), posterior cingulate cortex (PCC), and head of the caudate nucleus (HCN), obtained from the Religious Orders Study/Memory and Aging Project (ROSMAP). Subsequently, we inferred a disease progression trajectory for each brain region by applying unsupervised dimensionality transformation to the distribution of the subjects’ expression profiles. To interpret the MLP models, we employed an interpretable method for deep neural network models, obtaining SHapley Additive exPlanations (SHAP) values and identified the most significantly AD-implicated genes for gene co-expression network analysis. Our models demonstrated robust performance in classification and prediction across two other external datasets from the Mayo RNA-seq (MAYO) cohort and the Mount Sinai Brain Bank (MSBB) cohort of AMP-AD. By interpreting the models both mechanistically and biologically, our study elucidated subtle molecular alterations in various brain regions, uncovering shared transcriptomic signatures activated in microglia and sex-specific modules in neurons relevant to AD. Notably, we identified, for the first time, a sex-linked transcription factor pair (ZFX/ZFY) associated with more pronounced neuronal loss in AD females, shedding light on a novel mechanism for sex dimorphism in AD. This study lays the groundwork for leveraging artificial intelligence methodologies to investigate AD at the molecular level, which is not readily achievable from conventional analysis approaches such as differential gene expression (DGE) analysis. The transcription factor implicated in sex difference also underpins a new molecular mechanistic basis of women’s greater neurodegeneration in AD warranting further study.
Similar content being viewed by others

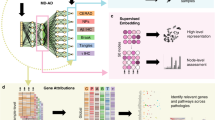

Introduction
Despite extensive research on Alzheimer’s disease (AD) for its underlying mechanism of onset, manifestation, and progression, the complex molecular events behind the disease spectrum remain incompletely understood1. Recently, large-scale high-throughput profiling of omics data such as RNA-seq has enabled the application of novel machine learning (ML) methods to dissect the gene expression profiles of postmortem brain tissues from large clinical AD cohorts. This opens the opportunities for using artificial intelligence (AI) to advance our understanding of the complicated molecular mechanism and seeking potential early treatment of the devastating disease2.
One of the challenges faced by the application of AI to multi-omic data is the lack of interpretability. Complex ML models, like Deep Neural Networks (DNNs), although with unparalleled predictive power, are often considered as “black box” models as their decision-making processes are not easily inspected by human investigators3. Existing literature has reported the use of different AI frameworks to uncover deep interrelationships between gene expression and AD neuropathologies4,5. However, due to the limited sample sizes, the interpretation of the models, specifically the DNNs, had to be oversimplified and relied on outcomes trained from datasets aggregated across multiple brain regions. Notably, these regions are known to be affected by AD neuropathology quite differently6. For example, Beebe-Wang et al.5 utilized a multi-task deep learning framework for AD neuropathology to examine 1758 gene expression samples altogether, which were collected from nine different brain regions in three studies, some from the same individuals. The models were interpreted with Integrated Gradients (IG)7. The resulting consensus scores were evaluated, and the top 50–100 ranked genes were reported and examined for functional enrichments. Their work also identified a sex-specific correlation between microglial immune activation and neuropathological changes. Nevertheless, the specific roles of these transcriptomic signatures in different brain regions remain unclear, and it’s possible that some region-specific signatures may be lost during this process. Additionally, since the models were trained using neuropathological phenotypes, their relevance to clinical manifestations requires further investigation. In another publication4, a prototype of DNN model was applied to classify AD vs control from gene expression data in AD cohort. However, the interpretation of the model was over-simplified, relying solely on the feature weights from the input to the first layer of the DNN for feature extraction and using a basic gene network analysis method. Achieving the full potential of modern AI techniques in genomics research necessitates a thorough evaluation of the model, and a precise and comprehensive interpretation of its mechanisms based on input features. Most recently, through the Accelerating Medicines Project for Alzheimer’s Disease (AMP-AD) Target Discovery Consortium and associated open-science consortia8, multidimensional molecular data from more than 2000 human brains and peripheral tissues from multiple AD cohorts have been made publicly available9. Coupled with comprehensive neuropathological and clinical characterizations, these data represent an unprecedented opportunity to advance our understanding of the molecular basis of AD.
Compared to traditional gene expression analysis methods like differential gene expression (DGE) analysis, we propose that explainable AI (XAI) has the potential to detect subtle, nonlinear molecular effects in disease-affected brain regions by leveraging large-scale molecular profiles from consortium-enabled clinical cohorts. In addition to corroborating previously known molecular regulators implicated in AD, this approach may lead to novel discoveries. Specifically, the new insights may enhance our understanding of the disease’s etiology such as the differential disease manifestation association with sex (sex dimorphism). Herein we report such efforts to build and interpret DNN models (Fig. 1) upon a simpler prototype4 using bulk RNA-seq data from three different brain regions (dorsolateral prefrontal cortex (DLPFC), posterior cingulate cortex (PCC), and head of the caudate nucleus (HCN)), profiled in the Religious Orders Study and Memory and Aging Project (ROSMAP) cohort10,11 from the AMP-AD open data platform. We substantially extended and improved the prototype4 by (1) extensively updating the framework to apply on larger sample datasets at 1090, 647, and 717 samples from the three brain regions respectively; (2) utilizing state-of-the-art model interpretation method, i.e. the Shapley Additive exPlanations (SHAP) values12 for feature extraction; and (3) applying a multiscale gene co-expression network analysis method to obtain novel biological insights based on the extracted features. To evaluate the generalizability of our proposed framework, we validated it using two independent AMP-AD datasets: the Mayo RNA-seq study cohort (MAYO)13 and the Mount Sinai Brain Bank (MSBB) study cohort14. The framework achieved superior performance in correlating expression profiles with both neuropathological and clinical traits and demonstrated unprecedented prediction accuracy. From model interpretation, we identified 1317, 1594, and 1643 genes implicated in AD in the three brain regions, respectively. We investigated the overlaps of these genes and compared them with the differentially expressed genes (DEGs) identified in the same AD versus control comparisons. We observed that our approach shows the potential to unveil non-linear gene regulation relationships that are obscured by 0 bulk-tissue profiling, which would otherwise only be observable in single-cell RNA-seq data. Based on these gene features, gene co-expression network analysis identified and derived region-consensus and -specific functional modules. We not only identified the transcriptomic modules associated with microglia activation shared among brain regions with many known regulators, but for the first time brought light on a region- and sex-specific transcription factor implicated for neuronal loss and linked with sexual dimorphism in AD. We believe that this work lays the foundation for the application of XAI approaches to high-dimensional molecular data and thus advances the study of AD etiology.

A Overview of the framework. Using the gene expression profiles from AD and control subjects and their diagnosis class as the input for supervised classification, the model was trained by a multilayer neural network. The trained network was passed forward to the profiles from the whole cohort with the resulting output manifold subject to unsupervised dimensionality transformation (UMAP) to obtain the pseudo-temporal trajectory and severity index (SI). SI was linearly correlated with phenotypic data for evaluation. Trained model was interpreted by SHAP explainer to obtain the most salient features (index genes, or IGs). Their co-expression relationship was examined for biological interpretation. The framework was applied to the three brain regions (DLPFC, PCC, and HCN) from ROSMAP cohort, respectively, and the derived data were compared across different brain regions. B Technical details of the workflow.
Results
Classification model architecture and performance
As illustrated in Fig. 1, the framework starts from a DNN classification model (supervised classification) on the RNA-seq data with two independent extension components (unsupervised dimensionality transformation and SHAP interpretation/feature extraction for biological interpretation). We utilized the datasets from three different brain regions (DLPFC, PCC, and HCN) of the ROSMAP cohort to train three models separately and applied the models on the data from the two independent cohorts (MAYO and MSBB) as external validations. Demographic information for each of the cohort (ROSMAP, MAYO, and MSBB) and the details of the phenotypical traits are reported in Supplementary Tables 1, 2. Detailed information about the definitions of the three classes (AD, OTHER, and CN subjects) in the ROSMAP cohort is described in “Materials and Methods”, with the number of AD and CN samples for each brain region used for classification model reported in Supplementary Table 3.
We first trained a DNN classification model of the two termini of the disease spectrum from the ROSMAP datasets, to separate the AD subjects from normal control participants (CN). Samples were randomly partitioned into training and testing sets, containing 80% and 20% of the samples, respectively. To determine the optimal classification model architecture, we conducted extensive experiments with various neural network architectures (Supplementary Table 3, Supplementary Fig. 1) after reviewing relevant literature15. Consistent with what’s reported in the literature, the final model chosen was an MLP for each brain region because of its best classification performance, with the respective hyperparameters in each model and classification metrics reported in Supplementary Table 3, and receiver operating characteristic (ROC) curves shown in Supplementary Fig. 1. Notably, the model trained from DLPFC (DLPFC model for simplicity, the same below) exhibited exceptional classification accuracy, achieving a test accuracy of 97.8% and a sensitivity of 100%, indicating its superior proficiency in accurately identifying AD samples. Conversely, the PCC model exhibited a comparably high testing accuracy of 96.0% and a sensitivity of 96.2%, highlighting its strong classification capabilities. The HCN model performed the least satisfactorily in comparison to the cortex dataset (DLPFC and PCC) models, showing a testing accuracy of 81.1% and a lower specificity of 56.3%. The distinction of the performance between these models also aligns with the variation in relevance to AD pathology among these brain regions, as the cortex regions are known to be highly associated with AD in contrast to subcortical regions16. We also compared the results obtained from other DNN methods as well as traditional machine learning methods such as support vector machine (SVM) and random forest (RF) and confirmed that MLP outperforms them considerably in the cortical regions (Supplementary Table 3 and Supplementary Fig. 1). This is likely due to its ability to capture complex, nonlinear relationships in high-dimensional gene expression patterns.
Model the whole AD spectrum as a pseudotemporal trajectory
Given that AD is a progressive disease, we reasoned that the application of a pseudotemporal trajectory approach might yield novel insight. We applied the dimensionality transformation technique-Uniform Manifold Approximation and Projection (UMAP)- to the latent features learned from the DNN supervised embeddings to produce a 3D projection for all the samples. The resulting UMAP space could reveal trajectories for the underlying continuous progression that reflects the underlying structure and illustrates class separability. As expected, we observed that the samples from AD/CN are mainly located at each of the two termini of a trajectory as two distinct clusters (Fig. 2A, Supplementary Fig. 2A, C). Adding the OTHER samples into the same space clearly indicated a continuous disease spectrum as well as a progression course along the trajectory (Fig. 2B, Supplementary Fig. 2B, D), although the trajectory derived from HCN is less smooth. We used a metric, denoted here as the Severity Index (SI), to stage each sample along the inferred trajectory—from the starting point (presumably representing the normal state) to its endpoint (advanced or terminal AD). We evaluated how accurately the trajectory models AD’s progression, by linearly correlating the SI to its respective neuropathological and clinical biomarkers. We reasoned that in the dimensionality transformation process (UMAP), the non-linear nature of AD progression has been modeled, and resulting pseudotime from the reduced space could be linearly correlated with the phenotypical traits (semiquantitative stagings such as ceradsc or braaksc, or logarithm scaled quantitative neuropathological assessments such as amyloid or tangles) to validate the modeling approach. We indeed observed that SI correlated strongly with all AD-specific biomarkers excluding diffuse plaques (P < 2.2E–16, R2 > 0.31, Fig. 2C, D, Supplementary Tables 4, 5), with most correlations still highly significant even after excluding those samples used in the training process (i.e. only considering OTHER samples), or within the AD samples only. As illustrated in the spider charts (Fig. 2C, D) comparing the P values (-logP) and coefficient of determination (R2) for all the phenotypical traits from the three regions, the regression metrics are the best for the model built from DLPFC followed by PCC, with HCN performing the least satisfactorily. All three models still considerably outperform the model built based on a subset of the DLPFC samples4.

A, B DLPFC without and with OTHER samples. C, D Spider plots showing the linear correlations of SIs with phenotypical traits for all three models.
Since UMAP can introduce variability due to random initialization, we tested whether different parameter settings would affect the resulting trajectories and their correlation with phenotypical traits. Our findings demonstrate that R2 and P values to associate SI with phenotypical traits remain stable across different parameter combinations, with the embeddings consistently forming progressive trajectories (Supplementary Fig. 2E, Supplementary Table 6). This suggests that our approach effectively preserves local data relationships for the correlation between SI and various phenotypic traits, as the transformation is applied to the one-dimensional output from supervised classification, rather than directly on high-dimensional data.
For validation, the three models were then applied individually to the harmonized transcriptomic data from both the MAYO and MSBB cohorts to derive their respective trajectories. Data from the MAYO cohort came from two different brain regions: temporal cortex (TCX) and cerebellum (CER). After projecting into the same 3D UMAP space, the subject distributions along the trajectories from the two different brain regions showed drastically different patterns (Fig. 3A, B, DLPFC model only). For TCX, it showed the distributions of different locations for AD versus CN subjects along the trajectory similarly to those from ROSMAP data, while this was not observed for CER. This is confirmed by the results obtained from linear regression of the SI versus pathological biomarkers (Braak and Thal scores, Fig. 3C, D). Only in the TCX samples were the SIs found to be significantly correlated with both Braak (P = 7.98E–14, 4.80E–9, and 7.40E–6 respectively) and Thal scores (P = 2.95E–7, 1.04E–5, and 5.13E–3, respectively). Again, when applied to the TCX dataset, the models explained a large amount of variance overall for both biomarkers, with R2 in the range of 0.40 to 0.53 for the three models, respectively. We also observed that the model trained on DLPFC data was the most predictive of biomarkers in the MAYO cohort, whereas the model based on HCN data showed the least predictive performance (Fig. 3C, D, Supplementary Table 7). Interestingly, the models derived from DLPFC and PCC also exhibited predictive capability for Braak score in the CER dataset (P = 4.94E–4 and 9.84E–3) although the proportions of variance explained (PVEs) are low (<0.09 in comparison with >0.29 in TCX data), indicating that tauopathy in AD is associated with widespread transcriptomic changes, including in regions comparatively spared from development of neurofibrillary tangles.

PA pathological aging, PSP progressive supranuclear palsy. A TCX mapped to DLPFC; B CER mapped to DLPFC. C, D Spider plots showing the linear correlations of SIs with phenotypical traits from all three models.
For the MSBB cohort, the models were applied to the gene expression profile of all four sampled regions [frontal pole (FP, Brodmann area BM10), superior temporal gyrus (STG, BM22), parahippocampal gyrus (PHG, BM36), and inferior frontal gyrus (IFG, BM44)] and all regions show similar albeit slightly different trajectories (Fig. 4A–D, DLPFC model only), with the SI consistently significantly correlated with all the neuropathological and clinical biomarkers (Braak score, PlaqueMean, CDR scale and CERAD score, Supplementary Table 8), for the models from DLPFC and PCC. Still, the model from HCN does not predict some of the biomarkers well when applied to certain brain regions, such as Braak or CERAD score in FP (Fig. 4E, F, Supplementary Table 8).

A–D Four regions (FP, STG, PHG, and IFG) mapped to DLPFC. E, F Spider plots showing the linear correlations of SIs with phenotypical traits from all three models.
Comparison of the models trained from and applied to different brain regions
As reported above, when comparing the models trained from three different brain regions in ROSMAP, neuropathology and cognitive impairment from AD are evidently better reflected in the transcriptomes of cortical regions. The model from HCN shows relatively poor performance in classification metrics, aligning with phenotypical traits within the cohort, and prediction in external datasets. In contrast, models from DLPFC and PCC show comparably excellent performances, especially considering that PCC was trained with a smaller number of samples. When comparing the SIs derived from the three regions for the same subjects, we also observed that DLPFC-PCC pairs are most significantly correlated with each other (n = 621, p = 1.07E–142, R2 = 0.65, no other covariates considered), followed by DLPFC-HCN (n = 670, p = 8.27E–118, R2 = 0.55), and PCC-HCN (n = 466, p = 1.46E–90, R2 = 0.58). We subsequently focused on the comparison of the models’ predictive performances from the DLPFC and PCC regions. When applied to external datasets, we observed that the model from DLPFC demonstrates the greatest predictive power and coefficient of determination R2 of traits with the transcriptome from the TCX region of the MAYO cohort, and IFG and STG regions of the MSBB cohort, all of which are located at the either prefrontal cortex (DLPFC and IFG) or temporal lobe (TCX and STG). Conversely, the model from PCC is more predictive of data from PHG of MSBB cohort (Figs. 3, 4), both of which are deemed as part of the hippocampocentric subdivision of the paralimbic zone17. This illustrates that nuanced molecular changes in different brain regions affected by AD could be captured by DNN methods.
Index genes derived from the model interpretation
We set out to interpret our models using one of the latest model interpreting methods, the SHAP values, to reveal which features (i.e., genes) have the most impact (whether positively or negatively) on a specific prediction. We averaged out the feature contributions to AD and CN, respectively, then took the absolute mean of the two to evaluate their importance to the classification. To robustly capture highly relevant genes, the training procedure was repeated 100 times using the same hyperparameters but different random number seeds, with the goal of simulating a “consensus network” and eliminating the randomness from a single or a few models. The process of averaging the SHAP values of all the features from each training to obtain the final SHAP metric and extract the list of the most salient genes based on the cutoff using the interquartile range (IQR) criterion ( > q0.75 + 1.5*IQR)18 is illustrated in Supplementary Fig. 3. The cutoff was set based on the distribution of the averaged final SHAP values from each gene, with a boxplot illustrating the outliers in the distribution as the most salient for their contributions (Supplementary Fig. 3B).
Following this procedure, we selected 1317, 1594, and 1643 genes for the models from three brain regions, respectively (Supplementary Table 9A–C). They were labeled as “index genes” (IGs) for the subsequent in-depth analysis. We compared the IGs with those DEGs identified from the same datasets (syn26967457), by the same contrast of neuropathological confirmed AD vs CN comparison. There are some overlaps, but still considerable differences between the IGs and DEGs for the same brain region, as shown in Supplementary Fig. 4A. In contrast to those DEGs, IGs are not constrained by their fold changes or P values in the contrast, as in fact, the logFC and P values for these IGs exhibit a consistently even distribution (Supplementary Fig. 4B, C), suggesting they are coordinately regulated at the molecular level.
We observed more overlaps between IGs of DLPFC and PCC as expected. All highly significant (P < 2.2E–16), the odds ratio (OR) of the overlaps between DLPFC and PCC (20.3) is higher than those between DLPFC and HCN (10.3) and PCC and HCN (15.5), confirming that the expression profiles of the two from cortical regions are more similar with each other than the subcortical region (HCN).
Co-expression modules of IG reveal molecular changes associated with AD from different brain regions
To gain novel biological insights, we derived co-expression network modules for the IGs based on the gene expression profiles of the whole cohort from the three regions respectively. For DLPFC, five distinct modules were resolved from the profile (Fig. 5A–E), ranging from 78 to 542 genes in each module. Functional annotations implicated them in different cellular processes from five major cell types (Fig. 5F, Supplementary Table 9A). Most of the genes in the modules from microglia and oligodendrocytes are upregulated, while those from neurons are downregulated (Supplementary Fig. 4B, C), indicating a consistent pattern of gliosis and neurodegeneration19. The wide range of gene regulation in astrocyte module also illustrates the cell type-specific heterogeneity from their diverse functions in AD20. In contrast, we obtained 14 and 18 modules ranging from 25 to 227 genes for PCC and HCN profiles respectively (Supplementary Table 9B, C), some of which are subsets of the five modules from DLPFC. The more fragmented module structure could be attributed to the facts that the RNA qualities for PCC are significantly lower than DLPFC (mean RIN = 5.64 vs 6.26, P = 4.82E–18, Mann–Whitney U test), while the model from HCN does not perform well. In addition, we have smaller numbers of samples to train the two models than DLPFC, so there are very few common networks shared among all three regions. We still observe sizeable submodules in common between the networks of DLPFC and PCC, especially for the networks from neurons, astrocytes, and microglia (Fig. 5A, D, E), with at least two out of three submodules in microglia, two submodules in astrocytes and two submodules neurons overlapped respectively (Supplementary Table 10).

A–E Five modules clustered by their cell type enrichment. Only hub genes are labeled. F Functional enrichment for each module.
We subsequently focused our analysis on the modules from DLPFC, for a comprehensive interpretation of the best model in our study to obtain the biological insights of molecular changes implicated in AD. We observed that the gene nodes are enriched for genetic risk loci21,22 from genome-wide association studies (GWAS) in the two glial modules (e.g. TREM2, MYO1E, PLCG2, CD33, HLA genes among others in microglia, P = 7.45E-6, OR = 8.75; and CR1, ADAMTS1, C2, C4A/B, IQCK among others in astrocytes, P = 2.87E-3, OR = 2.65, Supplementary Table 9A). Herein we report our major findings within the main text of this manuscript, with some additional results reported in the Supplementary Information.
The microglia module depicts an extensive picture of immune response in AD (Fig. 6A). In addition to the enrichment of GWAS loci, there is a high enrichment of disease-associated microglia (DAM) signature genes23 such as SPP1, ITGAX, CLEC7A, TMEM119, and TREM2 (P = 5.09E-12, OR = 20.36), as well as the TYROBP causal network in microglia (WP3625)24 (P = 5.28E–26, OR = 182.9) and the CD33/LAIR-1 inhibitory networks25. In the center of the networks, as well as from an overlapped module in PCC, is the hub gene C3AR1, recently identified as a major player mediating neuroinflammation and tau pathology in AD26,27. It is notable that many of these genes (e.g. PLCG2, MYO1E, VSIG4, LAIR1, and HLA genes) are not identified as DEGs in the same datasets used in our study by falling short of logFC or P value cutoff, although they have been reported as differentially expressed in single-nucleus RNA sequencing (snRNA-seq) data28,29, demonstrating the power of deep learning methods to uncover nuanced signals in convoluted data. Although some of these genes have been either extensively investigated for their roles in AD such as TREM230 or reported as DEGs in either bulk or snRNA-seq data analysis, we note that our study is the first to collectively illustrate the gene co-expression networks from these critical regulators. Notably, these findings are confirmed across multiple AD-affected brain regions.

A Microglia module. B A subset from the neuron module including the hub gene SVOP and a sex-linked submodule.
Sex-linked module and transcription factor (TF) in DLPFC neurons
We observed that the neuron module possesses several key gene hubs such as CACNG3, VGF, NPTX2, RPH3A, SVOP, and BDNF, recently reported to exhibit positive associations with global cognitive function and negative associations with neuropathology across various excitatory neuron subtypes in AD (Fig. 5D). They were found to be within a consensus signature significantly associated with global cognitive function in at least three different excitatory neuron subtypes, and prominently linked to both pre- and postsynaptic compartments, from a comprehensive analysis of snRNA-seq data from the prefrontal cortex (PFC) brain region within the ROSMAP cohort31. Again, they are not completely recapitulated by DGE analysis from bulk tissue RNA-seq data.
One prominent hub gene is synaptic vesicle 2 related protein (SVOP), a protein involved in synaptic vesicle transport. SVOP interconnects many of the aforementioned hubs, with a co-expression submodule primarily composed of sex-linked genes such as XIST, TTTY14, and KDM5D (Fig. 6B). It is connected to the submodule by the X-linked zinc finger transcriptional regulator ZFX, which is known to escape X chromosome inactivation (XCI)32,33. We assessed the transcriptional regulation of SVOP using the expression profile of the DLPFC neuron module and identified the candidate TFs that regulate SVOP expression (Supplementary Table 11). Among them, three TFs (SP1, MXI1, and ZFX) are directly connected to SVOP in the module (Fig. 6B), all of which would be expected to act as transcriptional repressors based on their inverse expression correlation to SVOP.
Since ZFX and ZFY are X-Y homolog pairs (gametologs) which share 96% overall similarity, with 99% similarity in the zinc finger domains and are assumed to have the same function, we examined the contribution of the two sex-linked genes to the expression of SVOP, for the whole cohort from DLPFC region. A simple linear regression shows that the expression of both genes is significantly correlated with SVOP expression, although ZFY at a much lesser degree (Fig. 7A, Supplementary Table 12). In addition, both correlations show considerable residual sex effects in the model. To account for their additive effects, we reprocessed the dataset with raw gene counts (syn22231797), adding an additional feature by combining ZFX + ZFY gene counts. Indeed, we observed that the cumulative expression of ZFX and ZFY completely explains the sex differences of SVOP expression by inverse correlation, with no residual sex effect, or additional diagnosis difference unaccounted for in the linear model (Fig. 7A, Supplementary Table 12). Nevertheless, the cumulative TF expression is still lower in males, due to the escape of XCI of ZFX in females and lower expression of ZFY in males in comparison with their homolog ZFX expression34. Consequently, SVOP is more significantly downregulated in AD females than their male counterparts (Fig. 7B). As SVOP plays a central role in the co-expression module associated with neuronal loss in AD, our research provides direct evidence of the molecular mechanisms underlying sex chromosome involvement and sexual dimorphism in AD.

A Regression between SVOP and ZFX, ZFY and their combined expression. B Boxplots showing ZFX, ZFY, their cumulation and SVOP expression stratified by sex.
To further validate the findings, we reprocessed the data from two additional brain regions in the ROSMAP cohort, as well as from the two independent cohorts (MAYO/MSBB). We observed a significant inverse correlation between the expression of SVOP and the combined expression of ZFX and ZFY across all analyzed brain regions (Supplementary Figs. 5–7, Supplementary Table 12). The downregulation of SVOP was particularly notable in female AD subjects in disease-affected areas. Interestingly, the correlation substantially reduced the previously observed sex differences in the individual relationships between ZFX or ZFY and SVOP. In several regions, such as DLPFC in ROSMAP and FP in MSBB, sex differences were completely accounted for (p > 0.05). Furthermore, differences related to AD diagnosis were no longer observed in all three ROSMAP regions and TCX from the MAYO cohort. Using a foundation model, we were also able to identify a binding site near the target gene SVOP for the TF ZFX as a repressor in excitatory neurons (Supplementary Fig. 8). Collectively, these results indicate that the ZFX/ZFY TF pair plays a significant role in the sex dimorphism of neurodegeneration in AD.
Discussion
In this study to exploit the application of XAI in the genomic research of AD to obtain novel insights into disease mechanism, we present a comprehensive interpretable deep learning framework on the RNA-seq data obtained from multiple postmortem brain regions of the ROSMAP cohort, a large clinical cohort of AD. We also applied the trained models to the transcriptomic data from two independent cohorts, the MAYO and MSBB cohort to validate its generalizability. Our models show excellent predictive power in aligning the transcriptomes with clinical and neuropathological traits in both internal and external validations, as indicated by the model metrics from the SI for delineating the progressive pseudotemporal trajectories in each dataset. This underscores the broader applicability of the framework in the study of neurodegenerative diseases such as AD as a continuum35. We have made substantial improvements in the current workflow upon the prototype4, which include innovative model interpretation and feature extraction method (SHAP), novel gene co-expression network analysis methods (MEGENA), as well as extensively tested DNN method coupled with UMAP dimensionality transformation of the learned representation. Applying this enhanced framework to larger datasets enabled us to uncover novel scientific insights, such as identifying transcription factor pairs associated with sex dimorphism in AD.
We tested multiple architectures in the DNN components of the framework and consistently observed, as supported by existing literature15, that MLP outperforms other DNN methods such as convolutional neural networks (CNN) and recurrent neural networks (RNN). This may be attributed to the fact that MLP, being a fully connected network, is particularly well-suited for analyzing gene expression data, which involves complex co-expression relationships. These relationships involve interactions among multiple genes (e.g., activation, inhibition, or enhancement of expression) and form a hierarchical network to regulate biological functions. These findings were consistent across various evaluation metrics (Supplementary Table 3) and ROC plots (Supplementary Fig. 1), further validating the robustness of the MLP approach. Additionally, advanced deep learning methods like CNNs and RNNs are less effective for our data due to its numerical matrix format, lacking spatial or sequential structure. CNNs excel in capturing spatial patterns in data like images, while RNNs handle sequential relationships in time-series or language tasks36. In contrast, MLPs are better suited for modeling the complex, non-linear relationships in gene expression data, making them the optimal choice for our study.
By individually modeling the transcriptomes from three distinct brain regions of the ROSMAP cohort, we demonstrate the capacity of deep learning methods to learn and capture subtle distinctions present in diverse brain regions affected by AD. The models successfully differentiate the non-specific subcortical region in AD, i.e., HCN. Moreover, they distinguish nuanced variations in the data from the two cortical regions (DLPFC vs. PCC), evident in the observable differences in predictive accuracy when applied to data from other relevant regions in an external cohort (MSBB, Fig. 4, Supplementary Table 8). This reiterates the notion that molecular changes in AD-affected brains are both specific and regional, underscoring the importance of considering these factors in comprehensive studies37.
The most pivotal insights derived from the study come from the IGs through model interpretation. By applying the SHAP method based on cooperative game theory to explain the outcome of the model, our approach provides a way to fairly allocate contributions of each feature, with both global and local interpretability. The summarized importance score would therefore offer an overview of feature importance across the entire dataset for AD vs control classification. Our framework consistently excels in extracting input features that go beyond the constraints of logFC or P values stemming from traditional DEG analysis, thus revealing nonlinear, coordinated, and cell type specific gene regulation from bulk tissue data. These signals are otherwise only available from deconvoluted or higher resolution omics data, such as single cell RNA-seq. It would eventually be desirable to apply the framework to such kind of data to obtain even more novel biological insights into disease etiology at the cell level.
The co-expression networks derived from the IGs from the two cortical regions, particularly DLPFC highlight the critical roles of gliosis and neurodegeneration in AD. In microglia, the networks corroborate many of the established activated pathways implicated in AD, such as the TYROBP causal network and the DAM signatures. In particular, the networks draw attention to the significance of the TREM2-DAP12-SYK pathway, which coordinates neuroprotective microglial responses in AD38,39. It will be worthwhile to further investigate the relationship between this pathway and the C3 and C3A receptor (C3AR1) signaling given their close co-expression pattern since there is limited study in this direction. The data highlights the neuroimmune axis as evidenced by MHC class II signaling transduction, which suggests an intricate interplay of adaptive and innate immune systems both within and outside the brain influencing the etiology and pathogenesis of AD40. It is worth noting that the networks also implicate the role of the C1Q/CD33/LAIR-1 inhibitory complex in AD, and it seems it is only present in DLPFC. Since the complex has been reported to dampen monocyte immune response41, an in-depth study is thus warranted to explore the functions of this complex and its influence the balance between immune activation and tolerance in AD, and whether this effect is brain region specific.
It has now been widely acknowledged that AD disproportionately affects women in both disease prevalence and rate of symptom progression, but the mechanisms underlying this sexual divergence are still being actively pursued. From transcriptional analyses, gene dysregulation in AD is particularly prominent in the neuronal cell populations, especially in females42. Notably, for almost all the hub genes identified in the neuronal module in this study such as CACNG3, VGF, NPTX2, RPH3A, SVOP, and CA10, their fold changes in DGE analysis are more significant in the females than males by sex stratified analysis (syn26967458), which suggests more severe neuronal damage in females. Conversely, the transcription factors predicted to repress the expression of SVOP (SP1, MXI1, and ZFX) all show more pronounced upregulation in females in DLPFC (Supplementary Fig. 9). Recently it has been reported that the human Y and inactive X chromosomes similarly modulate autosomal gene expression, with the homologous transcription factors – ZFX and ZFY acting in a mutually and cumulatively dose-dependent fashion43. Most importantly, they are prioritized as one of the genes on sex chromosomes most likely to contribute to male-female differences in common disease44. We observed highly correlated co-expression between their cumulative expression and that of SVOP, with the sum greater in females and more significantly upregulated in AD. Together with the two other transcription factors (SP1 and MXI1), their expressions explain over 70% variances of the expression of SVOP in the DLPFC data, indicating a highly probable orchestrated transcriptional regulation. We have extensively discussed the implication of this finding in sex dimorphisms of AD in the Supplementary Information.
In this study, we set out to apply an XAI framework to the gene expression data profiled by large clinical cohorts on the AD affected brain tissues to obtain novel biological insights. We not only identified the gene co-expression networks with some known regulators in the glia, but also presented novel preliminary data showing the ZFX/ZFY pair’s contribution to more severe neural loss in AD females. One limitation of our study is that the data collected from cohort-based autopsy samples such as AMP-AD might also be associated with selection bias with imbalanced age, sex, and/or ethnicity or racial groups45. We also applied the framework on the transcriptomes obtained from bulk-tissue, which could be affected by cell type proportion during disease progression. Considering the continuously expanding body of multi-omics data at different levels from more resources encompassing transcriptomics, proteomics, metabolomics, and epigenetics from both AD-affected brain regions and peripheral tissues such as whole blood, and the demonstrated capability for our approach to disentangle highly convoluted data, we anticipate these methods will offer broad utility in advancing our understanding of complex human diseases such as AD and offering new avenues for biomarker and drug target discovery for clinical use.
Materials and methods
RNA-seq datasets from AMP-AD consortium
All RNA-seq data were obtained from the AD Knowledge portal through Synapse (https://www.synapse.org/). Demographic information for each of the cohort (ROSMAP, MAYO, and MSBB) sampled in the RNA-seq study is reported in Supplementary Table 1. The processed, normalized data were obtained for each cohort, respectively, from the harmonized, uniformly processed RNA-seq data set across the three largest AMP-AD contributed studies (syn21241740). In the ROSMAP studies (syn3219045 and syn22695346), the brain tissue samples were collected from three different brain regions: dorsolateral prefrontal cortex (DLPFC, n = 1092), posterior cingulate cortex (PCC, n = 647), and head of caudate nucleus (HCN, n = 717). In Mayo RNA-seq study (syn5550404), brain tissue samples were collected from cerebellum (CER, n = 246) and temporal cortex (TCX, n = 259). The MSBB study (syn3159438) has over 1000 samples from the Mount Sinai/JJ Peters VA Medical Center Brain Bank, which were sequenced from 312 subjects from four brain regions including the frontal pole (FP, Brodmann area (BM) 10, n = 304), inferior frontal gyrus (IFG, BM 44, n = 297), superior temporal gyrus (STG, BM 22, n = 324) and parahippocampal gyrus (PHG, BM 36, n = 308), respectively. The harmonized processing of all the data from the three cohorts was accomplished using a common workflow (https://sage-bionetworks.github.io/sageseqr/).
The conditional quantile normalized46 log counts per million reads (logCPM) values from each data set (syn26967453, syn27024965, and syn27068756) were used in all the subsequent analyses.
Phenotypical data
The detailed definitions of phenotypic measurements used in the study, including clinical evaluation and postmortem neuropathological quantifications, together with their possible values of all three cohorts are reported in Supplementary Table 2.
All the clinical and pathological data for the ROSMAP cohort were obtained from the Rush Alzheimer’s Disease Center Research Resource Sharing Hub (https://www.radc.rush.edu/), upon approval of data-usage agreement. The details of the variables can be found in Supplementary Table 2 with the characterizations summarized in ref. 47.
For MAYO13 and MSBB cohorts14, subject clinical and pathological data were obtained from Synapse (syn27000373 and syn23277389 for Mayo samples and syn27000243 for all the MSBB samples). For MAYO cohort, the following data were used in the linear regression to validate the two phenotypes: Target variables: Braak = Braak stage; Thal = Thal amyloid stage. Dependent variables: ageDeath = age at death; sex = sex; race = racial group; apoe4 = apoe4 allele count; RIN = RNA integrity number; PMI = post-mortem interval. For MSBB cohort, the following data were used in the linear regression to validate the four phenotypes: Target variables: Braak = Braak stage; plaqueMean = plaque mean density; CDR = clinical dementia rating; CERAD = CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) score. Dependent variables: ageDeath = age at death; sex = sex; race = racial group; apoe4 = apoe4 allele count; RIN = RNA integrity number; PMI = post-mortem interval. The original CERAD score in the MSBB cohort was defined as: 1, Normal; 2, Definite Alzheimer’s disease; 3, Probable Alzheimer’s disease; 4, Possible Alzheimer’s disease. They were recoded to be semi-quantitative as follows: 1, Definite Alzheimer’s disease; 2, Probable Alzheimer’s disease; 3, Possible Alzheimer’s disease; and 4, Normal, to be consistent with the notion used in the ROSMAP cohort. APOE genotypes were obtained from whole genome sequencing harmonization (syn11707420) whenever it is missing in the original meta data file. Semiquantitative measurements (e.g. Braak stage, Thal phase, CERAD score, etc) were treated as quantitative. Quantitative measurements (e.g. amyloid or tangles) were log transformed.
Deep learning of the transcriptome from three brain regions in ROSMAP cohort
We extended the ML framework from our previous publication4 and applied the framework to the datasets in ROSMAP cohort for the three brain regions respectively. The framework consists of three major components: (1) supervised classification (deep learning); (2) unsupervised dimensionality transformation (UMAP); and (3) model interpretation (by SHAP explainer) for gene co-expression analysis (Fig. 1). Substantial extensions and improvements were made, specifically: (1) Instead of running a package designed for identifying multiple subtypes in cancer, we built a more generic classification model for each of the datasets, thus ensuring an easy-to-use DNN training process; (2) dimensionality transformation of the last layer of the predicted manifold embedding (previously the second last layer was used); (3) in previous work4, the feature weights from input to second layer were used to interrogate the importance of the contributing features. In this research, we leveraged SHAP explainer which comprehensively assessed the marginal contribution from one feature given the condition of all remaining features. SHAP is proved to be effective to disentangle the interplays among the features for improved interpretability48; and (4) In this research, we applied a multiscale embedded gene-coexpression analysis method (MEGENA) for in-depth network analysis.
For the DNN classification, we trained multi-layer perceptron (MLP) models using neuropathologically confirmed AD subjects and normal controls (CN), the two termini of the AD spectrum to maximally differentiate the two groups. Data collected from ROSMAP including cogdx, braaksc and ceradsc were used to define the class label for AD, control (CN) and OTHER groups:
-
1.
AD: cogdx = 4, braaksc ≥4 and ceradsc ≤2;
-
2.
CN: cogdx = 1, braaksc ≤3 and ceradsc ≥3;
-
3.
OTHER: all the other samples (including subjects of mild cognitive impairment, with mixed diagnoses with other dementias and comorbidities such as cardiovascular pathologies, etc.).
Total number of samples used in the training for each model, their diagnosis classifications and split by training/test sets for a fivefold cross validation, and the model configurations are reported in Supplementary Table 3. For the DLPFC dataset, visualization of gene expression of sex chromosomes (XIST as X-chromosome marker and UTY as Y-chromosome marker) revealed two samples with high expressions of both markers, and they were subsequently removed from the modeling process. The input The conditional quantile normalized46 log counts per million reads (logCPM) values from AD and CN samples were feature-wise z-transformed, and the mean and standard deviation (SD) of each feature were used for scaling for other samples/datasets used in validations.
After tuning the hyperparameters with learning rate ranging from [10–5, 10–4, 10–3, 10–2] and epoch ranging from [200, 300, 400, 500], for all the final model training we adopted the Adam optimizer49 with a learning rate set to 0.0001 (10–4). The training consisted of 300 epochs and for the purpose of selecting the most suitable model, a model checkpoint callback was used to store the weights of the best model based on the validation accuracy. The performance evaluation of these models was carried out using a comprehensive set of classification metrics, including test accuracy, sensitivity/recall, specificity, F1 score, area under the receiver operating characteristic curve (AUC) and precision50.
After each MLP model was trained, the data samples labeled as OTHER were scaled using the mean and SD values from the AD-CN samples used in the training process and added back to the dataset. Eventually, a manifold representation was generated through a forward pass utilizing the previously trained neural networks to all the samples of the whole cohort. To visualize the manifold, the dimensions of resulting representation of the final layer was further reduced to a three-dimensional UMAP space, by the “umap.UMAP.fit_transform” function in python, with the following parameters: n_components = 3, metric = “Euclidean”. Other parameters are tuned to be specific for each brain region as reported in Supplementary Table 3, together with the parameters we used for other deep/machine learning methods for the same task for comparison.
Applying the deep learning model to external data sets (MAYO/MSBB)
The harmonized, uniformly processed RNA-seq data sets from MAYO and MSBB were first sorted by the same gene order as the input data set of ROSMAP. Batch effects were then removed by the ComBat51 function in the R package sva52, on the common set of genes between the reference data matrix (ROSMAP) and the target data matrix (MAYO/MSBB) respectively using the empirical Bayes methods as described in ref. 51. The batch corrected input expression matrix subsequently was transformed to Z-score by scaling to the training dataset in the ROSMAP deep learning model. A manifold representation was obtained for all the samples in each cohort by forward pass of the trained network. Trajectories were obtained by carrying out the UMAP transformation of the existing embedding model from each set of ROSMAP data, by the “umap.UMAP.fit_transform” function using the same parameters as each model in python.
Model validation by correlation with phenotypic data
We derived an index, the Severity Index (SI), for staging the progression of AD from normal control to terminal disease based on the pseudotemporal trajectory in the UMAP embedding. SI was derived for each sample by applying the method of inferring pseudotimes for single-cell transcriptomics from the function ‘slingPseudotime’ as implemented in the R package Slingshot53. To evaluate the models built from three brain regions respectively, SIs were then linearly correlated with all the AD clinical and pathological biomarkers individually, in both the ROSMAP cohort and the two other independent cohorts (MAYO/MSBB), including the covariates PMI, RIN, apoe4 allele count, age at death, sex, race, and educ (ROSMAP only), using the following linear regression model:
For ROSMAP, we also included the following non-AD neuropathological measurements as additional covariates: r_pd, r_stroke, dlbdx, hspath_typ, arteriol_scler, tdp_st4, caa_4gp, cvda_4gp2, ci_num2_gct, and ci_num2_mct. Their detailed definitions can be found in Supplementary Table 2 and data collection is reported in Tasaki et al.54. The target dependent variables (biomarkers) are the AD neuropathological and clinical measurements, also reported in Supplementary Table 2. In MSBB cohort, duplicated samples were sequenced for the same individuals in some regions, and only one sample with the lowest rRNA rate was kept in the linear regression model. Regression statistics were obtained by the ‘lm’ function in R. The proportion of variance explained (PVE) for each predictor was obtained from the incremental sums of squares table by the ‘anova’ function in R on the model, using the order as reported.
Model interpretation
We applied the SHAP (SHapley Additive exPlanations)55 tool to interpret the MLP model trained for each brain region, for extracting gene features that explain the classification. The following steps were performed to derive a quantitative SHAP metric for each feature:
-
1.
The SHAP values for each feature \({x}_{i}\) were computed using the “shap.Explainer” function from the SHAP library in Python.
-
2.
For each feature in each sample, calculate the SHAP values for two diagnosis classes separately:
-
a.
Two sets of SHAP values (\({{SHAP}}_{{AD}}({x}_{i})\) and \({{SHAP}}_{{CN}}({x}_{i})\)) were derived separately for AD and CN samples, where i ∈ [1,…,Nfeatures], Nfeatures represents the total number of features.
-
b.
\({{SHAP}}_{{AD}}({x}_{i})\) represents the SHAP values for all AD samples for the \({i}^{{th}}\) feature.
-
c.
\({{SHAP}}_{{CN}}({x}_{i})\) represents the SHAP values for all CN samples for the \({i}^{{th}}\) feature.
-
a.
-
3.
For each class, compute the mean SHAP values for each feature. We computed the mean SHAP values for each feature by averaging the individual SHAP values across the samples in the AD and CN classes separately:
-
a.
For each feature \({x}_{i},\) computed the mean SHAP value for AD samples as Eq. (2), where \({N}_{{AD}}\) represents the total number of AD samples in the dataset.
$${{SHAP}\text{'}}_{{AD}}\left({x}_{i}\right)=\frac{1}{{N}_{{AD}}}\mathop{\sum }\limits_{j=1}^{{N}_{{AD}}}{{SHAP}}_{{AD}}({x}_{i},j)$$(2) -
b.
For each feature \({x}_{i},\) computed the mean SHAP value for CN samples as Eq. (3), where \({N}_{{CN}}\) represents the total number of CN samples in the dataset.
$${{SHAP}\text{'}}_{{CN}}\left({x}_{i}\right)=\frac{1}{{N}_{{CN}}}\mathop{\sum }\limits_{j=1}^{{N}_{{CN}}}{{SHAP}}_{{CN}}({x}_{i},j)$$(3) -
a.
-
4.
Compute the mean absolute SHAP Value of each feature for the model:
$${SHAP}({model})=\frac{1}{2}\left(|{{SHA}{P}^{{\prime} }}_{{AD}}\left({x}_{i}\right)|+|{{SHA}{P}^{{\prime} }}_{{CN}}\left({x}_{i}\right)|\right)$$(4) -
5.
Run an ensemble of 100 models, each with a different random seed and the same hyperparameters on all the AD-CN samples, with the goal of sampling a wide range of feature relevance across different model iterations. The mean SHAP values of all 100 models were then evaluated as the final SHAP value for each feature:
Eventually, we sorted the features by their final SHAP values calculated in Eq. (5) in descending order to identify the most influential features for the classification model while making predictions. The cutoff to extract features making significant contributions to the classification was set as 1.5 * interquartile range (IQR) above the third quartile of the distribution ( > q0.75 + 1.5*IQR). These gene features were eventually identified as the “index genes” in each brain region that explains AD vs control classification.
Gene network and functional analysis
Gene co-expression networks were built by MEGENA56, based on the expression values of the “index genes” from the residualized counts data after regressing out significant covariates, with three covariates (diagnosis, age and sex) added back, for the three brain regions (syn31141704) respectively. Common networks between those modules from different brain regions were identified by CoDiNA57, using the correlation coefficients of significant correlation pairs derived by MEGENA in each region. Common networks in another brain region were deemed to overlap with a DLPFC submodule by Fisher’s Exact test, using the union of the IGs from the two regions as the background.
Functional and cell type enrichment analysis for each module identified in DLPFC networks was performed using Metascape58, which uses a hypergeometric test and Benjamini-Hochberg P value correction to identify ontology terms that contain a statistically greater number of genes in common with an input list than expected by chance, using the whole transcriptome as background. Statistically significant enriched terms based on Gene Ontology59, KEGG60, Reactome61 and MSigDB62 were clustered based on Kappa-statistical similarities among their gene memberships. A 0.3 kappa score was applied as a threshold to identify enriched terms.
All the gene enrichment analyses were performed in R (version 4.0.0)63 by Fisher’s Exact Test (FET) on the overlaps between the gene sets of interest, using the whole transcriptome as background.
Transcriptional gene regulators were identified by Trena64, using the expression profile of the neuron co-expression module in DLPFC in combination with those transcription factors with known motifs65,66. Reprocessing of all the ROSMAP, MAYO, and MSBB data to include combined ZFX + ZFY gene counts was accomplished following the same workflow as previous reported (https://sage-bionetworks.github.io/sageseqr/), excluding four samples in ROSMAP and 14 samples from two subjects in MSBB with ambiguous sex markers expression. The conditional quantile normalized log counts per million reads (logCPM) values were used in the analyses. The transcription factor binding site on the target gene in excitatory neuron was identified by GET67, an interpretable foundation model designed to uncover regulatory grammars across 213 human fetal and adult cell types (https://huggingface.co/spaces/get-foundation/getdemo).
Data availability
The datasets analyzed during the current study are available on AMP-AD knowledge portal with synapse accession ID included in the main text. All the clinical and pathological data for the ROSMAP cohort were obtained from the Rush Alzheimer’s Disease Center Research Resource Sharing Hub (https://www.radc.rush.edu/).
Code availability
The underlying code for this study is available upon request from the corresponding author.
References
Golde, T. E. Alzheimer’s disease - the journey of a healthy brain into organ failure. Mol. Neurodegener. 17, 18 (2022).
Vadapalli, S., Abdelhalim, H., Zeeshan, S. & Ahmed, Z. Artificial intelligence and machine learning approaches using gene expression and variant data for personalized medicine. Brief Bioinform 23, https://doi.org/10.1093/bib/bbac191 (2022).
Santorsola, M. & Lescai, F. The promise of explainable deep learning for omics data analysis: Adding new discovery tools to AI. N. Biotechnol. 77, 1–11 (2023).
Wang, Q. et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Commun. 4, https://doi.org/10.1093/braincomms/fcab293 (2022).
Beebe-Wang, N. et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nat. Commun. 12, 5369 (2021).
Shakir, M. N. & Dugger, B. N. Advances in deep neuropathological phenotyping of Alzheimer disease: Past, present, and future. J. Neuropathol. Exp. Neurol. 81, 2–15 (2022).
Sundararajan, M, Taly, A. & Yan, Q. Axiomatic attribution for deep networks. Proceedings of the 34th International Conference on Machine Learning, Vol. 70, 3319–3328 (JMLR.org, 2017).
Hodes, R. J. & Buckholtz, N. Accelerating medicines partnership: Alzheimer’s disease (AMP-AD) knowledge portal aids Alzheimer’s drug discovery through open data sharing. Expert Opin. Ther. Targets 20, 389–391 (2016).
Greenwood, A. K. et al. The AD knowledge portal: A repository for multi-omic data on Alzheimer’s disease and aging. Curr. Protoc. Hum. Genet. 108, e105 (2020).
Bennett, D. A. et al. Overview and findings from the rush Memory and Aging Project. Curr. Alzheimer Res. 9, 646–663 (2012).
Bennett, D. A., Schneider, J. A., Arvanitakis, Z. & Wilson, R. S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 9, 628–645 (2012).
Rozemberczki, B. et al. The shapley value in machine learning. 31st International Joint Conference on Artificial Intelligence and the 25th European Conference on Artificial Intelligence, 5572–5579 (2022).
Allen, M. et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089 (2016).
Wang, M. et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci. Data 5, 180185 (2018).
Yu, H., Samuels, D. C., Zhao, Y. Y. & Guo, Y. Architectures and accuracy of artificial neural network for disease classification from omics data. BMC Genomics 20, 167 (2019).
Arango-Lasprilla, J. C. et al. Cortical and subcortical diseases: do true neuropsychological differences exist?. Arch. Clin. Neuropsychol. 21, 29–40 (2006).
Leech, R. & Sharp, D. J. The role of the posterior cingulate cortex in cognition and disease. Brain 137, 12–32 (2014).
Tukey, J. W. Exploratory Data Analysis. (Addison-Wesley, 1977).
Merluzzi, A. P. et al. Neurodegeneration, synaptic dysfunction, and gliosis are phenotypic of Alzheimer dementia. Neurology 91, e436–e443 (2018).
Monterey, M. D., Wei, H., Wu, X. & Wu, J. Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front Neurol. 12, 619626 (2021).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet https://doi.org/10.1038/s41588-022-01024-z (2022).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290 .e1217 (2017).
Haure-Mirande, J. V., Audrain, M., Ehrlich, M. E. & Gandy, S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol. Neurodegener. 17, 55 (2022).
Griciuc, A. & Tanzi, R. E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 34, 228–236 (2021).
Gedam, M. et al. Complement C3aR depletion reverses HIF-1alpha-induced metabolic impairment and enhances microglial response to Abeta pathology. J. Clin. Invest. 133, https://doi.org/10.1172/JCI167501 (2023).
Litvinchuk, A. et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 100, 1337–1353.e1335 (2018).
Wang, X. L. & Li, L. Cell type-specific potential pathogenic genes and functional pathways in Alzheimer’s Disease. BMC Neurol. 21, 381 (2021).
Sun, N. et al. Human microglial state dynamics in Alzheimer’s disease progression. Cell 186, 4386–4403.e4329 (2023).
Hou, J., Chen, Y., Grajales-Reyes, G. & Colonna, M. TREM2 dependent and independent functions of microglia in Alzheimer’s disease. Mol. Neurodegener. 17, 84 (2022).
Mathys, H. et al. Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer’s disease pathology. Cell 186, 4365–4385.e4327 (2023).
Schneider-Gädicke, A., Beer-Romero, P., Brown, L. G., Nussbaum, R. & Page, D. C. ZFX has a gene structure similar to ZFY, the putative human sex determinant, and escapes X inactivation. Cell 57, 1247–1258 (1989).
Tukiainen, T. et al. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017).
Godfrey, A. K. et al. Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 30, 860–873 (2020).
Aisen, P. S. et al. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimers Res Ther. 9, 60 (2017).
Goodfellow, I., Bengio, Y. & Courville, A. Deep Learning. (MIT Press, 2016).
Wang, M. et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 8, 104 (2016).
Wang, S. et al. TREM2 drives microglia response to amyloid-beta via SYK-dependent and -independent pathways. Cell 185, 4153–4169 .e4119 (2022).
Ennerfelt, H. et al. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 185, 4135–4152 .e4122 (2022).
Jorfi, M., Maaser-Hecker, A. & Tanzi, R. E. The neuroimmune axis of Alzheimer’s disease. Genome Med. 15, 6 (2023).
Son, M. et al. Evidence for C1q-mediated crosslinking of CD33/LAIR-1 inhibitory immunoreceptors and biological control of CD33/LAIR-1 expression. Sci. Rep. 7, 270 (2017).
Guo, L. et al. Sex specific molecular networks and key drivers of Alzheimer’s disease. Mol. Neurodegener. 18, 39 (2023).
San Roman, A. K. et al. The human Y and inactive X chromosomes similarly modulate autosomal gene expression. Cell Genomics https://doi.org/10.1016/j.xgen.2023.100462 (2023).
San Roman, A. K. et al. The human inactive X chromosome modulates expression of the active X chromosome. Cell Genom. 3, 100259 (2023).
Gauthreaux, K. et al. Different cohort, disparate results: Selection bias is a key factor in autopsy cohorts. Alzheimers Dement 20, 266–277 (2024).
Hansen, K. D., Irizarry, R. A. & Wu, Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216 (2012).
Bennett, D. A. et al. Religious orders study and rush memory and aging project. J. Alzheimer’s. Dis. 64, S161–S189 (2018).
Chen, H., Lundberg, S. M. & Lee, S. I. Explaining a series of models by propagating Shapley values. Nat. Commun. 13, 4512 (2022).
Kingma, D. P. & Ba, L. J. Adam: A Method for Stochastic Optimization. 3rd International Conference for Learning Representations, San Diego, CA, USA, (2015).
Powers, D. From precision, recall and F-factor to ROC, informedness, markedness & correlation. Mach. Learn. Technol 2, 37–63 (2008).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Tasaki, S., Gaiteri, C., Mostafavi, S., De Jager, P. L. & Bennett, D. A. The Molecular and Neuropathological Consequences of Genetic Risk for Alzheimer’s Dementia. Front Neurosci. 12, 699 (2018).
Shah, J. et al. Interpreting deep learning model predictions using Shapley values. INFORMS Annual Meeting (Anaheim, CA, USA, 2021).
Song, W. M. & Zhang, B. Multiscale embedded gene co-expression network analysis. PLoS Comput Biol. 11, e1004574 (2015).
Morselli Gysi, D. et al. Whole transcriptomic network analysis using Co-expression Differential Network Analysis (CoDiNA). PLoS One 15, e0240523 (2020).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Gene Ontology, C. et al. The Gene Ontology knowledgebase in 2023. Genetics 224, https://doi.org/10.1093/genetics/iyad031 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Gillespie, M. et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50, D687D692 (2022).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Team, R. C. R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria (2016).
Ament, S., Shannon, P. & Richards, M. TReNA: Fit transcriptional regulatory networks using gene expression, priors, machine learning. R package version 1.21.0 (2022).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020).
Kulakovskiy, I. V. et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 46, D252–D259 (2018).
Fu, X. et al. GET: a foundation model of transcription across human cell types. bioRxiv https://doi.org/10.1101/2023.09.24.559168 (2023).
Acknowledgements
The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). The RNA-seq Harmonization Study was supported by the NIA grants U01AG046152, U01AG046170, U01AG046139 and U24AG061340. Study data in ROSMAP cohort were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, IL, USA. Data collection was supported through funding by NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNA-seq), R01AG17917 (MAP), R01AG36836 (RNA-seq), the Illinois Department of Public Health and the Translational Genomics Research Institute. Additional phenotypic data were requested at https://www.radc.rush.edu. The data for MSBB cohort were generated from post-mortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr Eric Schadt from Mount Sinai School of Medicine. The MSBB study was led by Dr Nilufer Ertekin-Taner and Dr Steven G. Younkin, Mayo Clinic, Jacksonville, FL, USA, using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer’s Disease Research Center and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50AG016574, R01AG032990, U01AG046139, R01AG018023, U01AG006576, U01AG006786, R01AG025711, R01AG017216, R01AG003949, NINDS grant R01NS080820, CurePSP Foundation and support from Mayo Foundation. Study data include samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the NINDS (U24NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the NIA (P30AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. The authors acknowledge Research Computing at Arizona State University for providing computing resources that have contributed to the research results reported within this paper. Funding: This work is supported by National Institute on Aging (NIA) grant P30AG072980, the Arizona Department of Health Services (ADHS), and the state of Arizona (ADHS Grant No. CTR057001). Q.W. and B.R. are supported in part by NIA grant U01AG061835. J.S., T.W., and Y.S. are also supported in part by NIA grant RF1AG073424. Q.W. also receives support from NIA grant R21AG091567. The funding sources did not play a role in study design, the collection, analysis, and interpretation of data, writing of the report; or in the decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Trivedi, M.R., Joshi, A.M., Shah, J. et al. Interpretable deep learning framework for understanding molecular changes in human brains with Alzheimer’s disease: implications for microglia activation and sex differences. npj Aging 11, 66 (2025). https://doi.org/10.1038/s41514-025-00258-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41514-025-00258-5
This article is cited by
-
Targeting Microglial Activation to Modulate Neuroinflammation in Alzheimer’s Disease
NeuroMolecular Medicine (2025)
