
Abstract
Aneuploid cells are known to increase with age. Previously, we demonstrated an increased number of aneuploid fibroblasts isolated from aged mice due to chromosomal instability (CIN), which is caused by oxidative stress. It is unclear whether this phenomenon also occurs in human cells, which are more resistant to oxidative stress than mouse cells. Here, we found that fibroblasts from aged individuals exhibited an increase in aneuploid cells. The frequency of chromosome missegregation and micronuclei increased in these cells, indicating CIN. A DNA fiber assay revealed the presence of replication stress, accompanied by an increase in 53BP1 nuclear bodies and ultrafine bridges. Increased levels of reactive oxygen species derived from mitochondria, along with reduced mitochondrial membrane potential, imply that these cells experienced oxidative stress due to mitochondrial functional decline. Antioxidant treatment reduced the frequency of chromosome missegregation and micronuclei, suggesting that oxidative stress causes CIN. Oxidative stress also causes replication stress, which precedes CIN. Spindle microtubules were stabilized in fibroblasts from aged individuals, which was alleviated by antioxidant treatment. Taken together, these findings suggest that aging-related CIN in human fibroblasts is caused by oxidative stress associated with mitochondrial dysfunction, which induces replication stress that in turn causes CIN through microtubule stabilization. Although human fibroblasts are more resistant to the ambient oxygen environment than mouse fibroblasts, our findings showed that they undergo oxidative stress that causes CIN with age in a manner similar to mouse fibroblasts, revealing a conserved phenomenon in mammalian cells.
Similar content being viewed by others


Introduction
Genomic instability, a key characteristic of aging, manifests as a gradual build-up of genetic damage over an organism’s lifespan. This damage encompasses various forms, such as point mutations, translocations, gains and losses of chromosomes, telomere shortening, and gene disruption1. Among them, chromosomal gains and losses, known as aneuploidy, is known to increase with age2. While chromosome aberration in oocytes associated with maternal age is a well-known age-related chromosome abnormality3, an increase in aneuploid cells in somatic cells with aging has also been reported4. For example, it is known that Y chromosome is frequently lost in males with increasing age5,6. In the brain, mosaic aneuploidy increases with age and has been associated with neurodegenerative diseases7. With increasing age, mice also exhibit an increase in chromosome abnormalities, including aneuploidy8. Cancer is also characterized by genomic instability9, and aneuploidy is observed in most cancer cells10. Chromosomal instability (CIN), a state of frequent chromosome missegregation, typically accompanies aneuploidy in cancer cells11,12,13.
Recently, we reported that primary fibroblasts isolated from aged mice exhibit aneuploidy and CIN14. Fibroblasts from aged mice showed elevated levels of reactive oxygen species, along with a decline in mitochondrial function, suggesting oxidative stress. The rates of chromosome missegregation in cells from aged mice were reduced by treatment with antioxidants. We identified replication stress as a cause of CIN in aged mouse cells, which may promote CIN through microtubule stabilization. Our findings revealed the age-related onset of CIN and indicated a novel link between oxidative stress and CIN during aging. However, it is not known whether the same phenomenon occurs in human cells. While primary human fibroblasts proliferate for 50 or more population doublings under atmospheric oxygen environment, primary mouse fibroblasts rapidly enter a period of declining growth rate15, suggesting that mouse cells are more susceptible to oxidative stress than human cells16. In contrast to human cells that stop dividing with a normal karyotype in most of the cases17, mouse cells undergo immortalization with chromosomal abnormalities after the period of slowed growth18, raising a question on the assumption that age-related aneuploidy occurs via a common pathway in mouse and human cells.
To study aneuploidy and CIN with age in human cells, we observed fibroblasts isolated from young and aged individuals. We found that fibroblasts isolated from aged individuals exhibit aneuploidy and CIN. Similar to fibroblasts from aged mice, fibroblasts from aged individuals are under oxidative stress, which causes CIN through replication stress and microtubule stabilization. Our data show that the link between oxidative stress and age-related CIN is conserved between mouse and human cells despite the significant difference in their sensitivity to oxidative stress.
Results
Human fibroblasts isolated from aged individuals exhibit CIN
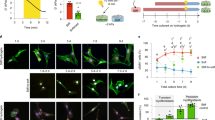
To study how chromosomal stability changes with age in human cells, we obtained human fibroblasts from the NIA Aging Cell Repository at the Coriell Institute for Medical Research. We selected cells derived from four young males and four aged males (Table 1). Of these, cells from two young individuals and four aged individuals were skin fibroblasts. We also selected two lung fibroblasts from fetal tissues to see the difference depending on the cellular origins. We first observed the proliferation of these cells. As shown in Fig. 1A and Supplementary Fig. 1A, the proliferation rate of fibroblasts from aged individuals was slower than that of fibroblasts from young individuals. In subsequent experiments, we used fibroblasts at a population doubling level (PDL) ranging from 17 to 35, during which the cells grew at a constant rate. We did not use cells at PDL 36 or higher, where cell growth was markedly suppressed. We then examined the chromosome numbers of these cell lines in metaphase spreads. To enrich mitotic cells, we treated cells with RO-3306, a CDK1 inhibitor, which arrest cells in G2 phase, and then washed out it to release cells synchronously in mitosis19. We found a significant increase in metaphases with abnormal chromosome numbers in fibroblasts from aged individuals (Fig. 1B), in line with the notion that aneuploid cells increase with age. Both the number of metaphases with fewer and more than 46 chromosomes increased, while the average number of chromosomes remained unchanged (Supplementary Fig. 1B, C), showing that despite the marked increase in aneuploid cells from aged individuals, the modal number did not change. To study whether CIN exists in fibroblasts from aged individuals as a cause of aneuploidy, we first observed the proportion of interphase cells with micronuclei, which are formed as a result of chromosome missegregation, in fixed cell samples. We found that fibroblasts from aged individuals showed a higher rate of the appearance of micronuclei (Fig. 1C). We next quantified the percentage of chromosome missegregation in fixed cell samples. Fibroblasts from aged individuals showed higher rates of lagging chromosomes and chromosome bridges than those from young individuals (Fig. 1D). These data suggest that fibroblasts from aged individuals exhibit CIN. We found that skin and lung fibroblasts from young individuals showed comparable levels of micronucleation and chromosome missegregation rates. These cells exhibited a similar phenotype in all subsequent experiments, so they were analyzed together. To study further CIN in fibroblasts from aged individuals, we performed live cell imaging and observed chromosome missegregation during mitosis. We confirmed that chromosome missegregation, both lagging chromosomes and chromosome bridges, were increased in fibroblasts from aged individuals (Fig. 1E, F). The rates of chromosome missegregation were higher in live cell imaging than in fixed cell samples. This difference may be due to the difference in detection sensitivity between continuous and snapshot observations, as well as the toxic effects of SiR-DNA, an intercalating dye used to visualize chromosomes in live cell imaging. We found that the total duration of prometaphase (from nuclear envelope breakdown (NEBD) to chromosome alignment) as well as the duration of mitosis (from NEBD to chromatin decondensation) were increased in fibroblasts from aged individuals (Fig. 1G). To see whether the activity of the spindle assembly checkpoint (SAC), which delays anaphase onset until all the chromosomes attach to spindle microtubules, differ in fibroblasts from young and aged individuals, we compared the duration of mitosis in the presence of nocodazole, a microtubule destabilizer, or Eg5 inhibitor III, which inhibits the activity of a motor protein Eg5, both of which arrest cells in mitosis by activating the SAC. As shown in Supplementary Fig. 1D, the duration of mitotic arrest was comparable between fibroblasts from young and aged individuals both in the presence of nocodazole and Eg5 inhibitor III, showing that the SAC activity is not altered in fibroblasts from aged individuals. We also observed the cellular response after prolonged mitotic arrest. When cells are arrested in mitosis for a prolonged period, they either die in mitosis (mitotic cell death) or enter the next cell cycle (mitotic slippage). It is known that whether cells undergo mitotic cell death or mitotic slippage after prolonged mitotic arrest varies not only between cell lines, but also within a population of the same cell line20. We found that the rate of mitotic slippage did not differ between fibroblasts from young and aged individuals in the presence of nocodazole as well as in the presence of Eg5 inhibitor III (Supplementary Fig. 1E).

A Growth of fibroblasts isolated from young and aged individuals. Repeated-measures ANOVA was used to judge the significance. B Chromosome numbers of fibroblasts isolated from young and aged individuals. 50–105 metaphase chromosome spreads were observed. Representative images are shown. Percentages of aneuploid cells were shown in the right graph. P-value was obtained using the Student’s t-test. C Micronucleation rates of fibroblasts isolated from young and aged individuals. Cells were fixed and stained with DAPI. 504-631 cells were analyzed for each cell. A representative image of a micronucleus is shown (arrowhead). P-value was obtained using the Welch’s t-test. D Chromosome missegregation rates of fibroblasts isolated from young and aged individuals. Cells were treated as in (C). Representative images of a lagging chromosome and a chromosome bridge are shown (arrowheads). 46–66 cells were analyzed for each cell. P-values were obtained using the Student’s t-test. E Mitotic progression of fibroblasts isolated from young and aged individuals. Cells were treated with SiR-DNA and subjected to live cell imaging. Images were taken every 2.5 min for 96 h, and mitotic progression of 22–61 cells for each cell was tracked and displayed in different colors depending on the mitotic phases. Cells that underwent chromosome missegregation are separately shown, and number of cells in each category is indicated. F Chromosome missegregation rates of fibroblasts isolated from young and aged individuals. Rates of lagging chromosomes and chromosome bridges shown in (E) are plotted. P-values were obtained using the Student’s t-test. G Mitotic duration of fibroblasts isolated from young and aged individuals. Time from nuclear envelope breakdown to chromosome alignment (duration of prometaphase) and time from nuclear envelope breakdown to cytokinesis (duration of mitosis) in cells shown in (E) are displayed as box and dot plots. P-values were obtained using the Mann–Whitney U test. In (B), (C), (D), (F), and (G), red, blue, or black-filled circles represent skin fibroblasts, while yellow-filled circles represent lung fibroblasts. Error bars represent S.D. Scale bars: 5 μm.
Since senescent cells are known to increase with age21, we examined their percentage using SPiDER-βGal, a fluorescent marker of senescence22. The percentage of cells positive for the SPiDER-βGal signal increased in fibroblasts from aged individuals, as expected (Supplementary Fig. 1F). Although the percentage of senescent cells was not so high (<5%) even in cells from aged individuals, it may be significant in tissues, considering that the accumulation of senescent cells in tissues is modest23. We also examined the percentage of cells with p16 signal, another marker of senescence24. The percentage of cells with p16 signal increased in fibroblasts from aged individuals, confirming the results of SPiDER-βGal (Supplementary Fig. 1G). Although only a small proportion of cells with micronuclei were positive for SPiDER-βGal or p16 signal, the presence of micronuclei and these signals were significantly correlated in cells from aged individuals, but not in cells from young individuals (Supplementary Fig. 1F, G), implying the relationship between micronucleation and cellular senescence. We also examined whether the appearance of micronuclei is related to the activation of the cGAS-STING pathway, which is an innate-immune pathway activated by cytoplasmic chromatin fragments25. When we observed the expression of IL-6, expression of which is activated by the cGAS-STING pathway, the IL-6 signal was significantly more frequently observed in cells containing micronuclei, although not all the cells with micronuclei were positive for the signal (Supplementary Fig. 1H). It is also known that myofibroblasts, which are differentiated from fibroblasts to facilitate tissue repair, increase in lung fibroblasts from aged individuals26,27. Quantifying the percentage of cells positive for α-smooth muscle actin (α-SMA), a myofibroblast marker, revealed an increase in fibroblasts from aged individuals (Supplementary Fig. 1I). Examining the relationship between micronucleation and differentiation into myofibroblasts revealed no correlation in fibroblasts from both young and aged individuals (Supplementary Fig. 1I), suggesting that micronucleation is unrelated to differentiation to myofibroblasts.
Human fibroblasts isolated from aged individuals are under replication stress
Next, we observed the presence of DNA damage in fibroblasts from young and aged individuals. The number of γ-H2AX foci, which represents the DNA damage response associated with DNA double strand breaks (DSBs)28, was increased in fibroblasts from aged individuals (Fig. 2A). This finding is consistent with previous reports29 and may be caused by both an increased rate of DNA damage and a decreased capacity for DSB repair30. We then observed cells with 53BP1 foci, which represent DSBs under processing31, and found that they were increased in fibroblasts from aged individuals (Fig. 2B). We noticed that these 53BP1 foci were large and few in number, suggesting that they are 53BP1 nuclear bodies formed around DNA lesions generated by replication stress32. It was reported that 53BP1 nuclear bodies are formed in G1 phase as a result of replication stress in the previous cell cycle. Consistent with the idea that fibroblasts from aged individuals are under replication stress, the percentage of cells with ultrafine bridges (UFBs), which are thin DNA threads connecting sister chromatids in anaphase and can result from replication stress33,34,35, was increased in fibroblasts from aged individuals (Fig. 2C). Furthermore, metaphase chromosome spreads showed an increase in chromosome breaks, which are indicative of structural chromosome aberrations that arise from pre-mitotic defects including replication stress, in fibroblasts from aged individuals (Fig. 2D). To directly assess DNA replication, we performed a DNA fiber assay, in which the progression of individual replication forks and the asymmetry between sister replication forks were measured36. Fibroblasts from aged individuals showed significantly slower fork rates than those from young individuals (Fig. 2E, Supplementary Fig. 2A). Furthermore, asymmetric sister fork progression was increased in fibroblasts from aged individuals (Fig. 2F, Supplementary Fig. 2B), indicating impaired replication fork progression. Collectively, these data suggest that fibroblasts from aged individuals are under replication stress. As we previously reported that fibroblasts from aged mice are also under replication stress14, replication stress may commonly increase with age in mammalian cells.

A Number of γ-H2AX foci. Cells were fixed and stained with an antibody against γ-H2AX. Representative images are shown. 15–24 cells for each cell were analyzed and displayed as box and dot plots in the right graph. Data from different cells are shown in different colors. P-value was obtained using the Mann-Whitney U-test. B Rates of cells with 53BP1 nuclear bodies. Cells were fixed and stained with an antibody against 53BP1. Representative images were shown (arrowheads). 95–138 cells for each cell were analyzed. P-value was obtained using the Welch’s t-test. C Rates of cells with ultrafine bridges. Cells were treated with RO-3306 for 12 h, released and fixed after 1 h, and stained with an antibody against PICH (ERCC6L). A representative image is shown (arrowheads). 46–66 cells were counted for each cell. P-value was obtained using the Student’s t-test. D Chromosome breaks were observed in 50–105 metaphase chromosome spreads for each cell. A representative image is shown (arrowhead). Percentages of cells with chromosome breaks were shown in the right graph. P-value was obtained using the Student’s t-test. E Replication fork speed. Cells were sequentially pulse-labeled with digoxigenin-conjugated deoxyuridine and biotin-conjugated deoxyuridine. Representative fibers are shown. Fork speeds were determined through the length of both labels (digoxigenin-dUTPs + biotin-dUTPs), according to the previous report that 1 μm is approximately equal to 3.5 kb of DNA72. 101–138 fibers per cell were quantified and shown as box and dot blots. P-value was obtained using the Mann–Whitney U test. F Replication fork asymmetry. Cells were treated as in (E). Representative fibers are shown. Fork asymmetry was determined by dividing longer biotin-dUTPs label on a bi-directional replication fork with shorter biotin-dUTPs label for 49-79 fibers per cell and shown as box and dot blots. P-value was obtained using the Mann–Whitney U test. In all the graphs, red, blue, or black-filled circles represent skin fibroblasts, while yellow-filled circles represent lung fibroblasts. Error bars represent S.D. DNA was stained with DAPI. Scale bars: 5 μm.
Human fibroblasts isolated from aged individuals are under oxidative stress
In a previous study, we reported that fibroblasts from aged mice are under oxidative stress, which promotes CIN14. To determine whether human fibroblasts from aged individuals are also under oxidative stress, we examined the level of reactive oxygen species (ROS) in human fibroblasts by detecting the CellRox Green signal. We found that the ROS level was higher in fibroblasts from aged individuals (Fig. 3A). The specificity of the signal was confirmed by its reduction when cells were treated with N-acetylcysteine (NAC), an antioxidant37,38 (Fig. 3A). As a major origin of ROS production, we observed the level of superoxide in mitochondria using MitoSOX Red and found that it was higher in fibroblasts from aged individuals (Fig. 3B). The MitoSOX Red signal was reduced when cells were treated with NAC, as expected. The reduction of CellROX and MitoSOX signals by NAC treatment even in cells from young individuals indicates that they are under oxidative stress in an atmospheric oxygen environment. It is known that increased ROS production is caused by mitochondrial functional decline, which is related to aging39,40. We then investigated mitochondrial function by analyzing the mitochondrial membrane potential by comparing the fluorescence signal ratios of aggregated versus monomeric JC-10. As shown in Fig. 3C, the ratio of aggregated to monomeric JC-10 was lower in fibroblasts from aged individuals, indicating that mitochondrial membrane potential was reduced. These data suggest that mitochondrial functional decline results in increased ROS production in fibroblasts from aged individuals, consistent with our previous results in mouse primary fibroblasts14. Interestingly, NAC treatment increased the mitochondrial membrane potential (Fig. 3C), possibly due to the protective effect of NAC on mitochondrial function, as reported previously41,42.

A Detection of ROS in the presence or absence of an antioxidant. Cells were treated with or without NAC for 48 h. Then cells were incubated with CellROX green reagent for 30 min before fixation and observed under microscope. Representative images were shown. Quantification of the ROS level in ROI around nuclei is shown in the right graph. Fluorescence intensity (A.U.) of 17–35 cells for each cell were quantified and displayed as box and dot plots. P-values were obtained using the Steel-Dwass multiple comparison test. B Detection of superoxide in the presence or absence of an antioxidant. Cells were cultured and treated as in (A). Then cells were incubated with MitoSOX Red reagent for 10 min and observed under microscope. Representative images were shown. Quantification of the superoxide level in ROI around cytoplasm is shown in the right graph. Fluorescence intensity (A.U.) of 27–60 cells for each cell was quantified and displayed as box and dot plots. P-values were obtained using the Steel-Dwass multiple comparison test. C Detection of mitochondrial membrane potential in the presence or absence of an antioxidant. Cells were cultured and treated as in (A). Then cells were incubated with JC-10 reagent for 30 min and observed under microscope. The signal representing JC-10 monomeric form (emission at 520 nm), and the signal representing JC-10 aggregated form upon polarization of mitochondrial membrane (emission at 590 nm) are shown. Representative images are shown. Quantification of the level of mitochondrial membrane potential, shown as the ratio of the emission at 590 nm to that at 520 nm, is shown in the right graph. The values of 33-98 cells for each cell were quantified and displayed as box and dot plots. The average ratio of mock-treated cells from young individuals was set as 1. P-values were obtained using the Steel-Dwass multiple comparison test. In all the graphs, red or blue-filled circles represent skin fibroblasts, while yellow-filled circles represent lung fibroblasts. Scale bars: 5 μm.
Antioxidant treatment ameliorates CIN in human fibroblasts
To examine the causal relationship between oxidative stress and CIN in fibroblasts from aged individuals, we observed micronucleation rates in the presence or absence of NAC. As shown in Fig. 4A, the percentage of cells with micronuclei was reduced in the presence of NAC in fibroblasts from both young and aged individuals, although it was not statistically significant in the former, showing that increased oxidative stress is involved in the increase in micronucleation rates. We also quantified the percentage of chromosome missegregation in fixed cell samples in the presence of NAC. We found that the rate of chromosome missegregation was significantly reduced by NAC treatment in fibroblasts from both young and aged individuals (Fig. 4B), further demonstrating that oxidative stress is a cause of CIN in human fibroblasts. The relationship between oxidative stress and CIN was also examined in live cell imaging in the presence or absence of NAC. We confirmed that cells with chromosome missegregation, both lagging chromosomes and chromosome bridges, were reduced in fibroblasts from young and aged individuals in the presence of NAC (Supplementary Fig. 3A, B). The duration of prometaphase and mitosis were reduced when cells were treated with NAC in fibroblasts from young and aged individuals (Supplementary Fig. 3A, C), reflecting improved mitotic progression.

A Micronucleation rates in the presence or absence of an antioxidant. Cells were treated with NAC for 48 h. Then cells were fixed and stained with DAPI. Rates of micronucleation were quantified for 469-788 cells for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. B Chromosome missegregation rates in the presence or absence of NAC. Cells were treated as in (A). Rates of lagging chromosomes and chromosome bridges were quantified for 72-103 cells for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. C Micronucleation rates under 3% O2 in the presence or absence of an antioxidant. Cells were cultured under 3% O2 and treated as in (A). Rates of micronucleation were quantified for 597-744 cells for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. D Chromosome missegregation rates under 3% O2 in the presence or absence of NAC. Cells were cultured as in (C). Rates of lagging chromosomes and chromosome bridges were quantified for 51-157 cells for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. In all the graphs, black-filled circles represent skin fibroblasts, while yellow-filled circles represent lung fibroblasts. Error bars represent S.D. n.s., not statistically significant.
The observation that the chromosome missegregation rate was reduced by NAC treatment even in fibroblasts from young individuals suggests that the atmospheric oxygen environment causes oxidative stress that leads to chromosome missegregation. To verify this possibility, we observed cells cultured under a 3% oxygen concentration, which is closer to in vivo environment. Cell proliferation was comparable between under 3% and 20% oxygen (Supplementary Fig. 3D), suggesting that human fibroblasts are resistant to oxidative stress in terms of cell proliferation, or they have adjusted to 20% oxygen environment during ex vivo culture. We found that micronucleation rates were reduced in fibroblasts from both young and aged individuals than in those cultured under an atmospheric oxygen concentration (Fig. 4C), suggesting that the atmospheric oxygen environment causes CIN through oxidative stress. Reduction of the micronucleation rates by NAC treatment was small and not statistically significant in both cells from young and old individuals (Fig. 4C), supposedly because they were reduced to the near basal levels in the 3% oxygen environment. Chromosome missegregation rates under the 3% oxygen environment were also lower in fibroblasts from both young and old individuals than in those cultured under an atmospheric oxygen environment (Fig. 4D). When these cells were treated with NAC, the rate of lagging chromosome was significantly reduced in cells from aged individuals, but not in cells from young individuals (Fig. 4D). The discrepancy in the effects of NAC treatment on micronucleation and lagging chromosome rates can be explained by the previous finding that only a fraction of cells with lagging chromosomes form micronuclei43. Thus, small differences in lagging chromosome rates may be obscured when measuring micronucleation rates. These data also suggest that fibroblasts from young individuals are under a basal level of oxidative stress in the in vivo oxygen environment that does not affect chromosomal stability, and the mechanisms of protection against excess free radicals in cells derived from aged individuals may be less effective44,45.
Antioxidant treatment ameliorates replication stress that is upstream of CIN in human fibroblasts
We next investigated the relationship between oxidative stress and replication stress in human fibroblasts. The number of γ-H2AX foci was reduced in fibroblasts from young and aged individuals after NAC treatment (Fig. 5A), which is consistent with previous reports that oxidative stress increases γ-H2AX foci46,47. We then observed 53BP1 foci and found that NAC treatment reduced the percentage of cells with 53BP1 nuclear bodies (Fig. 5B). Moreover, NAC treatment reduced the percentage of anaphase cells with UFBs (Fig. 5C), confirming that oxidative stress induces replication stress in human fibroblasts. NAC treatment reduced replication stress not only in fibroblasts from aged individuals, but also in those from young individuals, probably due to oxidative stress caused by the atmospheric oxygen environment, as shown in the previous section.

A Number of γ-H2AX foci in the presence or absence of NAC. Cells were treated with NAC for 48 h, fixed, and stained with an antibody against γ-H2AX. Representative images are shown. 33-49 cells were analyzed for each cell and displayed as box and dot plots in the right graph. P-values were obtained using the Steel-Dwass multiple comparison test. B Rates of cells with 53BP1 nuclear bodies in the presence or absence of NAC. Cells were treated with NAC for 48 h, fixed, and stained with an antibody against 53BP1. 526-770 cells were analyzed for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. C Rates of cells with ultrafine bridges in the presence or absence of NAC. Cells were treated with NAC for 48 h and stained with an antibody against PICH (ERCC6L). DNA was stained with DAPI. 72-103 cells were counted for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. D Rates of cells with 53BP1 nuclear bodies in cells supplemented with or without nucleosides (NUC). Cells were supplemented with nucleosides for 48 h, fixed, and stained with an antibody against 53BP1. 401-890 cells were counted for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. E Rates of cells with ultrafine bridges in cells supplemented with or without nucleosides. Cells were treated as in (D). Then cells were fixed and stained with an antibody against PICH (ERCC6L). DNA was stained with DAPI. 39-70 cells were counted for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. F Micronucleation rates of cells supplemented with or without nucleosides. Cells were treated as in (D). Then cells were fixed and stained with DAPI. 521-741 cells were counted for each cell. P-values were obtained using the Tukey–Kramer multiple comparison test. In all the graphs, red, blue, or black-filled circles represent skin fibroblasts, while yellow-filled circles represent lung fibroblasts. Error bars represent S.D. n.s., not statistically significant. Scale bars: 5 μm.
We then addressed the relationship between replication stress and CIN. It was reported that CIN is induced not only by defects in the mitotic process, but also by DNA damage in interphase caused by replication stress48,49, and we previously reported that replication stress causes CIN in fibroblasts from aged mice14. To validate the causal relationship between replication stress and CIN in human fibroblasts, we supplemented the cells with nucleosides, which partially rescue replication stress49. In fibroblasts from aged individuals, the percentages of cells with 53BP1 nuclear bodies as well as UFBs were reduced (Fig. 5D, E), confirming that replication stress was alleviated by nucleoside treatment. Under this condition, the percentage of cells containing micronuclei was reduced in fibroblasts from aged individuals (Fig. 5F), suggesting that replication stress is a cause of CIN in these cells. ROS level did not change upon nucleoside treatment, indicating that replication stress does not affect oxidative stress (Supplementary Fig. 4).
Microtubules stabilization is a cause of CIN in fibroblasts from aged individuals
For proper chromosome segregation, kinetochores on the sister chromatids must attach to microtubules from opposite spindle poles, referred to as bi-orientation50,51. Correction of erroneous kinetochore-microtubule attachments is crucial to ensure bi-orientation establishment for all chromosomes52. It was recently reported that replication stress causes CIN through stabilization of microtubules, which reduces the correction efficiency of erroneous kinetochore-microtubule attachments53. We previously reported that microtubules are stabilized in fibroblasts from aged mice, suggesting a link between microtubule stabilization and CIN14. To examine the stability of microtubules in human fibroblasts, we quantified the intensity of spindle microtubules resistant to a 10-min cold treatment on ice, which depolymerizes unstable microtubules54. The intensity of spindle microtubules was comparable between fibroblasts from young and aged individuals at room temperature (Supplementary Fig. 5A). However, spindle microtubule intensity after a 10-min cold treatment was significantly higher in fibroblasts from aged individuals (Fig. 6A), suggesting that microtubules were stabilized in these cells. The increase in the intensity of spindle microtubules was alleviated in the presence of NAC, indicating that oxidative stress is responsible for the microtubule stabilization (Fig. 6B). To clarify the relationship between replication stress and microtubule stabilization, we examined microtubule stability when replication stress was reduced by supplementation with nucleosides. We found that nucleoside treatment led to reduced microtubule stability in cells from aged individuals (Fig. 6C, Supplementary Fig. 5B), demonstrating that replication stress is a cause of microtubule stabilization. Next, we addressed whether increasing microtubule dynamics ameliorates CIN. To achieve this, we treated cells with UMK57, a potentiator of MCAK, a motor protein with microtubule depolymerizing activity55. As shown in Fig. 6D and Supplementary Fig. 5C, the intensity of spindle microtubules after a 10-min cold treatment was reduced by UMK57 treatment in cells from aged individuals, as expected. Under this condition, the percentage of cells containing micronuclei was reduced in cells from aged individuals, corroborating that microtubule stabilization is a cause of CIN (Fig. 6E). In contrast, the number of γ-H2AX foci as well as the percentage of cells containing 53BP1 nuclear bodies did not differ in the presence or absence of UMK57 (Fig. 6F, G), showing that replication stress is not downstream of microtubule stabilization. Consistent with the data, the frequency of cells with chromosome fragments did not change after UMK57 treatment (Supplementary Fig. 5D), suggesting that reduction in micronucleation rates was caused by a decrease in erroneous kinetochore-microtubule attachments rather than a decrease in chromosome fragments. Collectively, these data suggest that oxidative stress causes microtubule stabilization, which is a cause of CIN in human fibroblasts with age.

A Cells were synchronized by RO-3306 for 12 h, released and treated with MG132 for 3 h, and either fixed directly or after cold treatment for 10 min. Cells were stained with an antibody against α-tubulin, and relative fluorescence intensity on the spindle at 10 min compared with that at 0 min was quantified for 5–7 cells for each cell. The average value of four young or old individuals at the time 0 is set to 1. P-value was obtained using the Student’s t-test. B Cells were treated with NAC for 48 h. Relative fluorescence intensity was quantified as in (A) for 10-24 cells for each cell. C Cells were supplemented with nucleosides for 48 h. Relative fluorescence intensity was quantified as in (A) for 10-27 cells for each cell. D Cells were treated with UMK57 for 24 h. Relative fluorescence intensity was quantified as in (A) for 10-28 cells for each cell. E Cells were treated with UMK57 for 24 h, fixed, and stained with DAPI. 251-534 cells were counted for each cell. The average value of four young or old individuals at the time 0 is set to 1. F Cells were treated as in (D), fixed, and stained with an antibody against γ-H2AX. 49-70 cells were analyzed for each cell and displayed as box and dot plots. P-values were obtained using the Steel-Dwass multiple comparison test. G Cells were treated as in (D), and fixed and stained with an antibody against 53BP1. 261-416 cells were analyzed for each cell. H Schematic of the mechanism of CIN in fibroblasts from aged individuals suggested in this study. See text for details. In all the graphs, red, blue, or black-filled circles represent skin fibroblasts, while yellow-filled circles represent lung fibroblasts. In (B), (C), (D), (E), and (G), P-values were obtained using the Tukey–Kramer multiple comparison test. Error bars represent S.D. n.s., not statistically significant. Scale bars: 5 μm.
Discussion
In the present study, we found that aneuploid cells increase with age in human fibroblasts. This is due to an increase in chromosome missegregation, namely CIN. Our analysis revealed that oxidative stress caused by increased ROS due to mitochondrial dysfunction is responsible for CIN with age. It was suggested that oxidative stress induces replication stress, which may cause CIN via microtubule stabilization (Fig. 6H). This is similar to the mechanism we previously demonstrated in primary mouse fibroblasts, suggesting that aging-related CIN induced by oxidative stress is a common phenomenon in mammalian cells. The presence of replication stress in fibroblasts from aged individuals was demonstrated not only by established hallmarks (53BP1 nuclear bodies and UFBs), but also by direct measurement of replication fork progression using a DNA fiber assay, which was not shown in our previous study on cells from aged mice14, further strengthening our conclusion. Mitochondrial dysfunction, increased ROS production, and chromosome abnormalities in aged human fibroblasts were previously reported56,57. Analyzing the fibroblasts from the same donor at various ages would be informative to see the trends in the emergence and progression of chromosomal instability with age. It is also an important question for future research whether this phenomenon can be observed in cells other than fibroblasts from both genders. Connection between oxidative stress, replication stress, and premature aging has also been described in previous studies58,59.
In our previous study, we hypothesized that oxidative stress is a cause of CIN in primary mouse fibroblasts, since these cells immediately stopped proliferating under atmospheric oxygen conditions and exhibited an increase in chromosome missegregation14. High-oxygen culture conditions lead to increased ROS production as a byproduct of the electron transport chain in mitochondria60. In contrast, human fibroblasts can proliferate under atmospheric oxygen conditions and are considered resistant to oxidative stress16. This difference is thought to be related to differences in the response to DNA damage, although the details are poorly understood16. Despite the differences in the sensitivity to oxidative stress, our results show that the frequency of chromosome missegregation increases in human fibroblasts under atmospheric oxygen conditions, indicating that the appearance of CIN caused by oxidative stress is a common phenomenon even in human cells that are tolerant to oxidative stress. Mitotic defects caused by oxidative stress were also observed in human cells exposed to hydrogen peroxide61,62. It has been reported that lowering the oxygen concentration promotes cell proliferation and mitotic progression in human cells63,64. These findings suggest that the level of CIN observed in cells cultured under normal atmospheric oxygen conditions is overestimated compared to that in cells under physiological conditions in tissues. We confirmed that the chromosome missegregation rate in human fibroblasts was reduced under 3% oxygen concentration. However, fibroblasts from aged individuals show a higher rate of chromosome missegregation even under a 3% oxygen environment, which is consistent with previous reports of an increase in aneuploid cells with age2, and may suggest a causal relationship between oxidative stress and CIN with age in human cells. It is of note that although our data suggest that oxidative stress caused by elevated ROS is a consequence of mitochondrial functional decline with age, elevated ROS itself is also a cause of aging and senescence65, Cells isolated from aged individuals already have elevated ROS levels and reduced plasticity in response to stress, including oxidative stress, and this may be the cause of increased susceptibility to oxidative stress induced in culture at 20% O2.
NAC treatment improved all the phenotypes observed in fibroblasts from aged individuals we found in this study, including ROS levels, replication stress, and CIN, indicating that oxidative stress causes these phenotypes. Intriguingly, NAC treatment even improved mitochondrial function, as evidenced by an increase in mitochondrial membrane potential, as reported previously41,42, which would further contribute to reducing ROS levels. We also found that microtubule stabilization was ameliorated by NAC treatment, which was not shown in our previous study14, confirming the causal relationship between oxidative stress and microtubule stabilization. However, NAC treatment did not improve the parameters in fibroblasts from aged individuals to the level comparable to those in fibroblasts from young individuals, suggesting that oxidative stress does not explain all the changes seen in fibroblasts from aged individuals. A previous report showed a correlation between increased chromosome missegregation in fibroblasts of aged individuals and senescence-associated secretory phenotype (SASP), which is typically seen in senescent cells, and the downregulation of FoxM1, a transcription factor that plays a role in mitotic gene expression66. The relationship between FoxM1 repression and oxidative stress in the development of CIN requires further investigation.
As a cause of CIN, we found that microtubules were stabilized in fibroblasts from aged individuals, which was downstream of replication stress, as reported previously53. Microtubule stabilization in fibroblasts from elderly people and restoration of CIN by increasing microtubule dynamics was also observed in a previous study67. Replication stress leads to an increase in erroneous kinetochore-microtubule attachments due to microtubule stabilization, as well as chromosome fragmentation due to incomplete replication, resulting in CIN (Fig. 6H).
What are the consequences of the age-related CIN? Oxidative stress and the DNA damage it causes are known to induce cellular senescence68, which is one of the hallmarks of aging and related to aging-related disorders1. It has also been suggested that aneuploidization, resulting from mitotic defects in cells from elderly people, triggers the full senescence phenotype66. CIN is also known not only as a consequence of cancer, but also as a cause13. Because oxidative stress is a known cause of CIN in cancer69, the link we discovered between oxidative stress and CIN in cells from aged individuals could be connected to age-related oncogenesis. The higher sensitivity of mouse cells to oxidative stress compared to human cells may be related to their higher susceptibility to oncogenic transformation. Interestingly, when mouse embryonic fibroblasts with oncogene-induced CIN were treated with antioxidants, the CIN was reduced, implying a role of oxidative stress in CIN induction during neoplastic transformation70. Our finding that the modal number of chromosomes did not change in fibroblasts from aged individuals suggests that the aneuploid cells resulting from chromosome missegregation do not propagate to alter the karyotype of the cell population, supposedly due to the mechanism to prevent the proliferation of aneuploid cells, in which p53 plays a pivotal role71. When p53 is lost or inactivated, the age-related CIN may lead to oncogenesis by propagating aneuploid cells.
The finding that oxidative stress induces CIN in human cells as well as in mice during aging through a similar mechanism is important for understanding the background of aging and oncogenesis in humans. As a certain level of ROS is necessary for cellular homeostasis1, further study is required to regulate ROS level within a proper range in order to maintain chromosomal stability and prevent age-related disorders including cancer.
Methods
Cell culture and synchronization
The following human fibroblasts were obtained from the NIA Aging Cell Repository in the Coriell Institute for Medical Research (https://www.coriell.org): GM03349 (skin fibroblast, 10yo), GM08398 (skin fibroblast, 8yo), AG04434 (lung fibroblast, 19fw), AG06556 (lung fibroblast, 20fw), AG10884 (skin fibroblast, 87yo), AG11017 (skin fibroblast, 87yo), AG13129A (skin fibroblast, 89yo), and AG05248D (skin fibroblast, 87yo). Cells were cultured in Eagle’s Minimum Essential Medium with Earle’s salts and non-essential amino acids with 2 mM L-glutamine supplemented with 15% fetal bovine serum (FBS) at 37°C in 5% CO2. At confluence, the cells were trypsinized and counted by TC20 automated cell counter (Bio-Rad). Population doubling level (PDL) was calculated using the formula PDL = 3.33 × [log (number of cells harvested) – log (number of cells seeded)] + (doubling level at seeding). Cells at PDL ranging from 17 to 35, during which the cells grew at a constant rate, were used for experiments. To enrich mitotic cells, cells were treated with 2 mM of thymidine (Wako) for 24 h, followed by treatment with 10 μM of RO-3306 (Tokyo Chemical Industry, R0201) for 12 h, then released for indicated periods before fixation.
Reagents and antibodies
N-acetyl-L-cysteine (NAC; Merck, A7250) was used at 5 mM. Nocodazole (Merck, M1404) and Eg5 inhibitor III (Merck, 324622) were used at 1 μM. MG-132 (Merck, M8699) was used at 20 μM. UMK57 (AOBIOS, AOB8668) was used at 1 μM. For nucleoside supplementation, a mix of deoxycytidine (Tokyo Chemical Industry, D3583), deoxyadenosine (Tokyo Chemical Industry, D0046), thymidine (Tokyo Chemical Industry, T0233) and deoxyguanosine (Tokyo Chemical Industry, D0052) was applied at 20 μM for 48 h. Rabbit polyclonal antibodies for 53BP1 (NOVUS Biologicals, NB100-304) and PICH (ERCC6L; Proteintech, 15688-1-AP) were used for immunofluorescence at 1:1,000. Mouse monoclonal antibodies for α-tubulin (B5-1-2; Merck, T5168), p16 (2D9A12; Abcam, ab54210), IL-6 (E-4; Santa Cruz Biotechnology, sc-28343), alpha smooth muscle actin (α-SMA, 1A4; Abcam, ab7817), and phospho-Histone H2A.X (Ser139 (γ-H2AX), JBW301; Merck, 05-636) were used for immunofluorescence at 1:1,000.
Metaphase chromosome spreads
Cells were treated with 2 mM of thymidine for 24 h, followed by treatment with 10 μM of RO-3306 for 12 h, then treated with 20 μM of MG-132 and 2 μM of nocodazole for 2 h. Mitotic cells collected by a shake-off were swollen hypotonically by adding 4 volumes of tap water for 5 min, and then fixed with Carnoy’s solution (methanol: acetic acid = 3:1). The cell suspension was spotted onto slide glasses, dried, and mounted with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories, H-1200). Images were captured on an Olympus IX-83 inverted microscope (Evident) controlled by cellSens Dimension 1.18 (Evident) using a ×100 1.40 numerical aperture (NA) UPlanS Apochromat oil objective lens (Evident).
Immunofluorescence analysis
Immunofluorescence analysis was performed according to a previous report54. Briefly, cells were grown on a glass coverslip and fixed with 3% paraformaldehyde in PBS for 15 min at room temperature, and permeabilized with 1% Triton X-100 in PBS for 5 min. Fixed cells were incubated with primary antibodies for more than 12 h at 4°C, washed three times with PBS supplemented with 0.02% Triton X-100, and incubated with secondary antibodies coupled with Alexa-Fluor-488/594 (Thermo Fisher Scientific, 1:2,000) for 1 h at room temperature. Antibody incubations were performed in PBS supplemented with 0.02% Triton X-100. After final washes, cells were mounted with VECTASHIELD Mounting Medium with DAPI. Z-image stacks were captured in 0.2 or 0.27 μm increments on an Olympus IX-83 inverted microscope controlled by cellSens Dimension 1.18 using a ×100 1.40 or ×60 1.35 NA UPlanS Apochromat oil objective lens. Deconvolution was performed when necessary. Image stacks were projected and saved as TIFF files. All cells analyzed were selected from non-overlapping fields.
SPiDER-βGal staining
To observe the presence of micronuclei together with SA-β-gal activity, cells were treated with 1 μM SPiDER-βGal (Dojindo, SG02) for 15 min, and fixed with 3% paraformaldehyde in PBS for 15 min at room temperature. Then cells were stained with DAPI for 20 min. Images were captured on an Olympus IX-83 inverted microscope controlled by cellSens Dimension 1.18 using×60 1.35 NA Plan Apochromat oil objective lens (Evident).
DNA fiber assay
DNA fiber assay was performed as reported previously72. Briefly, cells were incorporated with 200 μM digoxigenin-dUTPs (Roche) for 10 min, followed by a gentle wash with fresh pre-warmed medium and the second incorporation of 200 μM biotin-dUTPs (Roche) for 10 min. After labeling, cells were trypsinized and fixed (acetic acid:methanol = 1:3). Then, the fixed cell suspension was dropped onto microscope slides and air-dried. To extend the DNA fibers, the slides are immersed in lysis buffer (0.5% SDS, 50 mM EDTA, 200 mM Tris–HCl, pH 7.0), then placed in a high-humidity chamber. After extension, the slides are washed, fixed, and dried. For detection, slides are treated with 1 x blocking reagent (Roche), followed by the addition of detection buffer containing Anti-Digoxigenin-Rhodamine, Fab fragments from sheep (Roche) and Streptavidin, Alexa Fluor® 488 Conjugate (Thermo Fisher Scientific) to visualize the labeled DNA forks. The slides are then washed and mounted with DAPI-supplemented medium. Images were captured on an Olympus IX-83 inverted microscope controlled by cellSens Dimension 1.18 using a ×100 1.40 numerical aperture (NA) UPlanS Apochromat oil objective lens.
ROS detection
Cells were grown in 3.5 cm glass bottom dishes (MatTek), and ROS was measured using CellROX Oxidative Stress Reagents (Molecular Probes, C10444), according to the manufacturer’s instructions. Briefly, the cells were stained with 5 μM CellROX green reagents with 50 μM verapamil hydrochloride (Fujifilm Wako Chemicals, 222-00781) for 30 min at 37 °C in 5% CO2 before imaging. Superoxide in mitochondria was detected using MitoSOX Red (Molecular Probes, M36008), according to the manufacturer’s instructions. Briefly, the cells were stained with 1 μM MitoSOX Red with 50 μM verapamil hydrochloride for 10 min at 37 °C in 5% CO2 before imaging. Images were captured on an Olympus IX-83 inverted microscope controlled by cellSens Dimension 1.18 using ×60 1.40 NA Plan Apochromat oil objective lens. Image stacks were projected and saved as TIFF files.
Mitochondrial membrane potential assay
Mitochondrial membrane potential was measured using JC-10 (AAT Bioquest, 22204) according to the manufacturer’s instructions. Briefly, the cells were grown in 3.5 cm glass bottom dishes and stained with 1× JC-10 with 50 μM verapamil hydrochloride for 30 min at 37 °C in 5% CO2 before imaging. Images were captured on an Olympus IX-83 inverted microscope controlled by cellSens Dimension 1.18 using ×60 1.40 NA Plan Apochromat oil objective lens. Image stacks were projected and saved as TIFF files.
Live cell imaging
Cells were grown in 12 or 6-well plates (Violamo) and treated with 100 nM SiR-DNA (Cytoskeleton, CY-SC007) and 2 μM verapamil hydrochloride for 4 h at 37 °C in 5% CO2 before imaging. Recordings were made every 2.5 min for 48 h using Celldiscoverer 7 live cell imaging system (Carl Zeiss) controlled by ZEN 2.6 (Carl Zeiss) at 37°C in 5% CO2 and 20% O2 using ×20 0.7 NA Plan Apochromat objective lens (Carl Zeiss). For observation of cells after mitotic arrest, cells treated with 1 μM nocodazole or Eg5 inhibitor III were observed every 15 min for 96 h. Images were analyzed using ZEN 2.6.
Statistical analysis
The Mann–Whitney U-test was used for comparison of dispersion, and a two-sided t-test was used for comparisons of average. A two-sided F-test validated the dispersibility of each category before the Student’s t-test. If the result of the F-test was an unequal variance, a significant difference between samples was validated by a two-sided Welch’s t-test. A one-way ANOVA test was used with the Tukey–Kramer post hoc test for comparisons between all groups showing normal distribution. The Kolmogorov–Smirnov test verified the normality of data distribution for each group before the one-way ANOVA test. If the result of the Kolmogorov–Smirnov test was a non-nominal distribution, the significant differences between all groups were validated by the Kruskal–Wallis test, which was used with Steel–Dwass post hoc test. A Chi-squared test was used for comparison between the measured value and theoretical value. All statistical analyses were performed with EZR73, which is a graphical user interface for R (R Core Team, R: A language and environment for statistical computing, https://www.R-project.org/, (2018)). More precisely, it is a modified version of R commander designed to add statistical functions frequently used in biostatistics. Samples for analysis in each dataset were acquired in the same experiment, and all samples were calculated at the same time for each dataset.
Data availability
All data generated during this study are included in the published article and supplementary files. Additional inquiries can be directed to the corresponding authors.
References
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Ricke, R. M. & van Deursen, J. M. Aneuploidy in health, disease, and aging. J. Cell Biol. 201, 11–21 (2013).
Nagaoka, S. I., Hassold, T. J. & Hunt, P. A. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat. Rev. Genet 13, 493–504 (2012).
Macedo, J. C., Vaz, S. & Logarinho, E. Mitotic dysfunction associated with aging hallmarks. Adv. Exp. Med Biol. 1002, 153–188 (2017).
Jacobs, P. A., Brunton, M., Court Brown, W. M., Doll, R. & Goldstein, H. Change of human chromosome count distribution with age: evidence for a sex differences. Nature 197, 1080–1081 (1963).
Pierre, R. V. & Hoagland, H. C. Age-associated aneuploidy: loss of Y chromosome from human bone marrow cells with aging. Cancer 30, 889–894 (1972).
Andriani, G. A., Vijg, J. & Montagna, C. Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 161, 19–36 (2017).
Lushnikova, T., Bouska, A., Odvody, J., Dupont, W. D. & Eischen, C. M. Aging mice have increased chromosome instability that is exacerbated by elevated Mdm2 expression. Oncogene 30, 4622–4631 (2011).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Weaver, B. A. & Cleveland, D. W. Does aneuploidy cause cancer?. Curr. Opin. Cell Biol. 18, 658–667 (2006).
Bakhoum, S. F. & Compton, D. A. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J. Clin. Invest 122, 1138–1143 (2012).
Gordon, D. J., Resio, B. & Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet 13, 189–203 (2012).
Tanaka, K. & Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim Biophys. Acta 1866, 64–75 (2016).
Chen, G., Li, Z., Iemura, K. & Tanaka, K. Oxidative stress induces chromosomal instability through replication stress in fibroblasts from aged mice. J. Cell Sci. 136, jcs260688 (2023).
Parrinello, S. et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5, 741–747 (2003).
Hornsby, P. J. Mouse and human cells versus oxygen. Sci. Aging Knowl. Environ. 2003, PE21 (2003).
Lansdorp, P. M. Repair of telomeric DNA prior to replicative senescence. Mech. Ageing Dev. 118, 23–34 (2000).
Todaro, G. J. & Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 (1963).
Vassilev, L. T. et al. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 103, 10660–10665 (2006).
Gascoigne, K. E. & Taylor, S. S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122 (2008).
McHugh, D. & Gil, J. Senescence and aging: causes, consequences, and therapeutic avenues. J. Cell Biol. 217, 65–77 (2018).
Doura, T. et al. Detection of LacZ-positive cells in living tissue with single-cell resolution. Angew. Chem. Int. Ed. Engl. 55, 9620–9624 (2016).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Dou, Z. et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017).
Plikus, M. V. et al. Fibroblasts: origins, definitions, and functions in health and disease. Cell 184, 3852–3872 (2021).
Paxson, J. A. et al. Age-dependent decline in mouse lung regeneration with loss of lung fibroblast clonogenicity and increased myofibroblastic differentiation. PLoS One 6, e23232 (2011).
Mah, L. J., El-Osta, A. & Karagiannis, T. C. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686 (2010).
Wang, C. et al. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8, 311–323 (2009).
White, R. R. & Vijg, J. Do DNA double-strand breaks drive aging?. Mol. Cell 63, 729–738 (2016).
Mirman, Z. & de Lange, T. 53BP1: a DSB escort. Genes Dev. 34, 7–23 (2020).
Lukas, C. et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 (2011).
Baumann, C., Korner, R., Hofmann, K. & Nigg, E. A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 128, 101–114 (2007).
Chan, K. L., North, P. S. & Hickson, I. D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409 (2007).
Chan, K. L., Palmai-Pallag, T., Ying, S. & Hickson, I. D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760 (2009).
Halliwell, J. A., Gravells, P. & Bryant, H. E. DNA fiber assay for the analysis of DNA replication progression in human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 54, e115 (2020).
Aruoma, O. I., Halliwell, B., Hoey, B. M. & Butler, J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med 6, 593–597 (1989).
Burgunder, J. M., Varriale, A. & Lauterburg, B. H. Effect of N-acetylcysteine on plasma cysteine and glutathione following paracetamol administration. Eur. J. Clin. Pharm. 36, 127–131 (1989).
Passos, J. F. et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347 (2010).
Vigneron, A. & Vousden, K. H. p53, ROS and senescence in the control of aging. Aging (Albany NY) 2, 471–474 (2010).
Zhou, J., Terluk, M. R., Orchard, P. J., Cloyd, J. C. & Kartha, R. V. N-Acetylcysteine reverses the mitochondrial dysfunction induced by very long-chain fatty acids in murine oligodendrocyte model of adrenoleukodystrophy. Biomedicines 9, 1826 (2021).
Zheng, J. et al. N-acetylcysteine, a small molecule scavenger of reactive oxygen species, alleviates cardiomyocyte damage by regulating OPA1-mediated mitochondrial quality control and apoptosis in response to oxidative stress. J. Thorac. Dis. 16, 5323–5336 (2024).
Cimini, D., Cameron, L. A. & Salmon, E. D. Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr. Biol. 14, 2149–2155 (2004).
Kozakiewicz, M., Kornatowski, M., Krzywińska, O. & Kędziora-Kornatowska, K. Changes in the blood antioxidant defense of advanced age people. Clin. Inter. Aging 14, 763–771 (2019).
Gorni, D. & Finco, A. Oxidative stress in elderly population: a prevention screening study. Aging Med. (Milton) 3, 205–213 (2020).
Katsube, T. et al. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem 156, 85–95 (2014).
Ye, B. et al. Dynamic monitoring of oxidative DNA double-strand break and repair in cardiomyocytes. Cardiovasc Pathol. 25, 93–100 (2016).
DePinho, R. A. & Polyak, K. Cancer chromosomes in crisis. Nat. Genet 36, 932–934 (2004).
Burrell, R. A. et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013).
Tanaka, K. Dynamic regulation of kinetochore-microtubule interaction during mitosis. J. Biochem 152, 415–424 (2012).
Tanaka, K. Regulatory mechanisms of kinetochore-microtubule interaction in mitosis. Cell Mol. Life Sci. 70, 559–579 (2013).
Tanaka, K. & Hirota, T. Chromosome segregation machinery and cancer. Cancer Sci. 100, 1158–1165 (2009).
Wilhelm, T. et al. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 10, 3585 (2019).
Iemura, K. & Tanaka, K. Chromokinesin Kid and kinetochore kinesin CENP-E differentially support chromosome congression without end-on attachment to microtubules. Nat. Commun. 6, 6447 (2015).
Orr, B., Talje, L., Liu, Z., Kwok, B. H. & Compton, D. A. Adaptive resistance to an inhibitor of chromosomal instability in human cancer cells. Cell Rep. 17, 1755–1763 (2016).
Kalfalah, F. et al. Inadequate mito-biogenesis in primary dermal fibroblasts from old humans is associated with impairment of PGC1A-independent stimulation. Exp. Gerontol. 56, 59–68 (2014).
Kalfalah, F. et al. Structural chromosome abnormalities, increased DNA strand breaks and DNA strand break repair deficiency in dermal fibroblasts from old female human donors. Aging (Albany NY) 7, 110–122 (2015).
de Magalhães, J. P. & Passos, J. F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 170, 2–9 (2018).
Barnes, R. P. et al. Telomeric 8-oxo-guanine drives rapid premature senescence in the absence of telomere shortening. Nat. Struct. Mol. Biol. 29, 639–652 (2022).
Baez, A. & Shiloach, J. Effect of elevated oxygen concentration on bacteria, yeasts, and cells propagated for production of biological compounds. Micro. Cell Fact. 13, 181 (2014).
D’Angiolella, V., Santarpia, C. & Grieco, D. Oxidative stress overrides the spindle checkpoint. Cell Cycle 6, 576–579 (2007).
Wang, G. F. et al. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic. Biol. Med 103, 177–187 (2017).
Kakudo, N., Morimoto, N., Ogawa, T., Taketani, S. & Kusumoto, K. Hypoxia enhances proliferation of human adipose-derived stem cells via HIF-1a activation. PLoS One 10, e0139890 (2015).
Stamatov, R. et al. Supra-second tracking and live-cell karyotyping reveal principles of mitotic chromosome dynamics. Nat. Cell Biol. 27, 654–667 (2025).
Shields, H. J., Traa, A. & Van Raamsdonk, J. M. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies. Front Cell Dev. Biol. 9, 628157 (2021).
Macedo, J. C. et al. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 9, 2834 (2018).
Barroso-Vilares, M. et al. Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO Rep. 21, e49248 (2020).
d’Adda di Fagagna, F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522 (2008).
Kudryavtseva, A. V. et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 7, 44879–44905 (2016).
Woo, R. A. & Poon, R. Y. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev. 18, 1317–1330 (2004).
Thompson, S. L. & Compton, D. A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 188, 369–381 (2010).
Luebben, S. W., Shima, N. & Kawabata, T. Methods for the detection of genome instability derived from replication stress in primary mouse embryonic fibroblasts. Methods Mol. Biol. 1194, 341–352 (2014).
Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 48, 452–458 (2013).
Acknowledgements
The authors thank members of the K.T. laboratory for discussions, and H. Sato for technical assistance. This work was supported by JSPS KAKENHI Grant Numbers, 22K19283, 23K23877; MEXT KAKENHI Grant Numbers, 23H04272; grants from the Takeda Science Foundation to K.T., JSPS KAKENHI Grant Numbers 20K16295, 23K05629; ACT-X, JST Grant Number JPMJAX2112; Yamaguchi Ikuei Foundation, the Pharmacological Research Foundation. Tokyo, and the Mochida Memorial Foundation for Medical and Pharmaceutical Research to K.I., JST the establishment of university fellowships towards the creation of science technology innovation Grant Number JPMJFS2102 to G.C., JST SPRING, Grant Number JPMJSP2114 to K.Z.
Author information
Authors and Affiliations
Contributions
K.Z. and K.T. designed the experiments. K.Z., G.C., and R.I. performed the experiments. K.Z., K.I., and K.T. wrote the manuscript. K.T. supervised the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, K., Chen, G., Ren, Y. et al. Human fibroblasts from aged individuals exhibit chromosomal instability through replication stress caused by oxidative stress. npj Aging 11, 107 (2025). https://doi.org/10.1038/s41514-025-00299-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41514-025-00299-w
