
Abstract
Bacteria commonly protect themselves from a variety of threats by forming biofilms, which are communities of bacteria that are tightly packed together within an extracellular matrix. Biofilm formation has generally been thought to protect bacteria from phage infection. The opportunistic pathogen Pseudomonas aeruginosa produces biofilm matrices that can contain three distinct exopolysaccharides that contribute to the difficulty in treating infected patients. Here, we demonstrate that two diverse P. aeruginosa phages have evolved to exploit this biofilm matrix to access the bacterial cells by both binding to and degrading a major biofilm exopolysaccharide, Psl. We examined the effect of these phages on biofilms in different in vitro biofilm models and found that both phages prevent bacterial surface attachment, but only one of the two phages can disrupt a mature biofilm under flow. The phages also rapidly lead to the emergence of bacterial strains that produce reduced amounts of Psl and are unable to adhere to surfaces. These phages may be useful therapeutically by driving bacteria away from producing biofilms and shifting P. aeruginosa cells into the more treatable planktonic growth state.
Similar content being viewed by others


Introduction
A major obstacle to the treatment of bacterial infections is the growth of bacteria in protective aggregates called biofilms, which occur in roughly 80% of human bacterial infections1. Within biofilms, bacteria are encased in a complex extracellular matrix that may be composed of a range of macromolecules such as exopolysaccharides, extracellular DNA, and proteins. This matrix serves as a barrier that can exclude a wide range of threats, ranging from antibiotics to immune molecules2. As a result, when pathogenic bacteria form biofilms in humans, these infections often become chronic and untreatable. The opportunistic pathogen Pseudomonas aeruginosa, a model for the study of biofilms, frequently causes biofilm-involved infections in a range of settings, including the lungs of people with cystic fibrosis, catheter or ventilator-associated infections, and chronic wounds. The P. aeruginosa biofilm matrix can vary in composition, with many clinical isolates of P. aeruginosa from the lungs of people with cystic fibrosis dominated either by alginate or by a mixture of the exopolysaccharides Pel and Psl3,4, as is the case for rugose small colony variants (RSCV). RSCVs are a hyper-biofilm-forming variant, which commonly arise in chronic infections and are especially recalcitrant to treatment5,6.
The biofilm matrix is generally thought to exclude bacteriophages (phages), the viruses that infect bacteria, thus limiting the utility of phage therapy for biofilm-involved infections7,8. However, few studies have examined these interactions at the cellular or molecular level. Phage propagation and characterization are typically performed using a double overlay assay in which bacteria are suspended in a gel-like agar matrix that is normally associated with bacterial motility9, rather than biofilm formation, and is thus not well suited to biofilm-phage interaction studies. Only a small handful of studies have investigated phages using in vitro biofilm models10,11,12,13. Commonly used static biofilm assays only represent one aspect of biofilm development and may miss some of the different effects that phages can exert on biofilms. For example, in a static biofilm assay as opposed to biofilms grown under flow (e.g., flow cell or flow tube biofilms), the phages do not have to contend with flow, which may wash away less adherent phages, but they are faced with relatively high concentrations of extracellular bacterial waste products14. Further, bacterial physiology, which matters for the efficacy of phage infection, varies depending on the biofilm model that is used in the experiment. For these reasons, it has been suggested that phages be tested against multiple biofilm models in order to gain a better understanding of phage-biofilm interactions15.
Interestingly, many phages are known to produce depolymerase enzymes as part of their tail fibers. Typically, depolymerases are identified through experiments in which phages are spotted on bacterial lawns grown on low-percentage agar nutrient plates, where they infect and lyse bacteria, resulting in cleared zones on the lawn. When phages contain depolymerases, a less turbid zone (the “halo”) may form outside of the main lysis area; this is often taken as indirect evidence of enzymatic activity. There are a handful of examples of phage depolymerases that target biofilm matrix exopolysaccharides, including those of Staphylococcal species16, Klebsiella pneumoniae17, and Pseudomonas putida18,19. Numerous studies have also characterized phage depolymerases that target other cell-associated extracellular sugars, such as lipopolysaccharides (LPS) or the capsule-associated exopolysaccharides (CPS)20,21. Increasing evidence indicates that several unrelated phages have evolved to specifically interact with the biofilms of P. aeruginosa. A Pseudomonas phage that targets the biofilm component alginate was described, and the specific depolymerase identified22. More recently, several studies have identified two distinct phages—a siphophage, Iggy, and a podophage of the family Bruynoghevirus—that both require the matrix component Psl for absorption23,24,25,26. These findings suggest that the biofilm matrix is a barrier to phage infection that some phages have evolved to overcome.
Indeed, given the diversity of both bacterial species and their phages, we have very little knowledge about how most phages interact with bacterial biofilms or whether biofilms do indeed represent a first line of defense against phage infection. Several studies, albeit with only a single bacterial and phage species, highlight the complexity of biofilm-phage interactions and provide motivation for further studies to probe these interactions. Specifically, in some instances, the spatially segregated, heterogeneous bacterial population present in biofilms supports bacterial co-existence with phages12. Studies using Escherichia coli biofilms that contain the functional bacterial amyloid called curli observed that the model T7 phage is trapped by curli, but depending on the location of the phages and the biofilm age, this interaction can both prevent T7 particles from accessing bacteria at the biofilm aggregate interior and also serve as a phage reservoir for newly arriving bacterial cells10,11.
Here, we describe the isolation of two P. aeruginosa phages—one podo- and one siphophage—that both require the exopolysaccharide Psl for infection. Notably, these phages are closely related to other recently described Psl-targeting phages23,24,25,26. We find that both Psl-dependent phages require Psl for adsorption to bacteria and degrade Psl to access the bacterial surface. Examining these phages in several different in vitro biofilm models reveals unexpected differences in their impact on biofilms. We find that both phages can prevent bacterial adherence to a surface and reduce biomass in a static biofilm model; however, only one of the phages can disrupt a mature biofilm under flow. Selective pressure of these phages leads to the rapid emergence of P. aeruginosa escape mutants with severely reduced Psl production and an inability to adhere.
Results
Isolation of two Psl-dependent P. aeruginosa phages
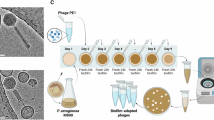
To identify phages that may have exopolysaccharide-degrading properties, we set out to test whether any phages in our collection formed haloes on an exopolysaccharide overproducing strain, PAO1 ∆wspF, wherein the deletion of the negative regulator wspF leads to the constitutive upregulation of the exopolysaccharides, Pel and Psl27. To our surprise, we found that two of our phages (PaStL1M and PaStL2M) displayed an increase in titer on the ∆wspF strain relative to wild-type PAO1 (Fig. 1A), with distinct plaquing morphology. Sequencing these phages revealed that each of our two phage stocks contained two unrelated phages, resulting in a total of four distinct phages. We thus renamed these as PaStL1-PaStL4, wherein the PaStL1M mixture contained PaStL1 (a new species of Bruynoghevirus) and PaStL3 (a new species of Yuavirus, family Mesyanzhinoviridae, subfamily Rabinowitzvirinae); and the PaStL2M mixture contained PaStL2 (a new species of Iggyvirus, subfamily Queuovirinae) and PaStL4, a new strain of Pbunavirus Ab28 (family Lindbergviridae). Transmission electron microscopy (TEM) of these phages confirmed morphology consistent with the sequencing data (Fig. 1B).

A Phage plaques of two phage stocks on PAO1 and PAO1 ∆wspF as determined via serial dilutions of the indicated phage stock that were spotted on plates made with the specified bacterial strain. B TEM images of the four phages. C Serial dilutions of each purified phage were spotted on a double overlay plate prepared with the indicated bacterial strain. D Bacterial growth was monitored via absorbance at OD600 after addition of the indicated phage at an MOI of 0.01.
We hypothesized that within each of the mixtures, one of the two phages had an increased capacity to form plaques on ∆wspF compared to the wild-type PAO1 strain, possibly because these phages utilize either the Psl or Pel exopolysaccharide as a receptor. To determine whether the phage with increased plaque formation on ∆wspF was dependent on either of these exopolysaccharides, we tested the phage mixtures on strains lacking genes encoding key early enzymes in the Psl and Pel biosynthetic pathways (Table S1), and found that plaques on ∆wspF ∆psl resembled those seen on PAO1, suggesting that the second, higher-titer phage in the mixture was Psl-dependent (Fig. S1A). We also tested plaquing on a clinical isolate, CF127, previously shown to produce high levels of Psl4, and its derivative CF127 ∆psl, and noted that plaques were visible on CF127, but no plaques occurred on CF127 ∆psl, suggesting that only the Psl-dependent phage in each case could replicate on CF127 (Fig. S1A). We thus purified the original PaStL1M and PaStL2M phage stocks using both ∆wspF ∆psl and CF127. This resulted in four isolated phages: two Psl-dependent phages (PaStL1 and PaStL2) that can form plaques on ∆wspF and CF127, but not PAO1, and two Psl-independent phages (PaStL3 and PaStL4) that can form plaques on all strains except CF127 and CF127 ∆psl (Fig. 1C). We note that in the course of performing this study, similar Psl-dependent phages were reported: Clew-126 and Bruynoghevirus LUZ2423 are related to PaStL1, while Knedl24,25—of the Iggyvirus genus—is related to PaStL2. These studies, together with our findings, suggest that there are two major genera of P. aeruginosa phages that use Psl as a receptor.
The fact that both PaStL1 and PaStL2 were unable to form plaques on PAO1 but were nonetheless present at high titers in the original mixtures suggested that they must be able to replicate in planktonic culture in the wild-type PAO1 strain that we had used for all enrichment and isolation steps. Indeed, when we added these phages at a low multiplicity of infection (MOI) of 0.01 to PAO1 and monitored bacterial growth under standard planktonic growth conditions, we observed a typical infection curve with a loss of bacterial density beginning around 2.5 h after phage addition (Fig. 1D). These data are consistent with PaStL1 and PaStL2 phages existing at a high titer in our phage stocks (evident in plaque assays with ∆wspF) but going undetected in initial plaque assays on PAO1. It is not clear why these phages can replicate in planktonic conditions but are not able to form plaques, as Psl levels are similar in planktonic and top agar growth conditions (Fig. S1B).
PaStL1 and PaStL2 bind and degrade Psl
We used an adsorption assay to assess whether phage attachment to bacterial cells was dependent on Psl. Specifically, phages were incubated with bacteria that did not express Psl (i.e., PAO1 ∆wspF ∆pel ∆psl; referred to as PAO1 ∆wspF ∆EPS) or expressed high levels of Psl via an arabinose-inducible promoter (i.e., PAO1 ∆wspF ∆pel pBAD-psl). The phages PaStL1 and PaStL2, which both required bacterial production of Psl for infectivity in a plaque assay, displayed high levels of Psl-dependent adsorption (Fig. 2A). In contrast, the presence of Psl did not impact adsorption of either PaStL3, which did not adsorb well to either strain, or PaStL4, in which 90% of the phage adsorbed to both strains. Together, these results support that PaStL1 and PaStL2 uniquely use Psl as a receptor for binding to the bacterial cells, while the two non-Psl-dependent, co-isolated phages do not require Psl for adsorption.

A Bacterial adsorption was measured following 10 min of incubation at room temperature with the indicated strain for each phage. The percentage of bound phage was calculated using 100% * (total-unbound) / total. B Psl degradation by each phage was determined by anti-Psl immunoblot.
Next, we determined whether the phages are able to depolymerize Psl, as has been observed for other phages that bind and degrade the surface glycan, o-antigen20. To do so, we incubated purified phage with cell-free supernatant preparations from the Psl-producing strain, PAO1 ∆wspF. Following 6 h of incubation at 37 °C, we measured Psl levels using an anti-Psl immunoblot (Fig. 2B). Incubation with phages PaStL1 and PaStL2 decreased Psl levels. In contrast, incubation with either PaStL3 or PaStL4 did not discernibly change Psl levels relative to the no-phage control. As is expected for depolymerization of Psl by an enzyme, heat inactivation of the phages prior to incubation with the cell-free supernatant abolished the ability of the phages to depolymerize Psl.
We surveyed the phage genomes for depolymerases using DePolymerase Predictor28 and identified putative depolymerases encoded in the genomes of both PaStL1 and PaStL2. These putative depolymerases had different sequences as well as strikingly different structures, as predicted by AlphaFold3 (Fig. S2). Notably, there were no similar depolymerases predicted that are shared between PaStL1 and PaStL2, suggesting that these two phages have independently evolved mechanisms for binding to and degrading Psl. These results support that both PaStL1 and PaStL2 bind and depolymerize Psl, but they likely do so via different depolymerases.
PaStL1 and PaStL2 clear static biofilms
Given the Psl-dependence of the phages, we predicted that they might be effective against biofilms. To test this, we grew biofilms statically in 96-well microtiter dishes for 24 h, removed any unattached biomass, and treated the remaining adherent biomass with the phages. We tested each of the phages individually as well as the two original phage mixtures, PaStL1M (i.e., PaStL1 and PaStL3) and PaStL2M (i.e., PaStL2 and PaStL4). As evidenced by the decrease in adherent biomass relative to the no-phage control, both Psl-dependent phages, PaStL1 and PaStL2, were more effective at clearing biofilms formed by PAO1 than their co-isolated counterparts, PaStL3 and PaStL4 (Fig. 3). Both PaStL1 and PaStL2 were similarly able to clear biofilms formed by PAO1 ∆wspF, and PaStL3 was unable to do so. Surprisingly, PaStL4 also reduced PAO1 ∆wspF adherent biomass, though to a lesser extent. The original phage mixtures showed differing results, with PaStL1M being similarly effective as PaStL1 alone, and PaStL2M being less effective than PaStL2 alone. However, the Psl-dependent phages had a reduced ability to clear biofilms formed by PAO1 ∆wspF ∆psl. Interestingly, when treated with the original phage mixtures, PaStL1M and PaStL2M, the phage-treated PAO1 ∆wspF ∆psl biofilms had increased adherent biomass relative to the respective no-phage control. This could be due to the release of extracellular DNA if the phages lysed a subset of the bacteria, or due to the phage-induced bacterial stress triggering an increase in biofilm formation, as has been previously described29. Overall, this static biofilm assay supports that, in addition to both PaStL1 and PaStL2 requiring Psl for infection (Fig. 1), both phages clear biofilms in a Psl-dependent manner.

Adherent biomass following incubation with phages was determined using a crystal violet assay. Data represent the means of results from 3 biological replicates (3 separate experiments, each with 4 wells per condition), and error bars represent standard deviation. The asterisks indicate a significant difference in the adherent biomass of the phage-treated biofilm relative to the no-phage control of the same strain (Welch’s t-test; P < 0.05), ns, not statistically significant.
Only PaStL2 clears mature biofilms cultured in flow cells
Given the role of Psl in P. aeruginosa surface attachment, we investigated whether the Psl-dependent phages, PaStL1 and PaStL2, could block bacterial surface attachment. To do so, we performed a bacterial attachment assay in microfluidic devices called flow cells. In this experiment, PAO1 ∆wspF bacteria that constitutively expressed the fluorescent protein GFP (PAO1 ∆wspF GFP+) were allowed to attach to the surface of a flow cell for 10 min while the flow was stopped prior to the addition of phage. Purified phages were added and incubated with the bacteria for an additional 30 min, and then flow was resumed. The flow cells were imaged by confocal microscopy, and the numbers of attached bacteria were quantified using BiofilmQ30. We observed that incubation with both Psl-dependent phages, PaStL1 and PaStL2, decreased bacterial attachment to the flow cell surface (Fig. 4A).

A The impact of phages on the initial attachment of PAO1 ∆wspF GFP+ bacteria to flow cells was evaluated by confocal microscopy. Phages were added to mature flow cell biofilms of PAO1 ∆wspF GFP+, and B 2 h or C 26 h post phage addition, confocal microscopy images of the biofilms were collected to determine whether the phages cleared the biofilms. Fluorescence due to GFP (bacteria) is shown in green, and HHL-TRITC (Psl) is shown in magenta. Typical images for each time point are shown. D The volume of bacteria in the flow cells for each time point and phage condition was determined using BiofilmQ, normalized to the volume of bacteria in the no-phage control at 2 h post phage addition. E The volume of Psl in the flow cells was similarly determined and normalized to the volume of Psl in the no-phage control at 2 h post phage addition. Data represent the means of results from 3 biological replicates (3 flow cells with 6 images collected per flow cell), and error bars represent standard deviation.
To test the impact of the phages on established biofilms, we cultured PAO1 ∆wspF GFP+ in flow cells for 72 h, and then phages were added and statically incubated with the biofilms for 30 min before resuming flow. In this experiment, in addition to monitoring the bacteria, we also monitored Psl levels by staining with the fluorophore-conjugated Psl-specific lectin, Hippeastrum hybrid lectin (HHL)-TRITC. Biofilms were stained and imaged 2, 10, 16, and 26 h following phage addition (Figs. 4B, C, and S3). Separately cultured biofilms were stained and imaged for each of the time points to ensure that staining with the Psl-specific lectin did not impact the results. Without phage incubation, typical biofilms formed with bacterial aggregates that extend into the flow cell channel ~60 µm and have peripherally localized Psl (Fig. 4B, C). At 2 and 10 h after the addition of phage, the flow cell biofilms appeared similar to one another, regardless of which phage had been added (Figs. 4B, and S3A). By 16 h after the addition of phage, biofilms that had been incubated with PaStL2 began to clear, and by 26 h, biofilms that had been incubated with PaStL2 were entirely cleared (Figs. 4C, and S3B). Biofilms incubated with PaStL1 did not clear, even by 26 h after the addition of phage. We quantified the overall biomass (GFP fluorescence) and Psl levels (HHL-TRITC fluorescence) using BiofilmQ, which allowed us to make two additional observations (Fig. 4D, E). First, for biofilms incubated with PaStL2, the Psl levels appeared to decrease more rapidly than the bacteria, suggesting that degradation of Psl precedes overall biofilm clearing. The second observation was that biofilms that were incubated with PaStL1 showed slightly increased levels of bacteria relative to biofilms that were not incubated with phage. This slight increase in biofilm bacteria may be attributed to the lysis of bacteria by the phage, resulting in elevated extracellular DNA levels and, consequently, increased bacteria within the biofilm aggregates.
We performed three additional flow cell controls, each in which we cultured flow cell biofilms for 72 h, added phage, and then monitored the effect 26 h later. In controls with the two non-Psl dependent phages, we found that neither PaStL3 nor PaStL4 cleared the flow cell biofilms or decreased Psl levels (Fig. S4). As an additional control, since the ∆wspF mutation alters expression of many genes beyond simply increasing Psl production27, we tested if PaStL2 could clear a flow cell biofilm produced by wild-type PAO1 cells. We found that PaStL2 also cleared PAO1 flow cell biofilms, indicating that the ∆wspF mutation was not necessary for clearing by PaStL2 (Fig. S5). Lastly, we tested whether Psl was required for clearing of flow cell biofilms by PaStL2, and we observed that PaStL2 was unable to clear PAO1 ∆wspF ∆psl biofilms (Fig. S6). As previously reported, PAO1 ∆wspF ∆psl flow cell biofilms appear different than PAO1 ∆wspF flow cell biofilms, even without phage infection, due to the reliance on Pel rather than Psl31. Together, these findings show that PaStL2 can clear both PAO1 and PAO1 ∆wspF flow cell biofilms, and that this ability is dependent on bacterial production of Psl.
PaStL2 propagates in biofilms grown under flow
Given the unexpected difference in the abilities of PaStL1 and PaStL2 to clear flow cell biofilms, we wondered if PaStL1 had a reduced ability, compared to PaStL2, to propagate in bacteria cultured under flow. To test this hypothesis, we cultured PAO1 ∆wspF GFP+ biofilms for 72 h in silicone tubing, which was cut to have a similar internal volume to the flow cells used in the prior experiment, and then statically incubated phage with the biofilms for 30 min before resuming flow. The biofilms were harvested 26 h later, and both colony-forming units (CFUs) and plaque-forming units (PFUs) were determined. CFUs were determined using the entire harvested biofilm, and PFUs were determined using the cell-free supernatant of the biofilm. To avoid confounding effects due to possible phage killing during the CFU assay, we determined CFUs indirectly by measuring the GFP fluorescence of the biofilms, which we correlated to CFUs via a standard curve of CFU versus GFP fluorescence that was obtained for PAO1 ∆wspF GFP+ bacteria that were cultured in the absence of phage (Fig. S7). Incubation of PaStL2, but not PaStL1, resulted in a decrease of the biofilm bacteria (p < 0.05), although the decrease in this growth system was not as dramatic as was observed in the flow cell experiment (Fig. 5A). Specifically, the CFUs for biofilms with PaStL1 added were ~5 × 109 CFUs/mL and with PaStl2 added, only around 1 × 109 CFUs/mL. A greater difference was observed for PFUs. For the biofilms treated with PaStL1, we recovered only ~5 × 104 PFUs/mL, and for those treated with PaStL2, we recovered 2 × 1010 PFUs/mL (Fig. 5B). To determine the dynamics of PaStL2 phage infection, we also harvested separately cultured biofilms at 2, 10, and 16 h post phage addition, and similarly determined both CFUs and PFUs (Fig. 5C). We observed that PaStL2 titers increased at 16 h, which preceded the decrease in biofilm bacteria CFUs. Given that the titer of the phage stock added to the biofilm was ~108 PFUs/mL, with a similar volume as the recovered cell-free supernatant of the biofilm (300 μL vs ~250 μL, respectively) these data support that PaStL2 is able to propagate within a biofilm cultured under flow, and PaStL1 is not.

A Colony forming units (CFUs/mL) were determined for mature flow tube biofilms formed by PAO1 ∆wspF GFP+ 26 h after the addition of phage. B Plaque forming units (PFUs/mL) within the supernatant of the tube biofilm were determined 26 h after the addition of phage. nd, none detected. C Both CFUs and PFUs were determined at 2, 10, 16, and 26 h after the addition of PaStL2, and the values were compared to those of the no-phage control. Data represent the means of results from 3 biological replicates, and error bars represent standard deviation.
P. aeruginosa evades PaStL1 and PaStL2 via loss of Psl
We noted that both in planktonic growth curves and on top agar, resistant bacteria typically emerged following infection with PaStL1 and PaStL2. We isolated individual colonies that emerged in planktonic growth of PaStL1 as well as within plaques of PaStL2 (Fig. 1C, D) and then retested these for sensitivity to both phages. A subset of these clones was sensitive to phage infection, suggesting a regulatory response to phage infection that reverted upon reculturing. However, many of these clones were persistently resistant and, in all cases, resistant to both PaStL1 and PaStL2. We hypothesized that these cells had likely lost the ability to produce Psl. To test this idea, we performed Psl immunoblots and found that all of the escape mutants produced much less Psl than either the PAO1 or ∆wspF strain (Fig. 6A).

A Psl production was measured with an anti-Psl immunoblot for each of the indicated strains. B Bacterial adherence as measured in a static biofilm assay for indicated control strains, in addition to the bacterial escape mutants. Bars represent the mean of 3 independent replicates with error bars representing standard deviation.
We also found that these escape mutants all had reduced ability to adhere to a surface as measured in a static biofilm assay (Fig. 6B). We sequenced a subset of these escape mutants and found that ∆wspF-PaStL1EA, which arose via resistance to PaStL1, had a mutation in pslA that led to a truncation of the protein, while surprisingly, ∆wspF-PaStL1B had only a substitution in PA1327, a predicted protease (Table 1). The escape mutants isolated from double overlays had all acquired a 7-base duplication in the wspR gene that encodes the regulator controlling the Wsp pathway, and in some cases, additional mutations as well (Tables 1, and S2). Taken together, these data demonstrate that both PaStL1 and PaStL2 provide a strong selective pressure that leads to the loss of Psl production in P. aeruginosa.
Discussion
Despite numerous studies that have pursued phage or phage cocktails as therapeutics for recalcitrant bacterial infections, including biofilm infections, there has been a general lack of mechanistic insight into interactions of phage with biofilms. In this study, we discovered two phages that require the P. aeruginosa biofilm matrix exopolysaccharide Psl for attachment to their bacterial hosts, similar to recently reported Psl-targeting phages23,24,25,26. We find that both phages, which, based on bioinformatic analyses, we predict use different depolymerase proteins to bind to and degrade Psl, prevent bacteria from establishing biofilms by eliminating the ability of the cells to attach to the surface. Investigating these phages using multiple biofilm models revealed unexpected differences in how two Psl-dependent phages affect biofilms, which was not reported in related studies that characterized similar phages23,24,25,26. While both PaStL1 and PaStL2, along with the non-Psl-dependent phage, PaStL4, can reduce adherent biomass in static biofilm-based assays to varying degrees (Fig. 3), only PaStL2 within our phage set can disrupt a fully formed biofilm that is cultured under flow (Fig. 4). We further demonstrate that both PaStL1 and PaStL2 strongly select for non-biofilm-forming bacteria that do not produce Psl and are unable to adhere to surfaces to establish biofilms, even in the absence of phage. These results demonstrate the utility of these phages in therapeutic applications where they could both disrupt biofilms, as well as drive bacteria into a planktonic growth state where they would be more amenable to treatment.
This study highlights the importance of considering phage isolation methodology when performing environmental phage isolations: phages will only replicate and form plaques on strains that are producing necessary receptors. Both Psl-dependent phages that we isolated were originally co-isolated with another non-Psl-dependent phage. This co-isolation was essentially what enabled us to discover these phages, as without the second phage present, our initial plaque assays on PAO1 would have been blank and falsely suggested that there was no phage present in our sample. The proteins and exopolysaccharides present on the cell surface are highly dynamic, as this first line of defense against many threats is critical to bacterial survival. Using conditions in which bacteria produce proteins and exopolysaccharides present during infections is critical for finding phages that are likely to be useful in therapeutic applications.
In addition to requiring Psl for adsorption to the bacterial cell, we found that both PaStL1 and PaStL2, but not PaStL3 or PaStL4, result in the degradation of Psl following their incubation with Psl-containing culture supernatants. We predict that both phages contain depolymerases, as predicted by bioinformatic analyses and analogous to the depolymerases that phages employ to degrade LPS and CPS. Unlike LPS and CPS in many bacterial species, P. aeruginosa Psl is generally believed to be chemically identical or similar across P. aeruginosa isolates due to the extremely high conservation of the Psl operon32. As such, Psl may serve as a better target for phage-based therapies as opposed to LPS or CPS, since bacteria cannot simply modify Psl to escape phage infection. Rather, as we observed in our experiments, bacteria that evolve to escape infection by PaStL1 or PaStL2 lose the ability to produce Psl altogether, which makes them unable to form biofilms, likely rendering them susceptible to more typical antibacterial therapeutics.
This study also highlights the need to evaluate phage-biofilm interactions in multiple biofilm models since phages can have dramatically different effects on different types of biofilms15. For example, we observed that both Psl-dependent phages, PaStL1 and PaStL2, cleared biofilms that were cultured statically in microtiter dishes. However, only PaStL2, and not PaStL1, could clear mature biofilms that were grown under flow growth conditions (i.e., flow cell biofilms, tube biofilms). While these are still simplified in vitro models, biofilms cultured under flow likely better mimic turbulent or dynamic conditions encountered in some infections (e.g., infections of catheters, heart valves, etc.), and thus, PaStL2 might be a better therapeutic candidate than PaStL1. The reason(s) for the different abilities of PaStL1 and PaStL2 to clear biofilms cultured under flow could include that PaStL1 does not bind as well as PaStL2 or perhaps the bacterial physiology in these biofilms is less amenable to PaStL1 propagation. Our results point toward both options as possibilities, as more PaStL2 adsorbed to Psl-producing bacteria compared to PaStL1, and PaStL2 was able to propagate in tube biofilms while PaStL1 did not, although additional experiments are required to say definitively.
Overall, in this study, we discovered two different phages that require Psl for infection and can degrade Psl and clear P. aeruginosa biofilms. In addition to providing candidates for phage therapy of chronic P. aeruginosa infections, the predicted Psl depolymerases could serve as anti-biofilm candidates on their own, similar to other hydrolases that degrade matrix exopolysaccharides and help to break apart biofilms, potentially broadening their potential impact beyond the narrow strain profile of these phages. This is an interesting avenue for future research, as currently, nearly all candidate hydrolases for anti-biofilm therapeutics come from either bacteria or fungi, and phage may be an untapped source of novel enzymes to break apart biofilms. Lastly, our study serves as a roadmap for future discovery of additional biofilm-targeting phages.
Methods
Bacterial strain growth
Planktonic cultures of P. aeruginosa were routinely grown on Lysogeny broth (LB) or TSB medium at 37 °C with constant shaking (225 rpm) unless indicated otherwise. Strains are listed in Table S1.
Phage isolation
Wastewater from a local treatment facility was centrifuged to remove particulates, then filtered with a 0.22 µm filter to remove bacterial cells. The resulting supernatant was mixed with concentrated LB broth for a final 1x concentration of LB and mixed with an overnight culture of PAO1 at 1:100. The suspension was incubated overnight at 37 °C with aeration. The following day, the culture was centrifuged to pellet bacteria and filtered, and the supernatant was spotted on double overlay plates prepared with 4 mL of 0.6% LB agar with 200 µL of an overnight PAO1 culture and poured onto a 1.2% LB agar plate. Plates were examined for clearing after 6–16 h, and for lysates that displayed clearing, the culture was serially diluted to determine an estimate of titer as described below with the spot titer assay, then plated for single plaque isolations using the full plate assay. Individual plaques were picked with a P10 pipette tip and added to 100 µL of a 1:10 dilution of an overnight PAO1 culture in LB and incubated overnight at 37 °C in a microcentrifuge tube. The resulting lysate was centrifuged and plaque purifications were performed 3x.
Phage titer assays
Phage concentration was determined by measuring plaque-forming units/mL (PFUs/mL) using either a spot titer or full plate assay. 200 µL of an overnight bacterial culture was mixed with 4 mL of melted 0.6% LB agar (55 °C) and poured on top of a 1.2% LB agar plate. Once solidified, 2–40 µL of a 10x serially diluted phage was spotted and allowed to dry before incubation of plates for 5–16 h at 37 °C. Alternatively, 100 µL of phage was added directly to the top agar. PFUs/mL were calculated by counting the number of plaques in the lowest serial dilution and then multiplying by the dilution factor.
Phage propagation and purification
P. aeruginosa phages were produced by back-diluting overnight cultures of PAO1 to an OD600 of 0.1 in LB, adding an aliquot of phage stock to a final MOI of 0.1, and then incubating the cultures for 5 h at 37 °C with constant shaking (225 rpm). After incubation, bacterial cells were separated from the supernatant by centrifugation (6000 × g for 20 min) at 4 °C. The supernatant was passed through a 0.22 µm filter and used directly for titering assays and adsorption experiments. For all biofilm experiments, the filtered lysate was further purified via ultracentrifugation through a 35% (w/v) sucrose cushion at 235,000 × g for 50 min. The supernatant was removed via decanting, and the pellet was suspended in 1 mL of 1x PBS. The sample was centrifuged at 6000 × g for 20 min and then dialyzed (8–10 kDa MWCO Float-A-Lyzer G2 Dialysis Device) (MilliporeSigma) against 1x PBS overnight at 4 °C. Afterwards, the sample was centrifuged at 17,000 × g for 10 min. The phage was then either used in assays directly or further subjected to size-exclusion chromatography (1x PBS; HiPrep 16/60 sephacryl S-500 HR column) (Cytiva). Phage-containing fractions were concentrated tenfold using a centrifugal filter (50 kDa MWCO) (MilliporeSigma).
Transmission electron microscopy
For analyses of phages at the ultrastructural level, samples were allowed to absorb onto freshly glow-discharged formvar/carbon-coated copper grids (200 mesh, Ted Pella Inc., Redding, CA) for 10 min. Grids were then washed two times in dH2O and negative stained with 1% aqueous uranyl acetate (Ted Pella Inc.) for 1 min. Excess liquid was gently wicked off and grids were allowed to air dry. Samples were viewed on a JEOL 1200EX transmission electron microscope (JEOL USA, Peabody, MA) equipped with an AMT 8 megapixel digital camera (Advanced Microscopy Techniques, Woburn, MA).
Static biofilm assay
Static biofilm formation was assessed by performing a crystal violet assay33. Biofilms were cultured in Nunc Bacti 96-well microtiter plates (Fisher Scientific) using TSB. 100 µL of mid-log culture was used to inoculate each well of the microtiter plate. The plates were statically incubated for 24 h at 37 °C. Non-adherent cells were removed, and wells were washed with 150 µL 1x PBS. Then 100 µL of phage (5 × 107 PFUs/mL, 1x PBS buffer) or buffer alone was added to each well and statically incubated for 4 h at 37 °C. Non-adherent cells were removed by pipetting, and the wells were washed three times with 150 µL of ddH2O. The 96-well plate was inverted and allowed to dry for 30 min before the addition of 150 µL of 0.1% (w/v) crystal violet (Fisher Scientific). The plate was statically incubated with crystal violet for 15 min. The crystal violet solution was removed, and the wells were washed three times with 150 L of ddH2O. The plate was inverted and allowed to dry overnight. Finally, the crystal violet was solubilized with 200 µL of 95% ethanol, and 100 µL was transferred to a fresh plate and the absorbance at 595 nm was measured (Varioskan LUX multimode microplate reader, Thermo Fisher Scientific).
Phage DNA sequencing
Phage DNA was extracted using a Norgen Phage DNA Extraction kit and short read Illumina sequencing was either performed by SeqCoast Genomics (Portsmouth, NH, USA) or libraries were prepared using Illumina Tagmentation reagents with a low input protocol34, and sequenced at the DNA Sequencing and Innovation Lab at the Edison Family CGS&SB Sequencing Center. Reads were de novo assembled in Geneious Prime (version 2025.1.1) and the TaxMyPhage35 tool was used to determine taxonomy. Phage genomic sequencing data is available at PRJNA1260455.
Adsorption assays
Overnight bacterial cultures were back-diluted into LB media supplemented with 1% (w/v) L-arabinose, and then grown overnight with shaking (225 rpm) at 37 °C. A 1-mL aliquot of the culture was centrifuged at 5000 × g for 10 min, the supernatant discarded, and the pellet suspended in 1 mL of 1x PBS. Phage was added to achieve an MOI of 0.01 and incubated for 15 min at room temperature with occasional inversion. 20 µL was removed for serial dilution; the remaining sample was centrifuged at 8000 × g for 3 min and a 20 µl aliquot of the supernatant was collected for serial dilutions. 20–40 µL of these dilutions were spotted on double overlay assays to determine relative titers.
Psl levels in planktonic and top agar conditions
An overnight culture grown at 37 °C in LB was subcultured 1:100 in LB and grown at 37 °C with shaking for 16 h, then normalized to an OD of 1.0 in a volume of 1 mL. For the top agar cultures, an overnight culture grown at 37 °C in LB was diluted 1:100 in 10 mL of LB 0.6% agar and poured into an empty petri dish and then grown at 37 °C for 16 h, then resuspended in 10 mL LB. The resulting slurry was then centrifuged at 1000 × g for 3 min to pellet agar but not bacterial cells. The turbid supernatant was collected and normalized to an OD of 1.0 in 1 mL. Psl levels were assessed via anti-Psl immunoblot.
Psl degradation assay
Overnight cultures of PAO1 ∆wspF and PAO1 ∆wspF ∆EPS in TSB were back diluted 100-fold and grown for 16 h at 37 °C with shaking (225 rpm). Cultures were normalized to OD600 of 1.0 and the centrifuged for 2 min at 15,060 rpm. The supernatant was then incubated with bacteriophage (10:1 supernatant to phage, 1 × 108 PFUs/mL) at 37 °C for 6 h. Immediately after incubation, samples were boiled at 98 °C for 5 min and cooled to 12 °C. The presence of Psl was assessed by anti-Psl immunoblot.
Anti-Psl immunoblot
To assess for the presence of Psl, samples were analyzed by anti-Psl immunoblot as previously described36. Samples were treated with proteinase-K (2 mg/mL) (Qiagen) at 60 °C for 1 h, 80 °C for 30 min, and then cooled down to 12 °C. After proteinase K treatment, 5 µL of sample was loaded onto a nitrocellulose membrane (Bio-Rad) (0.2 mm) and allowed to dry for 10 min. The membrane was blocked for 1 h with 5% (w/v) milk, 10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20 (TBST). Then the membranes were probed with anti-Psl primary antibodies (0.635 µg/mL WapR-016, 0.371 µg/mL WapR-001, 0.867 µg/mL Cam-003) (AstraZeneca) for 1 h37,38. Membranes were washed three times with TBST. The membranes were probed either with horseradish peroxidase conjugated goat anti-human antibody (Bio-Rad) (5 µL in 25 mL TBST) for 1 h or with IRDye 800CW goat anti-human IgG secondary antibody (LI-COR Biotech). Membranes were washed three times with TBST. Detection was performed with Supersignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific) and blots were imaged with AlphaImager HP (Alpha Innotech) or visualized with an Odyssey CLx imaging system (LI-COR Biotech).
Attachment assay
Flow cells were inoculated with PAO1 ∆wspF Tn7 Gm::P(A1/04/03)::GFP (referred to as PAO1 ∆wspF GFP+) bacteria that were grown to mid-log phase in TSB and then diluted to OD600 of 0.1 in 1% TSB. Bacterial cells were allowed to attach under static conditions in an inverted flow cell for 10 min and then incubated with 300 µL of phage (1 × 108 PFUs/mL) for 30 min. Then, non-attached cells were washed away by initiating media flow (40 mL/h) through the flow cell for 20 min. Flow was reduced to 3 mL/h prior to imaging. Attached cells were visualized on a Nikon Eclipse Ni-E confocal laser scanning microscope (20x objective), and six fields of view were captured (Ex 488 nm, Em 499–551 nm). Images were analyzed with BiofilmQ30. To extract 3D objects from the imported microscopy data, the biofilm images were segmented. To remove background fluorescence and fluorescence in between cells, the top-hat filter was set to 15 vox. Afterward, the Otsu thresholding method with a sensitivity of 0.25 was set. Cubes were used for the dissection method and were ~1 vox in length. The images for the GFP fluorescent channel were segmented with these parameters and then global biofilm properties were calculated to collect the bacterial volume.
Imaging of flow cell biofilms
Biofilms were cultured in flow cells as previously described39. The flow cells were made of polysulfone, and the microfluidic chamber was 1.125 inches long, 0.185 inches wide, and 0.162 inches deep. A microscope coverglass (24 × 60 mm, 0.175 mm thick) (Fisher Scientific) was fixed to the top of the chamber using clear adhesive sealant (Permatex silicone RTV 80050). After allowing the sealant to dry, the flow cells were autoclaved. Sterile flow cells were inoculated with bacterial cultures of PAO1 ∆wspF Tn7 Gm::P(A1/04/03)::GFP or as specified, that were grown to mid-log phase in TSB and then diluted to OD600 of 0.01 in 1% TSB. Bacterial cells were allowed to attach under static conditions in an inverted flow cell for 1 h before the start of media flow. Biofilms were grown in 1% TSB for 72 h at 25 °C under a constant flow rate (10 mL/h). After 72 h, flow was stopped, and the biofilm was statically incubated with 300 μL of phage in 1x PBS (1 × 108 PFUs/mL) for 30 min prior to resuming media flow. Biofilms were monitored 2, 10, 16, and 26 h after phage incubation. Biofilms were stained with 0.1 mg/mL HHL-TRITC (GlycoMatrix) to visualize Psl. After staining, flow cells were washed with medium at 10 mL/h for 5 min and visualized on a Nikon Eclipse Ni-E confocal laser scanning microscope using a 20X or 40X objective (GFP: Ex 488 nm, Em 499–551 nm; TRITC: Ex 561 nm, Em 571–625 nm). Images were analyzed with BiofilmQ30. To extract 3D objects from the imported microscopy data, the biofilm images were segmented. The images were scaled down 0.25x for pre-processing, and to remove background fluorescence and fluorescence in between cells, the top-hat filter was set to 15 vox. Afterwards, Otsu thresholding method with a sensitivity of 0.25 was set. Cubes were used for the dissection method and were ~1 vox in length. The images for each fluorescent channel were segmented with these parameters, and then global biofilm properties were calculated to collect the bacterial and Psl volume.
Flow tubes
Biofilms were cultured as tube biofilms as previously described with some modifications40. PAO1 ∆wspF Tn7 Gm::P(A1/04/03)::GFP bacteria were grown to mid-log phase in TSB and then diluted to OD600 of 0.01 in 1% TSB. This back-diluted culture then was used to inoculate a sterile 4.5-cm long Nalgene 50 platinum-cured silicon tubing (1/8 in interior diameter and ¼ in exterior diameter) (Thermo Fisher Scientific). Note that the actual tube length in which the biofilm grew after accounting for the lengths of the connecting luers was 3 cm. Bacterial cells were allowed to attach to the tubes under static conditions for 1 h before the start of media flow. Biofilms were grown in 1% TSB for 72 h at room temperature (~25 °C) under a constant flow rate (10 mL/h). After 72 h, flow was stopped, and the biofilm was statically incubated with 300 µL of bacteriophage (1 × 108 PFUs/mL) for 30 min. Then flow was resumed. Biofilms were collected at 2, 10, 16, and 26 h after phage incubation. The biomass inside of the tube was collected by using the handle of a sterile, disposable L-spreader (Fisher Scientific) to push out the biomass into a collection tube. The sample was vortex mixed for 15 s, and then fluorescence (Ex 488 nm, Em 520 nm) was recorded (Varioskan LUX multimode microplate reader) (Thermo Fisher Scientific). The sample was centrifuged for 2 min at 15,060 rpm. The supernatant was removed and used for plaque assays.
GFP standard curve for CFU calculation
To determine the CFUs in biomass collected from flow tubes incubated with phage, the correlation between GFP fluorescence (due to constitutive expression of GFP by the bacteria) and CFUs was analyzed to create a standard curve. To do so, overnight cultures of PAO1 ∆wspF Tn7 Gm::P(A1/04/03)::GFP that were grown in TSB were serially diluted, and then the fluorescence (Ex 488 nm, Em 520) was measured in a 96-well black plate (Thermo Fisher Scientific) with Varioskan LUX multimode microplate reader (Thermo Fisher Scientific). In parallel, CFUs of the samples were determined. The CFU values were plotted against GFP fluorescence to obtain a best line fit equation (y = 5.2545 × 107(x) – 1.3505 × 107), where y is CFUs, and x is fluorescence.
Bacterial escape mutant sequencing
Resistant mutants were used in titer assays to confirm resistance to PaStL1 and PaStL2. Resistant strains were further cultured and bacterial genomic DNA was extracted using a Qiagen Puregene Kit. Genomic DNA was sent to SeqCoast for short-read (Illumina) sequencing. Reads were mapped to PAO1 using Geneious Prime (version 2025.1.1) and analyzed for mutations using the Geneious Variant Finder tool, with the variant frequency cutoff set to 60% and coverage cutoff of 5. All mutations were compared with sequencing of the ∆wspF background strain, and only mutations unique to the escape mutants were further analyzed. All sequencing data were deposited to PRJNA1260455.
Data availability
All sequencing data have been deposited to NCBI under the BioProject accession number PRJNA1260455. All other data generated or analyzed during this study are included in this published article and its supplementary information files.
Code availability
All analyses were conducted using standard software packages, and no new code was written or developed for this study.
References
Flemming, H.-C. & Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 17, 247–260 (2019).
Karygianni, L., Ren, Z., Koo, H. & Thurnheer, T. Biofilm matrixome: extracellular components in structured microbial communities. Trends Microbiol. 28, 668–681 (2020).
Colvin, K. M. et al. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 7, e1001264 (2011).
Colvin, K. M. et al. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 14, 1913–1928 (2012).
Häußler, S., Tümmler, B., Weißbrodt, H., Rohde, M. & Steinmetz, I. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin. Infect. Dis. 29, 621–625 (1999).
Starkey, M. et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 191, 3492–3503 (2009).
Ferriol-González, C. & Domingo-Calap, P. Phages for biofilm removal. Antibiotics 9, 268–16 (2020).
Danis-Wlodarczyk, K. M., Wozniak, D. J. & Abedon, S. T. Treating bacterial infections with bacteriophage-based enzybiotics: in vitro, in vivo and clinical application. Antibiotics 10, 1497 (2021).
Ha, D.-G., Kuchma, S. L. & O’Toole, G. A. Plate-based assay for swarming motility in Pseudomonas aeruginosa. Methods Mol. Biol. 1149, 67–72 (2014).
Vidakovic, L., Singh, P. K., Hartmann, R., Nadell, C. D. & Drescher, K. Dynamic biofilm architecture confers individual and collective mechanisms of viral protection. Nat. Microbiol. 3, 26–31 (2018).
Bond, M. C., Vidakovic, L., Singh, P. K., Drescher, K. & Nadell, C. D. Matrix-trapped viruses can prevent invasion of bacterial biofilms by colonizing cells. eLife 10, e65355 (2021).
Simmons, E. L. et al. Biofilm structure promotes coexistence of phage-resistant and phage-susceptible bacteria. mSystems 5, 385–17 (2020).
Winans, J. B., Wucher, B. R. & Nadell, C. D. Multispecies biofilm architecture determines bacterial exposure to phages. PLoS Biol. 20, e3001913 (2022).
Rumbaugh, K. P. & Whiteley, M. Towards improved biofilm models. Nat. Rev. Microbiol. 23, 57–66 (2025).
Abedon, S. T., Danis-Wlodarczyk, K. M., Wozniak, D. J. & Sullivan, M. B. Improving phage-biofilm in vitro experimentation. Viruses 13, 1175 (2021).
Gutiérrez, D. et al. Role of the pre-neck appendage protein (Dpo7) from phage vB_SepiS-phiIPLA7 as an anti-biofilm agent in Staphylococcal species. Front. Microbiol. 6-2015, 1315 (2015).
Wu, Y. et al. A novel polysaccharide depolymerase encoded by the phage SH-KP152226 confers specific activity against multidrug-resistant Klebsiella pneumoniae via biofilm degradation. Front. Microbiol. 10-2019, 2768 (2019).
Cornelissen, A. et al. Identification of EPS-degrading activity within the tail spikes of the novel Pseudomonas putida phage AF. Spec. Issue Viruses Microbes. 434, 251–256 (2012).
Cornelissen, A. et al. The T7-related Pseudomonas putida phage φ15 displays virion-associated biofilm degradation properties. PLoS ONE 6, e18597 (2011).
Knecht, L. E., Veljkovic, M. & Fieseler, L. Diversity and function of phage encoded depolymerases. Front. Microbiol. 10-2019, 2949 (2020).
Latka, A., Maciejewska, B., Majkowska-Skrobek, G., Briers, Y. & Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 101, 3103–3119 (2017).
Glonti, T., Chanishvili, N. & Taylor, P. W. Bacteriophage-derived enzyme that depolymerizes the alginic acid capsule associated with cystic fibrosis isolates of Pseudomonas aeruginosa. J. Appl. Microbiol. 108, 695–702 (2010).
Billaud, M. et al. Complementary killing activities of Pbunavirus LS1 and Bruynoghevirus LUZ24 phages on planktonic and sessile Pseudomonas aeruginosa PAO1 derivatives. Antimicrob. Agents Chemother. 0, e00579–25 (2025).
Manner, C. et al. A genetic switch controls Pseudomonas aeruginosa surface colonization. Nat. Microbiol. 8, 1520–1533 (2023).
Maffei, E., Manner, C., Jenal, U. & Harms, A. Complete genome sequence of Pseudomonas aeruginosa phage Knedl. Microbiol. Resour. Announc. 13, e0117423 (2024).
Walton, B. et al. A biofilm-tropic Pseudomonas aeruginosa bacteriophage uses the exopolysaccharide Psl as receptor. eLife 13, RP102352 (2025).
Hickman, J. W., Tifrea, D. F. & Harwood, C. S. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl Acad. Sci. USA 102, 14422–14427 (2005).
Magill, D. J. & Skvortsov, T. A. DePolymerase Predictor (DePP): a machine learning tool for the targeted identification of phage depolymerases. BMC Bioinform. 24, 208 (2023).
LeRoux, M. et al. Kin cell lysis is a danger signal that activates antibacterial pathways of Pseudomonas aeruginosa. eLife 4, e05701 (2015).
Hartmann, R. et al. Quantitative image analysis of microbial communities with BiofilmQ. Nat. Microbiol. 6, 151–156 (2021).
Jennings, L. K. et al. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl Acad. Sci. 112, 11353 (2015).
Tabor, D. E. et al. Pseudomonas aeruginosa PcrV and Psl, the molecular targets of bispecific antibody MEDI3902, are conserved among diverse global clinical isolates. J. Infect. Dis. 218, 1983–1994 (2018).
O’Toole, G. A. Microtiter Dish Biofilm Formation Assay. J. Vis. Exp. 2437 https://doi.org/10.3791/2437 (2011).
Baym, M. et al. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE 10, e0128036 (2015).
Millard, A. et al. taxMyPhage: Automated taxonomy of dsDNA phage genomes at the genus and species level. PHAGE 6, 5–11 (2025).
Byrd, M. S. et al. Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol. Microbiol. 73, 622–638 (2009).
DiGiandomenico, A. et al. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J. Exp. Med. 209, 1273–1287 (2012).
Li, H. et al. Epitope mapping of monoclonal antibodies using synthetic oligosaccharidesuncovers novel aspects of immune recognition of the Psl exopolysaccharide of Pseudomonas aeruginosa. Chem. Eur. J. 19, 17425–17431 (2013).
Passos Da Silva, D. et al. The Pseudomonas aeruginosa lectin LecB binds to the exopolysaccharide Psl and stabilizes the biofilm matrix. Nat. Commun. 10, 2183 (2019).
Peterson, S. B. et al. Different methods for culturing biofilms in vitro. In Biofilm infections (eds Bjarnsholt, T., Jensen, P. Ø., Moser, C. & Høiby, N.) 251–266 (Springer, New York, NY, 2011).
Acknowledgements
This work was supported by an Office of the Vice Chancellor for Research Interdisciplinary Seed Grant to M.L. and C.R. and unrestricted funds from Washington University School of Medicine to M.L. The Psl-specific antibodies were obtained by C.R. from AstraZeneca (Dr. DiGiandomenico). We thank Wandy Beatty for assistance and training on TEM imaging. We thank Forrest Walker for feedback and discussion.
Author information
Authors and Affiliations
Contributions
M.L. and C.R. designed the research; K.A.S., L.C.A., A.Z. and A.J. performed research; K.A.S., M.L. and C.R. analyzed data; and M.L. and C.R. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Amyx-Sherer, K., Awasthi, L.C., Zheng, A. et al. Two unrelated Pseudomonas aeruginosa phages require the exopolysaccharide Psl for infection. npj Biofilms Microbiomes 11, 211 (2025). https://doi.org/10.1038/s41522-025-00841-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-025-00841-4
