
Abstract
Addressing antibiotic-resistant bacterial biofilm infections without promoting drug resistance is a pressing challenge. Pseudomonas aeruginosa is well known for causing biofilm-associated drug-resistant infections that often lead to treatment failure. In this study, we identified a previously uncharacterized membrane protein ferredoxin encoded by PA1551 using photoaffinity-based biomimetic probes based on our previous dual-acting antibiofilm compound 2-(heptanamido)methyl 3-hydroxy-1,6-dimethylpyridin-4(1H)-one (10d). The precision-targeted ferredoxin PA1551 exhibited excellent effectiveness in various model systems, suppressing bacterial biofilm and virulence, and enhancing the antibacterial effects of tobramycin (Tob, by about 200-fold) and ciprofloxacin (CIP, by 1000-fold) compared to single-dose antibiotic treatments in a mouse model of Pseudomonas aeruginosa wound infection. These results indicate that ferredoxin PA1551 can be used as target to design new antibiofilm drugs for the treatment of Pseudomonas aeruginosa infections, particularly challenging bacterial biofilms.
Similar content being viewed by others

Introduction
Antibiotics remain one of the most pivotal interventions in human medicine1,2,3,4. Effective and selective antibiotics have substantially prolonged the lifespan of humans, leading to a perception that bacterial infections have largely been controlled5,6. However, in recent years, the emergence of “superbugs” with broad-spectrum antibiotic resistance and the increasing prevalence of antibiotic-resistant biofilm infections have posed persistent global challenges7,8.
Pseudomonas aeruginosa (P. aeruginosa) is a common pathogen that can form biofilms, leading to the development of drug-resistant, hospital-acquired infections9,10. Biofilms formed by bacterial cells reduce the effectiveness of antimicrobials through various mechanisms, including reduced penetration of antimicrobials and persister cell formation11. The management of wound infections and lung infections is commonly complicated by the formation of biofilms12. It is estimated that 60% to 90% of delayed healing wounds and almost all lung infections are associated with biofilms13,14,15. Biofilms not only resist the host immune system and the killing effects of antibacterial drugs16,17 but also exhibit high resistance to antibiotic therapy, extending healing durations. This leads to difficult-to-treat infections accompanied by elevated levels of inflammatory factors18,19. Given the complex biofilm formation process, which regulates virulence and antibiotic effectiveness, it has been identified as a potential therapeutic target for managing wound and lung infections. Although the key regulatory proteins in the las, rhl and pqs quorum sensing systems involved in the regulation of biofilm formation have been extensively studied, the complex regulatory mechanism of P. aeruginosa biofilm formation has not been fully characterized, which is one of the reasons that no biofilm inhibitors have been marketed to date. Therefore, identifying precise targets that modulate biofilm formation is crucial for developing therapeutic strategies against drug-resistant bacteria, particularly for combating P. aeruginosa infections.
Our previous study discovered a dual-acting antibiofilm strategy based on 2-substituted 3-hydroxy-1,6-dimethylpyridin-4-ones20,21. A hit compound 2-(heptanamido)methyl 3-hydroxy-1,6-dimethylpyridin-4(1H)-one (10d) exhibited promising antibiofilm activity for both the standard laboratory P. aeruginosa strain PAO1 (IC50 = 6.6 μM) and several clinical multidrug-resistant P. aeruginosa strains20, which is able to interfere with iron uptake and the QS system of P. aeruginosa. Despite the unique biological activities of 10d, its exact target remains unknown. To investigate the protein target by which 10d exerts antibiofilm activity, we used affinity-based proteome profiling (ABPP) combined with bioimaging for profiling drug-target interactions in situ22,23. ABPP is a powerful method for the in situ investigation of noncovalent ligand-receptor interactions, and this technique plays a crucial role in various fields, such as chemical biology and drug discovery24,25. As illustrated in Fig. 1, we developed a series of chemical probes based on the scaffold of 2-(heptanamido)methyl 3-hydroxy-1,6-dimethylpyridin-4(1H)-ones (10d) for the study of biofilm formation at the proteomic level to identify biological drug-resistant targets via ABPP. These probes possessed diazirine photoreactive moiety and alkyne analytical handles for mediating copper-mediated azide-alkyne coupling (CuAAC) reactions. We evaluated the biological activity of all these probes in P. aeruginosa and used the most optimal probe for the ABPP experiment. Using these biomimetic probes, we identified an iron metabolism protein (ferredoxin PA1551) as a drug target for biofilm inhibitors against difficult-to-treat bacterial biofilms. We purified the full-length membrane protein ferredoxin PA1551 and validated its interaction using biophysical techniques. Furthermore, a PA1551-deficient mutant exhibited reduced virulence and altered biofilm phenotypic characteristics, supporting the antibacterial synergistic effect of targeting ferredoxin PA1551. In addition, in vivo assays confirmed that Δferredoxin (PA1551-deficient mutant) was effective in enhancing the antibacterial effects of tobramycin (Tob, by about 200-fold) and ciprofloxacin (CIP, by 1000-fold) compared with single-dose antibiotic treatments in a mouse model of P. aeruginosa infection. In summary, our results demonstrate the suppression of biofilm formation in P. aeruginosa by inhibiting the ferredoxin PA1551 of iron homeostasis. This research provides new insights for inhibiting pathogenic disease.

The ABPP methodology was used in this study.
Results
Compound 10d displays antibacterial synergistic efficacy on antibiotics against P. aeruginosa infection in vivo
To discover the potential target for inhibiting P. aeruginosa biofilm, compound 10d stands out for its promising antibiofilm activity and could be used to explore the possible novel functional target. Considering that wound infection is one of the major infections caused by P. aeruginosa, we initially examined whether 10d can enhance the effectiveness of commonly used antibiotics, Tob and CIP26 in an experimental open-wound mouse model of P. aeruginosa infection27. In this in vivo model, we created surface wounds in mice and inoculated the wound with 5 × 108 CFU of P. aeruginosa PAO1. Subsequently, the wounds were administered dropwise directly with physiological saline, 10d, Tob28 (0.5 mg/mL), Tob (0.0025 mg/mL), or the combination of Tob (0.0025 mg/mL) with 10d over a three-day period. We analyzed the bacterial load in each wound as a measure of the bacterial survival rate, and we monitored the status of mice for nine days. The results indicated that 10d exhibited significant synergistic effects with Tob in controlling P. aeruginosa infection and improved wound healing in the open-wound murine model.
In this model, lesions were significantly smaller in the 10d-Tob combination group than in the control group every day (Fig. 2C, D). Nine days after infection, all the wounds treated with Tob (0.5 mg/mL) and Tob (0.0025 mg/mL) in combination with 10d were nearly closed while that treated with 10d alone only achieved a 40% healing rate, suggesting that the in vivo efficacy of combined Tob (0.0025 mg/mL) and 10d was comparable to that of high-dose Tob (0.5 mg/mL) treatment alone. After three days of continuous treatment (Fig. 2A, B), there was no obvious bacterial elimination in the 10d-treated group compared to the control group. In contrast, the bacterial survival rate in the Tob (0.0025 mg/mL) + 10d combination group (2.3%) was significantly lower than that in the Tob (0.0025 mg/mL)-only group (16.8%) and the 10d-only group (99.9%). Moreover, the bacterial survival rate of the combination treatment group was comparable to that in the high-dose Tob (0.5 mg/mL)-only group (0%). This result indicated that 10d significantly improved the bactericidal effect of Tob by approximately 200-fold. Tissue immunohistochemistry section images revealed no apparent toxicity to the organs following three days of treatment with Tob alone, 10d alone or the combination of Tob and 10d when compared to the control group (Fig. 2E). What is more, 10d exhibited no obvious toxicity or effect on the body weight of the mice throughout the experiment (Supplementary Fig. 1A).

A The representative images of bacterial survival in the wound of mice on agar plate. B The survival rates of P. aeruginosa based on the standard plate counting assay after different treatments with control (saline), 10d, Tob (0.5 mg/mL), Tob (0.0025 mg/mL), and the Tob (0.0025 mg/mL) - 10d in P. aeruginosa PAO1 wound model, control was defined as 100%. C Visual observation of open-wound model of P. aeruginosa PAO1 infection treated with control (saline), 10d, Tob (0.5 mg/mL), Tob (0.0025 mg/mL), and the Tob (0.0025 mg/mL) - 10d. D Graphical representation of the open-wound model of P. aeruginosa PAO1 infection, the quantitative measurement of wound area within nine days, control was defined as 100%. E Histological hematoxylin and eosin (H&E) staining images of heart, liver, spleen, lung, and kidney tissue sections at day nine after different treatments are indicated. All experiments are performed in triplicate, and data are presented as mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
We conducted additional experiments to investigate the synergistic effects of 10d with CIP, another commonly used antibiotic for P. aeruginosa treatment. The wound was treated with physiological saline, CIP29 (1 mg/mL), CIP (0.01 mg/mL), CIP (0.001 mg/mL), the combination of CIP (0.01 mg/mL) with 10d, the combination of CIP (0.001 mg/mL) with 10d over a three-day period. In addition, we monitored the status of mice for nine days. Similar to the aforementioned findings, we observed that 10d enhanced the antibacterial effects of CIP by more than 100- to 1000-fold in an open-wound murine model of P. aeruginosa PAO1 infection (Fig. 3A, B). Lesions caused by the infection were significantly smaller in the 10d-CIP combination group than in the control group on every day, except on day 1 (Fig. 3C, D). Nine days after infection, all the wounds treated with CIP (1 mg/mL), the combination of CIP (0.01 mg/mL) with 10d, and the combination of CIP (0.001 mg/mL) with 10d were almost closed. This finding suggested that the in vivo efficacy of CIP (0.01 mg/mL)- 10d and CIP (0.001 mg/mL)- 10d combinations were comparable to that of the high-dose CIP (1 mg/mL) treatment alone. Furthermore, after three days of continuous treatment, the bacterial survival rate (0%) in the wound treated with the combination of CIP (0.01 mg/mL) and 10d was comparable to that in the wound treated with high-dose CIP (1 mg/mL) alone (0%) and was lower than that in the wound treated with low-dose CIP (0.01 mg/mL) alone (4.2%) (Fig. 3B). In addition, the bacterial survival rate (2.4%) in the CIP (0.001 mg/mL)- 10d combination group was lower than that in the CIP (0.001 mg/mL) alone group (8.2%). These results indicated that 10d significantly improved the antibacterial effect of CIP. Histological sections of tissues revealed no apparent organ toxicity following three days of treatment with CIP alone or the combination of CIP and 10d when compared to the control group (Fig. 3E). Furthermore, 10d exhibited no obvious toxicity or effect on the body weight of the mice throughout the experiment (Supplementary Fig. 1B).

A The representative images of bacterial survival in the wound of mice on agar plate. B The survival rates of P. aeruginosa based on the standard plate counting assay after different treatments with control (saline), CIP (1 mg/mL), CIP (0.01 mg/mL), CIP (0.001 mg/mL), CIP (0.01 mg/mL) - 10d, and CIP (0.001 mg/mL) - 10d in P. aeruginosa PAO1 infected wound model, control was defined as 100%. C Visual observation of open-wound model of P. aeruginosa PAO1 infection treated with control (saline), CIP (1 mg/mL), CIP (0.01 mg/mL), CIP (0.001 mg/mL), CIP (0.01 mg/mL) - 10d, and CIP (0.001 mg/mL) - 10d. D Graphical representation of the open-wound model of P. aeruginosa PAO1 infection, the quantitative measurement of wound area within nine days, control was defined as 100%. E Histological hematoxylin and eosin (H&E) staining images of heart, liver, spleen, lung, and kidney tissue sections at day nine after different treatments are indicated. All experiments are performed in triplicate, and data are presented as mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Compound 10d displays antibacterial synergistic efficacy on antibiotics against multidrug-resistant P. aeruginosa infection in vivo
To assess the clinical potential of compound 10d, its antibacterial synergistic effect on Tob and CIP was further evaluated in vivo against PA1121, a clinical MDR P. aeruginosa isolate. The results are presented in Fig. 4. Compared to the untreated group, administration of 10d alone (10 μM) did not reduce bacterial load, with bacterial survival at the wound site remaining as high as 100% (Fig. 4A, C). However, the combination of 10d with a low concentration of Tob (0.0025 mg/mL) reduced bacterial survival to 2.0%, a result comparable to that achieved with Tob monotherapy at 0.5 mg/mL (5.7%) and significantly superior to 0.0025 mg/mL Tob monotherapy (15.3%). This indicated that 10d also enhanced the activity of Tob by approximately 200-fold in the MDR strain PA1121. The improved outcome was further supported by accelerated wound healing observed in mice under the treatment of combination therapy (Fig. 4B, D). Similarly, 10d potentiated the antibacterial activity of CIP in PA1121 (Fig. 4E, G). The combination of 10d with CIP at 0.01 mg/mL CIP (1.4%) or 0.001 mg/mL CIP (2.7%) resulted in bacterial survival rates at mouse wounds comparable to those achieved with high-concentration CIP (1 mg/mL, 1.3%), both being lower than those observed with CIP alone at 0.01 mg/mL (6.7%) or 0.001 mg/mL (22.6%). Moreover, wound healing in mice treated with the combination therapy after nine days was superior to that with CIP alone (Fig. 4F, H). These results indicated that 10d can enhance CIP activity by 100- to 1000-fold in the MDR strain PA1121.

A, E The representative images of bacterial survival in the wound of mice on agar plate. B, F Graphical representation of the open-wound model of P. aeruginosa PA1121 infection, the quantitative measurement of wound area within nine days. C, G The survival rates of P. aeruginosa based on the standard plate counting assay after different treatments in PA1121 infected wound model, control was defined as 100%. D, H Visual observation of open-wound model of PA1121 infection treated with control (saline), 10d alone, antibiotics alone, and antibiotics in combination with 10d, control was defined as 100%. All experiments are performed in triplicate, and data are presented as mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
In summary, using an open-wound murine model of P. aeruginosa infection, we found that the biofilm inhibitor 10d significantly enhanced the antibacterial effects of Tob (about 200-fold) and CIP (1000-fold) in PAO1 and MDR strain PA1121. These results suggest that 10d is a promising candidate as an antibiotic adjuvant for the treatment of P. aeruginosa infections.
Design of chemical probes and evaluation of the antibiofilm effects of probes on P. aeruginosa
Our results revealed the promising antibacterial synergistic potential of biofilm inhibitor 10d for the treatment of P. aeruginosa infections. Next, we utilized affinity-based proteome profiling (ABPP) coupled with bioimaging for profiling drug-target interactions in situ. Over the past few years, Yao’s group has made several contributions to the field of ABPP by developing a set of minimalist diazirine photo-crosslinkers. These linkers, consisting of an alkyl diazirine, a reactive group, and a bioorthogonal handle, have not only substantially reduced the time and complexity involved in preparing photo-affinity probes but also considerably improved probe labeling efficiency. Diazirines, which remain stable under both acidic and basic conditions and are resistant to common nucleophiles and electrophiles, can be stored at room temperature, making them widely applicable in various labeling studies30,31.
Inspired by the promising therapeutic effect of biofilm inhibitor 10d in P. aeruginosa wound infection, we further synthesized a series of probes based on the 10d scaffold by introducing a diazirine photoreactive moiety and alkyne handles for copper-mediated CuAAC reactions (complete synthetic details are provided in Supplementary Scheme 1–10)20. Among these probes, the commercially available minimalist photocrosslinker L1 (Fig. 5A), which contains both diazirine and terminal alkyne, was selected for minimal modification of the affinity-based structural element, which is consistent with the fundamental principle that the ligand should closely resemble the natural ligand25,32. In the design of probe molecules, compound 10d-1 replaced the six-carbon chain of 10d, with the photocrosslinker L1 at the C-2 position to minimize ligand modification. In addition, previous studies examining structure-activity relationships have indicated that derivatives at the C-6 position of hydroxypyridinone exhibited minimal biofilm inhibitory effects20,21. This prompted us to focus on 2,6-disubstituted probe designs. Thus, we generated the affinity-based probe 10d-2 by introducing the photocrosslinker L1 at the C-6 position while preserving the six-carbon chain at the C-2 position of 10d. Initially, whether hydroxypyridinone derivatives could covalently modify their targets was unclear. Thus, we designed covalent probe molecules without a photoaffinity group. Among all three covalent probes, 10d-3, 10d-4, and 10d-5 corresponded to the parent drug 10d and the photoaffinity probe molecules 10d-1 and 10d-2, respectively. Furthermore, three matching negative probes NP, NP-1, and NP-2 were generated as controls in the following biological experiments to rule out any potential interference from the alkyne handle and photoaffinity group.

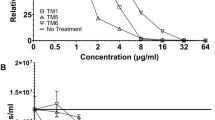
A The structure of photoaffinity probes, covalent probes, and negative probes. B Activity of probes on biofilm formation of P. aeruginosa PAO1. C Biofilm formation inhibition rates of different concentrations of 10d (1.25, 2.5, 5, 10, and 20 μM) for 24 h. D Biofilm formation inhibition rates of different concentrations of 10d-1 (1.25, 2.5, 5, 10, and 20 μM) for 24 h. All experiments are performed in triplicate, and data are presented as mean ± SD. aIC50 = concentration of inhibitor needed to inhibit biofilm formation by 50%. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
We performed a crystal violet staining assay (Fig. 5B) to examine the antibiofilm effects of these probes. Subsequently, we analyzed the capacity of these probes to inhibit the biofilms of P. aeruginosa PAO1. The antibiofilm effect of probe 10d-1 (IC50 = 7.5 μM) on P. aeruginosa was comparable to that of 10d (IC50 = 6.6 μM). Furthermore, to comprehensively evaluate the antibiofilm effects of probe 10d-1, we examined its effect on the biofilm formation of P. aeruginosa PAO1 strains at concentrations of 1.25, 2.5, 5, 10, and 20 μM. 10d was concurrently tested as a reference compound. As expected, probe 10d-1 (Fig. 5C, D), being the most suitable, significantly inhibited the biofilm formation of P. aeruginosa PAO1 in a dose-dependent manner. By contrast, the negative control probes (NP) displayed no biofilm inhibitory activity. In addition, the antibiofilm effects of these probes were also evaluated by using a metabolic dye named 2,3,5-triphenyltetrazolium chloride (TTC). TTC is a colorless metabolic dye that is reduced by dehydrogenases in living cells to produce a red formazan product, which can be used to identify live bacteria in biofilms33. As presented in Supplementary Table 1, probe 10d-1 displayed potent antibiofilm activity with an IC₅₀ of 4.59 μM, while other probes showed IC₅₀ values greater than 100 μM, further demonstrating that 10d (IC50 = 3.66 μM) and 10d-1 exhibited comparable antibiofilm activity. Collectively, these results indicate that 10d-1 is a potent probe for labeling intact bacteria for the in situ profiling of antibacterial synergistic target-drug interactions.
Affinity-based protein labeling and protein identification
Next, we investigated whether probe 10d-1 can be used for simultaneous imaging and covalent labeling of target proteins in vivo. Probe 10d-1 contains a diazirine photoreactive moiety and alkyne analytical handles for CuAAC reactions. To evaluate the live-cell imaging capability of probe 10d-1 for P. aeruginosa PAO1 cells, we added this probe directly to P. aeruginosa cultures, both with and without 10× the amount of (+)-10d as a competitor. Probe 10d-1, 10× competitive (+)-10d, and NP were added to P. aeruginosa cultures at a concentration of 10 μM. Subsequently, the CuAAC click reaction was performed with biotin azide under UV irradiation for the labeling of proteins. As shown in Fig. 6A and Supplementary Fig. 6A, photo labeling experiments with probe 10d-1 were highly successful (panel iii). Labeling bands were observed within the mass range of 25 to 55 kDa, and a strong band was visible in the mass range of 40 to 55 kDa. Weak fluorescence was detected in P. aeruginosa PAO1 cells treated with 10× competitive (+)-10d (panel ii). These results indicated the efficiency of labeling with 10d-1. When added at the same concentration as probe 10d-1, NP (panel i) also exhibited weakly fluorescently labeled proteins in live bacteria, and most of the proteins obtained were similar to those obtained for probe 10d-1. These “false positive” results might be attributed to structural similarities between NP and 10d-1, because both are small molecules containing minimal terminal alkyne-containing diazirine photocrosslinkers. Thus, NP easily formed covalent bonds with proteins and could exist stably. However, in terms of fluorescence intensity, NP labeled the P. aeruginosa PAO1 proteome considerably less selectively than 10d-1, resulting in several intense bands.

A Fluorescent scan of NP and 10d-1 labeled P. aeruginosa PAO1 proteins resolved by SDS-PAGE. B The most abundant protein hits (by score) were identified by ABPP experiments with probe 10d-1. C Fluorescent scan of 10d-1 (1, 10, and 20 μM) labeled P. aeruginosa N-GST-PA1551 proteins resolved by SDS-PAGE. D Effects of 10d-1 on biofilm formation in PA1551-deficient mutant strain. E PA1551-overexpressing strain. F ΔPA1551 strain with a plasmid carrying the PA1551 gene. G pilJ-deficient mutant strain. H PA4133-deficient mutant strain, and I iscS-deficient mutant strain. J Quantification of the binding affinity of 10d to PA1551 was performed using microscale thermophoresis (MST) assays (n = 3). All experiments are performed in triplicate, and data are presented as mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
The CuAAC click reaction of 10d-1 with biotin azide was performed under UV irradiation to label proteins. Subsequently, biotinylated proteins were captured using streptavidin agarose beads and identified via LC − MS/MS. To minimize the “false hits” resulting from nonspecific protein binding, protein bands appearing in LC − MS/MS results that were obtained with 10× competitive (+)-10d and NP were removed. The potential target proteins identified are summarized in Fig. 6C and Supplementary Fig. 2A–D. Notably, most of the downregulated proteins were associated with iron homeostasis. Among the top identified proteins, PilJ is localized at cell poles and is essential for surface-associated twitching motility34. Twitching motility allows P. aeruginosa to respond to stimuli by extending and retracting its type IV pili. Q9I3G6/PA1551 was identified as an uncharacterized protein; it contains a domain belonging to the iron-sulfur family of cytoplasmic membrane proteins, possibly ferredoxin35. The PA4133 gene is predicted to encode the cytochrome c oxidase subunit (cbb3-type), which belongs to the heme-copper oxidase superfamily and facilitates the coupling of oxygen reduction to proton translocation across the membrane36. IscS, an L-cysteine desulfurase (pyridoxal phosphate-dependent), is a master enzyme responsible for delivering sulfur to various partners involved in Fe-S cluster assembly, tRNA modification, or cofactor biosynthesis. IscS plays a crucial role in sulfur delivery for Fe-S cluster synthesis onto IscU, a Fe-S scaffold assembly protein, and other sulfur acceptor proteins37. These results were consistent with the aforementioned results of antibiofilm experiments, indicating that 10d can disrupt iron homeostasis.
Ferredoxin PA1551 may serve as a target to inhibit biofilm formation
Based on the findings of affinity-based protein labeling experiments, we speculate that an uncharacterized protein (PA1551) may play a key role in iron homeostasis (Supplementary Fig. 2D). The PA1551 gene is predicted to encode a ferredoxin cytoplasmic membrane protein, which we refer to as ferredoxin PA1551. The ferredoxin protein family comprises iron-sulfur (Fe-S) proteins that serve as electron carriers in various metabolic reactions and that connect biochemical pathways crucial for energy transduction, nutrient assimilation, and primary metabolism38. One of these subgroups is Fe-S ferredoxins, which are found in bacteria and often referred to as “bacterial-type” ferredoxins38,39,40.
To directly investigate the labeling of ferredoxin PA1551 by the 10d-1 probe, we expressed and purified recombinant N-GST-PA1551 in the E. coli strain BL21 Star (DE3). The purified N-GST-PA1551 protein appeared as a single band with a molecular mass of 79.6 kDa (Supplementary Fig. 3), consistent with the predicted molecular mass based on the DNA sequence. Next, we evaluated whether probe 10d-1 could be used for the simultaneous imaging and covalent labeling of the target protein. We incubated probe 10d-1 at concentrations of 1, 10, and 20 μM with P. aeruginosa cultures. Then, the CuAAC click reaction was performed with biotin azide under UV irradiation to efficiently label proteins. As shown in Fig. 6B and Supplementary Fig. 6, the in-gel fluorescence scanning results for 10d-1 at concentrations of 1, 10, and 20 μM demonstrated highly specific labeling of the 80-kDa N-GST-PA1551 protein (panel i-iii) in a dose-dependent manner. Quantification of the fluorescent band intensity in in-gel fluorescence scanning also showed that ferredoxin PA1551 is the biological target of compound 10d. To evaluate the binding of 10d with ferredoxin PA1551, we carried out a microscale thermophoresis (MST) assay using the full-length membrane protein PA1551 fusion protein with 10d. The results shown in Fig. 6J suggested that the dissociation constant (Kd) for the interaction between 10d and PA1551 was determined to be 56.96 nM, indicating a strong binding preference of 10d for PA1551 and supporting that PA1551 is the primary target of 10d.
To further validate the function of ferredoxin PA1551, we investigated the effects of the PA1551-deficient mutant and PA1551-overexpressing strain on iron homeostasis and biofilm formation in the presence of 10d. As shown in Fig. 6D, E, 10d did not affect the biofilm formation of the PA1551-deficient mutant or the PA1551-overexpressing strain at various concentrations (1.25, 2.5, 5, 10, and 20 μM). However, the complementation of the PA1551-deficient mutant with a plasmid carrying the PA1551 gene fully restored the biofilm inhibitory activity of 10d (Fig. 6F). Furthermore, 10d still led to a significant reduction of biofilm in the absence of pilJ, PA4133, and iscS genes (Fig. 6G–I). These results suggest that 10d disrupted biofilm formation by targeting ferredoxin PA1551.
To elucidate the mechanism of action between 10d and ferredoxin PA1551 at the molecular level, we built a computational model to identify the possible binding mode. The amino acid sequence of ferredoxin PA1551 was downloaded from Uniprot (Uniprot ID: Q9HGN8)41, and the protein structure was modeled using a local version of ColabFold42. ColabFold combines the fast homology search of MMseqs2 with AlphaFold2 to offer an accelerated prediction of unknown protein structures. The models were relaxed using amber force field after folding43. Despite the high-quality models, we still observed lower confidence (pLDDT < 50) in the loop region at the N terminus. Therefore, we conducted a molecular dynamics (MD) simulation to achieve the equilibrium of the N-terminal loop on a transmembrane model generated from the PPM server44. The entire structure reached equilibrium within 40 ns. To identify potential binding sites in the entire protein, a trajectory-based scan was conducted in the second half of the simulation (Supplementary Fig. 4A–C). We identified 43 site clusters from the equilibrium conformations (Fig. 7A). Among these, five clusters that appeared more than 75% of the time were inspected and filtered based on the size (>75) and docking results (Supplementary Table 2). Site 2, which was located around the luminal domain, could serve as a binding site, indicating that 10d is involved in Fe3+ reduction mechanisms (Fig. 7B). Given that cystine plays a crucial role in Fe biological reduction45, this site, which is located along the channel rich in cysteine residues on the luminal domain, may be a candidate site for reduction reactions. On the ligand side, a conformational search was performed to account for the flexibility of 10d before docking. All possible conformations and protonated states were considered. The docking results provided promising affinity data both topologically and energetically. To verify the stability of the predicted binding mode, MD simulation was performed on the transmembrane complex model built using PPM43, As shown in Fig. 7C, D, key interactions between 10d and THR282, ILE295, GLY296, and ALA298 were retained for most of the simulation. The MM/GBSA results based on the equilibrium trajectory supported the firm binding for the complex (ΔG = -64.96 Kcal/mol with STD 7.27). the site consisting of residues from Cys280 to Cys300 exhibited good compatibility for accommodating 10d-Fe3+, aligning with our speculation regarding the mechanism of ferredoxin PA1551.

A Binding site cluster density based on trajectory frames. B Location of site2 in the optimized ferredoxin PA1551 model. C RMSD of protein Cα and 10d. D 10d binds in the bottom of cystine rich channel.
Analysis of PA1551-deficient mutants and -overexpressing strains
Based on our exploration of the function of the previously uncharacterized cytoplasmic membrane protein (PA1551), we hypothesized that its involvement in the reduction of ferric iron is a critical process in iron homeostasis in P. aeruginosa (Fig. 8A). To determine whether ferredoxin PA1551 can reduce Fe3+ to Fe2+ and thus act as a reductase, we investigated the function of ferredoxin PA1551 in P. aeruginosa cells by performing validation experiments in vitro46. The reducing agent DTT can reduce ferric chloride to ferrous chloride. Ferrozine forms a magenta-red complex by chelating divalent iron ions, thus enabling the measurement of this reaction based on an increase in absorbance at 562 nm47. When ferric chloride was incubated with ferredoxin PA1551 and DTT, followed by the addition of ferrozine, we noted a gradual increase in absorbance at 562 nm over time. This result (Fig. 8B) indicated that both the protein and DTT acted in the same manner for reducing trivalent iron ions to divalent ferrous iron ions, suggesting the essential role of ferredoxin PA1551 in the reduction of trivalent iron ions in P. aeruginosa.

A Iron uptake in P. aeruginosa. B Reduction experiment of trivalent iron ions in vitro. 20 μM FeCl3 was incubated in 100 mM ammonium acetate buffer (pH=6.5) in the presence of 100 mM DTT or protein, 200 μM ferrozine. Experimental measurement of 562 nm absorbance. A0 = absorbance at 0 min and A = absorbance at time. C–E The production of iron-free pyoverdine was assessed in iron-supplemented ABTGC (0, 10 μM FeCl3), TSB medium. F–H The production of iron-free pyochelin was assessed in iron-supplemented ABTGC (0, 10 μM FeCl3), TSB medium. I Biofilm formation of P. aeruginosa PAO1, PA1551 deficient mutant and PA1551-overexpressing strains. J Pyocyanin production of P. aeruginosa PAO1, PA1551 deficient mutant and PA1551-overexpressing strain. K Rhamnolipid production of P. aeruginosa PAO1, PA1551 deficient mutant and PA1551-overexpressing strain. All experiments are performed in triplicate, and data are presented as mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
To investigate the role of ferredoxin PA1551 in the iron homeostasis of P. aeruginosa, we examined iron acquisition mechanisms in both the PA1551-deficient mutant and PA1551-overexpressing strains. P. aeruginosa has developed various systems for iron uptake, including those based on pyoverdine (Pvd) and pyochelin (Pch). To determine whether ferredoxin PA1551 interferes with iron homeostasis, we measured the levels of Pvd and Pch through fluorescence detection. These experiments were performed in TSB medium and ABTGC medium. The ABTGC medium is a variant of B-medium, supplemented with 0.1% MgCl2, 0.1% CaCl2, and 0.1% FeCl3, 10% A10, 0.2% glucose, and 0.2% casamino acids. It was prepared with 10 µM or 0 µM FeCl₃ to create iron- normal and iron-deficient conditions, respectively. As depicted in Fig. 8C–H, the PA1551-deficient P. aeruginosa strain produced higher levels of Pvd and Pch to counteract iron deficiency in the ABTGC medium containing 10 μM FeCl3 and in the TSB medium. However, the PA1551-deficient P. aeruginosa strain exhibited negligible effects on the production of Pvd in the ABTGC medium containing 0 μM FeCl3. These results suggest that by interacting with high-affinity Pvd and Pch iron acquisition systems, ferredoxin PA1551 plays a role in iron homeostasis, particularly when a certain amount of iron is already present in the environment. Ferredoxin PA1551 may serve as an electron carrier in this process. The inhibition of electron transfer by disrupting the function of ferredoxin PA1551 can lead to bacterial iron limitation even under iron-rich conditions.
To determine the role of ferredoxin PA1551 in the biofilm formation of P. aeruginosa, we examined the biofilm formation of the PA1551-deficient mutant and PA1551-overexpressing strains. As shown in Fig. 8I, biofilm formation was significantly abolished in the PA1551-deficient mutant. By contrast, biofilm formation was increased in the PA1551-overexpressing strain of P. aeruginosa.
Iron serves as an essential micronutrient for P. aeruginosa, facilitating not only bacterial growth but also playing an essential role in pathogenicity, including the formation of biofilms and the production of virulence factors. Therefore, we have hypothesized that the ferredoxin inhibition may lead to a reduction of virulence. To confirm this hypothesis and investigate how ferredoxin PA1551 regulates the biofilm formation of P. aeruginosa, we measured the expression levels of pyocyanin and rhamnolipids and performed motility assays using the PA1551-deficient mutant, PA1551-overexpressing strain, and wild-type strain. The results shown in Fig. 8J, K revealed that the deletion of PA1551 significantly reduced the expression of pyocyanin and rhamnolipids. However, no significant differences were observed between the PA1551-overexpressing strain and wild-type PAO1. The PA1551-deficient mutant did not display swarming motility compared with the wild-type and PA1551-overexpressing strain (Supplementary Fig. 5). In addition, we investigated the effects of ferredoxin PA1551 on bacterial swimming and twitching motility (Supplementary Fig. 5) and observed no change when compared with the wild-type strain. Because pyocyanin, rhamnolipids, and bacterial swarming motility are critical for the biofilm formation of P. aeruginosa, ferredoxin PA1551 may serve as an ideal target for developing novel antibiofilm therapeutics to combat P. aeruginosa biofilm-associated infections.
Ferredoxin PA1551 mutant exhibits strong antibacterial synergistic effects in vivo
Compared to the wild-type P. aeruginosa strain, the PA1551-deficient mutant strain exhibited a significant reduction in the secretion of various virulence factors and biofilms. This finding indicates that ferredoxin PA1551 plays a crucial role in maintaining typical virulence and biofilm formation in P. aeruginosa. Thus, we used an open-wound infection murine model to determine whether ferredoxin PA1551 can serve as a target for inhibiting biofilm to combat P. aeruginosa infection.
As shown in Fig. 9A, B, wounds infected with the Δferredoxin mutant displayed a lower bacterial survival rate (21.44%). However, no therapeutic effects were observed for wounds infected with PAO1 and treated with saline (control), indicating a significant reduction in the virulence of the PA1551-deficient mutant. Furthermore, the treatment of Δferredoxin-infected wounds with 0.0025 mg/mL Tob (4.0%) or 0.01 mg/mL (0%) and 0.001 mg/mL CIP (3.3%) resulted in the almost complete eradication of bacteria. These findings aligned with those for wounds infected with PAO1 and then treated with the combination of 10d and 0.0025 mg/mL Tob (2.3%) or the combination of 10d and 0.01/0.001 mg/mL CIP (0% to 2.4%), demonstrating that ferredoxin PA1551 plays a similar role as 10d in enhancing antibacterial effects. Similarly, compared with the control group (43.2%) and the wounds infected with Δferredoxin (74.7%), those infected with Δferredoxin and then treated with Tob or CIP exhibited high wound closure rates of 83.2% to 86.5%, emphasizing the importance of ferredoxin PA1551 in P. aeruginosa infection (Fig. 9C, D). In addition, we observed no substantial organ toxicity (Fig. 9E) or effects on the body weight of mice during the experiment (Supplementary Fig. 1C). These results demonstrated that the deficiency of ferredoxin PA1551 had a comparable effect to 10d in enhancing the antibacterial effects of Tob and CIP against P. aeruginosa infection. These results raise the possibility that human infections could be treated by targeting ferredoxin PA1551 in difficult-to-treat bacterial biofilms.

A The representative images of bacterial survival in the wound of mice on agar plate. B The survival rates of P. aeruginosa based on the standard plate counting assay after different treatments in PAO1 (or Δferredoxin) infected wound model, control was defined as 100%. C Visual observation of open-wound model of P. aeruginosa PAO1 (or Δferredoxin) infection treated with control (saline) (or 0.0025 mg/mL Tob, 0.01 mg/mL, and 0.001 mg/mL CIP). D Graphical representation of the open-wound model of P. aeruginosa infection quantitative measurement of wound area within nine days, control was defined as 100%. E Histological hematoxylin and eosin (H&E) staining images of heart, liver, spleen, lung, and kidney tissue sections at day nine after different treatments are indicated. All experiments are performed in triplicate, and data are presented as mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. ****p ≤ 0.0001.
Discussion
A major challenge in managing P. aeruginosa infections is the emergence of multidrug-resistant strains. This obstacle has prompted researchers to explore alternative, nonbiocidal methods to treat and eradicate bacterial infections38,48,49. The strategy of targeting biofilms is promising because it can reduce the use of antibiotics and curb the emergence and spread of antibiotic resistance.
Using chemical proteomics, the PA1551-deficient mutant, and the PA1551-overexpressing strain, we identified that ferredoxin PA1551 can serve as a target to inhibit biofilm. We determined that the uncharacterized cytoplasmic membrane protein (ferredoxin PA1551) is involved in ferrite reduction, which plays an essential role in iron homeostasis in P. aeruginosa50,51. In this study, we referred to ferredoxin encoded by PA1551 as ferredoxin PA1551. We propose that compound 10d52,53 (Fig. 10) forms a complex with Fe3+ outside the bacterial cell. This complex is then transported into the periplasm through the FpvA protein in the outer cell membrane of P. aeruginosa, where Fe3+ is reduced to Fe2+ by ferredoxin PA1551 in the inner membrane with the involvement of periplasmic proteins. Subsequently, the reduced Fe2+ is chelated by periplasmic proteins, which can carry ferrous iron to the transporter proteins, allowing Fe2+ to enter the cytoplasm. By inhibiting the action of ferredoxin PA1551, bacterial iron homeostasis, and quorum sensing can be disrupted, thus impeding biofilm formation. Our findings provide insights into the role of ferredoxin PA1551 in the antibiotic tolerance response, revealing a potential avenue to combat bacterial biofilm formation.

Compound 10d (a synthetic siderophore) loads iron into the periplasm, Fe3+ is reduced to Fe2+ by inner-membrane ferredoxin PA1551 in P. aeruginosa.
In conclusion, by using our chemical probes designed on the basis of biofilm inhibitor 10d, we identified ferredoxin PA1551 as a new target for antibiofilm agents to combat P. aeruginosa infection. We revealed that under normal bacterial growth conditions where an iron source is available, iron homeostasis mediated by ferredoxin PA1551 is critical for the formation of P. aeruginosa biofilms and the secretion of virulence factors, such as pyocyanin, rhamnolipids, Pvd, and Pch (siderophores). Furthermore, we used an open-wound infection murine model to demonstrate that ferredoxin PA1551 can serve as an antibacterial synergistic target to inhibit biofilm in the treatment of P. aeruginosa infections. The ferredoxin PA1551 mutant exhibited the same efficacy as 10d in improving the antibacterial effect of Tob and CIP by more than 200- to 1000-fold. Our findings indicate the potential of ferredoxin PA1551 as a target for inhibiting biofilm formation to combat P. aeruginosa infections.
Methods
Strain and cell culture conditions
Bacterial strains and plasmids used in this study are listed in Supplementary Table 3. E. coli strains were grown at 37 °C in either Luria-Bertani (LB) broth or agar. P. aeruginosa strains were grown at 37 °C in either LB, tryptic soy broth (TSB), ABTGC medium (B-medium54 (0.1% MgCl2, 0.1% CaCl2, 0.1% FeCl3) supplemented with 10% A10, 0.2% glucose and 0.2% casamino acids). PA1551 mutant was grown in LB medium and on LB agar supplemented with 100 µg /mL Gentamicin. PA1551-overexpressed was grown in LB medium and on LB agar supplemented with 100 µg /mL carbenicillin. Escherichia coli BL21 (DE3) carrying PA1551 cloned into the expression vector, pGEX-6P-1-1551 was grown in LB medium supplemented with 100 µg /mL ampicillin.
Plasmid construction
Primers55 used in this study are listed in Supplementary Table 4. To construct the knock-out plasmid for deletion, the PA1551 gene, the 1003-bp upstream fragment and the 1003-bp downstream fragment flanking PA1551 were amplified with primer pairs PA1551 up F/PA1551 up R and PA1551 low F/ PA1551 low R. The upstream and downstream PCR fragments were ligated by overlap PCR, and the resulting PCR products were inserted into the BamHI/HindIII sites of the suicide vector pK18. The gentamicin resistance cassette amplified from Gm was subsequently inserted into the same HindIII site to yield the knock-out plasmid pK18- PA1551.
Expression plasmid constructs were generated by standard methods and verified by DNA sequencing. The plasmids were transformed into E. coli by heat shock and P. aeruginosa strain by electroporation unless otherwise stated. To generate the PA1551 -overexpressing construct p20- PA1551, the encoding region of bswR was amplified with polymerase chain reaction (PCR) primers (Supplementary Table 4), digested with EcoRI and HindIII inserted into the corresponding site of pUCP20.
To construct pGEX-6P-1-PA1551, primer PA1551 F1/ PA1551 R1, PA1551 F2/ PA1551 R2, PA1551 F3/ PA1551 R3 were used to amplify the PA1551 gene. These PCR products of relevant genes were digested with BamHI/HindIII and inserted into the BamHI/HindIII sites of pGEX6P-1 resulting in plasmids pGEX6p-1-PA1551. The integrity of the insert in all constructs was confirmed by DNA sequencing.
For constructing pET-28a-1551 (full-length), the primer pair 1551 f and 1551r were used to amplify the PA1551 (full-length) fragment from the genomic DNA. Vector was digested with BamHⅠ and HindⅢ restriction enzymes and purified. The PCR products were digested and inserted into pET-28a to generate pET-28a-1551 (full-length) using the Seamless cloning kit. The resulting plasmid was transformed into E. coli DH5α, and the plasmid construct was verified by sequencing using primers that annealed to sites outside the multiple cloning site.
N-GST-PA1551 expression
A single bacterial colony of E. coli BL21 (DE3) carrying pGEX-6P-1-PA1551 strain was inoculated into 1 L of fresh LB medium flask supplemented with 100 ug/mL gentamycin. The culture was once again grown at 37 °C with good aeration on a rotary shaker (200 rpm) until OD600 reached 0.5-1. Protein expression was induced by the addition of IPTG to a final concentration of 1 mM. Following induction, the temperature was lowered to 16 °C, and growth (with vigorous aeration, shaking at 200 rpm) was continued overnight. The bacterial pellet expressing recombinant protein was collected by sedimentation (4000 rpm, 15 min, 4 °C). The supernatant was discarded and the cell pellet was washed with ice-cold 1×phosphate-buffered saline solution (PBS).
Purification of N-GST-PA1551 protein using AKTA pure protein purification layer system
GST-tagged recombinant proteins were purified as described56. Fractions containing protein were pooled and analyzed by SDS-PAGE (4% stacking and 12% resolving gel). The purification protein samples were stored at -80 °C.
Protein expression and purification: The plasmid pET-28a-1551 (full-length) was transformed into E. coli BL21 (DE3), enabling the expression of PA1551 (full-length) membrane protein containing a 6-His tag. PA1551 (full-length) was induced with 0.5 mM IPTG overnight at 16 °C. Cells were harvested and resuspended in lysis buffer composed of 30 mM HEPES pH 6.8, 150 mM NaCl, and 0.01% DDM (n-Dodecyl-β-D-Maltoside). Bacteria were lysed using probe sonication for a duration of 40 minutes, followed by the removal of cell debris through centrifugation at a speed of 15,000 rpm for 30 minutes. The supernatant was loaded onto a Ni-NTA column (L00250-50, GeneScript). The resin was washed with lysis buffer supplemented to 90 mM imidazole and subsequently eluted with lysis buffer supplemented to 300 mM imidazole. Where necessary, proteins were concentrated via filtration using centrifugation and a 10 kDa cut-off.
Microscale thermophoresis (MST) assay
The PA1551 (full-length)-His protein, at a 100 nmol/L concentration, was subjected to fluorescent tagging using a His-tag labeling kit (Nano Temper, MO-L008), adhering to the guidelines specified in the user manual. After the labeling was finalized, 10 μM of the fluorescently tagged ferredoxin PA1551 (full-length)-His protein was mixed with 10 M of PBST buffer, followed by the addition of 10d at a concentration of 1 mM. This mixture was then subject to serial dilution. The microscale thermophoresis assay was carried out on a NanoTemper Monolith NT.115 device, set to 20% LED power and medium MST power setting. Data collected from the assay were analyzed using the MO. Affinity Analysis software (X86).
In situ protein labeling
P. aeruginosa was inoculated in 5 mL LB and cultivated (200 rpm, 37 °C) until the culture reached OD600 to 0.05. Probe 10d-1 (10 μM), NP (10 μM), and 10d-1(10 μM)- 10d (50 μM) as the competitor were incubated for 24 h at 37 °C, with shaking (200 rpm). After 16 h incubation, the cultures were then centrifuged for 10 min at 4000 rpm and the cell pellets were washed with 2× PBS to remove excessive probe, followed by UV (365 nm) irradiation for 20 min on ice25. The cell pellets were resuspended in HEPES buffer (25 mM HEPES, 150 mM NaCl, 2 mM MgCl2) with a 1% protease inhibitor cocktail (APExBIO, EDTA-Free in DMSO). The suspension was repeatedly frozen and thawed 3 times followed by sonication for 1 h (15 s on and 15 s off). The precipitate was removed by centrifugation and the supernatant was adjusted to 1 mg/mL 1.6 mL of a freshly premixed click chemistry reaction cocktail (50 mM TAMRA-biotin-N3 from 50 mM stock solution in DMSO, 0.1 mM TBTA from 100 mM freshly prepared stock solution in DMSO, 1 mML TCEP from 1 M freshly prepared stock solution in deionized water, and 1 mM CuSO4 from 1 M freshly prepared stock solution in deionized water) was added. The reaction was incubated for 2 h with gentle mixing at r.t before being terminated by the addition of prechilled Me2CO (incubation at 20 °C for 1 h). Precipitated proteins were subsequently collected by centrifugation (10,000 rpm, 10 min at 4 °C). The supernatant was discarded and the pellet was washed twice with prechilled MeOH before redissolving in 2×loading buffer and heating for 10 min at 95 °C. Proteins were separated by SDS-PAGE (10% gel) and then visualized by in-gel fluorescence scanning.
Concentration-dependent N-GST-PA1551 labeling
Purified N-GST-PA1551 protein samples were thawed, and the protein concentration was measured. Samples adjusted to contain 0.5 mg/mL of N-GST-PA1551 in PBS, were incubated in the presence of probe 10d-1 (1 μM, 10 μM, and 20 μM). Cu2+-mediated azide-alkyne coupling (CuAAC) reactions were above. Proteins were separated by SDS-PAGE (10% gel) and then visualized by in-gel fluorescence scanning.
Large-scale Protein Identification by LC − MS/MS: StageTips were prepared according to a modified protocoFl57 with a C18 solid-phase extraction disc (3 M Empore). Large-Scale Protein Identification by LC − MS/MS, for subsequent MS analysis, the samples were resuspended (with sonication in a water bath) in 10 mL of H2O containing 0.5% FA.
Biofilm formation tested by crystal violet assay
The effect of compounds on biofilm formation in P. aeruginosa was measured by crystal violet assay. P. aeruginosa PAO1, ΔPA1551, PAO1(PA1551), ΔPA1551(PA1551), ΔpilJ, ΔPA4133, and ΔiscS were grown overnight in LB medium at 37 °C for 24 h. First, bacterial cultures were diluted in ABTGC medium to an OD₆₀₀ of 0.05 and were transferred to sterile 96-well flat-bottomed polystyrene microtiter plates (Corning/Costar, NY, USA). Next, compounds were added, and the plates were incubated at 37 °C for 24 h. Subsequently, supernatant cells were removed and the biofilms were washed three times with sterile phosphate-buffered saline (PBS), then MeOH as a fixative. After 30 min, the MeOH was removed, and the microtiter plates were dried at RT. Crystal violet (0.1% in H2O) was then added to dye the biofilms which were then incubated for 30 min. Crystal violet was removed, microtiter plates were rinsed three times with sterile PBS, and add 150 μL of acetic acid (33%) to dissolve the biofilm. The absorbance values were measured at 570 nm by a microplate reader. All compounds were dissolved in DMSO to make 100 μM stock solutions before transferring to 96-well plates for the experiment.
Biofilm formation tested by TTC assay
This experiment used the same culture medium and bacteria as the crystal violet staining method. A single colony of P. aeruginosa PAO1 was plated in LB medium one day in advance and cultured at 37 °C for 16 h. At the start of the experiment, the metabolic dye 2,3,5-triphenyltetrazolium chloride TTC (final concentration 0.05%), various concentrations of the compound, and bacteria (total concentration OD600 = 0.01) were added to each well. The plates were incubated overnight at 37 °C under static conditions to allow bacterial growth and biofilm maturation. The next day, non-adherent cells and spent culture medium were discarded, and adherent biomass was rinsed three times with distilled water. The metabolized TTC dye (red in the wells) was resuspended in 100 μL of methanol, gently mixed, and the absorbance at 500 nm was recorded on a microplate reader. Results represent the average of three independent biological replicates for each bacterial strain.
Rhamnolipid assay
Rhamnolipid was quantified according to the method of Koch et al, with modifications. A subculture was conducted by directly diluting the overnight culture 1:100 into fresh Minimal Medium (49.3 mM Na2HPO4, 50 mM KH2PO4, 4.8 mM MgSO4, 7.6 mM (NH4)2SO4, 0.6 mM CaCl2, 25 μM FeSO4, 0.162 μM (NH4)6Mo7O24, 38 μM ZnSO4, 14 μM MnCl2, 1.6 μM CuSO4, 0.86 μM CoCl2, 1.9 μM boric acid, 5.5 μM NiCl2, 6.72 μM EDTA, 0.6% glycerol in 18 MΩ deionized H2O). The cultures were grown for 24 h at 37 °C, with shaking (200 rpm). The final cell density was measured at 600 nm (OD600) using a microplate reader (Bio-Tek). Supernatants were collected by ̈centrifuging at 10000 rpm for 10 min and extracted with Et2O (twice). Organic fractions were concentrated to yield a white solid. It was resuspended in deionized H2O and added with 0.19% (w/v) orcinol in 50% H2SO4. The resulting mixture was incubated at 80 °C for 30 min to give a yellow-orange solution. After cooling to rt, the absorbance was measured at 421 nm and results were normalized with final OD600 values.
Pyocyanin assay
Overnight culture of P. aeruginosa PAO1, PA1551 deficient mutant, and PA1551-overexpressing strain were standardized to OD600 of 0.05 and diluted 100 times into 25 mL of LB Medium (Sangon Biotech, China) in 250 mL flask. The cultures were grown for 48 h at 37 °C, with shaking (200 rpm). The cultures were then centrifuged for 10 min at 10000 rpm and 8 mL of the supernatants were transferred into new tubes. Briefly, 8 mL of culture supernatant conducted as described above was extracted with chloroform at a ratio of 4:1, followed by extraction with 1 mL of 0.2 M HCl. The absorbance of the upper red phase was measured using the OD520. The data was normalized by dividing the OD520 reading with the final OD600 values.
Swarming motility assay
To monitor swarming, petri dishes were filled with 20 mL of LB medium supplemented with 0.5% (w/v) bacto agar (Becton, Dickinson and Co.), 0.5% (w/v) casamino acids(Becton, Dickinson and Co.) and 0.5% (w/v) glucose (Solarbio, China) in the presence of P. aeruginosa PAO1, PA1551 deficient mutant, and PA1551 overexpressing strains. Petri dishes were dried in a single stack for 1 h at RT and then dropped 1 μL PAO1 (OD600 = 1) onto the center of petri dishes and open the petri dishes to dry. The petri dishes were incubated for 16 h at 37 °C.
Swimming motility assay
To monitor swimming, petri dishes contained 20 mL of LB medium supplemented with 0.3% (w/v) bacto agar, 0.5% (w/v) casamino acids and 0.5% (w/v) glucose in the presence of P. aeruginosa PAO1, PA1551 deficient mutant, and PA1551 overexpressing strains. Petri dishes were dried in a single stack for 1 h at rt and then dropped 1 μL PAO1 (OD600 = 1) into the bottom of the petri dishes and open the petri dishes to dry. The petri dishes were incubated for 16 h at 37 °C.
Twitching motility assay
To monitor twitching, petri dishes were charged with 20 mL of LB medium supplemented with 1% (w/v) bacto agar, 0.5% (w/v) casamino acids and 0.5% (w/v) glucose in the presence of P. aeruginosa PAO1, PA1551 deficient mutant, and PA1551 overexpressing strains. Petri dishes were dried in a single stack for 1 h at room temperature and then dropped 1 μL PAO1 (OD600 = 1) into the bottom of the petri dishes and open the petri dishes to dry. The petri dishes were incubated for 16 h at 37 °C.
Pyoverdine and pyochelin assay
An overnight culture of P. aeruginosa PAO1 (grown in LB medium at 37 °C, 200 rpm) was diluted in ABTGC medium to a final optical density at 600 nm (OD600) of 0.02 (2.5 × 108 CFU/mL). The microtiter plate was incubated at 37 °C in a microplate reader (Bio-Tek) to measure the cell density (OD600), Pyoverdine fluorescence (excitation at 400 nm, emission at 447 nm), pyochelin fluorescence (excitation at 350 nm, emission at 430 nm). The experiment assay for all test compounds and controls was done in triplicate.
Reduction experiment of trivalent iron ions in vitro
20 μM FeCl3 was incubated in 100 mM ammonium acetate buffer (pH=6.5) in the presence of 100 mM DTT or protein, 200 μM ferrozine. Subsequently, the absorbance at 562 nm was monitored for 120 min. A0 = absorbance at 0 min and A = absorbance at testing time.
Validation of PA1551 protein for iron reduction in vitro
After the constructed GST PGEX6P-1 strain induced a large amount of protein expression, the bacteria were lysed and the supernatant was extracted. To a solution of 20 μM Ferric chloride in 200 μL of 100 mM ammonium acetate buffer, 100 mM DTT or the supernatant of GST PGEX6P-1 strain was added at 37 °C, then added separately 200 μM ferrozine in a 96-well plate. The 96-well plate was incubated at 37 °C in a microplate reader (BioTek, Synergy H1) to measure the absorbance at 562 nm, every 5 min for 1.5 h.
Computational methods
The sequence of Ferredoxin PA1551 from Uniprot41 was folded on a server with a local version of ColabFold42. Amber forcefield was used to relax the models. Top-ranked models were uploaded to PPM 2.0 server to predict the transmembrane region44. Apo protein with membrane system was prepared and constructed in Maestro protein preparation and Desmond system builder tools58,59. The orthorhombic simulation box was solvated of SPC water molecules, counterions, and an additional 0.15 M concentration of NaCl. A 100 ns molecular dynamics was performed using Desmond to settle the loops, details are as in our previous work if not specified. SiteMap was utilized on every 10 frames of the second 50 ns trajectory by a script60. All the sites identified were visually inspected. Top sites with preferable scores and sizes were all tested in Glide docking with the 10d conformations generated from LigPrep. The best 3 binding complexes were further validated in MD simulation, regular protein-ligand job settings were applied, and interaction and binding free energy analysis were performed to evaluate the affinity.
Wound infection experiment in mice
Ethical Statement. P. aeruginosa strain PAO1 was used for this animal experiment. Female 5-week-old BABL/C mice were purchased from SPF Biotechnology Co., Ltd with certificate number 20180302005. All animal experiments were performed under the animal care and use guidelines (IACUC approval number: Szwwbio-IACUC-20230930-01). During the whole experiment, the mice were kept at a constant temperature of 25 °C, with a 12 h light/night cycle, and provided with sufficient food and water without any animal cruelty. At the end of the experiment, the mice were all euthanized.
Firstly, mice were randomly divided into different groups of 5/group and treated with 4% chloral hydrate, then the hair was shaved off the back of the mice and a circular wound of 4-5 mm was created on the back of each mouse. The wound was inoculated with 5×108 CFU of PAO1 (or Δferredoxin) to establish a wound infection model for 24 h. The mice in different groups were then treated with different drugs (saline, 0.5 mg/mL Tob, 0.0025 mg/mL Tob, 0.0025 mg/mL Tob+10 μM 10d, 10 μM 10d, 1 mg/mL CIP, 0.01 mg/mL CIP, 0.001 mg/mL CIP, 0.01 mg/mL CIP + 10 μM 10d, 0.001 mg/mL CIP + 10 μM 10d). Mice were executed after three days of continuous dosing, wounded skin was cut off and homogenized, serially diluted and CFU counts were done on agar plates. In addition, histological sections were taken from the heart, liver, spleen, lung, and kidney of the mice.
Data availability
All data generated in this study are available upon request from the corresponding author.
References
Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12, 371–387 (2013).
Brown, E. D. & Wright, G. D. Antibacterial drug discovery in the resistance era. Nature 529, 336–343 (2016).
Blair, J. M. A. et al. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015).
Rossiter, S. E., Fletcher, M. H. & Wuest, W. M. Natural products as platforms to overcome antibiotic resistance. Chem. Rev. 117, 12415–12474 (2017).
Abouelhassan, Y. et al. Recent progress in natural-product-inspired programs aimed to address antibiotic resistance and tolerance. J. Med. Chem. 62, 7618–7642 (2019).
Huigens, R. W. et al. Inhibition of Pseudomonas aeruginosa biofilm formation with bromoageliferin analogues. J. Am. Chem. Soc. 129, 6966–6967 (2007).
Hancock, R. E. W. & Speert, D. P. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist. Updat. 3, 247–255 (2000).
Yayan, J., Ghebremedhin, B. & Rasche, K. Antibiotic resistance of Pseudomonas aeruginosa in pneumonia at a single university hospital center in germany over a 10-year period. PLoS ONE 10, e0139836 (2015).
Starkey, M. et al. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 10, e1004321 (2014).
Defoirdt, T. Quorum-sensing systems as targets for antivirulence therapy. Trends Microbiol. 26, 313–328 (2018).
Nguyen, D. et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986 (2011).
Flemming, H. C. & Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633 (2010).
Wu, Y. K., Cheng, N. C. & Cheng, C. M. Biofilms in chronic wounds: pathogenesis and diagnosis. Trends Biotechnol. 37, 505–517 (2019).
James, G. A. et al. Biofilms in chronic wounds. Wound Repair Regen. 16, 37–44 (2008).
Kolpen, M. et al. Bacterial biofilms predominate in both acute and chronic human lung infections. Thorax 77, 1015–1022 (2022).
Versey, Z. et al. Biofilm-innate immune interface: contribution to chronic wound formation. Front. Immunol. 12, 648554 (2021).
Sauer, K. et al. The biofilm life cycle: expanding the conceptual model of biofilm formation. Nat. Rev. Microbiol. 20, 608–620 (2022).
Martin, P. & Leibovich, S. J. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 15, 599–607 (2005).
Percival, S. L., McCarty, S. M. & Lipsky, B. Biofilms and wounds: an overview of the evidence. Adv. Wound Care 4, 373–381 (2015).
Liu, J. et al. Novel 2-substituted 3-hydroxy-1,6-dimethylpyridin-4(1H)-ones as dual-acting biofilm inhibitors of Pseudomonas aeruginosa. J. Med. Chem. 63, 10921–10945 (2020).
Liu, J. et al. New pqs quorum sensing system inhibitor as an antibacterial synergist against multidrug-resistant Pseudomonas aeruginosa. J. Med. Chem. 65, 688–709 (2022).
Ziegler, S., Pries, V., Hedberg, C. & Waldmann, H. Target identification for small bioactive molecules: finding the needle in the haystack. Angew. Chem. Int. Ed. Engl. 52, 2744–2792 (2013).
Kleiner, P. et al. A whole proteome inventory of background photocrosslinker binding. Angew. Chem. Int. Ed. 56, 1396–1401 (2017).
Pan, S. et al. Target identification of natural products and bioactive compounds using affinity-based probes. Nat. Prod. Rep. 33, 612–620 (2016).
Li, Z. et al. Design and synthesis of minimalist terminal alkyne-containing diazirine photo-crosslinkers and their incorporation into kinase inhibitors for cell- and tissue-based proteome profiling. Angew. Chem. Int. Ed. 52, 8551–8556 (2013).
Schütz, C. et al. A new pqsR inverse agonist potentiates tobramycin efficacy to eradicate Pseudomonas aeruginosa biofilms. Adv. Sci. 8, e2004369 (2021).
West, K. H. J. et al. Sustained release of a synthetic autoinducing peptide mimetic blocks bacterial communication and virulence in vivo. Angew. Chem. Int. Ed. Engl. 61, e202201798 (2022).
Thorn, C. R. et al. Liquid crystal nanoparticles enhance tobramycin efficacy in a murine model of Pseudomonas aeruginosa biofilm wound infection. ACS Infect. Dis. 8, 841–854 (2022).
Tian, S. et al. Self-targeting, zwitterionic micellar dispersants enhance antibiotic killing of infectious biofilms—an intravital imaging study in mice. Sci. Adv. 6, eabb1112 (2020).
Hatanaka, Y. & Sadakane, Y. Photoaffinity labeling in drug discovery and developments: chemical gateway for entering proteomic frontier. Curr. Top. Med. Chem. 2, 271–288 (2002).
Halloran, M. W. & Lumb, J. P. Recent applications of diazirines in chemical proteomics. Chemistry 25, 4885–4898 (2019).
Shi, H., Zhang, C. J., Chen, G. Y. & Yao, S. Q. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J. Am. Chem. Soc. 134, 3001–3014 (2012).
Haney, E. F., Trimble, M. J. & Hancock, R. E. W. Microtiter plate assays to assess antibiofilm activity against bacteria. Nat. Protoc. 16, 2615–2632 (2021).
DeLange, P. A., Collins, T. L., Pierce, G. E. & Robinson, J. B. PilJ localizes to cell poles and is required for type IV pilus extension in Pseudomonas aeruginosa. Curr. Microbiol. 55, 389–395 (2007).
Hazan, R. et al. Auto poisoning of the respiratory chain by a quorum-sensing-regulated molecule favors biofilm formation and antibiotic tolerance. Curr. Biol. 26, 195–206 (2016).
Xie, H. et al. Biochemical and biophysical characterization of the two isoforms of cbb3-type cytochrome c oxidase from Pseudomonas stutzeri. J. Bacteriol. 196, 472–482 (2014).
Fulton, R. L., Irons, J. & Downs, D. M. The cysteine desulfurase Iscs is a significant target of 2-aminoacrylate damage in Pseudomonas aeruginosa. mBio 13, e0107122 (2022).
Douafer, H., Andrieu, V., Phanstiel, O. T. & Brunel, J. M. Antibiotic adjuvants: make antibiotics great again!. J. Med. Chem. 62, 8665–8681 (2019).
Otaka, E. & Ooi, T. Examination of protein sequence homologies: IV. Twenty-seven bacterial ferredoxins. J. Mol. Evol. 26, 257–267 (1987).
Duée, et al.Refined crystal structure of the 2[4Fe-4S] ferredoxin from clostridium acidurici at 1.84 A resolution. J. Mol. Biol. 243, 683–695 (1994)..
Stover, C. K. et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 (2000).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
He, X. et al. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 114502 (2020).
Lomize, M. A. et al. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Chen, S. M. The electrocatalytic reactions of cysteine and cystine by water-soluble iron porphyrin, manganese porphyrin and iron(II) phenanthrolines. Electrochim. Acta 42, 1663–1673 (1997).
Ganne, G. et al. Iron release from the siderophore pyoverdine in Pseudomonas aeruginosa involves three new actors: FpvC, FpvG, and FpvH. ACS Chem. Biol. 12, 1056–1065 (2017).
Stookey, L. L. Ferrozine-a new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781 (1970).
Qu, D. et al. A new coumarin compound DCH combats methicillin-resistant Staphylococcus aureus biofilm by targeting arginine repressor. Sci. Adv. 6, eaay9597 (2020).
Zhao, X. L. et al. Glutamine promotes antibiotic uptake to kill multidrug-resistant uropathogenic bacteria. Sci. Transl. Med. 13, eabj0716 (2021).
Soldano, A. et al. Small molecule inhibitors of the bacterioferritin (BfrB)–ferredoxin (Bfd) complex kill biofilm-embedded Pseudomonas aeruginosa cells. ACS Infect. Dis. 7, 123–140 (2021).
Soldano, A., Yao, H., Chandler, J. R. & Rivera, M. Inhibiting iron mobilization from bacterioferritin in Pseudomonas aeruginosa impairs biofilm formation irrespective of environmental iron availability. ACS Infect. Dis. 6, 447–458 (2020).
Bonneau, A., Roche, B. & Schalk, I. J. Iron acquisition in Pseudomonas aeruginosa by the siderophore pyoverdine: an intricate interacting network including periplasmic and membrane proteins. Sci. Rep. 10, 120 (2020).
Imperi, F., Tiburzi, F. & Visca, P. Molecular basis of pyoverdine siderophore recycling in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 106, 20440–20445 (2009).
Fong, J. et al. Disulfide bond-containing ajoene analogues as novel quorum sensing inhibitors of Pseudomonas aeruginosa. J. Med. Chem. 60, 215–227 (2017).
Rybtke, M. T. et al. Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 78, 5060–5069 (2012).
Wang, T. et al. Type VI secretion system transports Zn2+ to combat multiple stresses and host immunity. PLoS Pathog. 11, e1005020 (2015).
Schrödinger, LLC. Schrödinger Release 2023-1: Protein Preparation Wizard; Epik (2023).
D. E. Shaw Research. Schrödinger Release 2023-1: Desmond Molecular Dynamics System (2023).
Schrödinger, LLC. Schrödinger Release 2023-1: Sitemap (2023).
Schrödinger, LLC. Schrödinger Release 2023-1: Glide (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 82173651 and 82473788).
Author information
Authors and Affiliations
Contributions
J.L. and A.R. contributed equally to this work. J.L. and A.R. wrote the manuscript and performed most of the experiments. W.C. designed the project and supervised the chemical experiments as well as revised the manuscript; L.Y. supervised the experiments of target identification and revised the manuscript; Z.M. performed chemical experiments. T.Z., C.Z., Y.C., S.Z., X.H., X.Z., T.J., Z.C., Z.L., and J.L. performed some chemical experiments or biological experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, J., Ren, A., Miao, Z. et al. Identification of ferredoxin PA1551 as an antibacterial synergistic target for biofilm inhibitors against Pseudomonas aeruginosa. npj Biofilms Microbiomes 12, 5 (2026). https://doi.org/10.1038/s41522-025-00871-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-025-00871-y
