
Abstract
Triple Negative Breast Cancer (TNBC) accounts for ~20% of all breast cancers and results in thousands of deaths every year. The median survival of TNBC patients sharply declines with the development of chemoresistance and metastatic disease. Although high expression of ETS1 in TNBC has been associated with aggressiveness, the mechanisms of ETS1 in TNBC therapy relapse are poorly understood. Here, we show that ETS1 is responsible for driving acquired drug resistance in the TNBC cell line models resistant to 5’-Fluorouracil and doxorubicin. Protein kinase, DNAPKcs (aka PRKDC) mediated phosphorylation of ETS1 at Serine 251 residue enhances protein stability by preventing ETS1’s degradation, thus enhancing ETS1-driven resistance mechanisms. Further, transcriptomic profiling of resistant cells and TNBC patients showed that phosphorylated-ETS1 could activate genes of the E2F, MYC and G2/M pathways, resulting in enhanced DNA synthesis and proliferation, leading to resistance. DNAPKcs inhibitors resulted in ETS1 degradation, inhibition of proliferation gene circuits and subsequent apoptosis in resistant TNBC cells. Phospho-S251 ETS1 and associated ETS1-driven proliferative gene signatures were observed in drug-resistant TNBC patients. Our findings suggest that DNAPKcs-mediated phosphorylation of ETS1 promotes chemoresistance in TNBC patients and can be targeted using DNAPKcs kinase inhibitors.
Similar content being viewed by others

Introduction
Triple Negative Breast Cancer(TNBC) represents almost 20% of all breast cancers and is considered one of the most aggressive and lethal cancers in women1. TNBC is known to be proliferative with faster recurrence and low overall survival2. The median survival of TNBC patients is reduced (~14-24 months) with the onset of therapy relapse and metastatic disease2,3,4,5. The absence of estrogen, progesterone and HER2 receptors in TNBC has made treatments reliant on chemotherapy6. Unfortunately, resistance mechanisms towards both conventional drugs and neoadjuvant combinations, including Anthracyclines, Taxanes, Platinum compounds, Taxol, Fluoropyrimidine, Olaparib, and TKI inhibitors, have been implicated in TNBC-therapy relapse7,8,9,10. In this regard, aberrantly expressed oncogenic transcription factors regulating various aspects of drug resistance can be potential therapeutic targets11,12. ETS1, an oncogenic transcription factor expressed highly in TNBC, has been implicated in transcriptional control of oncogenic processes like invasion, metastasis, proliferation and drug resistance13,14,15,16. ETS1 has proven roles in transcriptional remodelling of immune cells (T cells, NK cells) and fibroblasts polarization17,18. For example, ETS1 can transcriptionally activate regulatory genes like E4BP4, TXNIP, TBET, GATA3, HOBIT, and BLIMP1 for NK cell activation and differentiation17. ETS1 can drive fibroblast polarization by activating regulatory elements of the osteoclast differentiation factor receptor activator of nuclear factor-κB ligand (RANKL) and matrix metalloproteinases18. In this regard, post-translational modifications of ETS1, like phosphorylation, have been reported to control its stability and transcriptional functions19,20,21. For example, ERK-mediated phosphorylation at T38 and S41 of ETS1 enhances its transcriptional activities, backhandedly phosphorylated ETS1 dictates the genome-wide promoter occupancy over Ras/ERK inducible target genes, indicating phosphorylated ETS1 to be a genome-wide effector, while SRC-mediated phosphorylation was shown to control its protein stability19,20,21. PKCalpha-mediated phosphorylation of ETS1 can also activate transcriptional programs related to invasiveness in TNBC cells22. However, the cause of ETS1-mediated drug resistance in various cancers remains poorly understood, with limited clinical evidence16,23,24,25,26,27.
Heightened activity of DNA repair kinase, DNAPKcs, has been reported to have a poor prognosis and outcome in TNBC patients28. DNAPKcs has been reported to interact and subsequently phosphorylate various cancer-related transcription factors and promote their functions, including Oct-129, Fos and Jun30, MYC31, p5332. DNAPKcs holoenzyme was shown to interact and phosphorylate ETS1 protein33. However, the phosphorylation residue remained unidentified, and the functionality of the interaction over ETS1 transactivation was not well understood.
Here, we show that increased ETS1 expression is directly responsible for developing a resistance phenotype in TNBC. DNA repair kinase, DNAPKcs, can phosphorylate and stabilise ETS1 protein expression, increasing the growth of resistant cells. Blocking DNAPKcs-mediated phosphorylation results in proteasomal degradation of ETS1, leading to apoptosis and increased drug susceptibility. DNAPKcs phosphorylates the Serine 251 residue of ETS1, which was observed only in resistant TNBC cells or chemotherapy-relapsed TNBC patients. Transcriptomics profiling of resistance TNBC cells and patients indicated a strong correlation for cell proliferation pathways. ETS1 can transcriptionally activate critical genes of the E2F, MYC and G2/M pathways during acquired resistance, increasing DNA synthesis and proliferation. These pathway genes were activated in chemotherapy-resistant patients and resistant TNBC cell types treated with various drugs, indicating the existence of a unique ETS1-driven mechanism in chemoresistance. DNAPKcs inhibition blocked the activation of these genes and subsequently decreased DNA synthesis and proliferation in resistant cells. The findings indicated a drug resistance mechanism mediated via the DNAPKcs-ETS1 axis that drives cell proliferation networks, which can be further exploited therapeutically.
Results
Altered ETS1 expression promotes acquired drug resistance
ETS1 expression and transcriptional activity are associated with various aspects of TNBC progression like angiogenesis, metastasis, chemoresistance and low survival13,14,15,16. Since Fluoropyrimidine-like 5’Fluorouracil(5’FU) in combination with Adriamycin/Doxorubicin and Cyclophosphamide(CA) are widely used as the first line of treatment against TNBCs34,35,36, we have generated resistant TNBC cell line models (Res-MDA-MB231, Res-MDA-MB468, Res-MDA-MB453) with either 5’FU or Adriamycin/Doxorubicin, as described previously37,38,39,40. The IC50 of the resistant TNBC cells increased significantly compared to non-resistant parental cells (Fig. S1a, S1b). We observe an increase in IC50 and resistance index of our resistant cells (Res-MDA-MB231, Res-MDA-MB468) to other chemotherapeutics (Gemcitabine, Cisplatin, Doxorubicin/Adriamycin) used clinically for TNBC, indicating cross-resistance in our cell line models (Fig. S1c, S1d, S1e and S1f). To understand ETS1’s role in drug resistance, we assessed the expression of ETS1 and its bona fide downstream target genes, MMP3 and TWIST1, in Res-MDA-MB231, Res-MDAMB468 and Res-MDAMB453 cells41,42. We have observed increased protein but not transcript levels for ETS1, suggesting a possibility for post-translational regulation. We found elevated levels of MMP3 and TWIST1 as compared to the parental cells (Fig. 1a, b and S1g). We observed similar results for ETS1 and its target genes in MDA-MB231 cells resistant to Adriamycin and cyclophosphamide (CA) (Fig. S1h). MDA-MB231(R) or MDA-MB468(R) cells made resistant using Gemcitabine (GEM), Cisplatin (CDDP) or Doxorubicin (DOX) revealed increased levels of ETS1 protein in comparison to parental cell lines; MDA-MB231 (C) and MDA-MB468 (C), suggesting a role of ETS1 protein in the development of acquired resistance (Fig. S1i). Interestingly, we didn’t observe any change in ETS1 protein levels in parental MDA-MB231 cells subjected to DNA damage using various chemotherapeutic drugs, indicating the induced ETS1 protein level to be a feature in resistant TNBC cells (Fig. S1j). We observed increased colony formation in resistant TNBC cells compared to parental cells in the presence of 5’FU or Adriamycin (Figs. 1c, and S1k,S1l). Silencing ETS1 expression in resistant TNBC cells decreased colony forming ability in the presence of 5’FU, indicating a direct role of ETS1 in the regulation of resistant phenotype (Fig. S1m).

See also fig. S1. a Whole cell extract from parental and resistant MDA-MB231 and MDA-MB468 cells was immunoblotted against indicated proteins. b Relative transcript levels of genes from parental and resistance cell lines were analyzed using qRT-PCR. c Colony formation ability of indicated cells in the presence of 5’FU. The optical density of stained cells was measured at 595 nm. d Whole cell extract from resistant cells treated with indicated compounds for 48 h was immunoblotted using indicated antibodies. e Resistant cells were transfected with indicated siRNA for 48 h and subjected to immunoblotting against the indicated proteins. f (top panel) Representative immunoblots of the indicated proteins from resistance cells pre-treated with cycloheximide (CHX) and DMSO or DNAPKcs inhibitor for indicated time points. (bottom panel) Phase decay plot showing half-life (t1/2) of ETS1 protein normalized to actin, using images of immunoblots from three independent experiments per cell line. g Colony formation ability of indicated cells was assessed in the presence of DMSO or DNAPKcs inhibitor. The optical density of stained cells was measured at 595 nm. h Resistant cells were treated with increasing concentrations of 5’FU and DMSO or DNAPKcs inhibitor. Percent cell viability was plotted against drug concentration post 72 h of treatment. i Relative caspase3/7 activity of resistant cells treated with either DMSO or DNAPKcs inhibitor for the indicated time was measured. Experiments were performed as biological triplicates (mean ± SD). Two-way ANOVA with Sidak’s multiple comparison test was used for the statistical significance of 1b. For 1c, g, an unpaired t-test with Welch’s correction was applied. For 1i; one way ANOVA with Dunnett’s multiple comparison test was applied where ****p < .0001, ***p = 0.0001,**p = .001, *p = 0.01, n.s=not significant.
In response to chemotherapeutic agents-induced DNA damage, the repair activity of PIKK family kinases (ATM, ATR, DNAPKcs, MTOR) has been recently associated with acquired chemoresistance43,44,45,46,47. We envisage that repair kinases belonging to the PIKK family might regulate ETS1 expression and aid in TNBC resistance. Breast cancer cell lines of the TNBC lineage express high levels of ETS1 and PI3K-related kinases (PIKK), especially DNAPKcs and MTOR, which are associated with DNA repair (Fig. S1n). Since 90% of TNBCs represent Basal-like breast cancer48,49, we checked the co-expression pattern between ETS1 and PIKK expression in luminal and basal type breast cancer patients50. We observed positive co-expression for ETS1 and DNAPKcs in patients with basal subtype but not in Luminal grade breast cancer cases (Fig. S1o). We found similar correlation of DNAPKcs and ETS1 in basal subtype but no other subtypes from TCGA dataset (Fig. S1p, q) Interestingly, survival analysis using mean expression of ETS1 and PIKKs for PAM50 breast cancer subtypes51 undergoing chemotherapy indicated significantly poor survival in patients (ETS1-PIKK high) with basal phenotype only (Fig. S1r).
To understand PIKK-dependent modulation of ETS1 in resistant cells, we used a panel of DNAPKcs and MTOR inhibitors to check the effect on ETS1 expression. To inhibit PIKK activity specifically, we have used DNAPKcs inhibitors; NU7441 (1 μM)52, NU7026 (10 μM)53 and dual MTOR/DNAPKcs inhibitors CC115 (3 μM)54, Compound401 (5 μM)55,56. We found that kinase inhibitors effectively decreased the abundance of ETS1 protein and associated target genes MMP3 and TWIST1 in resistant TNBC cells (Fig. 1d and S2a). We could mirror the effect of kinase inhibitors using RNAi against DNAPKcs but not MTOR, indicating DNAPKcs to be a potential modulator of ETS1 expression and function in resistant TNBC cells (Fig. 1e). We didn’t observe any changes in ETS1 protein levels of parental TNBC cell lines during DNAPKcs abrogation, indicating the phenomenon to be associated with acquired resistance phenotype (Fig. S2b). We used NU7441 as a potent inhibitor of DNAPKcs (DNAPKcsinh) for further studies57. We observed a decrease in ETS1 protein levels in Res-MDA-MB231 cells upon treatment with DNAPKcs inhibitor, which became prominent in blocking new protein synthesis by cycloheximide, suggesting DNAPKcs inhibitor’s ability to alter ETS1 protein turnover in acquired resistance (Fig. 1f). While we failed to see any changes in ETS1 protein turnover of parental MDAMB231 cells when challenged with DNAPKcs inhibitor and cycloheximide, suggesting the possibility of DNAPKcs-independent regulation of ETS1 under basal conditions (Fig. S2c top). Further, in Res-MDA-MB231 cells, we didn’t observe any change in DNAPKcs protein levels after treatment with DNAPKcs inhibitor and cycloheximide, indicating a possible involvement of DNAPKcs kinase activity in regulating ETS1 protein stability (Fig. S2c bottom). We observed decreased colony formation in resistant TNBC cells treated with DNAPKcs inhibitor (Fig. 1g). DNAPKcs inhibitor was able to reduce IC50 of the resistant cells and induce caspase3/7 activity, resulting in apoptotic cell death (Fig. 1h, i). To understand if the resistant phenotype associated with ETS1’s protein stability and function is a general feature of TNBC cells, we treated parental MDA-MB231 and Res-MDA-MB231 cells with DNAPKcs inhibitor. As described above, we observed an increased protein level of ETS1 and its target genes in resistant cells, which were reduced in the presence of DNAPKcs inhibitor but not in the parental cells, indicating the DNAPKcs-ETS1 axis to be active in acquired resistance only (Fig. S2d). We observed increased DNAPKcs activity (phospho Serine 2056) and protein level in resistant cells, promoting ETS1 stability and function, leading to acquired resistance (Fig. S2d). Further colony formation ability of parental TNBC cells was not affected by siETS1, indicating ETS1-mediated acquired resistance as an evolving feature of resistant TNBC cells (Fig. S2e).
DNAPKcs-mediated phosphorylation regulates ETS1 stability and function
DNAPKcs can modulate its targets via interaction and subsequent phosphorylation58. Exogenously expressed DNAPKcs interacted with ETS1 present in Res-MDA-MB231 cells (Fig. 2a). While we could see modest interaction for endogenous ETS1 and DNAPKcs in parental TNBC cells as reported previously33, the interaction significantly increased in resistant cells (Fig. 2b). We speculate that low interaction at basal levels (Parental cells) might prevent DNAPKcs from phosphorylating and stabilising ETS1 protein, hence restricting the mechanism to acquired resistance phenotype. Next, we checked if this interaction resulted in the phosphorylation of ETS1 by DNAPKcs in resistant TNBC cells. Immunoprecipitated ETS1 protein from Res-MDA-MB231 cells indicated the presence of phosphorylated serine/threonine sites in ETS1, which diminished upon DNAPKcs inhibition (Fig. 2c). The phosphorylation of ETS1 remained unaffected by DNAPKcs inhibitor in the presence of a constitutively active T3950A DNAPKcs mutant59, indicating the phosphorylation to be mediated by DNAPKcs (Fig. 2c). To identify phosphorylation sites over ETS1, we interrogated phospho-proteome data of breast cancer patients50 with basal phenotype and found S251, T38 and S282 sites of ETS1 to be the most phosphorylated (Fig. 2d). We compared another human phosphoproteome analysis covering function-based phosphorylation and found T38, S251, S257, S282 and S285 sites of ETS1 to overlap with known regulatory sites from PhosphoSitePlus® (PSP)60,61 (Fig. S2f). Using a non-kinase-based algorithm62, we found sites S251 among Serine and T38 among Threonine have the highest probability score for phosphorylation (Fig. S2g). ETS1 protein sequence (NP_005229.1) queried against multiple kinase prediction algorithms indicated significant scores for S251 site phosphorylation mediated by DNAPKcs kinase (Fig. S2h). ETS1 peptides harbouring the serine/threonine sites subjected to DNAPKcs-dependent in vitro phosphorylation revealed significant phosphorylation (~40%) for the ETS1 peptide containing S251 residue (Fig. 2e). The phosphorylation was abolished when serine was replaced by alanine (S251A) (Fig. 2e). Although the T38 residue containing peptide is phosphorylated (<10%), we couldn’t observe phosphorylation at other residues (Fig. 2e). We purified full-length ETS1 from DNAPKcs null M059J cells to prevent phosphorylation of ETS1 by endogenous DNAPKcs present in TNBC cells. The full length purified ETS1 became phosphorylated in vitro at S251 residue in a DNAPKcs-kinase assay and was detected by an antibody against phosphorylated S251 ETS1 (Fig. S2i, j and Fig. 2f). DNAPKcs inhibition abrogated the phosphorylation of S251 from the kinase assay, indicating the specificity of the DNAPKcs-mediated phosphorylation site on ETS1 (Fig. 2f). We generated a phosphomimetic S251D (Serine → Aspartic acid) and a phospho-dead S251A (Serine→ Alanine) ETS1 mutant for understanding the effects of DNAPKcs inhibition over the mutant ETS1 protein during resistance. We checked the expression and function of exogenously expressed ETS1, S251D ETS1 and S251A ETS1 in T47D cell lines, and observed similar levels of ETS1 and its target gene activation as compared to Res-MDA-MB231 cells (Fig. 2g). To check DNAPKcs inhibitor’s effect over protein stability and transactivation functions of ETS1 and its phosphorylation mutants, we used MCF7 and T47D cells which express negligible amounts of ETS1 protein as compared to TNBC cell lines, thereby ruling out possible effects of endogenous ETS1 (Fig. S2k, S2l). The phosphomimetic S251D ETS1 harbouring MCF7 showed stable ETS1 expression in the presence of DNAPKcs inhibitor (Fig. 2h). Further, transfected S251D ETS1 mutant was able to rescue ETS1’s transactivation function in the presence of DNAPKcs inhibitor (Fig. 2i). We tested colony formation ability of T47D cells stably expressing ETS1 or its phosphorylation mutants (S251D, S251A) under combination of chemotherapy drugs (5’FU, Doxorubicin) and DNAPKcs inhibitor. While cells with ETS1 or S251A (phospho-dead) ETS1 showed a significant decrease in colony formation ability under the drug combination (5’FU or Doxorubicin with DNAPKcs inhibitor), S251D ETS1 phosphomimetic showed increased colony formation by overcoming drug pressure, indicating phosphorylated ETS1 is required and sufficient for acquired drug resistance (Fig. 2j). Moreover, exogenous expression of ETS1 S251D mutant was able to override DNAPKcs inhibitor-mediated ETS1 degradation which prevented caspase3/7 activation in Res-MDA-MB231 cells (Figs. 2k, l). Importantly, we were able to detect phosphorylated S251 ETS1 in chemo-refractory TNBC patients but not in treatment naïve patients, indicating a possible role of phosphorylated ETS1 in the development of resistance TNBC development (Fig. 2m). Together, these data indicate the DNAPKcs-mediated ETS1 phosphorylation event to be critical for developing acquired resistance.

See also Fig.S2. aRes-MDA-MB231 cells were transfected with either empty or DNAPKcs-FLAG plasmids, 48 h post-transfection, cell lysates were subjected to immunoprecipitations with either anti-ETS1 antibody (left panel) or anti-FLAG antibody (right panel) and immunoblots were performed as indicated. b MDA-MB231 or Res-MDA-MB231 cell lysates were subjected to immunoprecipitations with either anti-ETS1 antibody (left panel) or anti-DNAPKcs antibody (right panel) and immunoblots were performed as indicated. c Res-MDA-MB231 cells were transfected with plasmids and treated with compounds as indicated. MG132 was added for last 10 h. Res-MDA-MB231 cells lysate post-treatment was immunoprecipitated using ETS1 antibody and immunoblots were performed against indicated proteins. d Heatmap based on phosphorylation residue enrichment of ETS1 protein from TNBC patients. e in vitro phosphorylation of ETS1 peptides showing percentage ADP conversion against increasing concentration of DNAPKcs. f Purified ETS1-FLAG protein subjected to in vitro kinase assay in the presence of DMSO or DNAPKcs inhibitor was subjected to immunoblotting for the indicated proteins. g (top) whole cell lysates from resistant MDA-MB231 and ETS1 expressing T47D cells were subjected to Immunoblot against indicated proteins. (bottom) Transcript levels of ETS1 or ETS1 mutant expressing T47D cells were analysed using qrt-PCR. h Immunoblots of ETS1 and S251D ETS1 stable MCF7 cells post-treatment as indicated. i T47D(left) or MCF7 (right) cells were transfected as indicated for MMP9 promoter activity assay. DNAPKcs inhibitor was added for the last 24 h. Luciferase assay was performed 48 h post-transfection, and relative firefly luciferase activity was plotted. j Colony formation ability of T47D cells expressing ETS1 or ETS1 mutants was assessed in the presence of the indicated compounds. The optical density of stained cells was measured at 595 nm. k Whole cell lysate from Res-MDA-MB231 cells transfected with indicated plasmids and treated with DNAPKcs inhibitor as indicated was subjected to immunoblot for indicated proteins. l Relative caspase3/7 activity of Res-MDA-MB231 cells transfected with indicated plasmids and treated with DNAPKcs inhibitor was measured as indicated. m Representative phospho-S251 ETS1 staining (20x, 40x) in TNBC tumours (paired Naïve and Therapy relapse cases; n = 4). H&E was used as a counter stain. All experiments were performed as biological triplicates (mean ± SD). Two-way ANOVA with Dunnett’s multiple comparison test was used for 2 g and 2j. One-way ANOVA with Dunnett’s multiple comparison test was applied for 2i and 2 l, where****p < .0001, ***p = 0.0001,**p = .001, *p = 0.01, n.s=not significant.
Phosphorylation of ETS1 prevents its degradation via COP1
Since DNAPKcs inhibition alters the half-life(t1/2) of ETS1 protein, we used proteasomal or lysosomal inhibitors to interrogate the degradation mechanism in resistant TNBC cells. Proteasomal blocker MG132 was able to prevent ETS1 degradation and restore its transcriptional functions in contrast to pepstatin (Figs. 3a, b). We didn’t observe such changes in parental cells when cells were treated with combinations of DNAPKcs inhibitor and proteasomal or lysosomal blockers (Fig. S3a). COP1 has been shown to act as a putative E3 ligase for ETS1, while SRC kinase-mediated phosphorylation has been shown to stabilise ETS1 against COP1-mediated degradation20,63. In fact, SRC inhibitors and statin inhibitors are shown to deplete ETS1 in TNBC cells. However, we have not observed any significant change in ETS1 protein levels in the presence of SRC or statin inhibitors, indicating a possible bypass mechanism for ETS1 protein stability in resistant cells (Fig. S3b, c). We probed ETS1 immunoprecipitated lysates from resistant cells and detected COP1 interaction with ETS1 only during DNAPKcs inhibition, indicating the possible role of DNAPKcs phosphorylation in preventing COP1 binding (Fig. 3c). COP1 ablation using RNAi rescued ETS1 from DNAPKcs inhibitor-mediated proteasomal degradation (Fig. 3d). Moreover, RNAi against COP1 blocked caspase activation in Res-MDA-MB231 cells treated with DNAPKcs inhibitor (Fig. 3e). Interestingly, COP1 could bind more strongly with ETS1 in the presence of DNAPKcs inhibitor, and this binding was dramatically reduced in S251D ETS1 mutants (Fig. 3f). Together, these data suggest that DNAPKcs facilitated phosphorylation may act as a preventive measure against COP1-mediated degradation of ETS1 during chemoresistance in TNBC.

See also Fig. S3.a Res-MDA-MB231 and Res-MDA-MB468 cells were treated as indicated for 48 h. MG132 (10 μM) or Pepstatin (10 μM) were added for the last 10 h where indicated. Whole-cell lysates were immunoblotted against indicated proteins. b Res-MDA-MB231 cells were transfected with MMP9 promoter luciferase. DNAPKcs inhibitor was added for the last 24 h, while MG132 (10 μM) or Pepstatin (10 μM) were added for the last 10 h as indicated. Luciferase assay was performed 48 h post-transfection, and relative firefly luciferase activity was plotted. c Res-MDA-MB231 cells were treated as indicated for 48 h, harvested and immunoprecipitated using ETS1 antibody, and subsequently immunoblotted against indicated proteins. d Res-MDA-MB231 and Res-MDA-MB468 cells were transfected as indicated for 48 h. DNAPKcs inhibitor was added for the last 24 h. Whole-cell lysates were subjected to immunoblotting against indicated proteins. e Relative caspase3/7 activity of Res-MDA-MB231 cells transfected with indicated siRNAs and treated with DNAPKcs inhibitor was measured 72 h post-transfection as indicated. f MCF7 stable cells were treated as indicated for 48 h, harvested and immunoprecipitated using FLAG antibody, and subsequently immunoblotted against indicated proteins.
Phosphorylated ETS1 activates a proliferation gene network that promotes drug resistance
To understand the ETS1 high transcriptomic landscape in drug resistance, we performed RNA sequencing analysis of Res-MDA-MB231 cell lines made resistant to 5’FU and compared them with parental MDA-MB231 cells. Gene Set Enrichment Analysis (GSEA) using Molecular signature database (Hallmark gene sets) showed upregulation of E2F targets, MYC target and G2/M checkpoint pathway genes in resistant TNBC cells compared to parental cell lines (Fig. 4a). Additionally, we found enrichment of the same pathways in transcriptomic data sets from other TNBC cell lines (MDAMB468, SUM159, MDAMB231) resistant to various drugs (GSE230273, GSE206242, GSE256086, GSE199779), indicating that the pathways are critical for the development of resistance in TNBC (Figs. 4a, and S4a top and bottom). To understand the role of S251-ETS1 phosphorylation in regulating the transcriptome, we superimposed our transcriptomics data (Resistant versus parental MDA-MB231 cells) over TNBC patient transcriptome50 stratified by the phosphorylation status of S251-ETS1 (high versus low). Our resistant TNBC transcriptome indicated a strong correlation and similar pathway enrichment with patients having high S251-ETS1 phosphorylation, suggesting a critical role of altered ETS1 phosphorylation in regulating the transcriptomic landscape during drug resistance (Fig. S4b, c). Furthermore, GSEA analysis showed enrichment of similar pathways in patients with high phosphorylation status for S251 ETS1 (Fig. S4d). Further, RNA seq from T47D stable cell lines expressing either S251D or S251A ETS1 mutants exposed to 5’FU showed selectively enrichment for E2F, MYC and G2/M pathway genes in S251D ETS1 but not in S251A ETS1 mutant expressing cells, indicating the importance of a single posttranslational modification of ETS1 in promoting chemoresistance (Fig. 4b).

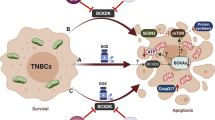
See also Fig. S4. a Enrichment plots from GSEA conducted with normalised differential expression from RNA seq of (top left) resistant-MDA-MB231 over parental MDA-MB231 cells, (top right) resistant-SUM-159 over parental SUM-159 cells, (bottom left) resistant MDA-MB 468 over parental MDA-MB468 cells. Plots represent statistically significant enrichment (FDR < 0.25) of indicated hallmark pathways. b GSEA plots using normalized differential expression from RNA seq of S251D-ETS1 and S251A-ETS1 expressing stable T47D cells treated with 5’FU. Plots represent statistically significant enrichment (FDR < 0.25) of indicated hallmark pathways. Heatmap of selected genes from enriched pathways in RNA seq of c Resistance and parental MDA-MB231 cells, d Patient stratified by phospho-S251 ETS1 scores. e Relative transcript levels of indicated genes from Res-MDA-MB231 cell lines transfected with siRNAs were analysed using qRT-PCR. f Relative transcript levels of genes from parental MDA-MB231 and resistance MDA-MB231 cell lines post-treatment were analysed using qRT-PCR as indicated. g Relative transcript levels of genes from biopsy tissue samples collected from the treatment relapsed subjects before (Naïve) and after chemotherapy were analysed using qrt-PCR. Experiments were performed as biological triplicates (mean ± SD, n = 3). h Exon level expression and fold-change (Log2 FC) of indicated genes from ETS1 knockdown (ETS1ShRNA) cells were matched with enriched ETS1-ChIP signals over their promoters from GSE109896, GSE109884. i) Representative FACS profile for EdU staining post DNAPKcs inhibitor treatment as indicated. Alexa-488 labelled Edu-stained proliferating cells were measured in the FITC channel and plotted for three independent experiments, SD ± 3. j Model: ETS1 stabilised by DNAPKcs-mediated phosphorylation at S251 promotes resistance phenotype by activating proliferation-related genes in TNBC (Created with BioRender.com). For 4e, f, g; Two way ANOVA with Sidak’s or Dunnett’s multiple comparison test was applied for the statistical significance, For 4i, One way ANOVA with Dunnett’s multiple comparison test was applied where ****p < .0001,***p = 0.0001,**p = 0.001, *p = 0.01, n.s=not significant.
We observe a characteristic increase in the expression of selected genes associated with the cell cycle (MCM4, SMC6, AURKB, PTTG1, KIF4A), DNA repair and nucleotide metabolism (RRM2, PRPS1, SMC6, MSH2, POLD1) in the above data sets (Fig. 4a–d). Interrogation of chemorefractory-metaplastic TNBC patient samples64 (GSE165407) revealed concurrent high expression of these genes associated with resistance (Fig. S4e). We analysed ETS1 silenced transcriptomics datasets (GSE59020, GSE101833, GSE109886)65,66,67 and found an inverse profile for the same pathways, indicating ETS1 as a direct modulator for cell proliferation circuits (Fig. S4f). Further, silencing ETS1 in Res-MDA-MB231 or Res-MDA-MB468 cells decreased the expression of all the genes (Figs. 4e and S4g).
Suppressing ETS1 activity using DNAPKcs inhibitor reduced the expression of these genes, underpinning the significance of phosphorylated ETS1 in resistance (Fig. 4f). ETS1 S251D in T47D cells were able to rescue the expression of these genes in the presence of DNAPKcs inhibitor, indicating phosphorylated ETS1 to drive the proliferation gene circuit during resistance (Fig. S4h). We could follow the activation of these genes in paired samples (naïve versus treated) from chemorefractory TNBC patients, indicating previously unknown ETS1-driven gene networks that might be responsible for therapy relapse via a possible bypass of cell cycle blockage (Fig. 4g). To understand the ETS1 occupancy at promoters of these genes and their subsequent mRNA expression, we matched ETS1 Chip Sequencing data with the exon-level expression of this gene upon ETS1 knockdown (GSE109896, GSE109884). We found ETS1 binding at promoters of its known targets (FLI1, EZH2) alongside these genes, while ETS1 knockdown exhibited depletion of expression across exons of all genes, indicating possible ETS1-mediated transcriptional activation of these genes (Figs.4h and S4i). We also found enrichment of ETS1 at promoters of these genes in another ChIP seq data(GSE138516)68, which got depleted on silencing ETS1 (Fig. S4j). We report increased incorporation of nucleotides in resistant cells, which was reversed in the presence of DNAPKcs inhibitor, indicating active DNA synthesis in resistant cells (Fig. 4i). The data suggest that DNAPKcs promotes ETS1-mediated activation of the cell proliferation network during resistance development.
In conclusion, our study indicates the role of DNAPKcs in phosphorylating and stabilizing ETS1 protein, increasing proliferation and drug resistance phenotype. Aborting ETS1 or DNAPKcs functions decreased the proliferation and colony formation ability of resistant cells. We identified DNAPKcs-mediated phosphorylation in the Serine-251 residue of ETS1 as critical for the stability and oncogenic functions of ETS1 in resistant cells. DNAPKcs inhibition resulted in proteasomal degradation of ETS1 in a COP1 E3 ligase-dependent manner, resulting in apoptosis and dampened colony formation ability of resistance cells (Fig. 4j). Interestingly, S251 ETS1 phosphorylation was also observed in chemotherapy-relapsed TNBC patients, indicating the possibility of an evolving post-translational modification mark that regulates the transcriptional functions of ETS1 during acquired resistance. Together, our studies indicate DNAPKcs inhibition as a potential vulnerability in the case of TNBC acquiring drug resistance.
Discussion
Being the third most commonly used drug for treating solid tumours, including colorectal, gastric, pancreatic and breast cancers, resistance against 5’FU and its derivatives confers a challenge to patient burden69,70. In fact, preoperative chemotherapy involving 5’Fluorouracil, doxorubicin and cyclophosphamide has the highest rate of complete pathological response (45%) of basal subtypes34,36,71,72. Additionally, somatic copy number variations reported in 5’FU treated TNBC patients are associated with lower recurrence rates73. 5’FU derivative capecitabine in combination with cisplatin showed a significant increase in disease-free survival and overall survival for metastatic TNBC patients74,75. Mechanisms of 5’FU resistance in colorectal, gastric and pancreatic cancer cells have been reported, including stemness, increased drug transporter expression, metabolic reprogramming, angiogenesis and altered activity of rate-limiting enzymes, but the mechanism in TNBC cells is poorly understood76. Although oncogenic ETS factors have been identified as a promising drug target in many cancers, a few ETS1-targeted drug studies against TNBC have been reported63,77,78. Our study on 5’FU-resistant TNBC cells shows cross-resistance towards other drugs and increased proliferation activity associated with acquired drug resistance. Cross-resistance to anthracyclines, taxanes and platins, observed in resistant TNBC tumours9,79,80,81, has been connected with various mechanisms like increased expression of transcription factors [RB182, FOXO383], drug transporters [ABCB1, ABCG2/BCRP]83,84,85 or alterations of biochemical pathways like PI3K/Akt86, DNA repair/E2F/G2-M pathways87. ETS1 has been shown to directly regulate several of these drug transporters16,88, transcription factors89,90 and biochemical pathways91,92, indicating a possible regulatory role in cross-resistance mechanisms. Our observation of increased levels of ETS1 protein in TNBC cells made resistant by other drugs indicates the existence of a specific ETS1-driven mechanism that can possibly be targeted for therapy (Fig. 1 and S1).
Since TNBC patients benefit more from neoadjuvant chemotherapy, which mainly includes DNA-damaging agents, our and other studies will help understand DNA repair mechanisms during resistance development and may provide better targeting strategies for resistant cancers93,94,95. We observed a high expression of DNAPKcs in association with ETS1, which predicts poor survival specific to the basal subtype (Fig. 1). Here, we identified DNAPKcs-mediated phosphorylation of ETS1 as a vulnerability for drug-resistant TNBC. DNAPKcs inhibitors in TNBC-specific studies are mostly reported to exhibit synergism with DNA-damaging drugs52,96,97, and recent trials on solid tumours encouraged the use of specific DNAPKcs inhibitors as a clinical intervention either as monotherapy or in combinations (NCT02644278, NCT04750954, NCT02516813, NCT03724890, NCT03907969, NCT03579628). DNAPKcs-mediated phosphorylation has been implicated in the transcriptional regulation of various metabolic genes, nuclear receptors, EMT, metastasis and oncogenic pathway regulators, establishing it as a major regulator in various cancers58. Like many substrates, the identified serine 251 (S251) phosphorylation site over ETS1 is a non-canonical DNAPKcs phosphorylation site through which the kinase might elicit protumorigenic functions58,98. Interestingly, we could detect S251 ETS1 phosphorylation in TNBC patients with chemotherapy relapse but not in naïve patients, indicating S251 to be an evolving phosphorylation during acquired resistance (Fig. 2). Although the patient phosphoproteomics study50 indicates an inherent presence of phosphorylated S251 (S251 high) in treatment naïve patients, we do not observe any S251 phosphorylation in immunohistochemistry of our naïve TNBC patients (Fig. 2). We conceive that S251 phosphorylation status is associated with increased proliferation and therapy relapse and can be used as a predictive biomarker in future. Moreover, the S251D ETS1 phosphomimetic mutant but not the S251A phospho-dead mutant was able to stabilise and activate ETS1 oncogenic signatures irrespective of resistance status, indicating a strong role of S251 phosphorylation in promoting drug resistance.
Our study indicates inhibition of ETS1 phosphorylation promotes proteasomal degradation via COP1, a culin RING ligase (CRL), reversing the resistance phenotype. Phosphorylation of CRL substrates is known to alter their binding ability towards CRL E3s, resulting in altered degradation status99. COP1 has been reported to mediate ubiquitination and proteasomal degradation of ETS family members ETS1, ETS2, ETV1, ETV4, and ETV520,100. While truncated mutants of ETV1/4/5 are implicated in prostate cancer progression, SRC kinase-mediated phosphorylation of ETS1/2 hinders COP1 binding in breast cancer20,100. We do not observe any effect of the SRC inhibitor over ETS1 stability in our resistance lines, indicating an SRC phosphorylation-independent mechanism in resistance cells (Fig. S3). We speculate that ETS1 protein phosphorylation by DNAPKcs at S251 might alter ETS1 protein conformation, thereby overruling SRC-mediated phosphorylation/binding events at S282/285 residues, resulting in sustained stability and transcriptional activity101,102. Surprisingly, impaired COP1 binding was observed in the presence of S251D ETS1 phosphomimetic protein, indicating a possibility of S251 phosphorylation-induced conformational change at the C-terminal region of ETS1, which might explain the substrate selectivity of COP1(Fig. 3)103.
Most deaths in TNBC are due to evolving resistance mechanisms and subsequent metastasis, fostering the need for understanding the gene networks responsible for chemo-resistance and the development of new therapeutics104,105. Our study indicates that ETS1-dependent networks encompassing E2F, MYC, and G2/M pathway genes are responsible for active proliferation in our resistant cells. Interestingly, these pathways were also identified in previous meta-analysis data regarding target gene identification and had a significant association with TNBC patient outcomes106,107,108. Additionally, the transcriptome of our acquired resistant cells strongly correlated with phospho-S251 high-TNBC patient samples. We found ETS1 phosphorylation at Serine 251 in chemoresistant TNBC patients and cells that not only stabilises ETS1 protein but also activates a previously unknown network of proliferative genes (Fig. 4). Surprisingly, calcium-dependent phosphorylation across Serine 251, 257, 282, and 285 of ETS1 has been reported to cooperatively inhibit ETS1’s DNA binding and transcriptional function109,110. While this inhibitory phosphorylation module can’t act on singular phosphorylation event109,111, the C-terminal Serine-rich intrinsically disordered region (Ser282, Tyr283, Phe286, Tyr288, and Tyr291) of ETS1 implicate that both hydrophobic and electrostatically induced conformational changes are required to affect ETS1 DNA binding affinity and transcriptional activity102,112,113. Additionally, protein-protein interactions with other ETS1 binding partners are known to counteract the auto-inhibitory module114. This might explain the transactivation of MMP9 promoter via phosphorylated ETS1, hence indicating S251’s independent function during chemoresistance (Fig. 2). Transcriptomic analysis of S251D versus S251A phosphorylation-specific mutants of ETS1 indicates selective activation of ETS1-mediated gene signatures observed in resistant TNBC cells and patients. Our analysis of non-metaplastic and metaplastic (chemo-refractory) TNBC patients indicates a similar enrichment signature towards cell proliferation, DNA repair, and nucleotide metabolism pathway genes (Fig. 4 and S4). We observed a significant induction of ETS1-driven genes in paired samples (before versus after chemotherapy) from therapy-relapsed TNBC patients, indicating previously unknown ETS1-driven gene networks that might be responsible for therapy relapse (Fig. 4 and S4). In conclusion, our study identifies an evolving phosphorylation of the ETS1 mediated by DNAPKcs during chemoresistance development, which can provide new avenues of therapies and detection for resistant TNBC.
Methods
No statistical methods were used to predetermine the sample size. Experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment. Patient samples were coded, and the experimenter was blinded towards sample identity.
Cell lines and culture conditions
MDAMB231, MDAMB468, MDAMB453, MCF7 and T47D cells were obtained from NCCS, Pune, India. M059J (ATCC: CRL-2366™) cells were obtained from ATCC, Manassas, VA. T47D and MCF7 cells were cultured in RPMI medium (ThermoFisher Scientific), and MDAMB231, MDAMB468 and MDAMB453 cells were cultured in DMEM medium (ThermoFisher Scientific). M059J cells were cultured in DMEM/F-12 medium (ThermoFisher Scientific). The medium was supplemented with 10% fetal bovine serum, and cells were maintained at 37 °C in 5% CO2 for all experiments. The identity of all cell lines was confirmed by STR analysis. All cell lines were determined to be mycoplasma-free using the MycoFluor Mycoplasma detection kit (Thermo Fisher Scientific).
For the generation of drug-resistant cell lines, MDAMB231 or MDAMB468 cells were treated with an increasing amount of 5’Fluorouracil (Sigma) starting from 3.5 µM, as described previously38,40. Media containing 5’FU was replenished every 72 h until full confluency, cells were harvested, and the 5’FU dose was doubled every 2-3 weeks for six months, up to a final concentration of 20 μM. Resistance cells were designated as Res-MDA-MB231 and Res-MDA-MB468. For CDDP, Gemcitabine and Doxorubicin cells were treated as described previously for six months with sequential passaging39,115,116,117,118.
Plasmids, cloning and transfections
ETS1_pet28a plasmid was a gift from Peter Hollenhorst (Addgene plasmid # 131663), and DNAPKcs plasmids were a gift from Katheryn Meek (Addgene plasmid # 83317, #83320)59,119,120. MMP9 promoter (~1.6 kb) was amplified from MDAMB231 cells genomic DNA and cloned into pGL4.24[luc2P/minP] plasmid (Promega). Human FLAG-tagged ETS1 and DNAPKcs-FLAG were subcloned into the pcDNA3.1+ plasmid (Invitrogen). Site-directed mutagenesis was performed on ETS1FLAG pcDNA3.1+ constructs using a Q5 SDM kit (New England Biolabs) per the manufacturer’s instructions. All clones were confirmed using Sanger sequencing using a 3500xL genetic analyzer (Applied Biosystems). For exogenous transgene expression studies, plasmid DNA was transfected using Lipofectamine 3000 (Thermo Fisher Scientific) as per the manufacturer’s instructions. We used G418 (600 µg/ml) to select stably expressing T47D cells.
Chemicals, siRNAs and transfections
See Supplementary Table 1 for details of compounds, assay kits, primers and siRNAs used in this study. All compounds were dissolved as per the manufacturer’s instructions. 5’ Fluorouracil, Doxorubicin, CDDP (cis-Diammineplatinum(II) dichloride), cycloheximide (CHX), MG-132, DNAPKCS inhibitors NU7441, NU7026, CC115 and compound 401 were dissolved in DMSO, while Gemcitabine, Cyclophosphamide were dissolved in water or PBS (Phosphate Buffered Saline). MTT (3-(4,5- Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide), Crystal Violet were dissolved in PBS as per manufacturer’s instruction. Coomassie stain was prepared as per the manufacturer’s instruction. All siRNAs and primers used in the study were purchased from Qiagen or synthesized from vendors. All siRNAs were transfected using RNAi-Max (ThermoFisher Scientific) as per the manufacturer’s instructions.
Gene expression
For real-time PCR analysis, total RNA from cells and tissues were extracted using PureLink™ RNA Mini Kit (Thermo Fisher Scientific) and cDNA was made using High-Capacity cDNA reverse transcription kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. Real-time PCR was performed in a QuantStudio 5 instrument (Applied Biosystems). Tissue samples were homogenized in Trizol reagent (Thermo Fisher Scientific) before processing through the PureLink™ RNA Mini Kit (Thermo Fisher Scientific). qPCR was performed using the primer sets listed in the supplementary Table 1 using Power Up SYBR Green (Thermo Fisher Scientific). Relative gene expression was assessed using three biological replicas and data are shown as the mean ± SD. 18sRNA was used as an internal control for all samples.
TNBC patient samples
The study protocol Institutional Ethical Committee/All India Institute of Medical Sciences/Kalyani/2022-002 (IEC/AIIMS/Kalyani/2022-002) was reviewed and approved by the IEC (Institutional Ethics Committee) of All India Institute of Medical Sciences (AIIMS), Kalyani, India. All methods were performed in accordance with the relevant guidelines, regulations and the Declaration of Helsinki. Informed consent was obtained from all subjects for the study. Biopsy tissue samples were collected at the Department of Surgery, All India Institute of Medical Sciences (AIIMS), Kalyani, India. Patients diagnosed with TNBC (ER, PR, HER2 status) were kept under the inclusion criteria. Chemotherapy included conventional AC (Adriamycin and cyclophosphamide) followed by paclitaxel. For histological analysis, patient tumour samples were fixed in formalin and paraffin-embedded. Tumour sections were stained with Hematoxylin and Eosin (H&E). IHC staining was performed using Phospho-ETS1 (S251) primary antibodies overnight at 4 °C. Images were captured by a Nikon 80i research microscope (20 & 40×).
Immunoblotting, immunoprecipitation, and cycloheximide analysis
Total lysates from cells were prepared using cell extraction buffer (ThermoFisher Scientific) supplemented with protease inhibitor (complete mini EDTA free) and Phos-STOP tablets (Merck). Immunoblotting was performed using standard SDS-PAGE electrophoresis and transferred to a PVDF membrane (Millipore Merck). Antibodies used were: anti-ETS1, anti-PRKDC/DNAPKcs, anti-MMP3, anti-COP1 and anti-phospho-serine/threonine from Abcam, anti-phosphoS2056 PRKDC/DNAPKcs, anti-SRC and Anti-Twist1 antibodies were from Cell Signaling Technology, anti-Actin, anti ETS1 and anti-Phospho S251-ETS1 from Thermo Fisher Scientific, anti-FLAG (Oct-A probe) was from Santa Cruz Biotechnology, anti-mTOR, anti-phospho-S251 ETS1 were from G-biosciences (Immunotag). Immunoblots shown are representative of at least three independent biological replicas.
Immunoprecipitations were performed from 500 μg cell extracts using Pierce IP kit (ThermoFisher Scientific) as per manufacturer’s instructions. Briefly, cells were lysed using lysis buffer supplemented with protease inhibitor cocktail (complete mini EDTA free) and Phos-STOP and incubated with antibody overnight. The following day, pre-cleared magnetic beads were added to lysate and the magnetic bead-bound immune complex was incubated for 1 h at room temperature. Samples were washed and eluted from the beads for analysis by immunoblotting.
Cycloheximide (CHX) chase assays were performed as described previously121. Briefly, cells were treated with 75 µM cycloheximide and DMSO or DNAPKcs inhibitor. Cells were harvested 15 to 480 min after CHX treatment, and lysates were prepared as described above. Samples were analyzed by immunoblotting for three independent biological replicas. Images were analyzed using Image studio lite Version 5.0 (Li-COR Bioscience, Lincoln, NE). The intensity profiles and exponential decay for t1/2 were calculated using Prism 8.0.2 (GraphPad Software, Inc., San Diego, CA) using decay plot.
In vitro kinase assays
Peptides with 95% purity were used for kinase assay, and their sequence are RGKLGGQDSFE: (S251), IESYDSCDRLTQ: (S257), DMECADVPLLTP: (T38), FNSLQRVPSYD : (S282), RGKLGGQDAFE: (S251A). For purification of ETS1, a FLAG-tagged ETS1 cDNA was expressed in PRKDC null M059J cells. Cells were lysed, and ETS1 was purified using an anti-FLAG M2 Affinity Gel system (Sigma) according to the manufacturer’s instructions and detected in gel using Coomassie stain (Himedia Laboratories). ADP-Glo kinase assay (Promega) was performed following the manufacturer’s instructions. Briefly, PRKDC/DNAPKcs kinase enzyme was incubated in serial dilution (starting concentration of DNAPKcs 200 ng/μl) with 1 mg/ml peptide substrate. For kinase assay involving full length ETS1, 50 μg ETS1 protein was incubated in serial dilution of DNAPKcs enzyme (starting concentration of DNAPKcs 200 ng/μl) in the presence or absence of DNAPKcs inhibitor. This reaction mixture was incubated at room temperature with kinase activation buffer and ATP for 90 min. The reaction was terminated by adding ADP-Glo reagent. Finally, luminescence was recorded using a varioskan lux reader (Thermo Scientific) after adding a kinase detection reagent.
EdU staining and flow cytometry
Replication was traced in cells using the Click It Edu kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. Briefly, cells were incubated with EdU component A for 3 h, fixed and permeabilized before performing a click chemistry reaction to label DNA with alexa488 dye. Following labelling, cells were subjected to flow cytometry (FACSVerse cell analyser, BD Biosciences), and data was analyzed using FACSuite software. For all FACS assays, three biological replicas were used, and data were shown as mean ± SD.
Functional assays
For colony formation assays, cells were seeded in equal density for all treatments and the assay was performed as described previously122. Colonies were stained with crystal violet and dissolved in 10% acetic acid; absorbance was measured at 595 nm. For IC50 experiments, the MTT assay was performed as described previously123. Briefly, cells were seeded in equal density (~2000 cells/well) in 96 well plates the day before drug treatment. An MTT assay was performed 72 h post-treatment, and absorbance was measured at 570 nm.
Cells were transfected as indicated for luciferase assays. Post-experiment, luciferase activity was determined using the Dual Glo Luciferase Reporter assay system (Promega) as per the manufacturer’s instruction. The difference in transfection efficiency across samples was normalized by co-transfecting pRL-TK, which expresses Renilla luciferase. Caspase activity was measured using the Caspase Glo 3/7 assay system (Promega) per the manufacturer’s instruction. Briefly, cells were lysed at room temperature using the reagent and analyzed for luminescent signals using a varioskan lux plate reader (Thermo Scientific). For all functional assays, three biological replicas were used and data were shown as mean ± SD.
RNA sequencing
RNA sequencing was performed from total RNA. The following samples were used: Control MDAMB231, Resistance MDAMB231, S251D ETS1 T47D stable, S251A ETS1 T47D stable. RNA quality was checked using Agilent Tape Station 4150, and RNA sequencing was performed using Novaseq 6000 (Illumina) to generate 2 × 150 bp paired-end (PE) reads. Sequence qualities were analysed using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc).
Data analysis
For RNA sequencing analysis, the sequences were subsequently aligned using HISAT2124 and hg38 as the reference genome. Subsequent BAM output files were subjected to read counting using Feature Counts125. NOISeq126, an R package, was used to analyze differential gene expression. The read counts were TMM normalised, and five replicates were simulated with 0.02 variability among simulated data. A fold change value of ±2 and probability > 0.8 was selected as the criteria for a gene to be classified as differentially expressed.
ChIP-Seq and RNA-Seq datasets65 were downloaded from NCBI GEO. ChIP-Seq reads were aligned to the human hg38 reference genome, and peak calling was performed using MACS2127. RNA-Seq samples were also aligned to the hg38 genome. Gene quantification was carried out using feature Counts, referencing GENCODE human release 47128. Counts were generated at the exon level, using the multi-mapping and overlapping features option to track expression levels across exons. Differential gene expression analysis was performed using DESeq2129. Finally, ChIP-Seq peak files, bigWig tracks, and the log2 fold change (log2FC) values from the DESeq2 output were visualized on the UCSC Genome Browser130 to explore ETS1 binding patterns in relation to transcriptional changes. For ChIP Seq analysis, Sequence qualities were analyzed using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). ChIP-Seq data (GSE138516) were aligned using Bowtie2131, with reference genome hg38. MACS2 was used for peak calling. Visualization of peak regions around genes was done using a UCSC browser132.
For TNBC patient transcriptome and ETS1 Phosphoproteome, normalized data used was provided by authors upon request50. For PCA analysis and correlation matrix, basal-type breast cancer (TNBC patient) gene expression data was downloaded from the GDC portal133. Phosphoproteome data from Mertins et al. 50 was used to classify the basal type data from high to low. The raw read counts downloaded from GDC were in STAR-aligned format. Therefore, the MDA-MB-231 control and treated samples were aligned using STAR134, generating raw counts. Since the data from GDC and our data were sequenced under different conditions, the batch effect was corrected using the remove batch effect function in Limma135. The subsequent data was normalized using upper-quartile normalization and log-transformed for visualization. Highly correlated features between MDA-MB-231 control and treatment samples were removed.
Heat map analysis: For CCLE analysis, data was curated from https://www.ebi.ac.uk/gxa/experiments/E-MTAB-2770/Results, with a cutoff of 0.5 FPKM135. Data was arranged as per ETS1 expression and the heat map was plotted after Log2 transformation using Morpheus software [https://software.broadinstitute.org/morpheus/. For the patient transcriptome, data for selected genes were z-score normalised and heatmap was generated using R-studio’s pheatmap package. For correlograms, the z-score normalised values were plotted using corrplot package. For heatmaps of selected genes from cell lines and patients’ transcriptome (BioProject:PRJNA1122549, GSE256086, GSE199779), log2 normalized values were plotted using multi-experiment viewer (MeV) software136.
Gene expression clustering data for TCGA-BRCA were downloaded from the National Cancer Institute’s GDC Data Portal137. The dataset was annotated to include only the Basal, HER2-enriched, Luminal A, and Luminal B breast cancer subtypes. A heatmap of the annotated dataset comparing Basal vs. Luminal A subtype was generated using the pheatmap package in R. Subtype-specific correlograms (Basal, HER2, Luminal A, and Luminal B) were subsequently produced using the corrplot package.
For Kaplan Meier plots, KM plotter tool was used51. Mean expression of gene probes was considered for pam50 breast cancer subtype (n = 1195) for plotting the overall survival of patient cohort undergoing chemotherapy.
For non-kinase-based score-wise prediction of S/T/Y phosphorylation sites of ETS1, the PhosphoSVM program was used62.
Gene Set Enrichment Analysis was performed using the program GSEA version 4.1.0 (http://www.broadinstitute.org/gsea/index.jsp)138. The following datasets were used in this study for ETS1 knockdown GSE59020, GSE101833, and GSE109886, for patients pertaining to Mertins et al. 50; data was provided by authors upon request, for metaplastic TNBC patients data GSE16540764, for doxorubicin resistance MDAMB231 data GSE256086, for pacilitaxel resistance data GSE199779, for resistance MDAMB231 and control cells and T47D cells expressing S251D ETS1 or S251A ETS1 data was deposited in SRA. Hallmark gene set from molecular signature database (MSigDB v5.1; http://software.broadinstitute.org/gsea/msigdb) was used with a permutation of 1000. Weighted or classic enrichment statistics and mean div normalization mode were used for all analyses. A pre-ranked gene list based on the S251 phosphorylation score of ETS1 was used against the Hallmark database to analyze Mertins et al. data. Pathways showing Normalized Enrichment Scores with FDR q value < 0.25 was considered significant.
Statistics analysis
Collected data were tabulated using Microsoft Excel and analyzed using GraphPad Prism version 8.3.02 (GraphPad Software, CA, United States). One-way analysis of variance (One way-ANOVA) was performed in Prism (GraphPad) using Dunnett’s multiple comparisons test to correct for multiple comparisons to the same control group; ap-value < 0.05 was considered significant. Brown Forsythe Welch corrections were performed for variation in SD. Two-way analysis of variance (2-way-ANOVA) was performed in Prism (GraphPad) using Sidak’s multiple or Dunnett’s comparisons test to correct for multiple comparisons to the same control group; a p value < 0.05 was considered significant. All experiments were performed in triplicate and mean ± SD was used to represent data.
Data availability
Resistance and parental MDAMB231 cell line RNA seq data accompanying this study have been deposited into SRA (Sequence Read Archive) under BioProject: PRJNA1122549(https://dataview.ncbi.nlm.nih.gov/object/PRJNA1122549?reviewer=ndui5fkrtd1v83reg4hklehica). S251D ETS1 and S251A ETS1 expressing T47D cell line RNA seq data accompanying this study have been deposited into SRA (Sequence Read Archive)underPRJNA1161002(https://dataview.ncbi.nlm.nih.gov/object/PRJNA1161002?reviewer=dvjh0gnafsrqe8880vpuoc2h2c). For GSEA, heatmaps, and ChIP seq analysis, the details of data sets are provided in supplementary table 2 in the supplementary file.
Code availability
All codes are freely available. NOISeq126, a R package, was used to analyze differential gene expression for our RNA seq data. For ChIP Seq analysis, peak calling was performed using MACS2 package127 and resulting bedGraph files were converted to bigWig format for visualization. PCA was calculated using the corrr package in R [Kuhn M et al (2022). corrr: Correlations in R.] and plotted using Plotly [https://plot.ly]. A correlation matrix and correlograms were generated using the corrplot package in R [Wei, T., and Simko, V. (2017). R Package “Corrplot”: Visualization of a Correlation Matrix]. For heatmaps, data for selected genes were z-score normalised and heatmap was generated using R-studio’s pheatmap package.
References
Foulkes, W. D., Smith, I. E. & Reis-Filho, J. S. Triple-negative breast cancer. N. Engl. J. Med. 363, 1938–1948 (2010).
Malorni, L. et al. Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term follow-up. Breast Cancer Res. Treat. 136, 795–804 (2012).
Grinda, T. et al. Real-world clinical and survival outcomes of patients with early relapsed triple-negative breast cancer from the ESME national cohort. Eur. J. Cancer 189, 112935 (2023).
Kassam, F. et al. Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin. Breast Cancer 9, 29–33 (2009).
Lin, N. U. et al. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer 113, 2638–2645 (2008).
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016).
Lei, Z. N. et al. Understanding and targeting resistance mechanisms in cancer. MedComm (2020) 4, e265 (2023).
Li, Y. et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 15, 121 (2022).
Nedeljkovic, M. & Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 8, https://doi.org/10.3390/cells8090957 (2019).
Silva, D. & Mesquita, A. Evolving Evidence for the Optimization of Neoadjuvant Therapy in Triple-Negative Breast Cancer. Breast Cancer (Auckl.) 16, 11782234221107580 (2022).
Henley, M. J. & Koehler, A. N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 20, 669–688 (2021).
Kohno, K. et al. Transcription factors and drug resistance. Eur. J. Cancer 41, 2577–2586 (2005).
Dittmer, J. The role of the transcription factor Ets1 in carcinoma. Semin Cancer Biol. 35, 20–38 (2015).
Kim, G. C. et al. Upregulation of Ets1 expression by NFATc2 and NFKB1/RELA promotes breast cancer cell invasiveness. Oncogenesis 7, 91 (2018).
Li, Y. et al. ETS1 is a prognostic biomarker of triple-negative breast cancer and promotes the triple-negative breast cancer progression through the YAP signaling. Am. J. Cancer Res. 12, 5074–5084 (2022).
Zhang, Y. et al. ETS1 is associated with cisplatin resistance through IKKalpha/NF-kappaB pathway in cell line MDA-MB-231. Cancer Cell Int. 18, 86 (2018).
Luchtel, R. A., Zhao, Y., Aggarwal, R. K., Pradhan, K. & Maqbool, S. B. ETS1 is a novel transcriptional regulator of adult T-cell leukemia/lymphoma of North American descent. Blood Adv. 6, 5613–5624 (2022).
Taveirne, S. et al. The transcription factor ETS1 is an important regulator of human NK cell development and terminal differentiation. Blood 136, 288–298 (2020).
Foulds, C. E., Nelson, M. L., Blaszczak, A. G. & Graves, B. J. Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol. Cell Biol. 24, 10954–10964 (2004).
Lu, G. et al. Phosphorylation of ETS1 by Src family kinases prevents its recognition by the COP1 tumor suppressor. Cancer Cell 26, 222–234 (2014).
Nelson, M. L. et al. Ras signaling requires dynamic properties of Ets1 for phosphorylation-enhanced binding to coactivator CBP. Proc. Natl. Acad. Sci. USA 107, 10026–10031 (2010).
Lindemann, R. K. et al. Protein kinase Calpha regulates Ets1 transcriptional activity in invasive breast cancer cells. Int J. Oncol. 22, 799–805 (2003).
Kato, T. et al. ETS1 promotes chemoresistance and invasion of paclitaxel-resistant, hormone-refractory PC3 prostate cancer cells by up-regulating MDR1 and MMP9 expression. Biochem Biophys. Res. Commun. 417, 966–971 (2012).
Khanna, A., Mahalingam, K., Chakrabarti, D. & Periyasamy, G. Ets-1 expression and gemcitabine chemoresistance in pancreatic cancer cells. Cell Mol. Biol. Lett. 16, 101–113 (2011).
Vishnoi, K. et al. Ets1 mediates sorafenib resistance by regulating mitochondrial ROS pathway in hepatocellular carcinoma. Cell Death Dis. 13, 581 (2022).
Wei, J., Zhou, Y., Jiang, G. Q. & Xiao, D. Silencing of ETS1 reverses adriamycin resistance in MCF-7/ADR cells via downregulation of MDR1. Cancer Cell Int. 14, 22 (2014).
Zhong, J., Zhang, J., Yu, X., Zhang, X. & Dian, L. Olmutinib Reverses Doxorubicin Resistance in ETS1-Overexpressing Leukemia Cells. Med Sci. Monit. 26, e924922 (2020).
Asleh, K. et al. Proteomics-derived basal biomarker DNA-PKcs is associated with intrinsic subtype and long-term clinical outcomes in breast cancer. NPJ Breast Cancer 7, 114 (2021).
Schild-Poulter, C., Shih, A., Yarymowich, N. C. & Hache, R. J. Down-regulation of histone H2B by DNA-dependent protein kinase in response to DNA damage through modulation of octamer transcription factor 1. Cancer Res 63, 7197–7205 (2003).
Abate, C. et al. Dimerization and DNA binding alter phosphorylation of Fos and Jun. Proc. Natl. Acad. Sci. USA 90, 6766–6770 (1993).
Iijima, S., Teraoka, H., Date, T. & Tsukada, K. DNA-activated protein kinase in Raji Burkitt’s lymphoma cells. Phosphorylation of c-Myc oncoprotein. Eur. J. Biochem. 206, 595–603 (1992).
Lees-Miller, S. P., Sakaguchi, K., Ullrich, S. J., Appella, E. & Anderson, C. W. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell Biol. 12, 5041–5049 (1992).
Choul-li, S., Drobecq, H. & Aumercier, M. DNA-dependent protein kinase is a novel interaction partner for Ets-1 isoforms. Biochem Biophys. Res. Commun. 390, 839–844 (2009).
Colleoni, M. et al. Classical cyclophosphamide, methotrexate, and fluorouracil chemotherapy is more effective in triple-negative, node-negative breast cancer: results from two randomized trials of adjuvant chemoendocrine therapy for node-negative breast cancer. J. Clin. Oncol. 28, 2966–2973 (2010).
Martin, M. et al. Doxorubicin in combination with fluorouracil and cyclophosphamide (i.v. FAC regimen, day 1, 21) versus methotrexate in combination with fluorouracil and cyclophosphamide (i.v. CMF regimen, day 1, 21) as adjuvant chemotherapy for operable breast cancer: a study by the GEICAM group. Ann. Oncol. 14, 833–842 (2003).
Naoi, Y. et al. Prediction of pathologic complete response to sequential paclitaxel and 5-fluorouracil/epirubicin/cyclophosphamide therapy using a 70-gene classifier for breast cancers. Cancer 117, 3682–3690 (2011).
Ciocan-Cartita, C. A. et al. New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells. J. Exp. Clin. Cancer Res. 39, 241 (2020).
Minegaki, T. et al. Histone deacetylase inhibitors sensitize 5-fluorouracil-resistant MDA-MB-468 breast cancer cells to 5-fluorouracil. Oncol. Lett. 16, 6202–6208 (2018).
Paramanantham, A. et al. Doxorubicin-Resistant TNBC Cells Exhibit Rapid Growth with Cancer Stem Cell-like Properties and EMT Phenotype, Which Can Be Transferred to Parental Cells through Autocrine Signaling. Int.J. Mol. Sci.22,https://doi.org/10.3390/ijms222212438(2021).
Takahashi, K. et al. Establishment of a 5-fluorouracil-resistant triple-negative breast cancer cell line. Int J. Oncol. 43, 1985–1991 (2013).
Jayaraman, G. et al. p300/cAMP-responsive element-binding protein interactions with ets-1 and ets-2 in the transcriptional activation of the human stromelysin promoter. J. Biol. Chem. 274, 17342–17352 (1999).
Millien, G. et al. ETS1 regulates Twist1 transcription in a Kras(G12D)/Lkb1(-/-) metastatic lung tumor model of non-small cell lung cancer. Clin. Exp. Metastasis 35, 149–165 (2018).
Bukhari, A. B. et al. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Invest. 129, 1329–1344 (2019).
Lee, K. J. et al. Exploiting DNA repair defects in triple negative breast cancer to improve cell killing. Ther. Adv. Med. Oncol. 12, 1758835920958354 (2020).
Li, L. Y., Guan, Y. D., Chen, X. S., Yang, J. M. & Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front Pharm. 11, 629266 (2020).
Mehlich, D. et al. MLK4 regulates DNA damage response and promotes triple-negative breast cancer chemoresistance. Cell Death Dis. 12, 1111 (2021).
van Waardenburg, R. & Yang, E. S. Targeting DNA repair pathways to overcome cancer drug resistance. Cancer Drug Resist 4, 837–841 (2021).
Cheang, M. C. et al. Defining breast cancer intrinsic subtypes by quantitative receptor expression. Oncologist 20, 474–482 (2015).
Prat, A. et al. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br. J. Cancer 111, 1532–1541 (2014).
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
Gyorffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 19, 4101–4109 (2021).
Ciszewski, W. M., Tavecchio, M., Dastych, J. & Curtin, N. J. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 143, 47–55 (2014).
Veuger, S. J., Curtin, N. J., Richardson, C. J., Smith, G. C. & Durkacz, B. W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res 63, 6008–6015 (2003).
Tsuji, T. et al. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 8, 74688–74702 (2017).
Ballou, L. M., Selinger, E. S., Choi, J. Y., Drueckhammer, D. G. & Lin, R. Z. Inhibition of mammalian target of rapamycin signaling by 2-(morpholin-1-yl)pyrimido[2,1-alpha]isoquinolin-4-one. J. Biol. Chem. 282, 24463–24470 (2007).
Griffin, R. J. et al. Selective benzopyranone and pyrimido[2,1-a]isoquinolin-4-one inhibitors of DNA-dependent protein kinase: synthesis, structure-activity studies, and radiosensitization of a human tumor cell line in vitro. J. Med. Chem. 48, 569–585 (2005).
Leahy, J. J. et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 14, 6083–6087 (2004).
Dylgjeri, E. & Knudsen, K. E. DNA-PKcs: A Targetable Protumorigenic Protein Kinase. Cancer Res. 82, 523–533 (2022).
Douglas, P. et al. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol. Cell Biol. 27, 1581–1591 (2007).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
Ochoa, D. et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol. 38, 365–373 (2020).
Dou, Y., Yao, B. & Zhang, C. PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids 46, 1459–1469 (2014).
Jung, H. H. et al. Statins affect ETS1-overexpressing triple-negative breast cancer cells by restoring DUSP4 deficiency. Sci. Rep. 6, 33035 (2016).
Shah, V. V. et al. Mammary-specific expression of Trim24 establishes a mouse model of human metaplastic breast cancer. Nat. Commun. 12, 5389 (2021).
Gluck, C. et al. Molecular dissection of the oncogenic role of ETS1 in the mesenchymal subtypes of head and neck squamous cell carcinoma. PLoS Genet. 15, e1008250 (2019).
Plotnik, J. P., Budka, J. A., Ferris, M. W. & Hollenhorst, P. C. ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res. 42, 11928–11940 (2014).
Tomar, S. et al. ETS1 induction by the microenvironment promotes ovarian cancer metastasis through focal adhesion kinase. Cancer Lett. 414, 190–204 (2018).
McCarter, A. C. et al. Combinatorial ETS1-dependent control of oncogenic NOTCH1 enhancers in T-cell leukemia. Blood Cancer Discov. 1, 178–197 (2020).
Cameron, D. A., Gabra, H. & Leonard, R. C. Continuous 5-fluorouracil in the treatment of breast cancer. Br. J. Cancer 70, 120–124 (1994).
Ghafouri-Fard, S. et al. 5-Fluorouracil: A Narrative Review on the Role of Regulatory Mechanisms in Driving Resistance to This Chemotherapeutic Agent. Front. Oncol. 11, 658636 (2021).
Ayers, M. et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J. Clin. Oncol. 22, 2284–2293 (2004).
Rouzier, R. et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 11, 5678–5685 (2005).
Gross, E. et al. Somatic copy number changes in DPYD are associated with lower risk of recurrence in triple-negative breast cancers. Br. J. Cancer 109, 2347–2355 (2013).
Li, Q. et al. A phase II study of capecitabine plus cisplatin in metastatic triple-negative breast cancer patients pretreated with anthracyclines and taxanes. Cancer Biol. Ther. 16, 1746–1753 (2015).
Li, Y. et al. Adjuvant addition of capecitabine to early-stage triple-negative breast cancer patients receiving standard chemotherapy: a meta-analysis. Breast Cancer Res. Treat. 179, 533–542 (2020).
Sethy, C. & Kundu, C. N. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: Implication of DNA repair inhibition. Biomed. Pharmacother. 137, 111285 (2021).
Tetsu, O. & McCormick, F. ETS-targeted therapy: can it substitute for MEK inhibitors?. Clin. Transl. Med. 6, 16 (2017).
Wu, M. et al. Targeting ETS1 with RNAi-based supramolecular nanoassemblies for multidrug-resistant breast cancer therapy. J. Control Release 253, 110–121 (2017).
Echeverria, G. V. et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci.Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aav0936 (2019).
Eckstein, N. Platinum resistance in breast and ovarian cancer cell lines. J. Exp. Clin. Cancer Res. 30, 91 (2011).
Petrosyan, V. et al. Identifying biomarkers of differential chemotherapy response in TNBC patient-derived xenografts with a CTD/WGCNA approach. iScience 26, 105799 (2023).
Marangoni, E. et al. Capecitabine Efficacy Is Correlated with TYMP and RB1 Expression in PDX Established from Triple-Negative Breast Cancers. Clin. Cancer Res. 24, 2605–2615 (2018).
Aldonza, M. B., Hong, J. Y., Alinsug, M. V., Song, J. & Lee, S. K. Multiplicity of acquired cross-resistance in paclitaxel-resistant cancer cells is associated with feedback control of TUBB3 via FOXO3a-mediated ABCB1 regulation. Oncotarget 7, 34395–34419 (2016).
Chen, Y. N. et al. Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J. Biol. Chem. 265, 10073–10080 (1990).
Guo, B. et al. Cross-resistance studies of isogenic drug-resistant breast tumor cell lines support recent clinical evidence suggesting that sensitivity to paclitaxel may be strongly compromised by prior doxorubicin exposure. Breast Cancer Res. Treat. 85, 31–51 (2004).
Memar, M., Farazmandfar, T., Sabaghian, A., Shahbazi, M. & Golalipour, M. Transcriptome Profiling of Cisplatin Resistance in Triple-negative Breast Cancer: New Insight into the Role of PI3k/Akt Pathway. Curr. Mol. Med. 23, 559–568 (2023).
Anurag, M. et al. Proteogenomic Markers of Chemotherapy Resistance and Response in Triple-Negative Breast Cancer. Cancer Discov. 12, 2586–2605 (2022).
Wang, J., Zeng, X., Luo, T., Jin, W. & Chen, S. Involvement of V-Ets erythroblastosis virus E26 oncogene homolog 2 in regulation of transcription activity of MDR1 gene. Acta Biochim. Biophys. Sin. (Shanghai) 44, 752–758 (2012).
Dean, K. C., Huang, L., Chen, Y., Lu, X. & Liu, Y. An Rb1-dependent amplification loop between Ets1 and Zeb1 is evident in thymocyte differentiation and invasive lung adenocarcinoma. BMC Mol. Biol. 16, 8 (2015).
Li, K. et al. Ets1-Mediated Acetylation of FoxO1 Is Critical for Gluconeogenesis Regulation during Feed-Fast Cycles. Cell Rep. 26, 2998–3010 e2995 (2019).
Forough, R. et al. Transcription factor Ets-1 regulates fibroblast growth factor-1-mediated angiogenesis in vivo: role of Ets-1 in the regulation of the PI3K/AKT/MMP-1 pathway. J. Vasc. Res. 43, 327–337 (2006).
Singh, A. K., Swarnalatha, M. & Kumar, V. c-ETS1 facilitates G1/S-phase transition by up-regulating cyclin E and CDK2 genes and cooperates with hepatitis B virus X protein for their deregulation. J. Biol. Chem. 286, 21961–21970 (2011).
Carey, L. A. et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 13, 2329–2334 (2007).
Schmid, P. et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med 379, 2108–2121 (2018).
von Minckwitz, G. et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 30, 1796–1804 (2012).
Fok, J. H. L. et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 10, 5065 (2019).
Revia, S. et al. Peposertib, a DNA-PK Inhibitor, Enhances the Anti-Tumor Efficacy of Topoisomerase II Inhibitors in Triple-Negative Breast Cancer Models. Int. J. Mol. Sci .25, https://doi.org/10.3390/ijms25105120 (2024).
Dylgjeri, E. et al. A Novel Role for DNA-PK in Metabolism by Regulating Glycolysis in Castration-Resistant Prostate Cancer. Clin. Cancer Res 28, 1446–1459 (2022).
Chen, Y. et al. Phosphorylation regulates cullin-based ubiquitination in tumorigenesis. Acta Pharm. Sin. B 11, 309–321 (2021).
Vitari, A. C. et al. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature 474, 403–406 (2011).
Joshi, M. K. et al. Substrate binding to Src: A new perspective on tyrosine kinase substrate recognition from NMR and molecular dynamics. Protein Sci. 29, 350–359 (2020).
Kasahara, K., Shiina, M., Higo, J., Ogata, K. & Nakamura, H. Phosphorylation of an intrinsically disordered region of Ets1 shifts a multi-modal interaction ensemble to an auto-inhibitory state. Nucleic Acids Res. 46, 2243–2251 (2018).
Uljon, S. et al. Structural Basis for Substrate Selectivity of the E3 Ligase COP1. Structure 24, 687–696 (2016).
Early Breast Cancer Trialists et al. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 379, 432–444 (2012).
O’Reilly, D., Sendi, M. A. & Kelly, C. M. Overview of recent advances in metastatic triple negative breast cancer. World J. Clin. Oncol. 12, 164–182 (2021).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Dutta, D., Sen, A. & Satagopan, J. Sparse canonical correlation to identify breast cancer related genes regulated by copy number aberrations. PLoS One 17, e0276886 (2022).
Mares-Quinones, M. D. et al. Identification of modules and key genes associated with breast cancer subtypes through network analysis. Sci. Rep. 14, 12350 (2024).
Cowley, D. O. & Graves, B. J. Phosphorylation represses Ets-1 DNA binding by reinforcing autoinhibition. Genes Dev. 14, 366–376 (2000).
Pognonec, P., Boulukos, K. E., Gesquiere, J. C., Stehelin, D. & Ghysdael, J. Mitogenic stimulation of thymocytes results in the calcium-dependent phosphorylation of c-ets-1 proteins. EMBO J. 7, 977–983 (1988).
Pufall, M. A. et al. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309, 142–145 (2005).
Ning, S. et al. The auto-inhibition mechanism of transcription factor Ets-1 induced by phosphorylation on the intrinsically disordered region. Comput. Struct. Biotechnol. J. 20, 1132–1141 (2022).
Perez-Borrajero, C. et al. The Biophysical Basis for Phosphorylation-Enhanced DNA-Binding Autoinhibition of the ETS1 Transcription Factor. J. Mol. Biol. 431, 593–614 (2019).
Garvie, C. W., Pufall, M. A., Graves, B. J. & Wolberger, C. Structural analysis of the autoinhibition of Ets-1 and its role in protein partnerships. J. Biol. Chem. 277, 45529–45536 (2002).
Vojtek, M. et al. Pd(II) and Pt(II) Trinuclear Chelates with Spermidine: Selective Anticancer Activity towards TNBC-Sensitive and -Resistant to Cisplatin. Pharmaceutics 15, https://doi.org/10.3390/pharmaceutics15041205 (2023).
Wang, B. et al. Cisplatin-resistant MDA-MB-231 Cell-derived Exosomes Increase the Resistance of Recipient Cells in an Exosomal miR-423-5p-dependent Manner. Curr. Drug Metab. 20, 804–814 (2019).
Yang, X. L., Lin, F. J., Guo, Y. J., Shao, Z. M. & Ou, Z. L. Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways. Onco. Targets Ther. 7, 1033–1042 (2014).
Zhao, Y., Jiang, H., Gu, M., Zu, C. & Zheng, X. Gemcitabine resistance in triple-negative breast cancer cells can be reverted by Drosophila melanogaster deoxyribonucleoside kinase in the nucleus or cytosol. Oncol. Lett. 20, 247 (2020).
Cui, X. et al. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell Biol. 25, 10842–10852 (2005).
Selvaraj, N., Kedage, V. & Hollenhorst, P. C. Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells. Cell Commun. Signal 13, 12 (2015).
Kao, S. H. et al. GSK3beta controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene 33, 3172–3182 (2014).
Franken, N. A., Rodermond, H. M., Stap, J., Haveman, J. & van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 1, 2315–2319 (2006).
Kumar, P., Nagarajan, A. & Uchil, P. D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb Protoc 2018, https://doi.org/10.1101/pdb.prot095505 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Tarazona, S. et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 43, e140 (2015).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Mudge, J. M. et al. GENCODE 2025: reference gene annotation for human and mouse. Nucleic Acids Res. 53, D966–D975 (2025).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Perez, G. et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 53, D1243–D1249 (2025).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Nassar, L. R. et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 51, D1188–D1195 (2023).
Grossman, R. L. et al. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 375, 1109–1112 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378 (2003).
Heath, A. P. et al. The NCI Genomic Data Commons.Nat.Genet.53, 257–262 (2021)
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
The authors acknowledge Bose Institute, Kolkata, for providing funds and facilities to carry out experiments. The University of Kalyani and Burdwan University, West Bengal, are acknowledged for providing experimental facilities. N.S. was funded by DBT- Ramalingaswami Fellowship (Award sanction No: BT/RLF/Re-entry/27/2016) from the Department of Biotechnology, India. A.D. worked under a fellowship from SERB-DST (Award sanction No: ECR/2018/000595) awarded to N.S. from the Department of Science and Technology, India. M.S. and S.D. worked under fellowships [Award No: 231620139663 (M.S.), 231610193664 (S.D.)] from CSIR-UGC, India. The authors acknowledge members of the laboratory for helpful discussion.
Author information
Authors and Affiliations
Contributions
A.D. and N.S. designed the study, A.D., S.D., M.S., and R.K. performed the experiments, analysed the data and helped with writing and editing. N.S. was responsible for Conceptualisation, Funding acquisition, Investigation, Project administration, Resources, Supervision, writing and editing. IA and P.K.V. were responsible for the investigation, data curation, and visualisation related to bioinformatics analysis. A.H. provided the patient samples and helped with the proper identification of the samples. S.D. & S.K.H. were responsible for IHC-related work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dhara, A., Aier, I., Das, S. et al. Enhanced ETS1 stability by DNAPKcs orchestrates transcriptional changes during chemoresistance in triple negative breast cancer. npj Breast Cancer 11, 114 (2025). https://doi.org/10.1038/s41523-025-00782-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00782-3
