
Abstract
Only 15% of young-onset breast cancers have identifiable hereditary germline pathogenic variants (PVs) in an established breast cancer susceptibility gene. However, it is believed that a significant proportion of these breast cancers have additional monogenic or rare risk variants that require identification. To uncover novel cancer susceptibility genes, we performed germline whole-exome/genome sequencing of samples from 564 patients with young-onset breast cancer (aged <40 years), as well as samples from 4032 female controls. The identified candidate variants were further genotyped in 6,967 independent breast cancer cases across all age groups. We identified two PVs that were significantly associated with the risk of hormone receptor-negative young-onset breast cancer: POLH p.K589T (OR = 3.65, 95% confidence interval [CI] = 1.28–10.4, P = 0.0095) and RAD51 p.M1fs (OR = 2.15, 95% CI = 1.15–4.02, P = 0.014). When BRCA1/2 PV carriers were excluded from the analysis, only RAD51 p.M1fs retained a significant association. Whole-genome sequencing of tumor samples carrying these germline risk variants revealed that they harbored mutational signatures indicative of a deficiency of homologous recombination. These findings suggest that hereditary POLH p.K589T and RAD51 p.M1fs are candidate variants associated with an increased risk of hormone receptor-negative breast cancer.
Similar content being viewed by others

Introduction
Breast cancer is the most frequently diagnosed cancer among females worldwide, and approximately 30% of cancers in females under the age of 40 are breast cancers1,2. In most countries, breast cancer is the primary cause of cancer-related mortality among females. According to the SEER database, 5.6% of all invasive breast cancers are diagnosed in females aged <40. The incidence of invasive breast cancer in young females (aged <40 years) has increased since the early 2000s. Younger age at diagnosis is correlated with high-risk characteristics of breast cancer, such as hormone receptor (HR)-negativity and high-grade histology, as well as with poor prognosis3. Young-onset breast cancer is more often familial or hereditary than late-onset breast cancer, and 15% of cases of this type of cancer carry germline pathogenic and likely pathogenic variants (PVs) in cancer susceptibility genes4,5,6. For females harboring highly penetrant PVs in genes such as BRCA1/2 and TP53, risk-reducing bilateral mastectomies are an option7,8. In carriers of mutations in homologous recombination-associated genes, including BRCA1/2 and PALB2, tumors show a characteristic mutational signature derived from homologous recombination deficiency (HRD) and display sensitivity to poly(ADP-ribose) polymerase (PARP) inhibitors9. These findings highlight the importance of genetic testing in young-onset breast cancer to facilitate disease prevention and treatment selection.
In the past decade, many genome-wide association studies have been conducted to identify common variants that affect the risk of developing breast cancer; however, few causal variants have been identified. Genetic linkage and targeted sequencing studies identified loss-of-function (LoF) variants and several rare missense mutations in ATM, BARD1, BRCA1, BRCA2, CHEK2, RAD51C, RAD51D, PALB2, and TP5310. The contribution of rare genetic variants to complex traits and diseases has been investigated by whole-exome sequencing (WES) or whole-genome sequencing (WGS), leading to the identification of novel causal variants and genes11. However, only approximately half of breast cancers involve common susceptibility variants12,13,14,15. Thus, young-onset breast cancer, in which genetic predisposition may play an important role, warrants further study. Rare variants that remain unidentified may partly explain the genetic risk in younger patients with breast cancer. It is also unclear whether rare variants reported in Caucasians can also be detected in Asian populations.
Studies of germline variants are generally limited to the assessment of the biological role of germline variants in tumorigenesis, and somatic mutations or mutational signatures are not evaluated. Carriers of PVs associated with high penetrance frequently develop related tumors via second-hit somatic mutations, resulting in biallelic inactivation16. These tumors also exhibit somatic and clinical hallmarks of dependence on the germline allele, such as early age of onset and a low number of required somatic oncogenic driver mutations. Integrated germline analysis and somatic tumor profiling are important for assessing the contribution of germline variants to tumorigenesis.
In this study, we performed germline WES or WGS in 564 Japanese females with young-onset breast cancer and 4032 non-cancer controls to identify candidate susceptibility variants. The identified candidate variants were genotyped in an independent cohort of 6,967 breast cancers in all age groups. In addition, WGS was performed on tumor samples with candidate risk variants to determine whether these tumors had genomic characteristics associated with HRD.
Results
Characteristics of study participants
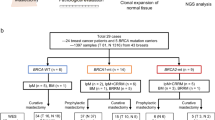
The study included 564 patients (aged <40 years) with young-onset breast cancer who were evaluated by WES or WGS (the young-onset breast cancer cohort), 6967 all-generation patients with breast cancer who underwent TaqMan assays (the all-generation breast cancer cohort), and 4032 Japanese non-cancer controls from the National Cancer Biobank Network (NCBN), all of whom were evaluated by WGS (Fig. 1). The median ages at diagnosis for the young-onset breast cancer cohort (aged <40 years) and all-generation breast cancer cohort were 36 and 56 years, respectively, and 23.4% and 15.4% of the cases were HR-negative, respectively. Patients with a family history of breast and/or ovarian cancer accounted for 19% of the young-onset breast cancer cohort and 4.5% of the all-generations cohort (Table 1).

STROBE flow chart.
Germline PVs in 26 known cancer susceptibility genes
First, germline PVs in 26 established cancer susceptibility genes included in the Myriad myRisk Hereditary Cancer Test were evaluated in the young-onset breast cancer cohort. PVs of the 26 cancer susceptibility genes were found in 110 (19.5%) patients (Fig. 2). Among these patients, the median age at diagnosis was 34 years, 67.3% (66/98) were HR-negative, and 34.3% (37/108) had a family history of breast and/or ovarian cancer. PVs of BRCA 1/2 (n = 71, 12.6%), PTEN (n = 7, 1.2%), BARD1 (n = 6, 1.1%), PALB2 (n = 5, 0.9%), and TP53 (n = 5, 0.9%) were frequently observed. These PVs were mutually exclusive, whereas three patients had two PVs, with CHEK2, NF1, and MSH6 overlapping with the BRCA2 PVs.

The 113 germline pathogenic variants were identified in 110 young-onset breast cancer patients. Each column corresponds to a patient. The upper panel shows the patient’s clinical characteristics. IDC invasive ductal carcinoma, HR hormone receptor.
Identification of novel risk variants in young-onset breast cancers
To identify novel candidate risk variants, we evaluated germline variants of 692 genes associated with cancer susceptibility and/or with DNA repair functions based on previous reports (Supplementary Data S1)17,18,19,20. According to our selection criteria, we identified one pathogenic missense and two LoF variants that were possibly associated with the risk of HR-negative young-onset breast cancer (Supplementary Fig. 1). The first risk variant was a pathogenic missense variant, p.K589T (rs121908565), in POLH (polymerase eta), observed in 3 of 114 HR-negative young-onset breast cancers and 31 of 4,032 NCBN controls (2.6% vs. 0.77%, OR = 3.46, 95% CI = 1.05–11.4, P = .03). The second risk variant was a frameshift variant, p.74Sfs*8 (rs541992483) in BPIFB4 (BPI fold-containing family B, member 4). This variant was found in 3 of 114 HR-negative young-onset breast cancers and 32 of 4032 controls (2.6% vs. 0.79%, OR = 3.35, 95% CI = 1.02–11.01, P = .035). The third variant was the frameshift variant pM1fs (rs55714242) in RAD51 (RAD51 recombinase), found in 10 of 114 HR-negative young-onset breast cancers and 146 of 4,032 controls (8.8% vs. 3.6%, OR = 2.49, 95% CI = 1.29–4.79, P = .0048) (Supplementary Data S2(A)). Forty-two patients with young-onset breast cancer carried one of the three candidate risk variants, and their median age at diagnosis was 37 years. Sixteen (39%, 16/41) were HR-negative, and five (12%, 5/41) had a family history of breast and/or ovarian cancer. Eight patients (19%, 8/42) also carried germline PVs in BRCA1, BRCA2, and MLH1 (Supplementary Fig. 2). To investigate the mutual exclusion between the candidate variants and known high-penetrance PVs such as BRCA1/2, we performed an analysis in which patients with BRCA1/2 PV were excluded. The estimated OR of candidate variants decreased, and only RAD51 pM1fs was associated with HR-negative breast cancers (Supplementary Data S2(B)). Although genograms were not available for carriers with these risk variants in the young-onset breast cancer cohort, our review of patients referred to the National Cancer Center Hospital for genetic counseling who underwent germline WGS identified a 38-year-old POLH p.K589T carrier with BRCA1/2-negative breast cancer who had a family history of breast and pancreatic cancer (Supplementary Fig. 3).
Frequency of rare coding variants identified in the European population
Recently, whole-exome-based association studies have identified many novel rare coding variants associated with breast cancer risk in persons of European ancestry11. We examined the frequency of PVs in the 12 genes identified in this study (MAP3K1, LZTR1, MMP26, ATRIP, BAP1, KCND2, CUL9, CFAP126, GPR37, TGM7, ZFYVE19, and SEC62) in our cohort. However, only two patients with young-onset breast cancer had PVs in LZTR1, and no PVs were detected in any of the other genes (Supplementary Data S3).
Validation of candidate variants across all-age breast cancer cases
To further assess age-specific associations between candidate variants and breast cancer, we genotyped POLH and RAD51 variants in 6,967 patients with breast cancer across all age groups. The BPIFB4 variant was not genotyped because the corresponding TaqMan PCR assay could not be designed. Compared with the same 4,032 controls described above, POLH p.K589T and RAD51 p.M1fs variants were significantly associated with HR-negative young-onset breast cancers (POLH: OR = 3.65, 95% CI = 1.28–10.4, P = .0095; RAD51: OR = 2.15, 95% CI = 1.15–4.02, P = .014), but not with HR-negative non-young-onset breast cancers. Additionally, POLH p.K589T also showed nominal associations with young-onset breast cancers irrespective of HR status (OR = 1.98, 95% CI = 1.03–3.79, P = .036), and with HR-negative breast cancers regardless of age (OR = 2.12, 95% CI = 1.01–4.46, P = .04; Fig. 3, Supplementary Data S4).

Allele frequencies of the POLH and RAD51 variants in the case and control groups are shown. POLH p.K589T and RAD51 p.M1fs variants were significantly associated with HR-negative young-onset breast cancers (POLH: OR = 3.65, 95% CI = 1.28–10.4, P = .0095; RAD51: OR = 2.15, 95% CI = 1.15–4.02, P = .014). HR hormone receptor, YO young-onset.
Tumor characteristics of POLH and RAD51-mutated breast cancers
We next evaluated the whole-genome landscape of tumor samples with germline risk variants of POLH and RAD51 to determine whether these tumors exhibited HRD-associated mutational signatures. Among the 43 females with breast cancers harboring POLH (n = 10) or RAD51 (n = 33) germline risk variants for which fresh frozen tumor tissues were available, 12 patients (four POLH and eight RAD51) with triple-negative breast cancer (TNBC, estrogen and progesterone receptor <10%) or young-onset disease were selected for WGS analysis. Two patients, one with a POLH-variant and another with a RAD51-variant, had concurrent germline or somatic LoF mutations in BRCA1. All three POLH-mutated breast cancers without concurrent BRCA1 mutations were classified as HRD using the HRDetect and CHORD algorithms (Fig. 4). One of the three POLH-mutated breast cancers showed loss of heterozygosity (LOH) at the variant locus. Of the seven RAD51-mutated breast cancers without concurrent BRCA1 mutations, one displayed LOH and was classified as having HRD. Circos plots of the genomic structures of representative HRD cases with POLH or RAD51 variants are shown in Fig. 5. A POLH-mutated TNBC with predicted BRCA1-type HRD has a large number of tandem duplications, whereas a RAD51-mutated TNBC with predicted BRCA2-type HRD has a large number of deletions. Finally, we examined the presence of LOH in additional tumors from carriers of POLH (n = 21) or RAD51 (n = 87) variants. LOH was found in six (28.5%) and four (4.6%) tumors from patients with POLH or RAD51 variants, respectively (Supplementary Data S5).

Oncogenic or pathogenic somatic mutations, and mutational signatures identified in 12 tumors from patients carrying POLH (n = 4) or RAD51 (n = 8) variants. The phenobar provides information on clinicopathological features, HRDetect, and CHORD. SBS single base substitution, IDC invasive ductal carcinoma, HR hormone receptor, TN triplenegative, ER estrogen-receptor, HG histological grade.

Circos plots of POLH (upper panel) and RAD51 (lower panel) mutated breast cancers are shown. The first outer circle represents the chromosomes. The second circle shows the base substitutions. Circles with short green linings represent insertions; circles with short red lines represent deletions. The third circle shows the major copy number changes (green, gain). The fourth circle represents the minor allele copy number (red, loss). The central lines represent rearrangements.
Discussion
Breast cancers diagnosed in young females are more often familial or hereditary than in older females, but the causative genes are unknown in most cases. In this study, we found that germline variants in POLH and RAD51 moderately increased the risk of young-onset HR-negative breast cancer.
Young-onset breast cancer is associated with a high frequency of germline PVs. PVs in BRCA1/2 are the most frequent, accounting for 10–14% of young-onset breast cancer regardless of ethnicity6,21,22. In this study, 12.6% of the young-onset breast cancer cohort had BRCA1/2 PVs, which is consistent with the results of previous reports. When the analysis was expanded to 26 known cancer susceptibility genes, 19.5% had germline PVs, which is also consistent with a study of 35,000 cases in a multi-ethnic population (13–18%)4. However, among rare coding variants other than those of known breast cancer susceptibility genes identified in European populations, only two PVs in LZTR1 were found in Japanese young-onset breast cancer. These results suggest that the contribution of low-to-moderate breast cancer susceptibility genes to young-onset breast cancer may differ according to ethnicity.
RAD51 is a well-established central protein in DNA double-strand break repair and is regulated by several proteins, including BRCA2, PALB2, and RAD51 paralogs23. Following double-strand breaks, RAD51 binds to DNA and forms a nucleoprotein filament that invades the homologous double helix. RAD51 paralogs, including RAD51C and RAD51D, contribute to the stabilization and elongation of RAD51 filaments24. Although RAD51C and RAD51D are established susceptibility genes for breast cancer, the impact of RAD51 on breast cancer risk remains to be elucidated. Although the single-nucleotide polymorphism −135 G > C in RAD51 was reported to be associated with an increased risk of breast cancer in BRCA2 carriers25, no large case-control or genome-wide association study has demonstrated that RAD51 is a breast cancer susceptibility gene in the general population26,27,28,29. By contrast, few studies have evaluated the contribution of POLH to breast cancer risk. POLH is involved in DNA damage repair via its translesion synthesis activity and is a causative gene for xeroderma pigmentosum (XP) variant disease. XP is associated with an increased risk of skin cancers, central nervous system tumors, hematologic malignancies, and gynecological cancers, but not breast cancers30. In the GENESIS study that examined 113 DNA repair-related genes in 1,207 French patients with BRCA1/2-negative familial breast cancer and 1,199 controls, POLH PVs were not associated with an increased risk of breast cancer31,32. Given that allele frequencies of POLH and RAD51 are highest in East Asian populations according to the gnomAD database, the contribution of POLH and RAD51 to breast cancer risk may be specific to these populations.
In addition to providing evidence that POLH and RAD51 variants are associated with breast cancer risk, we observed that tumors harboring POLH variants exhibited genomic characteristics consistent with HRD. POLH contributes to homologous recombination by interacting with BRCA2 and PALB2 to form a D loop33,34. Our findings from WGS analysis of tumors suggest that POLH variants play a critical role in tumorigenesis. In the latest ClinVar, POLH p.K589T has been reclassified as a variant of uncertain significance. However, others classified this variant as pathogenic because it was originally identified in a Japanese family with XP variant disease, and cells with the POLH p.K589T mutation display decreased recovery of DNA synthesis after irradiation35. Furthermore, the gene expression level of POLH in breast-mammary tissue was 6.99 TPM in the GTEx database (Supplementary Data S2), supporting the idea that decreased POLH gene expression by the LoF variant may have a high functional impact. Interestingly, LOH was found in only one-quarter of patients with POLH variants and HRD, indicating that a single-allele variant may contribute to carcinogenesis, similar to POLE36. By contrast, the HRD phenotype was observed in tumors from RAD51 variant carriers only in the presence of LOH. Although the RAD51 p.M1fs variant is annotated as likely benign in ClinVar primarily because of its relatively high allele frequency in the general population, it causes a frameshift at the initiation codon and eliminates the canonical start site, likely resulting in a LoF effect on the RAD51 protein.
Given the unfavorable prognosis associated with young-onset breast cancer and the limited therapeutic options available for HR-negative breast cancer, there is an urgent need to develop early cancer detection methods and novel treatment strategies. The present study identified POLH p.K589T and RAD51 p.M1fs variants in 1.5% and 4.8% of Japanese young-onset breast cancers, respectively. However, POLH is currently not included in widely used gene panel tests, and identifying POLH variants is thus difficult.
This study had several limitations. First, although germline whole exome/genome analysis was performed, we focused on the 692 selected genes. This approach may have underestimated the presence of potential candidate risk variants. Second, information on breast cancer subtypes was unavailable for a significant number of cases. Third, it remains unclear whether the POLH variant functions as a modifier that enhances breast cancer risk in the presence of high-penetrance PVs or as an independent low- to moderate-risk variant. Given that the allele frequency of the POLH variant was less than 1%, the statistical power of the subgroup analysis was limited. Further investigations are necessary to address this question. Lastly, this study lacked an independent cohort to evaluate the association between candidate variants and the risk of young-onset breast cancer. However, young-onset breast cancer is a rare occurrence, accounting for approximately 5% of all breast cancers. Consequently, this study represents one of the largest case-control studies conducted on homogenous racial populations, and it thus provides valuable insight into this particular issue.
In conclusion, PVs in POLH and RAD51 may contribute to susceptibility to HR-negative young-onset breast cancer. If further large-scale studies confirm their association with the risk of young-onset breast cancer, these genes should be incorporated into gene panel tests to predict heritable risk.
Methods
Study cohorts
The young-onset breast cancer cohort comprised 567 patients diagnosed with breast cancer aged <40 years who were collected from three institutions: National Cancer Center (NCC) Hospital, NCC Hospital East, and Biobank Japan. Biobank Japan is a multi-institutional, hospital-based registry that collects DNA and clinical information from patients with various common diseases, including breast cancer, from all over Japan37,38. The all-generation breast cancer cohort included 7,025 patients with breast cancer from all age groups from five institutions: NCC Hospital, NCC Hospital East, Kanagawa Cancer Center, Yamanashi Prefectural Center Hospital, and Fukushima Medical University. After excluding males with breast cancer and those who did not have sufficient sequence depth for targeted sequencing6,39, 7,531 female cases (including 858 young-onset breast cancer cases) were analyzed. The controls were recruited from the NCBN40,41, and comprised 4,032 females aged ≥16 years, none of whom had a history of cancer. A STROBE flow chart of the study is shown in Fig. 1.
Sequencing of germline variants
Genomic DNA was extracted from leukocytes and non-cancerous breast cancer tissues using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) or the Allprep DNA Mini Kit (Qiagen, Hilden, Germany). For 183 breast cancer patients, WES was performed using the Agilent SureSelect Human All Exon V4 or V5 platform and the Illumina Nextera Exome Kit and Nextera DNA Library Prep for Enrichment according to the manufacturer’s instructions. Sequencing of the 75 bp paired-end reads was performed using HiSeq2500 (Illumina, San Diego, CA, USA) at a depth of approximately 100×. For 384 breast cancer patients, WGS libraries were prepared using the TruSeq DNA PCR-Free Library Prep Kit (Illumina). Sequencing of the 150 bp paired-end reads was performed using NovaSeq6000 (Illumina) at a depth of approximately 30×. FASTQ data from 235 cases in Biobank, Japan, were downloaded from NBCD (JGAS000114). The resulting FASTQ data were subjected to genome mapping and variant calling using our in-house data analysis pipeline. Genome mapping was performed using Parabricks v3.1.3 (NVIDIA), which delivers the high-speed analysis recommended by the Genomic Analysis Toolkit (GATK) with GPU acceleration42. Genome Reference Consortium Human Build 38 was used as the reference sequence. The pipeline used in this study implemented algorithms equivalent to those of Burrows–Wheeler Aligner (v0.7.15)43 and GATK (v4.1.0). Duplicates from mapped reads were flagged, and realignment and base quality score recalibration were performed. Mapped data were outputed in BAM format44. Variant calls were converted into the gVCF format for joint calling. Genotyping of candidate variants was performed using GATK HaplotypeCaller45, with the hard-filtering setting suggested by GATK.
Germline variant classification
Two gene sets were evaluated, namely 26 established cancer susceptibility genes (APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDK4, CDKN2A, CDH1, CHEK2, EPCAM, NBN, NF1, MLH1, MSH2, MSH6, MUTYH, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53) included in the Myriad myRisk Hereditary Cancer Test4, and 692 genes associated with cancer predisposition and DNA repair reported in previous studies17,18,19,20 (Supplemental Data S1). Germline variants were considered pathogenic if they met the following criteria [(1) + (2) + (3) or (1) + (2) + (4) or (1) + (2) + (5)]: (1) global minor allele frequency (MAF) < 0.05 in ExAC46 and/or Tohoku Medical Megabank Organization47; (2) variant allele frequency (VAF) ≥30% and ≤70%; (3) null variants (nonsense, frameshift indel, and splice-site variants) and missense variants classified as “pathogenic” or “likely pathogenic” in ClinVar48 (https://www.ncbi.nlm.nih.gov/clinvar/); (4) high-impact LoF variants such as stop-gain, stop-loss, start-loss, frameshift, splice acceptor gain or loss, and splice donor gain or loss defined by SnpEff v4.349; and (5) splice variants with a delta score >0.5 annotated by SpliceAI50. Finally, annotations of each variant were reviewed by an expert panel. Pathogenic variants were validated by Sanger Sequencing51.
Selection of candidate susceptibility genes and risk variants in young-onset breast cancers
Candidate risk variants were selected according to the following criteria: (1) variants classified as pathogenic according to the criteria listed above; (2) variants detected in ≥5 cases; and (3) variants with odds ratio (OR) ≥1.5 and P value < 0.05 against NCBN female controls. The selected candidate risk variants in the all-generation breast cancer cohort were further genotyped using TaqMan SNP Genotyping Assays.
Detection of genomic alterations by WGS
DNA was extracted from the tumor and matched normal tissues and subjected to library preparation. Tumor sequencing was performed using NovaSeq6000 (Illumina) at an approximate depth of 120×, whereas matched normal tissue sequencing was performed at an approximate depth of 30×. The resulting reads were aligned to the hg38. Somatic SNVs were called using mutect2 (gatk version 4.1.2.0)52, and small indels were called using mutect2 and strelka253. The detected variants were annotated using OncoKB54, ClinVar48, and SnpEff49, and oncogenic variants were defined as those annotated as oncogenic or likely oncogenic in the OncoKB database54. These variants were validated using the Integrative Genomics Viewer (IGV)55. Single base substitution signatures were estimated using the Fit Multi-Step (FitMS) algorithm introduced in previous studies56,57. Breast-specific signatures detected in the Genomics England (GEL) cohort were assigned to each sample. Structural variants were detected using Manta (version 1.6.0)58, and rearrangement signatures were extracted from Signature.tools.lib59. SVs were classified into 32 SV types based on size, topology, and junction clustering as previously described, and were fit to 20 rearrangement signatures derived from 3,107 cancers57. Allele-specific copy number, tumor purity, and ploidy were estimated by facets (version 0.6.2)60. Loss of heterozygosity (LOH) was considered to be present when the total copy number of a gene was one and the minor copy number was zero.
HRD prediction
HRD status was predicted by R package Classifier of HOmologous Recombination v2.0 (CHORD)61 and HRDetect59, as previously described. HRD status by CHORD and HRDetect was determined as HRD when the predictive score was >0.5 and >0.7, respectively. CHORD also distinguished BRCA1-type or BRCA2-type HRD based on 1–100 kb duplications.
Detection of copy number alterations using the TaqMan assay
Copy number alterations were detected by real-time genomic PCR using the TaqMan copy number assay and ABI 7900HT real-time PCR system (Thermo Fisher Scientific, MA, USA). The two genes, POLH (NM_006502.3) and RAD51 (NM_002875.5), and all TaqMan probes, including POLH (ID Hs00165713_cn), RAD51 (ID Hs00114987_cn), and RNase P (cat. no. 4403328), were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Genomic data were analyzed using the ABI PRISM 7900HT Sequence Detection Software CopyCaller v2.1 (Thermo Fisher Scientific) for copy number analysis. LOH was determined by CopyCaller according to the manufacturer’s instructions.
Statistical analysis
The Mann-Whitney U test was used for continuous variables, and Fisher’s exact test was used for categorical variables. Case-control association analyses were performed using Fisher’s exact test to calculate the OR and 95% confidence interval (CI) for each variant. In addition, PLINK1.06 was used for the statistical analysis of association studies. All tests were two-tailed and the significance level was set at α = .05. The Bonferroni correction was applied for association analysis between the two genotyped variants and breast cancer (P < .025 = 0.05/2). Association analyses were prespecified for the overall cohort and for subgroups defined by age (≥40 vs. <40 years) and HR status (positive vs. negative). Statistical analyses were performed using STATA (version 15.1; StataCorp, College Station, TX, USA) and GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA).
Ethics approval
This study was approved by the Institutional Review Board of all participating institutions: NCC Hospital (2015-278, 2017-353, and 2019-229), NCC Hospital East (2015-278 and 2017-353), Kanagawa Cancer Center (2017-74), Yamanashi Prefectural Center Hospital (1709-28), Fukushima Medical University (29275). All patients provided written informed consent to participate. This study involving human material and data has been performed in accordance with the Declaration of Helsinki.
Data availability
Genome data are available from the National Bioscience Database Center (NBDC) Human Database (research ID: JGAS000114). Other genome data that support the findings of this study and further information are available from the corresponding author upon reasonable request.
Code availability
The code used for the data analysis presented in this manuscript utilizes publicly available software packages with no customization. The code can be provided upon reasonable request.
References
Cathcart-Rake, E. J. et al. Breast cancer in adolescent and young adult women under the age of 40 years. JCO Oncol. Pr. 17, 305–313 (2021).
Dyba, T. et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 157, 308–347 (2021).
Partridge, A. H. et al. Subtype-dependent relationship between young age at diagnosis and breast cancer survival. J. Clin. Oncol. 34, 3308–3314 (2016).
Buys, S. S. et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 123, 1721–1730 (2017).
Tung, N. et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J. Clin. Oncol. 34, 1460–1468 (2016).
Momozawa, Y. et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 9, 4083 (2018).
Yadav, S. et al. Contralateral breast cancer risk among carriers of germline pathogenic variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2. J. Clin. Oncol. 41, 1703–1713 (2023).
Daly, M. B. et al. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc Netw. 19, 77–102 (2021).
Gruber, J. J. et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat. Cancer 3, 1181–1191 (2022).
Breast Cancer Association, C. et al. Breast cancer risk genes - association analysis in more than 113,000 women. N. Engl. J. Med 384, 428–439 (2021).
Wilcox, N. et al. Exome sequencing identifies breast cancer susceptibility genes and defines the contribution of coding variants to breast cancer risk. Nat. Genet 55, 1435–1439 (2023).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Lilyquist, J. et al. Common genetic variation and breast cancer risk-past, present, and future. Cancer Epidemiol. Biomark. Prev. 27, 380–394 (2018).
Sakoda, L. C., Jorgenson, E. & Witte, J. S. Turning of COGS moves forward findings for hormonally mediated cancers. Nat. Genet 45, 345–348 (2013).
Loveday, C. et al. Analysis of rare disruptive germline mutations in 2135 enriched BRCA-negative breast cancers excludes additional high-impact susceptibility genes. Ann. Oncol. 33, 1318–1327 (2022).
Srinivasan, P. et al. The context-specific role of germline pathogenicity in tumorigenesis. Nat. Genet 53, 1577–1585 (2021).
Rahman, N. Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014).
Akhavanfard, S. et al. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat. Commun. 11, 2206 (2020).
Rotunno, M. et al. A systematic literature review of whole exome and genome sequencing population studies of genetic susceptibility to cancer. Cancer Epidemiol. Biomark. Prev. 29, 1519–1534 (2020).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Copson, E. R. et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 19, 169–180 (2018).
Waks, A. G. et al. Somatic and germline genomic alterations in very young women with breast cancer. Clin. Cancer Res. 28, 2339–2348 (2022).
Groelly, F. J. et al. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 23, 78–94 (2023).
Grundy, M. K., Buckanovich, R. J. & Bernstein, K. A. Regulation and pharmacological targeting of RAD51 in cancer. NAR Cancer 2, zcaa024 (2020).
Antoniou, A. C. et al. RAD51 135G->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am. J. Hum. Genet. 81, 1186–1200 (2007).
Jia, G. et al. Genome- and transcriptome-wide association studies of 386,000 Asian and European-ancestry women provide new insights into breast cancer genetics. Am. J. Hum. Genet. 109, 2185–2195 (2022).
Mueller, S. H. et al. Aggregation tests identify new gene associations with breast cancer in populations with diverse ancestry. Genome Med. 15, 7 (2023).
Le Calvez-Kelm, F. et al. RAD51 and breast cancer susceptibility: no evidence for rare variant association in the Breast Cancer Family Registry study. PLoS One 7, e52374 (2012).
O’Brien, K. M. et al. A family-based, genome-wide association study of young-onset breast cancer: inherited variants and maternally mediated effects. Eur. J. Hum. Genet. 24, 1316–1323 (2016).
Nikolaev, S., Yurchenko, A. A. & Sarasin, A. Increased risk of internal tumors in DNA repair-deficient xeroderma pigmentosum patients: analysis of four international cohorts. Orphanet J. Rare Dis. 17, 104 (2022).
Girard, E. et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int. J. Cancer 144, 1962–1974 (2019).
Martens, M. C., Emmert, S. & Boeckmann, L. Xeroderma Pigmentosum: Gene Variants and Splice Variants. Genes (Basel). 12, 1173 (2021).
Buisson, R. et al. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 6, 553–564 (2014).
McIlwraith, M. J. et al. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell 20, 783–792 (2005).
Itoh, T. et al. Xeroderma pigmentosum variant heterozygotes show reduced levels of recovery of replicative DNA synthesis in the presence of caffeine after ultraviolet irradiation. J. Invest Dermatol. 115, 981–985 (2000).
Palles, C. et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 45, 136–144 (2013).
Hirata, M. et al. Cross-sectional analysis of BioBank Japan clinical data: a large cohort of 200,000 patients with 47 common diseases. J. Epidemiol. 27, S9–S21 (2017).
Kubo, M. & Guest, E. BioBank Japan project: epidemiological study. J. Epidemiol. 27, S1 (2017).
Momozawa, Y. et al. Low-frequency coding variants in CETP and CFB are associated with susceptibility of exudative age-related macular degeneration in the Japanese population. Hum. Mol. Genet. 25, 5027–5034 (2016).
Omae, Y., Goto, Y. I. & Tokunaga, K. National center biobank network. Hum. Genome Var. 9, 38 (2022).
Kawai, Y. et al. Exploring the genetic diversity of the Japanese population: insights from a large-scale whole genome sequencing analysis. PLoS Genet. 19, e1010625 (2023).
Franke, K. R. & Crowgey, E. L. Accelerating next generation sequencing data analysis: an evaluation of optimized best practices for Genome Analysis Toolkit algorithms. Genomics Inf. 18, e10 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, 2014 (2021).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Nagasaki, M. et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6, 8018 (2015).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e524 (2019).
Yazaki, S. et al. Impact of germline variants on breast and ovarian cancer risk in Japanese women: an original cohort study and meta-analysis. EBioMedicine 116, 105758 (2025).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Chakravarty, D. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 1, 1–16 (2017).
Robinson, J. T. et al. Variant review with the integrative genomics viewer. Cancer Res. 77, e31–e34 (2017).
Degasperi, A. et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat. Cancer 1, 249–263 (2020).
Degasperi, A. et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science. 376, abl9283 (2022).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Nguyen, L. et al. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 11, 5584 (2020).
Acknowledgements
The authors wish to thank physicians and staff members at the National Cancer Center and other hospitals for their assistance and support. We thank all of the subjects for participating in the study, as well as Maiko Matsuda, Yoko Shimada, Sadaaki Takata, Hiromichi Nakajima, Hikari Kiyohara, Akira Hirota, Mao Uematsu, Misao Fukuda, and Nobuyuki Takahashi for assisting with sample collection. We acknowledge the staff of the Laboratory for Genotyping Development in RIKEN, the RIKEN-IMS Genome Platform, the NCBN Controls WGS Consortium, and the BioBank Japan project. We would like to express our gratitude to Bioedit for assisting with English language editing. This research was supported in part by Japan Agency for Medical Research and Development (AMED) (JP15ck0106096 and 25ck0106879h0003 to T.Ko., 19cm0106605h0003 and 23ama221520h0001 to K.Sh., and JP19kk0305010 to Y.Mo.), Health Labour Sciences Research Grant (202108001B to K.Sh.), the Japan Society for the Promotion of Science (JSPS) KAKENHI Early-Career Scientists JP18K16292 (to Y.H.), a Grant-in-Aid for Scientific Research (B) 20H03668 and 23H02955 (to Y.H.), 17H06162 (to H.N.), 20H03695 (to K.Sh.), and 16H06277 (CoBiA: to Y.Mo.), a Grant-in-Aid for the Genome Research Project from Yamanashi Prefecture (to M.O. and Y.H.), BRIDGE (programs for bridging the gap between R&D and the ideal society (Society 5.0) and generating economic and social value to K.Sh.), National Cancer Center Research, and Development Fund (2022-A-20, 2023-J-2, NCC Biobank, and NCC Core Facility to K.Sh.).
Author information
Authors and Affiliations
Consortia
Contributions
S.Y., T.Ko., and K.Sh. conceptualized the study and developed the methodology. S.Y., R.K., Y.Mo., T.Yo., T.Ya., S.S., C.Y., K.H., M.S., Y.H., H.A., R.H., C.S., A.Sh., T.S., K.Su., M.Y., K.Sun., M.Hi., Y.Y., T.Kog., T.Mu., S.F., Y.Mi., K.Ta., K.M., Y.Mur., H.N., K.To.,Y.K., NCBN Controls WGS Consortium, Biobank Japan Project, M.O., T.Oh., A.Su., T.On., Y.N., T.Yam., K.Y., T.Ko., and K.Sh. provided patient samples for the study. Y.Mo., S.T., A.O., Y.Shim., and K.Sh. performed the experiments. S.Y., R.K., and H.A. curated the data. Y.Shir., M.T., A.M., K.Hi., E.F., K.K., M.Ho., and A.K. supervised the analyses and contributed to data interpretation. S.Y. and K.Sh. performed the formal analysis. S.Y. and K.Sh. wrote the original draft of the manuscript. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Masayuki Yoshida reported receiving personal fees from Roche Japan, Agilent Technologies, Chugai Pharma, Ono Yakuhin, MSD, and Daiichi Sankyo. He has participated on a Data Safety Monitoring Board or Advisory Board for Daiichi Sankyo. Kenichi Harano reported receiving grants from AstraZeneca, Chugai, Daiichi Sankyo, MSD, and Takeda, and personal fees from AstraZeneca, Chugai, Eisai, MSD, Taiho, and Takeda. He has participated on a Data Safety Monitoring Board or Advisory Board for AstraZeneca, Chugai, Daiichi Sankyo, Taiho, and Takeda. Takashi Yamanaka reported receiving personal fees from Daiichi Sankyo, Eli Lilly Japan, AstraZeneca, Chugai, Pfizer Japan, Kyowa Kirin, and Taiho. Kazunoshin Tachibana reported receiving grants from Chugai, Eisai, Taiho, Takeda, MSD, Daiichi Sankyo, Eli Lilly, Asahi Kasei, Nihon Kayaku, Kyowa Kirin, Astellas, and Maruho, and personal fees from Chugai, AstraZeneca, Pfizer, Eisai, Daiichi Sankyo, Eli Lilly, MSD, Kyowa Kirin, Teijin, Taiho, PDR Pharma, and Exact Sciences. Chikako Shimizu reported receiving personal fees from Chugai and has participated on a Data Safety Monitoring Board or Advisory Board for Daiichi Sankyo. Akihiko Shimomura reported receiving grants from Chugai Pharmaceutical, AstraZeneca, and Eisai, and personal fees from AstraZeneca, Daiichi Sankyo, Pfizer, Eli Lilly, MSD, Chugai Pharmaceutical, Nihon Medi-Physics, Taiho Pharmaceutical, and Exact Sciences. Takahiro Kogawa reported receiving grants from Eli Lilly, AstraZeneca, and Guardant Health; consulting fees from Daiichi Sankyo and Astellas Pharma; and personal fees from Daiichi Sankyo, Ono Pharma, Gilead Sciences, Astellas Pharma, Eisai, AstraZeneca, Taiho Pharma, and Chugai Pharma. He has received payment for expert testimony from Astellas Pharma and support for attending meetings from Pfizer and Eisai. He has participated on a Data Safety Monitoring Board or Advisory Board for Daiichi Sankyo, Ono Pharma, Gilead Sciences, Oncotherapy Sciences, Eisai, AstraZeneca, and Taiho Pharma. Tohru Ohtake reported receiving grants from Chugai, Eisai, Taiho, Takeda, Asahi Kasei, Daiichi Sankyo, Eli Lilly, Nihon Kayaku, and Kyowa Kirin, and personal fees from Chugai, Pfizer, AstraZeneca, Eisai, Daiichi Sankyo, Eli Lilly, Kyowa Kirin, Novartis, FUJIFILM Toyama Chemical, Johnson & Johnson, Asahi Kasei, Exact Sciences, Otsuka, and MSD. Tatsuya Onishi reported receiving grants from Daiichi Sankyo and Bayer Pharma, and personal fees from Daiichi Sankyo. Toshinari Yamashita reported receiving grants from Chugai, Taiho, Nippon Kayaku, Eli Lilly, Daiichi Sankyo, Pfizer, AstraZeneca, Seagen, MSD, Kyowa Kirin, Ono, Gilead Sciences, and Eisai, and personal fees from Chugai, Eisai, Daiichi Sankyo, Taiho, Nippon Kayaku, AstraZeneca, Kyowa Kirin, Pfizer, Eli Lilly, Novartis Pharma, and MSD. Yoichi Naito reported receiving grants from AbbVie, Ono, Daiichi Sankyo, Taiho, Pfizer, Boehringer Ingelheim, Eli Lilly, Eisai, AstraZeneca, Chugai, and Bayer, and personal fees from AstraZeneca, Eisai, Ono, Guardant, Takeda, Eli Lilly, Novartis, Pfizer, Chugai, PDR Pharma, Nihon Kayaku, Taiho, Bristol Myers Squibb, Bayer, Daiichi Sankyo, and MSD. Kan Yonemori reported receiving grants from MSD, Daiichi Sankyo, Merck Biopharma, AstraZeneca, Taiho, Pfizer, Novartis, Takeda, Chugai, Ono, Sanofi, Seagen, Eisai, Eli Lilly, Genmab, Boehringer Ingelheim, Kyowa Hakko Kirin, Nihon Kayaku, and Haihe; personal fees from Pfizer, Eisai, AstraZeneca, Eli Lilly, Takeda, Chugai, Fuji Film Pharma, PDR Pharma, MSD, Boehringer Ingelheim, Ono, Daiichi Sankyo, Bayer, Janssen, Astellas, Bristol Myers Squibb, Novartis, and Sanofi; and has participated on a Data Safety Monitoring Board or Advisory Board for Eisai, AstraZeneca, Sanofi, Genmab, Gilead, OncXerna, Takeda, Novartis, MSD, and Henlius. All remaining authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yazaki, S., Kitadai, R., Momozawa, Y. et al. Germline variants of the POLH and RAD51 genes are candidate variants associated with risk of hormone receptor-negative young-onset breast cancer. npj Breast Cancer 11, 133 (2025). https://doi.org/10.1038/s41523-025-00848-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00848-2
