
Abstract
Up to 30% of women with no evidence of disease after curative intent breast cancer treatment will relapse and succumb to metastatic disease. Liquid biopsy of circulating tumor DNA (ctDNA) has been shown to identify patients with relapse prior to imaging. Tumor-informed ctDNA panels are expensive and require patient tumor tissue for customization. We aimed to assess tumor-agnostic methylated DNA markers (MDMs) to detect recurrent metastatic breast cancer in comparison to cancer-free patients and those with no evidence of disease. We discovered and down-selected candidate MDMs from a series of tissue-based experiments on primary breast cancer tissue from a balance of molecular subtypes compared to matched non-cancer patient breast tissues. We then used Target Enrichment Long-probe Quantitative Amplified Signal assays to quantify 16 MDMs from cfDNA extracted from peripheral blood samples from 60 metastatic breast cancer patients (59 females, 1 male), 60 age-matched females with no prior cancer, and 60 age-matched females without evidence of metastatic disease at least 9 months after curative intent treatment of primary breast cancer. Among the 16 MDMs, ten had single-marker areas under the curve (AUCs) above 0.90, with the highest at 0.96, whereas in the same patients, three protein markers (CA153, CEA, CA125) performed with an AUC of 0.86, 0.84, and 0.75, respectively. The results indicate that a multi-marker methylation panel can detect metastatic breast cancer patients.
Similar content being viewed by others

Introduction
One in ten to one in three patients with breast cancer (BC) experience recurrence; this can be either locally (in the breast) or distant (other organs)1,2,3. However, in the absence of clinical signs and symptoms, current guidelines do not endorse the routine use of imaging (beyond surveillance breast imaging) for the monitoring of asymptomatic breast cancer patients after therapy with curative intent, particularly for those whose disease was early stage4. This recommendation stemmed from older literature, showing little utility to insensitive serologic testing and high downstream resource utilization after false-positive cross-sectional imaging5,6. Currently, molecular testing for circulating tumor DNA (ctDNA) in blood is a rapidly expanding area of scientific focus, which has applications in both early detection and recurrence monitoring7,8. ctDNA, unlike protein markers such as CA125 and CA19-9, originates from the tumor itself and, as such, reflects specific alterations and molecular events inherent to the tumor. ctDNA levels in blood are also reflective of tumor9,10,11 burden and clinical outcome, which potentially makes it an ideal analyte for assessing molecular residual disease12. Detection of ctDNA in early-stage breast cancer, using tumor informed assays or targeted next generation sequencing (NGS) panels is associated with increased risk of breast cancer recurrence9,10,11. However, tumor informed assays require a tissue sample from each patient and have high costs of goods and services.
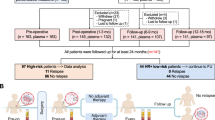
Breast cancer, like most solid epithelial cancers, is a heterogenous and complex disease and is characterized by the accumulation of numerous genetic and epigenetic alterations. Epigenetic modifications, such as DNA methylation, play a crucial role in early cancer development and progression13. We have shown previously that broadly informative aberrantly methylated tumor-agnostic DNA markers (MDMs) from peripheral blood can be used to detect minimal residual disease or early recurrence in patients with colorectal cancer who completed curative-intent therapy14. Despite the multitude of previous investigations of methylated genes in breast cancer, testing for early detection of recurrence in a tumor-agnostic setting is lacking. Here, we identify a panel of breast cancer MDMs from unbiased next-generation methylation sequencing on tissue samples, followed by targeted assay validation on independent tissues, and pilot application using plasma samples from cohorts of women with metastatic breast cancer (MBC), healthy benign controls, and those in remission from stage I to III breast cancer with no evidence of disease (NED) (Fig. 1).

Experimental outline of case and control samples used for discovery of methylation markers, biological validation, and the plasma pilot.
Results
Tissue sequencing
Reduced representation bisulfite sequencing (RRBS) uses endonuclease digestion and DNA fragment size selection to enrich regions of high CpG density in the genome (e.g., CpG islands) that tend to include numerous gene promoter and regulatory regions15,16. RRBS was performed on 86 invasive BC tissue specimens exhibiting the following clinical subtypes: 22 (25.6%) triple-negative (TNBC), 19 (22.1%) HER2+, 25 (29.1%) Luminal A, and 20 (23.3%) Luminal B, including 14 (16.3%) of these tissues collected from BRCA1 (N = 6) and BRCA2 (N = 8) mutation carriers. The control cohort included 27 benign breast tissue controls from women of similar age distribution as the cases. Nine of these were from BRCA mutation carriers and 18 were non-BRCA carriers (Table S1). In addition, 18 buffy coat (blood leukocyte fraction) samples from women free from cancer were included to ensure that any MDMs selected would have minimal white blood cell (WBC)-derived cell-free DNA (cfDNA) background signal in blood.
Each sample yielded 3–4 million CpGs with at least 10X deduplicated coverage (average 48X). Employing an internally developed software pipeline, 327 genomic regions differentially methylated regions (DMRs) were selected based on significant differential methylation between cancers and tissue controls (methods). Most of them mapped to gene regulatory sequences and were less than 1 Kb in length. DMRs were ranked using performance metrics, as previously described17. At the subtype level, 49 regions were significant for TNBC, 76 for Luminal A, 40 for Luminal B, and 123 for HER2+. In addition, 50 BRCA1+ and 46 BRCA2+ regions were identified. All subtypes had generous overlap between regions, but some regions were specific to the BC subtype (Table S2).
Given methodological constraints and sample requirements of the quantitative methylation-specific PCR (qMSP) platform we used for subsequent marker validation and the limited quantities of DNA for each study participant, it was not feasible to test all 327 regions. Instead, a candidate DMR subset was chosen based on individual performance metrics (see “Methods”). Regions with elevated methylation signal (>1%) in leukocyte-derived cfDNA, the major source of cfDNA in plasma, were discarded. Selections included DMR representation from each of the four major BC subtypes as well as BRCA+ carrier status. These steps resulted in a reduced panel of 53 MDMs.
MDM validation and selection for plasma testing
Tissue Technical Validation of qMSP assays: qMSP assays were designed for each selected region and assay performance was verified using commercial controls and additional aliquots of the previously sequenced patient samples. Logistic regression of β-actin (ACTB) normalized individual marker results yielded similar performance levels to the corresponding sequencing metrics in approximately 30% of the assays. Marker performance varied by cancer subtype compared to benign tissue with an AUC > 0.9 obtained for 7 of 53 DMRs in the TNBC cohort; 18 of 53 for HER2+, 17 of 53 for Luminal A; 18 of 53 for Luminal B. In addition, for the aggregate of cancer subtypes, 8 of 53 MDMs yielded an AUC > 0.9. When non-cancer buffy coat samples were used as the control group instead of benign tissue, 41 of 53 MDMs had AUC > 0.9 for all BCs (not shown).

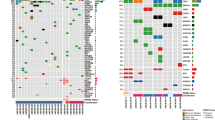
The samples are grouped left to right: control tissues, DCIS (stage 0), and invasive breast cancer case samples sorted secondarily by clinical subtype, stage, and grade. Benign controls are missing for grade, stage, and subtype, whereas DCIS are all stage 0 with grade indicated as either high or low. Markers are grouped by the top 12 as those used in the plasma pilot, and within the groups in the order of AUC comparing cancer case tissue DNA from benign control DNA as listed in Supplementary Table 2. Note MDM number axis does not include MDM13-16.
Independent Tissue Validation of MDMs: To validate performance, the 53 qMSP markers were assayed on an independent set of tissues which included 29 (22.8%) TNBC, 33 (25.9%) HER2+, 43 (33.9%) Luminal A, and 22 (17.3%) Luminal B, and 31 early non-invasive ductal carcinomas in situ (DCIS, 10 low-grade, 21 high-grade). Controls consisted of 39 benign breast tissues. Of these, 15 (28%) markers had an AUC > 0.90 for predicting the invasive cancer samples relative to controls (Fig 2 Table S2). The performance of markers within subgroups was further evaluated for their sensitivity to detect cancer at the 90th percentile of the 39 controls. Overall performance was strongest in HER2+ tissue samples with 19 (36%) of the markers displaying 90% sensitivity (Table S2).
Plasma Marker Selection: For plasma-based assays, TELQAS oligos (F/R primers and flap/invader probes) were designed for the 53 DMRs (assuming fully methylated CpGs) and retested the tissue samples on that platform. TELQAS was selected for this step to facilitate multiplex assay of MDMs from relatively small quantities of cfDNA18 at high dilution (1:10,000 analytical sensitivity)17. Results closely correlated with those from the qMSP assays (Fig. 3). We evaluated a subset of 28 MDMs (those with elevated AUCs across the BC subtypes and others with demonstrated complementarity) on cfDNA isolated from pooled normal plasma to assess background methylation levels. Subsequently, 16 MDMs were selected for the final panel.

Each column contains a patient plasma sample, and each row represents a candidate MDM, which are sorted top to bottom by their AUC performance in both Benign and NED versus MBC cases as listed in Table 2. Protein markers CA153, CEA, and CA125 are included for comparison on the same patients. Missing grade, stage, or subtype values are in white.
Application of breast cancer MDMs in plasma of women with and without breast cancer
Plasma (6 mL) was collected in LBgard (Biomatrica, San Diego, CA) tubes from 60 patients with MBC, 60 patients with stage I-III BC in remission with NED recurrence 6 months to 3 years post-treatment, and 60 healthy benign controls (normal mammogram in the past 12 months and no history of BC) (Table 1). The median follow-up time from sample collection was 2.7 (range, 1.0–3.5) years in the NED cohort, and 2.4 (range, 0.6–5.2) years for the Benign cohort. The subsequent on-bead cfDNA isolation and bisulfite treatment was designed for maximum recovery and near 100% conversion efficiency17.
All 16 MDMs showed increased copy-normalized methylation levels in the MBC cohort compared to both the NED and benign cohorts (Fig. 3). Eleven of 16 MDM markers had an AUC of 0.9 or higher for the MBC compared to combined controls (Benign plus NED), with a consistent performance in both control sets (Table 2). The protein markers (CA153, CEA, CA125) were among the lowest performing markers, with AUCs of 0.86, 0.84, and 0.75, respectively. A random forest (RF) model using all 16 MDMs discriminated MBC from Benign controls with an overall AUC of 0.97. The sensitivity of predicting MBC status from the 95th percentile of the Benign controls was 0.95, including correct MBC status for all 3 BRCA1/2 mutation carriers. In addition, the model predicted NED samples as controls with 98% accuracy using a cut-off based on the 95th percentile of the Benign controls. Similarly, an overall AUC of 0.97 was obtained for predicting MBC relative to combined NED and Benign controls, with 0.95 sensitivity at the 95th percentile of the combined controls.
Discussion
We have shown that methylation markers discovered from tissue-extracted DNA are reproducible in plasma samples with high prediction accuracy to differentiate MBC patients from benign non-cancer subjects and previous cancer patients in clinical remission (NED). We report multiple methylation markers with AUCs above 0.9, and further, we show evidence that a multi-marker panel has an AUC and sensitivity at or above 0.95 for MBC. Eleven of the 16 MDMs occur within genes which have documented associations with cancer, of which most are directly involved in tumorigenic signaling via canonical regulatory pathways, with 2 MDMs specifically implicated in breast cancer19,20.
Other investigators have explored plasma methylation markers in breast cancer using a range of gene panels and assay methods. Using the MethyLight assay, Shan et al. reported on a six-marker panel (FN, P16, hMLH1, HOXD13, PCDHGB7 and RASSF1a) that had a sensitivity of 79.6% and AUC of 0.727 for breast cancer patients relative to healthy controls21. Zhang and colleagues assessed the AnchorIRIS assay and identified a 4-marker panel (TLR5, TOP, FEZF2, TSHZ3) that was able to differentiate early breast cancers from healthy controls with an AUC of 0.9422. A meta-analysis of 74 studies of DNA methylation for hormone-receptor positive breast cancer noted little overlap in DNA methylation markers across panels23, making it difficult to compare studies in this field directly.
A limitation of the current study is an imbalance of clinical molecular subtypes in the plasma pilot experiment, where the NED has 94.9% Luminal and 5.1% HER2+ cancer patients, where the MBC subtypes are more representative of breast cancer population rates with 15% HER2+, 80% Luminal, and 5% TNBC matching to 14%, 79% and 11%, respectively, from seer.cancer.gov/statfacts (accessed, 8/6/2024). This imbalance in subtype distributions between cases and controls could potentially result in an over optimistic estimate of discriminate accuracy when compared to real-world applications. We anticipated subtype-dependent DNA methylation patterns24, and deliberately selected markers in the MDM marker panel to account for this. However, the markers selected generally underperformed in both Luminal A and triple negative subtypes in the BV set, which were over-, and underrepresented in the plasma sets, respectively. Notably, the selected markers performed stronger in HER2+ in the BV set, a subtype that was under-represented in the plasma sets. Nonetheless, the 16-marker panel performed strongly for individual markers and in a multi-marker prediction model and appears suitable for further development.
The next step is to assess if the MDM panel can detect breast cancer recurrence prior to clinically detected recurrence. A methylation-based panel assay may offer advantages when compared to either a somatic targeted NGS panel or tumor informed evaluated in this molecular residual disease setting. While highly sensitive, tumor-informed assays fail approximately 10% of the time due to inadequate DNA yield or unavailability of primary tissue. Tumor-informed assays also have a longer turnaround time and are more costly. Targeted NGS panels that evaluate somatic mutations in blood may be confounded by the presence of clonal hematopoiesis of indeterminate potential (CHIP), which occurs in up to 15% of early breast cancers prior to systemic therapy25.
The plasma study herein shows that a multi-marker MDM panel can capture breast cancer methylation patterns in MBC patients and holds promise for early detection of recurrence through a routine blood draw. A future experiment that is powered to compare performance of a multi-marker panel across serial samples in the adjuvant setting will be the next step in making an active surveillance test with high prediction accuracy a reality.
Methods
Patient sample collection and clinical information
The study was conducted according to the guidelines of the Declaration of Helsinki. All study procedures and use of patient specimens were overseen by the Mayo Clinic Institutional Review Board in accordance with the US Common Rule 45 CFR 46, under which informed consent was waived for use of archival tissue specimens (protocol 16-001291) and written signed informed consent was obtained for use of peripheral blood samples (20-000529, 14-002919 [NCT06217874], 1815-04, 18-002366, and clinical trial NCT06192875). Recruitment was carried out blind to race, ethnicity, and gender; demographics that are reported in our patient characteristics tables.
Tissue Cohort: Fresh frozen breast tissues (invasive tumor, DCIS, and normal breast tissue) were acquired from the Mayo Clinic Breast Cancer Registry and the Biospecimen Resource for Breast Disease Study. These tissues were collected from clinical procedures performed on women ages 26–83, during the years 2001–2014. Patients with breast cancer had tumor size, grade, and clinical subtype abstracted from the electronic medical record. Clinical stage from tumor, nodal, metastatic invasive cancer status were defined by the AJCC breast staging, 8th edition26. Clinical molecular subtypes included human epidermal growth factor receptor positive (HER2+), triple negative (TNBC), luminal A (LA), and luminal B (LB). DCIS cases were either ER+ or ER- and split between those with HGD or LGD. Additionally, invasive cancer tissue from BRCA1+ and BRCA2+ carriers were included in the study. Normal breast tissue and histologically normal tissue from BRCA carriers were collected from reduction mammoplasty and prophylactic mastectomy procedures, respectfully. Non-neoplastic buffy coat samples were collected from healthy women enrolled in our internal NOMAD blood biobank.
Plasma Cohort: For plasma MRD testing, patients were prospectively enrolled into 3 cohorts: (1) Healthy benign controls (adult females with a normal mammogram in past 12 months with no history of breast cancer [NCT06192875]), (2) NED [stage I-III breast cancer with no clinical evidence of recurrence 9 months to 3 years post-treatment (adjuvant endocrine therapy allowed)], and (3) MBC [NCT 06217874] (MBC collected either at initial diagnosis of untreated metastatic disease (N = 10) or at time of radiographic progression (N = 50), whose samples were from 1 to 43 years after their primary breast cancer treatment). Blood samples were collected in 2 × 10 mL LBgard® tubes (Exact Sciences Corp., San Diego CA) and 1 serum separator tube at a single timepoint for each study participant. Serum CA 15-3 and CEA were performed at the Mayo Clinic reference laboratory from the same blood draw. All case and control patients provided written informed consent for use of their plasma and clinical data, and the study was conducted in accordance with Mayo IRB guidelines.
Sample processing
Tissue blocks were reviewed by Mayo pathologists to confirm histologic classifications and guide macro-dissection. The Mayo Clinic Pathology Research Core provided 10-micron sections from which genomic DNA was purified using the QIAmp Mini protocol (Qiagen, Valencia, CA). The DNA was cleaned up with AMPure XP beads (Beckman-Coulter, Brea CA) and quantified by PicoGreen fluorescence (Thermo-Fisher, Waltham MA). DNA integrity was assessed using real time qPCR. Buffy coat samples were processed similarly. Cell-free DNA (cfDNA) was isolated from 6 mL of double spun LBgard plasma using an internally developed automated method (Exact Sciences). Briefly, plasma was initially subjected to proteinase K treatment, followed by lysis with detergent and chaotropic reagents. Silica-coated binding beads and lysis buffer containing isopropyl alcohol were added to each sample for cfDNA capture. All samples were subjected to multiple rounds of washing on the Hamilton STARlet Liquid Handling System (Hamilton Company, Reno NV). The beads were then dried, and the cfDNA eluted in buffer.
Sequencing and qMSP
RRBS libraries were prepared from tissue and buffy coat DNA as previously described27. Sequencing was performed on the Illumina HiSeq 2500 instrument (Illumina, San Diego CA) at the Mayo Clinic Medical Genomics Facility. Reads were unidirectional for 101 cycles and processed for methylation content using SAAP-RRBS28. qMSP primers were designed using MethPrimer29 and synthesized by IDT (Integrated DNA Technologies, Coralville, IA). Reactions were optimized using converted methylated and unmethylated commercial gDNA controls (Zymo Research, Irvine, CA) to ensure specific and robust amplification. Sample DNA was bisulfite-treated using the EZ-96 DNA Methylation kit (Zymo) and amplified using SYBR Green detection on Roche 480 LightCyclers (Roche, Basel, Switzerland). Results were normalized to an ACTB reference to control for genomic input.
Plasma testing
Cell-free DNA was bisulfite converted using an automated STARlet method. Briefly, samples were initially denatured with sodium hydroxide. Ammonium bisulfite was added to each sample for deamination. Samples were subsequently bound to silica-coated binding beads and washed several times prior to desulphonation. Sample washing was repeated, and purified converted cfDNA was eluted in buffer. The MDM panel was tested using the TELQAS platform (Exact Sciences)17, a multiplexed PCR method which amplifies both the genomic targets and their corresponding FRET signals additively. Oligonucleotides (forward invasive primer, reverse primer, and flap/invader probes) were designed to anneal to multiple methylated CpG dinucleotides within the breast cancer MDM panel, a methylated reference gene, and a zebrafish spike-in processing control gene. 10uL of bisulfite-treated DNA was used in tri-plex format, FAM, HEX, Quasar 670 (Hologic, Marlborough, MA), in which two markers plus a genomic reference gene were amplified and quantified. TELQAS reactions were performed on a 7500 Fast Dx Real-Time PCR Instrument (Applied Biosystems, Waltham, MA).
Statistical and analysis methods
Discovery: DMRs were identified using proprietary software as previously described27. The statistical significance of DMRs was evaluated and ranked using the p-value from an overly dispersed logistic regression of percent methylation. DMRs were further filtered based on Area Under the Receiver Operating Characteristic Curve (AUC), methylation ratios of cases over controls, and absolute differences in methylation between cases and controls. Comparisons included (1) pooled BC cases vs benign tissue (2) BC cases with benign breast tissue vs buffy coat control samples and (3) individual subtypes for invasive breast cancer cases vs benign breast tissue.
Tissue Validation and Plasma Testing: Distributions of individual markers were examined using boxplots and marker intensity maps. Areas under the receiver operating characteristics curve (AUC) were generated for each marker to assess accuracy. Sensitivity analyses were performed to assess marker performance by subgroups, where we report the percent in the case group whose marker value exceeds the 90th percentile from the control set. Sample size considerations for biological, tissue-based validation were based on minimizing the width of a 95% confidence interval (95% CI) for sensitivity and specificity. With an assumed specificity of 95%, a control set of 29 would provide a 95% CI no wider than ±10%. To achieve a 95% CI that was no wider than ±7% for a target sensitivity of 90%, a minimum of 84 samples was required. Sample size estimates for the plasma-based clinical pilot were based on detecting an AUC of 0.70 from the null AUC of 0.50. With 60 MBC cases and 60 NED and 60 Benign, there was greater than 80% power to detect this difference using a one-sided test at a 5% significance level. RF was applied to the full 16 MDMs to predict MBC cases versus control sets of NED plus Benign in one model, and just NED controls in a second model. We report the overall AUC of the RF and the sensitivity of predicting case status based on the 95th percentile of the control set.
Data availability
Methylated DNA marker data are subject to restrictions under a Sponsored Research and Intellectual Property Licensing Agreement between Mayo Clinic and Exact Sciences. The authors oversee data sharing of all datasets and will review requests from qualified investigators that initiate requests for clinical data. All requests will be reviewed for scientific merit and feasibility. In addition to required approval from the Mayo Clinic Institutional Review Board, a Data Use Agreement will be put in place by Mayo Clinic Legal Contracting Administration with any academic group or scientists before any transfer of data. Investigators receiving the data will be required to abide by the conditions of these agreements.
Code availability
Code and methods used in marker discovery have been described above and are subject to restrictions under a Sponsored Research and Intellectual Property Licensing Agreement between Mayo Clinic and Exact Sciences. Other calculations were performed using open-source code in R version 4.2.2 for sample size (base R), and plasma marker evaluation; specifically for background marker adjustment (mgcv package version 1.8-41), AUC calculations (base R), visualization (base R), and multi-marker performance (randomForest package version 4.7-1.1).
References
Dent, R. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 13, 4429–4434 (2007).
Early Breast Cancer Trialists’ Collaborative, G Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365, 1687–1717 (2005).
Saphner, T., Tormey, D. C. & Gray, R. Annual hazard rates of recurrence for breast cancer after primary therapy. J. Clin. Oncol. 14, 2738–2746 (1996).
Gradishar, W. J. et al. Breast cancer, version 3.2024, NCCN clinical practice guidelines in oncology. J. Natl Compr. Cancer Netw. 22, 331–357 (2024).
Henry, N. L. et al. Promoting quality and evidence-based care in early-stage breast cancer follow-up. J. Natl. Cancer Inst. 106, dju034 (2014).
Keating, N. L., Landrum, M. B., Guadagnoli, E., Winer, E. P. & Ayanian, J. Z. Surveillance testing among survivors of early-stage breast cancer. J. Clin. Oncol. 25, 1074–1081 (2007).
Dawson, S. J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209 (2013).
Lennon, A. M. et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 369, https://doi.org/10.1126/science.abb9601 (2020).
Lipsyc-Sharf, M. et al. Circulating tumor DNA (ctDNA) and late recurrence in high-risk, hormone receptor–positive, HER2-negative breast cancer (CHiRP). J. Clin. Oncol. 40, 103–103 (2022).
Magbanua, M. J. M. et al. Clinical significance and biology of circulating tumor DNA in high-risk early-stage HER2-negative breast cancer receiving neoadjuvant chemotherapy. Cancer Cell 41, 1091–1102.e1094 (2023).
Radovich, M. et al. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: preplanned secondary analysis of the BRE12-158 randomized clinical trial. JAMA Oncol. 6, 1410–1415 (2020).
Wan, J. C. M. et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat. Rev. Cancer 17, 223–238 (2017).
Douglas Hanahan, R. A. W. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Zhu, M. et al. Plasma assay of cell-free methylated DNA markers of colorectal cancer: a tumor-agnostic approach to monitor recurrence and response to anticancer therapies. Cancers 15, https://doi.org/10.3390/cancers15245778 (2023).
Meissner, A. et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877 (2005).
Baylin, S. B. & Jones, P. A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 8, https://doi.org/10.1101/cshperspect.a019505 (2016).
Kisiel, J. B. et al. Hepatocellular carcinoma detection by plasma methylated DNA: discovery, phase I pilot, and phase II clinical validation. Hepatology 69, 1180–1192 (2019).
Thierry, A. R. Circulating DNA fragmentomics and cancer screening. Cell Genom. 3, 100242 (2023).
Boimel, P. J. et al. Contribution of CXCL12 secretion to invasion of breast cancer cells. Breast Cancer Res. 14, R23 (2012).
Shi, W. et al. SphK2/S1P promotes metastasis of triple-negative breast cancer through the PAK1/LIMK1/Cofilin1 signaling pathway. Front. Mol. Biosci. 8, 598218 (2021).
Shan, M. et al. Detection of aberrant methylation of a six-gene panel in serum DNA for diagnosis of breast cancer. Oncotarget 7, 18485–18494 (2016).
Zhang, X. et al. Circulating cell-free DNA-based methylation patterns for breast cancer diagnosis. npj Breast Cancer 7, 106 (2021).
de Ruijter, T. C. et al. Prognostic DNA methylation markers for hormone receptor breast cancer: a systematic review. Breast Cancer Res. 22, 13 (2020).
Fleischer, T. et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat. Commun. 8, 1379 (2017).
Morganti, S. et al. Prevalence, dynamics, and prognostic role of clonal hematopoiesis of indeterminate potential in patients with breast cancer. J. Clin. Oncol. https://doi.org/10.1200/JCO.23.01071 (2024).
Amin, M. B. et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Kisiel, J. B. et al. New DNA methylation markers for pancreatic cancer: discovery, tissue validation, and pilot testing in pancreatic juice. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-14-2469 (2015).
Sun, Z. et al. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics 28, 2180–2181 (2012).
Li, L. C. & Dahiya, R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18, 1427–1431 (2002).
Acknowledgements
DNA sequencing costs and TELQAS reagents were supported through a sponsored research agreement between Exact Sciences and Mayo Clinic. This study was supported in part by NIH grants from the National Cancer Institute, including R37CA214679, R35CA253187, a Specialized Program of Research Excellence (SPORE) in breast cancer to Mayo Clinic (P50 CA116201), and the Breast Cancer Research Foundation. This work utilized shared resources of the Mayo Clinic Comprehensive Cancer Center (P30 CA015083) from the National Cancer Institute. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health. Presented in abstract form at the San Antonio Breast Cancer Symposium on December 7, 2022. [https://doi.org/10.1158/1538-7445.SABCS22-P2-11-06].
Author information
Authors and Affiliations
Contributions
K.V.G.: Writing—original draft, conceptualization. J.P.S.: Writing— original draft, visualization. S.W.S.: Software, writing—editing and review. W.R.T.: Methodology, writing—original draft. D.W.M.: Formal analysis, writing—editing and review. P.H.F.: Project administration, resources. M.C.O.: Investigation. M.J.R.: Investigation. M.E.D.: Investigation. A.M.G.: Project administration. N.L.L.: Resources. K.A.D.: Project administration. K.N.B.: Data curation. M.W.K.: Investigation. H.T.A.: Validation, writing—editing and review. K.J.R.: Writing—editing and review. J.E.O.: Resources, writing—editing and review. F.J.C.: Conceptualization, writing—editing and review. J.B.K.: Funding acquisition, supervision, conceptualization, writing—editing and review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing non-financial interests and the following competing financial interests: M.W.K. and H.T.A. are employees of Exact Sciences. S.W.S., W.R.T., D.W.M., and J.B.K. are inventors of Mayo Clinic intellectual property under license to Exact Sciences.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Giridhar, K.V., Sinnwell, J.P., Slettedahl, S.W. et al. Plasma assay of methylated DNA markers detects recurrent metastatic breast cancer. npj Breast Cancer 12, 2 (2026). https://doi.org/10.1038/s41523-025-00866-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00866-0
