
Abstract
High-throughput search for new crystal structures is extensively assisted by data-driven solutions. Here we address their prospects for more narrowly focused applications in a data-efficient manner. To verify and experimentally validate the proposed approach, we consider the structure of higher tungsten borides, WB4.2, and eight metals as W substituents to set a search space comprising 375k+ inequivalent crystal structures for solid solutions. Their thermodynamic properties are predicted with errors of a few meV/atom using graph neural networks fine-tuned on the DFT-derived properties of ca. 200 entries. Among the substituents considered, Ta provides the widest range of predicted stable concentrations and leads to the most considerable changes in mechanical properties. The vacuumless arc plasma method is used to perform synthesis of higher tungsten borides with different concentrations of Ta. Vickers hardness of WB5-x samples with different Ta contents is measured, showing increase in hardness.
Similar content being viewed by others
Introduction
The efficient and accelerated development of new materials for civil and industrial applications has long been a goal of materials scientists. The rapid development of computational methods and artificial intelligence enables the prediction of the crystal structure of new materials1,2,3,4,5,6,7,8 and their properties9,10,11,12,13. Another big challenge is posed by seeking improvements in the properties of materials by alloying or doping14,15,16,17,18,19. However, these are only the first steps towards an automated and physically informed search for new materials with desired predefined properties.
On the other hand, the traditional approach devoted to “blind” experimental search for optimal chemical modifications is resource- and time-consuming, and not efficient. In turn, computational studies also become challenging or even intractable for large model cells due to the enormously large number of possible structure realizations of material with disorder. For this reason, machine learning emerges as a powerful tool20 in this field, since it allows us to predict a target property using just a piece of hardly derived values for training. Recent years, the most successful solutions in this area have been based on geometric graph neural networks (GNNs)21. This type of data-driven solution opens up perspectives for implementing transfer learning routines for small data applications22 by using density functional theory (DFT) data from the available datasets, such as Materials Project23, AFLOWLib24, etc. For a comprehensive description of GNNs, we refer to the detailed review by Duval et al. 25.
GNNs became directly applicable for composition/configuration spaces of materials26, heterogeneous systems27,28, and even composition-structure-property relationships for low-dimensional materials29,30. These studies can provide a foundation for a comprehensive research into the properties of known materials with the addition of defects and chemical substitutions, screening reaction pathways, and searching for stable defects alternating target properties of materials of interest. Moreover, such hybrid approaches have recently been applied to searching for new materials in vast chemical search space.
Following this “one-to-many” concept, Merchant et al.5 reported more than 400 k potentially new compounds generated using random subsampling of chemical substitutions in known structure types. Subsequent analysis of the obtained theoretical predictions has clearly shown a need for incorporation of expertise in materials synthesis and crystallography during predictions31. In turn, validation of these theoretical predictions in the lab by Szymanski et al.32 has also been questioned from the experimental perspective33. Finally, such an impressive number of potentially new materials could be caused by the absence of any documented competing phases for ternary, quaternary, and more complex compositions, as highlighted by Bartel34. In this regard, it also seems a gimmick to compare the promising candidates with the MP dataset instead of more numerous AFLOWLib records obtained using a similar data augmentation protocol.
On the other hand, the huge potential of hybrid approaches has recently been demonstrated in a more targeted area of materials, namely solid electrolytes, by applying a sequence of filtration procedures35. Strictly speaking, in many specific applications concerning smaller search spaces, hybrid DFT/GNN approaches require less data and computational resources to provide sufficient quality of predictions. This is, for sure, maintaining the tradeoff between the transferability of a certain model and its generalization ability. For instance, for four Sc-rich bimetallic systems36, the hybrid approach provided root-mean-squared errors of formation energies of ca. 10 meV/atom. Nevertheless, it was shown that the corresponding GNN predictions resulted in a high false positive rate for potentially new candidates, which a well-defined set of competing phases might cause.
In this work, we showcase the effectiveness of combining transfer learning routines, domain-inspired heuristics for training data selection, and machine learning-assisted screening of complete composition/configuration spaces for the design and discovery of new materials. Our approach targets a small number of DFT-derived properties for model training, clearly addresses potential issues caused by the fundamental incompleteness of available datasets, and represents a more thorough method for thermodynamic stability assessment of potentially new candidates in a certain class of functional materials. Perspectives of our approach are shown for the case of predicting optimal chemical modifications of the higher tungsten boride, WB5-x37,38,39, by other transition metals. We then successfully prove our prediction in the lab by vacuumless arc plasma synthesis and characterization of obtained samples of doped higher tungsten boride.
The paper is organized as follows. First, the search space for the title compounds is defined and considered. Next, the developed GNN-based workflow for the calculation of formation energies of modified higher tungsten boride structures is described. After high-throughput screening for stable compositions, DFT calculations of the mechanical properties of the predicted structures during the screening are presented. Obtained information is the base for the experimental synthesis of powders and ceramic samples of W-Ta substituted higher tungsten boride. The next part is devoted to Vickers microhardness measurements and a comparison of the results obtained for the synthesized samples with theoretical predictions. The final part is a discussion of the developed approach and its perspectives of use in the field of materials discovery.
Results
Search space
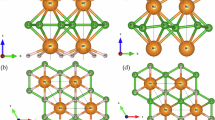
To set up the space for computational search, we start from a 1 × 1 × 2 supercell of the WB4.2 crystal structure38,39,40 shown in Fig. 1a. A complete set of symmetrically inequivalent substituent arrangements in model cells—composition/configuration space (CCS)—is built using the Supercell41 program (see details in “Methods” Section “Supercell approach”). For each metal X (Ti, V, Cr, Zr, Nb, Mo, Hf, or Ta) and all possible substitution levels, this space comprises 46,996 (W, X)B4.2 structures. Their spatial symmetries, reduced compared to the original structure due to substitution defects, are additionally determined before DFT relaxations. Such meta-information does not require DFT-based evaluation of the entire search space and is therefore accessible at low computational costs.

a Crystal structure of the 1x1x2 supercell of WB4.2 (20 formula units per cell). The image is obtained using the OVITO program69; b Heatmap of the number of inequivalent structures for sample metal X of the complete CCS via W substitutions and space groups determined prior to DFT relaxations. The empty slots correspond to the absence of certain composition-symmetry combinations. The coloring map corresponds to the logarithm of the number of inequivalent structures (values in the heatmap annotation).
The resulting space groups in the built CCS turn out to be severely unbalanced as shown in Fig. 1b (more details in Supplementary Fig. 1 and Table 2). Thus, the two highest symmetries—the P63/m and \(P\overline{6}\) space groups—correspond to 260 structures (less than 0.6% of the built CCS) in the entire substitution range. On the contrary, 86.8% of the generated structures are of the lowest P1 symmetry and cover the substitution range of 10–90%. This observation clearly shows the potential of exploiting the symmetry of generated disordered structures to stratify training data, minimizing the randomness impact (further arguments in “Methods” Section “Supercell approach”).
Subsequently, an entire search space of 375,961 structures is generated from the built CCS by replacing sample metal X with one of the eight metals (Ti, V, Cr, Zr, Nb, Mo, Hf, or Ta).
Workflow performance
To estimate thermodynamic properties in the built search space within a reasonable time frame, we develop an approach resting upon combining DFT calculations and surrogate models for structure-to-property predictions as shown in Fig. 2a. At the first setout, we (1) select a minimum subset of the entire search space, (2) evaluate target properties of its entries using DFT, and (3) train GNNs models on them. In inference mode, the models are tested (4) on “out-of-domain” entries of the search space not represented in the training data. Thus, their target properties are additionally evaluated using DFT. By comparing the model performance, the test parts with worst quality predictions are selected. According to the test results, a part of the search space is added to the training set of the model for the next stage (5). Within each of the subsequent stages, steps 2–5 are repeated until the required accuracy is achieved. The resulting model can then be used for reliable and fast screening over the entire search space.

a Flowchart of the iterative process of training/validation data extension. The numbers denote steps being taken. The arrows correspond to the steps taken in this work (solid) and ability to further extend training data if necessary (dashed). The green (down) and red (up) arrows indicate smaller and larger prediction errors, respectively, compared to the overall scores at a certain stage. For the stage#1 and stage#2 models, the overall RMSEs obtained on the b first and c second held-out tests, and those for each of the individual parts of them. The RMSE scores represented by the dotted lines correspond to the stage#2 models used for the second held-out test generation (“ME (aflowsmall-FE)”) and inference over the entire search space (“ME (aflow-FE),” “FE (aflow-FE),” and “RE (aflow-RE)”).
In this work, the proposed approach has required only two stages and ca. 265 DFT relaxations for both training/fine-tuning and testing purposes. Below is a detailed description of the above steps.
Step 1. To build an approach that is as efficient as possible in terms of the data requirements, we start with a minimum dataset. To set it for a sample substituent X, we randomly select just one CCS entry with a substitution step of 10%. These structures expectedly possess the lowest P1 symmetry for substitutions of 20–80%, whereas Pm is obtained for the structures with 10 and 90% (see Fig. 1b).
Subsequently, 81 structures are generated from the chosen CCS entries by substituting X with one of the eight considered metals, including the original structure without any substitutions and eight fully substituted versions. Thus, the situation when different substituents possess different arrangements at the same substitution level is avoided by the one-factor-at-a-time method.
Step 2. We calculate relaxed structure energies (REs), formation energies (FEs), and mixing enthalpies (MEs) using DFT (further details in “Methods” Section “DFT computations”) and consider them as target properties for the surrogate models.
Step 3. For the structure-to-property predictions in this work, the Allegro architecture42 is used. At the first stage, we test performance of 11 individual models trained/fine-tuned on 73 structures of the introduced minimum dataset (except for eight half-substituted structures). Such a number of models is related to the multiple target properties and several options for pre-training (details in “Methods” Section “Transfer learning and data splits” and Supplementary Fig. 5).
In turn, a particular model is named after the target properties it is trained/fine-tuned to predict and pre-training used (if applicable). For instance, the “ME (aflow-FE)” tag corresponds to the Allegro model pre-trained on the FEs from AFLOWLib24 and fine-tuned on the MEs evaluated in this work. Since we use only one architecture in this work, “Allegro” is omitted in the notations for short.
Step 4. The half-substituted structures, referred to as “l.s.-hW” (low symmetry, half-substituted W), are held as a part of test data.
To select additional test structures, we analyze the changes in target properties caused by substituent content variation. Within the minimum dataset, Ti leads to the highest variation among considered metals (see Supplementary Fig. 4). For this reason, high symmetry Ti-W-B structures are selected as another part of the test, referred to as “h.s.-TiW” (high symmetry, Ti-substituted W). At the substitution levels of 40, 50, and 60%, a total of 11 structures are used (4, 3, and 4 structures of \(P\overline{6}\) symmetry, respectively). We additionally consider 6 random (low symmetry) CCS entries at 30% substitution level and intermediate levels of 25% and 35% referred to as “l.s.-TiW” (low symmetry, Ti-substituted W).
All introduced parts together are considered as the first held-out test, which serves for checking performance of the stage#1 models on the intermediate compositions, high symmetry structures, and various substituent arrangements.
Step 5 (stage#2). The obtained root-mean-square errors (RMSEs) of predictions are shown in Fig. 2b. The “h.s.-TiW” part corresponds to the highest errors observed (the median RMSE of ca. 10 meV/atom). For this reason, Ti-substituted structures possessing the P63/m (all) and \(P\overline{6}\) space groups (with a step of 10%) are added to the training dataset of the next stage avoiding data leakage from stage#1.
Steps 2–3 (stage#2) are qualitatively the same to those of stage#1. The DFT relaxations are carried out for the additional dataset only. The model training/fine-tuning is performed by using the whole extended dataset.
Step 4 (stage#2). The stage#2 models can be firstly checked on the first held-out test and expectedly provide the best performance on “h.s.-TiW” (see Fig. 2b). However, these models also demonstrate a better performance (the median RMSE of less than 1 meV/atom, see Fig. 2b) for the “l.s.-hW” and “l.s.-TiW” structures.
Regarding the overall scores on the first held-out test (see Supplementary Fig. 6), the “ME (aflowsmall-FE)” model is the most precise. Based on its predictions, the most energetically favored and unfavored structures at 20, 50, and 80% substitution levels are chosen for eight metals (48 structures, 6 structures per substituent) as individual parts of the second held-out test. The RMSE scores obtained for the second held-out test by using the models of both stages are given in Fig. 2c.
Intriguingly, due to addition of high symmetry Ti-substituted structures to the training data, the stage#2 models outperforms those of stage#1 for other substituents as well. This is definitely in line with the mentioned improvement in their performance for the “l.s.-hW” test. Both of these observations may be related to better distinguishability of composition-property and structure-property relationships. Indeed, the extended training data also contains information about the influence of Ti arrangements on the target properties in addition to the type and content of the substituent at stage#1.
Based on the overall RMSEs on the second held-out test (see Supplementary Fig. 7), the “ME (aflow-FE)” model provides the lowest error of 2.22 meV/atom. The “ME (aflowsmall-FE)” model used previously becomes the second most precise. In general, the models pre-trained on “aflowsmall” are found to be inferior to those pre-trained on “aflow” (see Fig. 2c and Supplementary Fig. 7). The “FE (aflow-FE)” and “RE (aflow-RE)” models provide the next best scores of 2.69 and 2.84 meV/atom, respectively, and can be further used to check the self-consistency of the inferences between the models trained on different target properties.
Step 5 (stage#3). The highest RMSEs of the stage#2 models are obtained for Cr-, Hf-, and Zr-substituted systems. Following the approach proposed, corresponding parts of the entire search space can be used for the subsequent training data extensions (see Fig. 2a, dashed arrows). However, the RMSE values of top 4 models remain less than 5 meV/atom for the test structures corresponding to these substituents (see Supplementary Fig. 7). These errors turn out to be much smaller than the variation in the target thermodynamic properties. Therefore, no further extension of the training data is carried out.
Screening results
The most precise stage#2 models can then be used to estimate FEs over the entire search space. To find potentially new compositions promising for experimental testing, we use the standard convex hull approach34 resting upon FEs from AFLOWLib and followed by their refining and addition of missing competing phases (more details in “Methods” Section “Convex hull analysis and refinement”). The results of convex hull analysis before refinements are given in Supplementary Fig. 8. As shown in Fig. 3, Nb-, Ta-, and Mo-substituted systems possess negative Ehull values among all substituents studied and are further considered.

The Ehull dependencies via fraction of W substituted by a Nb, b Ta, and c Mo. The AFLOWLib-derived convex hulls (red dotted lines) and their refined/updated versions (red dash dotted lines) represent WB4.2—XB4.2 cross-sections of the corresponding 3D convex hulls. The DFT-derived energies are shown as crosses for the structures of the minimum dataset (light gray) and the second held-out test (dark gray). For the GNN-based model predictions, the energy ranges are given by the lowest and highest values for a certain W substitution.
With refinements related to the computational scheme, the corresponding deviations of reference FEs do not exceed 5 meV/atom (Fig. 3a, b). In contrast, consideration of just one undocumented competing phase—the recently discovered MoB4.243 structure—qualitatively changes the predictions. This leads to ca. 15 meV/atom lower convex hull energies compared to those available from AFLOWLib (Fig. 3c). Reducing the reference energy results in a significant portion of the built Mo-W-B CCS above the hull, whereas all its entries are below the convex hull obtained from AFLOWLib. This observation clearly illustrates the obstacles to searching for new materials in vast chemical spaces based only on faith in the completeness of the existing databases.
As Fig. 3 shows, the usage of random subsamples of complete CCS during searching for potentially new substituted higher W borides may provide qualitatively different results. For instance, the minimum dataset, which initiates the developed workflow, erroneously results in the absence of any promising compositions for the Nb-W-B systems, as well as Ehull values close to zero for Mo-W-B. For the same reasons, a narrower range of promising compositions can be observed for Ta-W-B.
Despite the fact that the “ME (aflow-FE)”, “FE (aflow-FE)”, and “RE (aflow-RE)” models predict similar FE ranges, the most precise “ME (aflow-FE)” model is used further to search for the most energetically favored structures within the composition ranges below the hulls. The space group statistics for the structures selected using the ME (aflow-FE) predictions are given in Table 1.
It is worth noting here that no meta-information (e.g., symmetry of a certain entry) is used as a constraint or guide for the selection of energetically favorable structures. However, a considerable fraction of them have symmetry higher than Pm, which is not represented in the training data at either stage#1 or stage#2 for Nb, Ta, and Mo as substituents. Moreover, it can be concluded that the generalization of composition-structure-property relationships makes the developed DFT/GNN-based approach highly selective since the higher symmetries are also rare in the entire search space.
Assessments of mechanical properties
The screening provides insights into the stability of the mixed WB4.2 structure with W substitutions by transition metals. Further study of the mechanical properties of these structures is devoted to those showing the highest predicted stability, i.e., the lowest formation energies. We selected the following substituents and their content in WB4.2: Nb (5–40%), Ta (5–85%), and Mo (5–95%) (see Fig. 3).
For each of the compositions, the elastic tensor is calculated, which is then used to evaluate the bulk and shear moduli with the Voigt-Reuss-Hill averaging scheme. The elastic tensor is used to calculate the Pugh-Pettifor44 criterion for all mixtures of W1-yXyB4.2. The results of the calculations are compared to those of other known transition metal borides in Fig. 4a. It can be observed that the chemical modification of WB4.2 changes the overall brittleness of the structure. The zoomed region, shown in Fig. 4b, clearly identifies the changes in mechanical properties that possess after W substitution. The W-Ta and W-Nb substitutions are less ductile than pure WB4.2 (black cross in Fig. 4b), with the exception of W0.95Ta0.05B4.2 and W0.95Nb0.05B4.2, which exhibit similar values. Increasing the Mo content in the WB4.2 leads to an increase in ductility, see green squares in Fig. 4b. Overall, Mo-substituted systems are more brittle than MoB5 but more ductile than WB5. This indicates that Mo makes W1-yXyB4.2 more ductile and less susceptible to fracture.

a Pugh-Pettifor criterion, a relation between \(\frac{{C}_{12}-{C}_{44}}{B}\) and the Pugh ratio (\(\frac{G}{B}\)). b Zoomed region showing how brittleness varies with the type and concentration of metal substituent. c Ashby plot in coordination of Vickers hardness and fracture toughness. Vickers hardness is calculated using Chen’s model45, while fracture toughness is calculated using Niu–Oganov model46. d Zoomed region corresponding to the considered chemical modification of higher tungsten boride.
The bulk and shear moduli are used to calculate Pugh ratio44 and Vickers hardness (Hν) using Chen’s empirical model45. To calculate the fracture toughness (KIC), the Niu-Oganov46 model is used. Also, the Vickers hardness and fracture toughness are calculated using the Mazhnik-Oganov models47 as shown in Supplementary Fig. 10. For further details, please refer to the Supplementary eqs. 7–10.
Obtained results are presented in Fig. 4c, d. It can be observed that tantalum substitutions in concentrations ranging from 5 to 20% exhibit higher hardness and fracture toughness. Hardness and fracture toughness decrease significantly when Ta content is above 20%. The addition of Nb from 5 to 20% does not affect the fracture toughness with respect to WB4.2, but we observe an increase in hardness (from 37.5 to 40 GPa). Substitution of 5% W with Mo leads to slightly lower values of hardness and fracture toughness than for WB4.2. However, the higher the Mo content, the lower the hardness and fracture toughness values we obtain (see Fig. 4d).
According to these results, we conclude that the addition of about 20% Ta leads to the greatest increase in Vickers hardness of the higher boride compared to other substituents. However, further increase in Ta content results in the destabilization of the higher borides, as the TaB4.2 structure is unstable, and thus, hardness will decrease. This observation is validated by performing the synthesis of the higher tungsten boride modified by tantalum.
Synthesis of predicted compounds
For experimental synthesis, we have selected the predicted compounds with W-Ta substitutions as they show larger variation in mechanical properties and lower Ehull values. To determine the optimal experimental regime for the synthesis of required compounds, experiments are conducted in three series, as detailed in “Methods” Section “Synthesis and experimental characterization”. The X-ray diffraction patterns of the experimental series are presented in Supplementary Fig. 10. A preliminary series of experiments is conducted to determine the optimal parameters for synthesis. These parameters are used in further experiments, which involve a current of 150 A with a dwell time of 30 s, an initial powder mass of 1.5 g, and a fixed atomic ratio of WTa:B at 1:15.
Based on the information obtained, we carry out the fourth series of experiments with a different W:Ta ratio in order to synthesize W1-yTayB5-x powders (with y = 0.2, 0.3, 0.4, 0.5, and 0.6). The X-ray diffraction patterns of the synthesized powders with results of refinement based on simulated structure models are shown in Supplementary Fig. 12. The EDX images of the obtained powders are shown in Supplementary Figs. 13–15. For further investigations, we have selected three compositions—W0.8Ta0.2B5-x, W0.6Ta0.4B5-x, and W0.4Ta0.6B5-x—to be used for sintering cylindrical tablets with a diameter of 12.7 mm using the sintering parameters described in “Methods” Section “Synthesis and experimental characterization”. To characterize the structure of prepared ceramic samples we have used XRD and XPS analysis, which are shown in Fig. 5. Careful analysis of XRD patterns gives the information about potential coexistence of several phases in the samples, namely (W,Ta)B5-x, TaB2, WB5-x, and traces of WB2 (usually exists together with WB5-x37,38,39), see Fig. 5a. Presence of TaB2 is identified by the reflections at 27.2, 33.8, and 43.8०. It is clear to see that the amount of TaB2 increases with an increase of Ta concentration in the sample, i.e., the intensities of all mentioned reflections increase. The WB2 reflections are also observed as a minor phase (see circles in Fig. 5a). It may be suggested that Ta incorporates into WB5-x lattice according to the presence of low-intensity reflections at 19.75 and 21.7०, as these reflections do not correspond to any known Ta-B phases. The intensities of the main reflections (24.2, 34.5, 45.1०) change with respect to TaB2 amount, indicating the formation of samples with higher Ta content. The splitting of each reflection belonging to either WB5-x or (W,Ta)B5-x may indicate the formation of a mixture of WB5-x and (W,Ta)B5-x phases.

a X-ray diffraction patterns of sintered ceramics from samples with compositions W0.8Ta0.2B5-x, W0.6Ta0.4B5-x, and W0.4Ta0.6B5-x. High-resolution XPS spectra for b B1s and c Ta4f regions of samples with different Ta contents.
However, XRD analysis cannot fully confirm the formation of the ternary phase as Ta may be located separately from the tungsten boride structure, and tantalum may not be bonded to boron in this compound. For these purposes, the XPS analysis is performed, and the spectra for Ta4f and B1s regions are shown in Fig. 5b, c. The XPS spectra corresponding to the W4f, C1s, and O1s regions are shown in Supplementary Fig. 15.
The Ta4f spectra are deconvolved into 4 components, see Fig. 5b. Among them, that characterizes the presence of a Ta-B/Ta-C bond48,49 corresponds the Ta4f binding energies (BEs) of 24.50–25.06 eV highlighted in Fig. 5b. Other components with BEs(Ta4f) of 26.35–26.40 eV, BE(Ta4f7/2) 26.88–27.30 eV and BE(Ta4f5/2) 29.08–29.40 eV, indicate the presence of a Ta-O bond within the sub-oxide48 and in Ta2O550,51. As one can see, the intensity of this component increases as the Ta concentration increases in the sample. This may indicate that Ta either incorporates into the structure of higher tungsten boride or forms separate diboride phase, rather than presence of a separate metallic Ta-based phase. This is also confirmed by the XRD data in Fig. 5a.
The XPS spectra of B1s region (see Fig. 5c) contains five peaks with BEs of: 187.56–187.90 eV, characterizing the presence of the Me-B bonds (Me = W, Ta)49,52. The component with BEs of 188.99–189.38 eV corresponds to the B-B bond52,53, while BEs of 190.55–191.68 eV correspond to the B-O bond within interfacial sub-oxide54. Other regions with BEs of 193.65–193.84 and 195.08–195.21 eV characterize the presence of the B-O bond within BxOy I and BxOy II oxides49.
The most important regions are devoted to Me-B and B-B bonds are highlighted in Fig. 5c. The samples with two highest Ta contents show increased intensity of the Me-B peaks, indicating the formation of boride phases. At the same time, the intensity of B-B peak decreases as concentration of tantalum increases from 20% to 40%, and then increases for 60% of tantalum. This may indicate either the formation of higher borides as there more B-B bonds should be formed compared to diboride, or formation of free boron as TaB2 becomes a major phase as can be seen from the XRD data (see Fig. 5a). As a result, one can conclude the formation of multi-phase system containing (W,Ta)B5-x compound together with TaB2, WB2, and WB5-x.
For this multi-phase system, the mechanical properties, namely Vickers nanohardness, is measured. Fig. 6a shows the dependence of nanohardness of ceramic samples on the load. At a load of 0.01 kg, ceramic samples show the Vickers nanohardness values of 48.92 ± 5.15, 44.18 ± 8.35, and 44.28 ± 4.79 GPa, respectively, for W0.8Ta0.2B5-x, W0.6Ta0.4B5-x, and W0.4Ta0.6B5-x. The hardness for W-Ta substituted samples exceeds the value obtained for WB5-x-WB2 sample at the same load (34.95 ± 4.79 GPa). When the load is increased to 0.05 kg and higher, the hardness for all W-Ta-B samples generally remains higher compared to pure WB5-x-WB2. The dependence of the hardness on the Ta concentration is shown in Fig. 6b, where the hardness estimated by empirical Chen’s model is shown for comparison. Such an increase in hardness is caused by the presence of multi-phase system containing TaB2, which is harder than WB2 in a single crystal form. Thus, the addition of tantalum in the sample should lead to an increase of hardness.

Measured Vickers microhardness of WB5-x-WB2, W0.8Ta0.2B5-x, W0.6Ta0.4B5-x, and W0.4Ta0.6B5-x ceramic samples. a Dependence of microhardness on the load; b dependence of microhardness on the Ta concentration in the structure for three load values of 0.01, 0.05, and 0.1 kg. Hardness values calculated by using empirical Chen’s and Mazhnik–Oganov’s models are shown by dashed lines.
Discussion
In this work, we have proposed and tested the approach of predicting new functional materials by evaluating thermodynamic properties in large composition/configuration spaces of chemical modifications. This approach is based on a combination of a neural network and smart data sampling routines aimed at developing efficient data-driven solutions in terms of the amount of DFT-derived data. The proposed approach is used to predict the optimum concentration of transition metals for chemical modification of higher tungsten boride, which is of practical interest from the point of view of mechanical properties. Our search made it possible to identify three perspective novel systems—W-Ta-B, W-Nb-B, and W-Mo-B. One of these systems is studied experimentally. To summarize, we would also like to note some features that distinguish the proposed approach from numerous high-throughput studies aimed at searching for new materials.
Firstly, the study of composition/configuration spaces in depth, rather than in breadth, allowed potentially new materials to be explored in that area of the search space where random sampling of training/validation or inference structures would be unlikely to yield similar results for statistical reasons. Thus, the particular importance of high-symmetry configurations is demonstrated both in terms of their presence in the training dataset and their thermodynamic favorability in the inference mode. On the other hand, it is clearly shown that the number of such configurations in the search space for crystal structures with chemical substitutions can be vanishingly small.
Secondly, the proposed method of sequentially increasing the training set allowed us to achieve impressive efficiency of the approach in terms of the training data preparation (while this task is the most time-consuming for AI-based approaches). As a result, it is possible to predict thermodynamic properties in an array of ca. 375 thousand structural configurations using the result of DFT calculations for ca. 200 entries. The predictions corresponded to the test errors of less than 3 meV/atom according to the performance of the winner models. On this track, we have clearly demonstrated that composition-property and structure-property correlations can be independently captured by modern AI models, which opens excellent prospects for scaling up the proposed approach within the commonly used one-to-many concept in materials science.
Thirdly, the predictive study carried out in our work is supplemented by experimental confirmation of the results. In a wide range of predicted compositions, experimental synthesis and mechanical property evaluations are performed for the W-Ta-B system. Among the potentially new compositions obtained during screening, this possesses the largest variation in mechanical properties and the lowest Ehull values. Considering the demonstrated influence of any undocumented competing phases on the stability assessments, it seems a much easier task to fill such gaps in data and avoid high false positive rate of computation-based “discoveries” for the experts in particular fields of materials research.
These points convince us that we are on the right track and the approach is valid. We propose a calculation scheme, which can be applied not specific to borides, but appeals to the properties inherent in any crystal, hence, we believe that the approach can be easily extended to other families of functional materials in experimental labs all over the world. The combination of such an efficient predictive method with vacuumless arc plasma synthesis technique allowed us to indicate the potential of this method to search for new materials. However, more accurate synthesis and analysis should be done to undoubtedly conclude about the formation of new ternary phase. The selection of other methods different from the vacuumless plasma technique will allow to carry out quick validation of theoretical predictions. This also works in reverse; efficient material prediction algorithms that can operate in a narrow search space are needed for the synthesis and laboratory testing of new functional materials.
However, the implications and importance of this work extend far beyond the results shown herein and may be interesting for the wide audience of this research—the experimentalist and experimental groups, who are interested in a particular class of functional materials or a certain structure types rather than high-throughput screening results over the vast chemical spaces.
Methods
Supercell approach
We use the supercell approach to model substitutional defects and search for potentially new (W,X)B4.2 compositions starting from the original WB4.2 crystal structure38,39. The multiplicities of the W sites (6, 2, and 2) and the chosen supercell size (1 × 1 × 2) result in 325 possible input combinations for the Supercell program41. To obtain them, we vary occupancy factors of the W sites listed in Supplementary Table 1 from 1 (non-modified structure) to 0 (complete substitution of W by metal X). For each of these combinations, a set of disordered structures is first generated in a purely combinatorial manner. For one metal, a total of 1,048,576 structures can be obtained at this stage.
Supercell then searches for symmetric equivalence within all possible arrangements of substituting atoms, which allows it to merge symmetrically equivalent structures (for more details, see the original work41) and use inequivalent entries only for CCS generation. Merging reduces CCS complexity to 46,996 inequivalent entries in the low-symmetry P1 representation. To determine their actual space groups, we use the pymatgen.symmetry package and spglib library for Python55. Merging equivalent structures, Supercell also assigns a “weight” to each CCS entry. This value shows the degree of degeneracy of a particular unique structure and equals the number of its merged copies.
A joint analysis of the obtained space groups and weights shows that the entries of the highest P63/m (176) space group possess the lowest weights of 1 (no symmetrically equivalent copies). The \(P\overline{6}\) structures, in turn, have weights of 2 or 4, and so on. Finally, all entries with the lowest P1 symmetry possessed the highest weights of 24. Thus, subsampling of a particular search space that is based on the random selection of positions being substituted is unlikely to provide high-symmetry structures not only for their smaller number (see Fig. 1b), but also because of their lower weights. A visual representation of the symmetry/weight relations is provided in Supplementary Fig. 1.
The chosen supercell size allows us to vary the substitution level with a step of 5%, which corresponds to an increase in the substituent content by one atom per cell. The use of larger (doubled, quadrupled, etc.) supercells results in higher discretization of search spaces. For this reason, the larger supercells can provide additional potentially new structures and compositions. However, these findings should not invalidate the results obtained for the smaller supercells due to the backward inclusion of simpler spaces into enhanced ones in case of disordering of a certain structure type.
Transfer learning and data splits
The Allegro architecture42 is used in this work for evaluations of the disordered crystal structure properties. Due to the strict locality (no message passing), this model has recently been able to predict formation energies of chemically disordered CsPbI3 phases with high accuracy (RMSEs of ca. 0.7 meV/atom)56, when trained on a wittingly small dataset. As mentioned, we use several models at each of the aforementioned stages regarding a certain target property and initialization of trainable parameters, including those with and without pre-training.
At each stage of the developed workflow, three models—RE (no pre-training), FE (no pre-training), and ME (no pre-training)—are trained from scratch on the target properties derived in this work. For pre-training purposes, we set up two datasets using the AFLOWLib24 database (as downloaded on 3 March 2023). The first dataset (referred to as “aflowsmall”) comprises 4126 structures with W-B, X-B, and X-W-B compositions, where X is one of the studied substituents. We additionally set its extended version (referred to as “aflow”) with 10,026 entries by adding all available X1-X2-B combinations. Both datasets provide DFT-derived REs and FEs. Thus, the pre-training routine results in 4 models—aflowsmall-RE, aflowsmall-FE, aflow-RE, and aflow-FE—which can be further fine-tuned on the properties derived in this work—REs and FEs (4 models) or MEs (4 models). Thus, eight pre-trained and fine-tuned models are considered at each stage of the developed workflow in addition to those with no pre-training used. A visual representation of the flowchart described is given in Supplementary Fig. 5.
All GNN predictions made and analyzed in this work correspond to the model states (training epochs) that provide the lowest loss on the validation subsets. As a general rule, validation subsets are chosen in a stratified manner with respect to substituent type, substituent content, and space groups available, which is of particular interest for small training datasets. Further details are discussed in the Supplementary Materials (Chapter 2). In turn, the structures from the held-out tests and entire search space are analyzed in the inference mode of deployed models. This way, we can be sure that our results are not intentionally biased to the test samples and can control for overfitting at each stage.
It can be seen from Fig. 3c that models other than the top 4 can result in RMSE values that are an order of magnitude higher. The highest overall errors are obtained without pre-training (5.20–7.26 meV/atom) and for the models pre-trained on AFLOWLib REs and fine-tuned on the MEs derived in this work (7.88–9.83 meV/atom). Further details can be found in Supplementary Fig. 7. This observation points at a necessity of comprehensive performance analysis in particular tasks, when using transfer learning resting upon datasets providing different target properties in a combination with small datasets for fine-tuning. Apparently, the choice of target energies may have an even greater impact on resulting performance than the size of the pre-training dataset, as evidenced by the fact that “ME (aflowsmall-FE)” model outperforms “FE (aflow-FE)” and “RE (aflow-RE)” on both test sets introduced.
Convex hull analysis and refinement
The convex hull approach is commonly used to assess the thermodynamic stability of potentially new structures by evaluations of their related Ehull energies above the reference fragments of the hull connecting competing phases. For each of the W-X-B systems studied, the convex hull is a three-dimensional surface built using thermodynamic properties of known structures with WmXnBl compositions. For clarity, two dimensions correspond to a simplex of ternary composition—regular W-X-B triangle on plane. In turn, the reference energies represent the third dimension. The chemical modifications studied in this work are, in turn, points on the WB4.2—XB4.2 segment within the simplex. For this reason, we represent the Ehull dependencies as 2D plots, keeping in mind that each of them is a cross-section of the corresponding 3D hull.
For the convex hull refinements, we first address the possible impact of differences in the computational circuit used in this work and that of AFLOWLib (DFT pseudopotentials, energy cutoffs, convergence control parameters, etc.). To do so, we reoptimize WB3, TaB2, and NbB2 phases, defining faces of the convex hulls for the composition of interest. This results in changes in the reference FEs by a few meV/atom and corresponds to the inconsistency of the refined and AFLOWLib-derived convex hulls in Fig. 3 (except for the right-hand side in Fig. 3c). Such scales are certainly not capable of changing most of the conclusions drawn for the AFLOWLib-derived convex hulls. In turn, when only one MoB4.243 phase undocumented in the AFLOWLib database is considered, dramatic reduction in the number of potentially new Mo-W-B entries of the search space is obtained as discussed above.
DFT computations
Our calculations are based on the density functional theory57,58 within the generalized gradient approximation (the Perdew−Burke−Ernzerhof functional59) and the projector augmented wave (PAW) method60,61 as implemented in the VASP code62,63,64. To perform a relaxation of considered supercells, the 5 × 5 × 3 Monkhorst-Pack grid65 of k-points is implied to integrate the Brillouin zone. A plane-wave cutoff of 500 eV is used, and electronic energies are converged to 10−5 eV.
The energy of formation (FE) and the enthalpy of mixing (ME) are calculated using the DFT-PBE energies of elemental single crystals according to the following formulas
where Etot is the total energy of considered supercell, NW and NB, EW and EB are the number of tungsten and boron atoms, energies of elemental tungsten and boron crystals, respectively, Ndop and Edop are the number of substituent atoms and energy of elemental crystal of dopant atom.
The ME is calculated with energies of each individual MeB4.2 in a compound using the following formula:
where NWB4.2 and EWB4.2 are the number of tungsten pentaboride structural units in the compound and the energy of tungsten pentaboride single crystal, respectively. NMeB4.2 and EMeB4.2 number of individual structure units of MeB4.2 in the compound and the energy of MeB4.2 structure, Nborides is the total number of structural units in the considered compound.
Second-order elastic constants are calculated with ElasTool66 using the optimized high-efficiency strain-matrix set (OHESS) with ±5% amplitude for each mixture. The elastic constants are used to compute Voigt-Reuss-Hill (VRH) values67 of the bulk (BVRH) and shear (GVRH) moduli. BVRH and GVRH need to determine Young’s modulus, Poisson’s ratio, Vickers hardness (Hν), and Pugh-Pettifor criterion44 for Ta-, Nb-, and Mo-included compounds. Vickers hardness (Hν) is calculated based on Chen’s45 and Mazhnik–Oganov’s47 models.
Synthesis and experimental characterization
Experimental studies on obtaining (W,Ta)B5-x are conducted on a laboratory arc-melting setup68. A rectifier-inverter converter with a working current of up to 200 A served as the direct current source. Graphite electrodes are connected to the poles of the power source. A solid graphite rod with an 8 mm diameter is used as the anode, and a graphite crucible with a cylindrical cavity, 40 mm deep and 30 mm in diameter, served as the cathode. The arc discharge is initiated by brief contact between the anode and the bottom of the cathode cavity, and the positioning of the anode is achieved using a linear electric drive controlled by a reactor program.
For powder synthesis, commercial powders of tungsten (>99.97%), tantalum (>99.9%), and X-ray amorphous boron, no less than 99%, with an average particle size not exceeding 20 µm are utilized. An excess of boron in a mass ratio of 15:1 to the metal is used for the synthesis of (W,Ta)B5-x. Initially, the powders are mixed in a Retsch pm 100 planetary mill for 1 h, with a WC milling jar of 50 ml and 9 WC balls of 12 mm diameter, rotating at a speed of 300 rpm. The initial mixture is placed in a crucible (20 mm height and 20 mm diameter) with graphite paper linings at the bottom and walls. Subsequently, the small crucible is placed into a larger crucible (40 mm height and 30 mm diameter), and to prevent displacement of the small crucible, a piece of graphite paper is inserted between the small and large crucibles.
To determine the optimal experimental regime for synthesis of required compounds, we conduct several experimental series with different parameters for synthesis.
-
The first series examined the influence of the type of mixing of individual W and Ta metals with boron on the characteristics of the synthesized W0.8Ta0.2B5-x powder. We prepare either a preliminary mixed powder between W and Ta or place W and Ta separately in a crucible. Synthesis is carried out by electric arc heating with a current of 150 A and 30 s dwell.
-
The second series investigated the effect of the arc current on the characteristics of the synthesized W0.8Ta0.2B5-x powder obtained by electric arc heating with currents of 100, 150, and 220 A with 30 s dwell.
-
The third series explored the influence of synthesis time on the characteristics of the synthesized W0.8Ta0.2B5-x powder obtained by electric arc heating with a current of 150 A for 20, 30, and 40 s.
In these experimental series, the mass of the initial powder is 1.5 g, with a fixed atomic ratio of WTa:B at 1:15. The obtained materials are analyzed using X-ray diffraction (Shimadzu XRD 7000 s, CuKα radiation). X-ray diffraction patterns of synthesized compounds during the described series are presented in Supplementary Fig. 10.
Based on the information obtained, we carry out the fourth series of experiments with a different W:Ta ratio in order to synthesize the WyTazB5-x compound (with y:z = 0.8:0.2, 0.7:0.3, 0.6:0.4, 0.5:0.5, and 0.4:0.6). Conditions for synthesis are the following: current of 150 A with 30 s dwell, the mass of the initial powder is 1.5 g, with a fixed atomic ratio of WTa:B at 1:15. The size of synthesized nanoparticles is measured manually based on SEM images.
After a sufficient amount of WTaB5-x is produced, the powder is milled in a SPEX SamplePrep high-activity vibrating ball mill for 1 h in a Si3N4 grinding jar with two Si3N4 balls and sintered by spark plasma sintering (SPS). A GT Advances Technologies SPS 10-4 equipment is used for sintering. Simultaneous pressing and sintering of the powder material is performed in vacuum during the fabrication of the bulk ceramic sample. Sintering of 3 cylindrical tablets with a diameter of 12.7 mm is carried out under the following sintering parameters: sintering temperature 1500 °C, heating rate 100 °C/min, isothermal 5 min dwell, pressure 50 MPa. The obtained materials are analyzed by X-ray diffractometry (Shimadzu XRD 7000 s, CuKα-radiation). SEM imaging of (W,Ta)B5-x ceramics is performed on a Hitachi TM 3000 scanning electron microscope. The imaging is performed in backscattered electron mode in the “Compo” mode, which is a compositional contrast in which lighter chemical elements appear darker and heavier elements appear brighter.
XPS measurements are performed using a Thermo Fisher Scientific XPS NEXSA spectrometer with a monochromated Al K Alpha X-ray source working at 1486.6 eV. The spectra are recorded at pass energy 200 and 50 eV with a step of 1 and 0.1 eV, respectively. The flood gun is used for the charge compensation. Additionally, if necessary, the charge effect is corrected using the C1s peak at 285.0 eV as a reference. The obtained spectra are processed using Avantage Thermo Fisher and CasaXPS software, and interpreted based on the literature data. Prior to XPS studies, the samples are not subjected to additional purification or activation in a reducing atmosphere, therefore, due to interaction with air oxygen and atmospheric pollutants, high levels of C and O are observed on the samples surface.
Nanohardness is measured using a DUH-211S Shimadzu dynamic hardness tester with a Berkovich tip at loads of 0.01, 0.5, and 0.1 kg according to ISO 14577-1 (Annex A).
Data availability
The data obtained and used in this study are available upon request.
References
Computational Materials Discovery. (Royal Society of Chemistry, https://doi.org/10.1039/9781788010122 (2018).
Oganov, A. R., Pickard, C. J., Zhu, Q. & Needs, R. J. Structure prediction drives materials discovery. Nat. Rev. Mater. 4, 331–348 (2019).
Kim, S., Noh, J., Gu, G. H., Aspuru-Guzik, A. & Jung, Y. Generative adversarial networks for crystal structure prediction. ACS Cent. Sci. 6, 1412–1420 (2020).
Long, T. et al. Constrained crystals deep convolutional generative adversarial network for the inverse design of crystal structures. npj Comput. Mater. 7, 66 (2021).
Merchant, A. et al. Scaling deep learning for materials discovery. Nature 624, 80–85 (2023).
Glass, C. W., Oganov, A. R. & Hansen, N. USPEX—evolutionary crystal structure prediction. Comput. Phys. Commun. 175, 713–720 (2006).
Pickard, C. J. & Needs, R. J. Ab initiorandom structure searching. J. Phys. Condens. Matter. 23, 053201 (2011).
Podryabinkin, E. V., Tikhonov, E. V., Shapeev, A. V. & Oganov, A. R. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys. Rev. B 99, 064114 (2019).
Mansouri Tehrani, A. & Brgoch, J. Hard and superhard materials: a computational perspective. J. Solid State Chem. 271, 47–58 (2019).
Rajan, A. C. et al. Machine-learning-assisted accurate band gap predictions of functionalized MXene. Chem. Mater. 30, 4031–4038 (2018).
Mortazavi, B. et al. Accelerating first-principles estimation of thermal conductivity by machine-learning interatomic potentials: a MTP/ShengBTE solution. Comput. Phys. Commun. 258, 107583 (2021).
Podryabinkin, E. V. et al. Nanohardness from first principles with active learning on atomic environments. J. Chem. Theory Comput. 18, 1109–1121 (2022).
Mazheika, A. et al. Artificial-intelligence-driven discovery of catalyst genes with application to CO2 activation on semiconductor oxides. Nat. Commun. 13, 419 (2022).
Zhu, C., Xu, L., Liu, M., Guo, M. & Wei, S. A review on improving mechanical properties of high entropy alloy: interstitial atom doping. J. Mater. Res. Technol. 24, 7832–7851 (2023).
Feng, Y., Yuan, X., Zhou, M. & Gao, L. Improvement of mechanical properties and elastocaloric effect in Ag doped Ni-Mn-In magnetic shape memory alloys. J. Alloy. Compd. 944, 169143 (2023).
Male, J. P. et al. Dislocations stabilized by point defects increase brittleness in PbTe. Adv. Funct. Mater. 31, 2108006 (2021).
Chepkasov, I. V. et al. Origin of brittle behavior of doped PbTe-based thermoelectric materials. Appl. Phys. Lett. 124, 022104 (2024).
Zuo, Y. et al. Enhanced hardness and conductivity motivated by Ni and Co doping in β-MoB2. Int. J. Refract. Met. Hard Mater. 110, 105997 (2023).
Zhao, F. et al. Improving the hardness of tungsten boride-based composites through titanium doping. Mater. Lett. 357, 135649 (2024).
Peterson, G. G. C. & Brgoch, J. Materials discovery through machine learning formation energy. J. Phys. Energy 3, 022002 (2021).
Fung, V., Zhang, J., Juarez, E. & Sumpter, B. G. Benchmarking graph neural networks for materials chemistry. npj Comput. Mater. 7, 1–8 (2021).
Xu, P., Ji, X., Li, M. & Lu, W. Small data machine learning in materials science. npj Comput. Mater. 9, 42 (2023).
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Curtarolo, S. et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Duval, A. et al. A Hitchhiker’s guide to geometric GNNs for 3D atomic systems. Preprint at https://arxiv.org/abs/2312.07511v2 (2023).
Yuan, X. et al. Active learning to overcome exponential-wall problem for effective structure prediction of chemical-disordered materials. npj Comput. Mater. 9, 12 (2023).
Chanussot, L. et al. Open catalyst 2020 (OC20) dataset and community challenges. ACS Catal. 11, 6059–6072 (2021).
Kolluru, A. et al. Open challenges in developing generalizable large-scale machine-learning models for catalyst discovery. ACS Catal. 12, 8572–8581 (2022).
Huang, P. et al. Unveiling the complex structure-property correlation of defects in 2D materials based on high throughput datasets. npj 2D Mater. Appl. 7, 6 (2023).
Kazeev, N. et al. Sparse representation for machine learning the properties of defects in 2D materials. npj Comput. Mater. 9, 113 (2023).
Cheetham, A. K. & Seshadri, R. Artificial intelligence driving materials discovery? Perspective on the article: Scaling deep learning for materials discovery. Chem. Mater. 36, 3490–3495 (2024).
Szymanski, N. J. et al. An autonomous laboratory for the accelerated synthesis of novel materials. Nature 624, 86–91 (2023).
Leeman, J. et al. Challenges in high-throughput inorganic materials prediction and autonomous synthesis. PRX Energy 3, 011002 (2024).
Bartel, C. J. Review of computational approaches to predict the thermodynamic stability of inorganic solids. J. Mater. Sci. 57, 10475–10498 (2022).
Chen C. et al. Accelerating Computational Materials Discovery with Machine Learning and Cloud High-Performance Computing: From Large-Scale Screening to Experimental Validation. J. Am. Chem. Soc. 146, 200009–20018 (2024).
Eremin, R. A. et al. Hybrid DFT/data-driven approach for searching for new quasicrystal approximants in Sc-X (X = Rh, Pd, Ir, Pt) systems. Cryst. Growth Des. 22, 4570–4581 (2022).
Kvashnin, A. G. et al. New tungsten borides, their stability and outstanding mechanical properties. J. Phys. Chem. Lett. 9, 3470–3477 (2018).
Kvashnin, A. G. et al. WB5–x: synthesis, properties, and crystal structure—new insights into the long-debated compound. Adv. Sci. 7, 200775 (2020).
Pak, A. Ya. et al. Efficient synthesis of WB5–x–WB2 powders with selectivity for WB5–x content. Inorg. Chem. 61, 6773–6784 (2022).
Lech, A. T., Turner, C. L., Mohammadi, R., Tolbert, S. H. & Kaner, R. B. Structure of superhard tungsten tetraboride: A missing link between MB2 and MB12 higher borides. Proc. Natl. Acad. Sci. USA 112, 3223–3228 (2015).
Okhotnikov, K., Charpentier, T. & Cadars, S. Supercell program: a combinatorial structure-generation approach for the local-level modeling of atomic substitutions and partial occupancies in crystals. J. Cheminform. 8, 17 (2016).
Musaelian, A. et al. Learning localequivariant representations for large-scale atomistic dynamics. Nat. Commun. 14, 579 (2022).
Rybkovskiy, D. V., Kvashnin, A. G., Kvashnina, Y. A. & Oganov, A. R. Structure, stability and mechanical properties of boron-rich Mo-B phases: a computational study. J. Phys. Chem. Lett. 11, 2393–2401 (2020).
Senkov, O. N. & Miracle, D. B. Generalization of intrinsic ductile-to-brittle criteria by Pugh and Pettifor for materials with a cubic crystal structure. Sci. Rep. 11, 4531 (2021).
Chen, X.-Q., Niu, H., Li, D. & Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 19, 1275–1281 (2011).
Niu, H., Niu, S. & Oganov, A. R. Simple and accurate model of fracture toughness of solids. J. Appl. Phys. 125, 065105 (2019).
Mazhnik, E. & Oganov, A. R. A model of hardness and fracture toughness of solids. J. Appl. Phys. 126, 125109 (2019).
Wang, L. et al. One step transformation of waste polyvinyl chloride to tantalum carbide@carbon nanocomposite at low temperature. J. Am. Ceram. Soc. 102, 6455–6462 (2019).
Rajaji, U. et al. Fabrication of a novel tantalum boride/vanadium carbide modified screen-printed carbon electrode for voltammetric determination of pimonidazole in bio-fluids. Microchim. Acta 191, 112 (2024).
Rahaman, S. Z. et al. Excellent resistive memory characteristics and switching mechanism using a Ti nanolayer at the Cu/TaOx interface. Nanoscale Res. Lett. 7, 345 (2012).
Hu, Y. et al. Study on the Electrical, Structural, Chemical and Optical Properties of PVD Ta(N) Films Deposited with Different N2 Flow Rates. Coatings 11, 937 (2021).
Guo, F. et al. A class of metal diboride electrocatalysts synthesized by a molten salt-assisted reaction for the hydrogen evolution reaction. Chem. Commun. 55, 8627–8630 (2019).
He, D. et al. Tailoring the mechanical and tribological properties of B4C/a-C coatings by controlling the boron carbide content. Surf. Coat. Technol. 329, 11–18 (2017).
Magnuson, M. et al. Reactive magnetron sputtering of tungsten target in krypton/trimethylboron atmosphere. Thin Solid Films 688, 137384 (2019).
Togo, A. & Tanaka, I. Spglib: a software library for crystal symmetry search. Sci. Technol. Adv. Mater. Methods 4, 2384822 (2018).
Eremin, R. A. et al. Graph neural networks for predicting structural stability of Cd- and Zn-doped γ-CsPbI3. Comput. Mater. Sci. 232, 112672 (2024).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. B 140, A1133–A1138 (1965).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396–1396 (1997).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 16, 11169–11186 (1996).
de Wijs, G. A., de Boer, P. K., de Groot, R. A. & Kresse, G. Anomalous behavior of the semiconducting gap in WO3 from first-principles calculations. Phys. Rev. B 59, 2684–2693 (1999).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Monkhorst, H. J. & Pack, J. D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 13, 5188–5192 (1976).
Liu, Z.-L., Ekuma, C. E., Li, W.-Q., Yang, J.-Q. & Li, X.-J. ElasTool: an automated toolkit for elastic constants calculation. Comput. Phys. Commun. 270, 108180 (2022).
Zuo, L., Humbert, M. & Esling, C. Elastic properties of polycrystals in the Voigt-Reuss-Hill approximation. J. Appl. Crystallogr. 25, 751–755 (1992).
Pak, A. Ya., Shanenkov, I. I., Mamontov, G. Y. & Kokorina, A. I. Vacuumless synthesis of tungsten carbide in a self-shielding atmospheric plasma of DC arc discharge. Int. J. Refract. Met. Hard Mater. 93, 105343 (2020).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2009).
Acknowledgements
The authors thank the hardware and software facilities of the “Christofari” heterogeneous platform (Sber, Moscow, Russia) used for calculations with GNN models. DFT calculations were carried out using Skoltech supercomputer Zhores. Experiments on the vacuumless synthesis of higher tungsten boride were carried out with support from the Ministry of Science, Higher Education of the Russian Federation in part of the Science program (Project FSWW-2025-0003).
Author information
Authors and Affiliations
Contributions
A.G.K., R.A.E., A.A.O., and S.A.B. initiated the project, R.A.E., I.S.H., A.V.K., and V.D.L. carried out the development and application of the data-driven approach, prediction of thermodynamic properties accounting for the known competing phases, selection of candidate materials, running models, and data management during the pre-training, fine-tuning, and inference stages, A.A.K. and A.Y.P. performed synthesis of powders and ceramics of chemically modified higher tungsten borides, did the experiments on microscopy characterization and hardness measurements. Y.Z.V. performed XPS measurements and analysys. N.A.M. and A.G.K. performed theoretical calculations and analysis of the results obtained. N.A.M., R.A.E., and A.G.K. wrote the original draft of the manuscript. The manuscript has been written through contributions of all authors. All the authors provided critical feedback and helped shape the research. All the authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Matsokin, N.A., Eremin, R.A., Kuznetsova, A.A. et al. Discovery of chemically modified higher tungsten boride by means of hybrid GNN/DFT approach. npj Comput Mater 11, 163 (2025). https://doi.org/10.1038/s41524-025-01628-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-025-01628-z
