
Abstract
Developing inverse design methods for functional materials with specific properties is critical to advancing fields like renewable energy, catalysis, energy storage, and carbon capture. Generative models based on diffusion principles can directly produce new materials that meet performance constraints, thereby significantly accelerating the material design process. However, existing methods for generating and predicting crystal structures often remain limited by low success rates. In this work, we propose a novel inverse material design generative framework called InvDesFlow-AL, which is based on active learning strategies. This framework can iteratively optimize the material generation process to gradually guide it towards desired performance characteristics. In terms of crystal structure prediction, the InvDesFlow-AL model achieves an RMSE of 0.0423 Å, representing an 32.96% improvement in performance compared to existing generative models. Additionally, InvDesFlow-AL has been successfully validated in the design of low-formation-energy and low-Ehull materials. It can systematically generate materials with progressively lower formation energies while continuously expanding the exploration across diverse chemical spaces. Notably, through DFT structural relaxation validation, we identified 1,598,551 materials with Ehull < 50 meV, indicating their thermodynamic stability and atomic forces below 1e-4 eV/Å. These results fully demonstrate the effectiveness of the proposed active learning-driven generative model in accelerating material discovery and inverse design. To further prove the effectiveness of this method, we took the search for BCS superconductors under ambient pressure as an example explored by InvDesFlow-AL. As a result, we successfully identified Li2AuH6 as a conventional BCS superconductor with an ultra-high transition temperature of 140 K, and also discovered several other superconducting materials that surpass the theoretical McMillan limit and have transition temperatures within the liquid nitrogen temperature range. This discovery provides strong empirical support for the application of inverse design in materials science.
Similar content being viewed by others

Introduction
The discovery of new materials1,2 plays a vital role in advancing technological progress and providing essential solutions to global challenges, e.g., photovoltaic and battery materials3, high-temperature superconductors4,5, novel catalysts and degradable materials6, semiconductor materials7, and biocompatible materials8. Traditional material discovery faces limitations such as long experimental cycles, uncertain research paths, and lacking clear exploration directions. While high-throughput screening methods based on density functional theory (DFT)9 could accelerate the discovery process, they still demand costly computational resources. In recent years, technologies like large language models10,11, geometric graph neural networks12,13, and generative AI14,15 have demonstrated significant advantages in material generation and screening, further driving new material discoveries. However, current generative models often fail to produce stable materials based on DFT calculations, remain limited to a narrow subset of elements, or can only optimize a very limited set of properties. For example, generative AI faces limitations in material stability and functionality2,16,17,18, where generated materials may fail in practical applications due to thermodynamic instability or inaccurate functional property predictions. Moreover, discriminative AI struggles with overfitting issues, particularly on small datasets, resulting in poor generalization ability across different material systems (e.g., predicting the critical temperature Tc of superconductors9). Theoretically viable materials may also prove impractical in experiments due to synthesis challenges. Future research needs to address these challenges to shift from generating “feasible materials” to producing “manufacturable materials”.
The current approaches for controlling the generation of functional materials primarily include adapter modules for conditional control2 and latent variable decoding of material properties17,19,20,21,22. Due to the large datasets available for some functional materials, such as formation energy, magnetic density, band gap, or bulk modulus, these generative AI methods perform well. However, for high-temperature superconductors, synthesizable materials, and stable materials, the data is scarce, and the labeling costs are high. In this context, the aforementioned generative AI methods fail to account for these challenges in utilizing minimal data for inverse materials design. It is common knowledge that models trained with Adapter Modules do not perform as well as those trained with full-parameter training. Inspired by reinforcement learning, we believe that active learning can maximize model performance23. Active learning’s iterative learning cycle allows the model to actively select the most valuable data for improving its performance, significantly enhancing model efficacy, particularly in scenarios where data labeling is expensive or time-consuming.
In this study, we present InvDesFlow-AL, a material inverse design generation framework based on active learning, which is capable of directing the generation of target functional materials and continuously optimizing the generation outputs through a step-by-step iterative process. Such an approach can adapt to a wide range of downstream tasks by altering the training dataset, thereby achieving efficient inverse material design (as shown in Fig. 1). To this end, we have combined a generative model that progressively refines atomic types, coordinates, and periodic lattices with an online learning strategy. By selecting more valuable data from the model’s generated results for labeling and training, we have significantly enhanced the performance of the generative model.

a Active learning-based diffusion model for designing functional materials. The core of active learning lies in selecting the most valuable data for models to enhance their performance. It primarily involves three strategies: diversity sampling, expected model change, and query-by-committee. b Steps of InvDesFlow-AL consist of the following four stages: first, a pre-trained crystal generation model is constructed. Second, this model is fine-tuned on functional materials. Third, the fine-tuned generator is used to generate candidate crystal structures. Finally, a QBCs-based multi-objective function is applied to select the most informative data, which is then used to further fine-tune the generative model. c Applications of InvDesFlow-AL. Stable crystal generation, discovery of high-Tc superconductors, crystal structure prediction, identification of ultra-high temperature ceramics, and guidance for experimental synthesis.
Compared with DiffCSP16, InvDesFlow-AL has reduced the root mean square error (RMSE) in crystal structure prediction tasks to 0.0423 Å, representing a 32.96% improvement in performance over current methods. In terms of generating materials with low formation energy and low Ehull (energy above the convex hull, indicating thermodynamic stability) targets, InvDesFlow-AL has successfully identified 1,598,551 materials with an Ehull < 50 meV. All these materials have undergone structural relaxation, with interatomic forces less than 1 × 10−4eV/Å (achieving DFT precision). As a proof of concept, InvDesFlow-AL has generated the material with the highest transition temperature in the current conventional superconducting system (Li2AuH6, 140 K). Moreover, this method can also design materials under various property constraints, such as ultra-high-temperature ceramics. Finally, we summarized the current limitations of InvDesFlow-AL in terms of algorithms, data, and scientific theory, and proposed corresponding improvement strategies. We also discussed its potential applications in areas such as electrode materials, hydrogen storage materials, and bioinspired materials, as well as its prospects for facilitating experimental synthesis and industrial deployment.
Results
Active learning-based diffusion model
InvDesFlow-AL is an active learning-based diffusion model for designing target functional inorganic crystal materials across the periodic table. The diffusion model generates samples by reversing a fixed corruption process. Following the conventions of existing generative models for crystal matrials2,16,19, we define a crystal material by its unit cell, which includes atom types A (e.g., chemical elements), coordinates X, and periodic lattice L (see the Methods section). For each component, we define a corruption process that takes into account its specific geometry and has a physically motivated limiting noise distribution. Unlike previous generative models, we also incorporate an active learning strategy that actively selects the most valuable data for labeling and training to enhance its performance. Common strategies for identifying the most valuable data include: the diversity sampling (DS) strategy, which ensures that the selected samples represent different regions of the data distribution; the expected model change (EMC) strategy, which selects samples that have the greatest impact on the model parameters; the query-by-committee (QBC) strategy, which trains multiple models to form a “committee” and selects the most valuable samples.
To train the proposed InvDesFlow-AL, we utilized the Alex-MP-20 dataset2, which was released by MatterGen and contains 607,683 crystalline materials, along with 381,000 inorganic materials1 from the GNoME dataset. These datasets encompass both ordered and disordered crystal structures and cover a wide range of inorganic materials, including conductors, semiconductors, insulators, and magnetic materials (see Fig. 1). The pretrained model employs an active learning-based diversity sampling strategy to cover different regions of the inorganic materials distribution. It is capable of unconditionally generating crystal structures with high stability, uniqueness, and novelty. Furthermore, the model can be fine-tuned to generate materials with specific functional properties. InvDesFlow-AL has been successfully applied and demonstrated its feasibility in three tasks: generation of low formation energy materials, generation of high-temperature superconducting materials, and crystal structure prediction. In these tasks, we implemented multiple rounds of fine-tuning (Fig. 1) for the generative model. The initial fine-tuning utilized crystal data from target functional materials, while subsequent iterations of fine-tuning employed crystal data generated by the model itself. We regard this iterative fine-tuning process as an implementation of the expected model change strategy. For different tasks, InvDesFlow-AL uses different QBC methods to select suitable crystal data. These QBCs are designed as multi-objective functions and are used to evaluate the most valuable data.
The pre-trained crystal generation mode of InvDesFlow-AL is trained on a large-scale inorganic materials dataset, demonstrating strong performance in material novelty (Supplementary Fig. S4) and excellent stability (Fig. 2). Compared to MatterGen2, which adopts adapter-based fine-tuning for inverse design of functional materials, InvDesFlow-AL leverages full-parameter fine-tuning across the entire architecture. This approach demonstrates better adaptability to downstream tasks, as extensively validated in domains such as large language models24,25 and computer vision26. For crystal representation, InvDesFlow-AL adopts fractional coordinates anchored to lattice vectors as the fundamental basis, which explicitly preserves crystalline periodicity and enables more intuitive handling of crystal symmetry. This methodology ensures higher efficiency and stability during both model training and sampling processes, a conclusion further substantiated by subsequent crystal structure prediction tasks.

a InvDesFlow-AL employs a crystal generation model to generate new crystal structures, followed by formation energy prediction using FormEGNN. A lower threshold is applied to retain newly generated materials for fine-tuning the generative model. After five iterations, a progressive decrease in formation energy is observed. b InvDesFlow-AL adopts the same strategy to generate materials with low Ehull. Through multiple rounds of generation, a total of 1,610,600 new crystal structures with Ehull < 50 meV have been obtained, expanding the chemical space exploration to a broader range of atomic species. c Crystal structures containing 2, 3, ..., and up to 7 elements generated by InvDesFlow-AL.
Generation of low formation energy materials
The pursuit of synthesizing materials with low formation energy (Eform) and minimal energy above the convex hull Ehull constitutes the central objective of InvDesFlow-AL. The formation energy, quantifying the thermodynamic stability of a compound relative to its elemental constituents, serves as a fundamental indicator of synthesizability—only materials with negative Eform are thermodynamically viable under equilibrium conditions. Energy above hull Ehull, conversely, measures a material’s metastability by evaluating its energetic proximity to the convex hull in phase space. A low Ehull (typically < 50 meV/atom) signifies resilience against decomposition into competing phases, a critical prerequisite for practical synthesis and operational durability.
In this section, we focus on the ability of the InvDesFlow-AL to generate materials with low formation energy (Eform) and minimal energy above the convex hull Ehull. To achieve thermodynamically favorable (low Eform), structurally stable (interatomic forces < 1e-4 eV/Å), and compositionally novel candidates, we propose an active learning framework integrating EMC and QBC strategies.
The pretrained model is fine-tuned on the GNoME dataset1, focusing exclusively on crystals with Eform < −0.5 eV/atom to establish thermodynamic stability priors. The fine-tuned generator synthesizes novel crystal structures, filtered by compositional uniqueness against existing materials databases (e.g., Materials Project27). Generated candidates undergo atomic-scale structural relaxation using the DPA-2 interatomic potential28, which achieves DFT-level accuracy. Structures failing force convergence criteria (∣∣F∣∣ > 1e − 4 eV/Å) are systematically discarded. The formation energy prediction model (which we introduce here as FormEGNN) was developed in InvDesFlow-1.018. It predicts Eform for relaxed structures, with a dynamically adjusted threshold to retain only the most stable candidates for subsequent generator retraining. The iterative refinement process is driven by EMC-guided data selection, where a committee consisting of DPA-2 and FormEGNN evaluates candidate materials using a multi-objective scoring function. “Methods” section provides a detailed explanation of this multi-objective function. This closed-loop paradigm progressively biases the generator toward materials with low formation energy and relaxed structures while maintaining structural diversity.
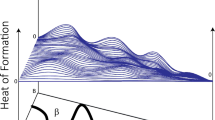
InvDesFlow-AL iteratively generated and fine-tuned the model five times. The average formation energies of the generated crystals in these five iterations were μ = −1.14, −2.03, −2.93, −3.56, −3.77, with the number of generated structures being 80,707, 95,580, 97,379, 136,784, and 166,663, respectively, resulting in a total of 577,113 generated crystals. The dataset has been open-sourced. All these structures achieved DFT relaxation accuracy. As shown in Fig. 2a, the formation energy of the generated crystals decreases with increasing iterations.
InvDesFlow-AL generates low-hull crystal structures through iterative optimization. During each iteration, the Bohrium platform is utilized to query the Ehull values of materials, and crystals with lower Ehull values are selected to fine-tune the model. As more stable structures are discovered during exploration, both Ehull and Eform are dynamically updated, with the reference energies adjusted accordingly to ensure consistent and accurate evaluation.
We have performed 10 rounds of fine-tuning in total, generating 1,598,551 materials with formation energies below 50 meV (see Supplementary Fig. S5). As shown in Fig. 2b, to generate lower-Ehull crystal structures, the elemental composition distribution of the generated crystals by InvDesFlow-AL differs significantly from those in the Materials Project database. InvDesFlow-AL exhibits a strong preference for generating materials with higher atomic diversity, many of which are high-entropy alloys that do not exist in existing databases. While binary and ternary chemical spaces will likely be exhaustively explored by chemists in the near future, despite the experimental challenges in synthesizing multi-component materials, we believe that these complex multi-element crystals will make groundbreaking contributions to future advanced materials discovery, Fig. 2(c) shows the multicomponent crystal structures generated by InvDesFlow-AL. All generated crystal structures will be fully open-sourced to the community.
Generation of high-temperature superconducting materials
In this section, we apply InvDesFlow-AL to the discovery of high-temperature superconducting materials. High-temperature superconductors hold significant importance for achieving efficient power transmission, controlled nuclear fusion energy generation, magnetic resonance imaging, and superconducting quantum computing. Superconducting materials surpassing critical temperature thresholds—including the McMillan limit (40 K), liquid nitrogen temperature regime (77 K), and even room temperature—attract distinct levels of interest from physicists. For instance, hydrogen-based systems like H3S (200 K)29 and LaH10 (260 K)30,31 with exceptionally high transition temperatures have drawn extensive research attention. Subsequent discoveries reported in ref. 32, including YH18 (183 K), AcH18 (206 K), LaH18 (271 K), and ThH18 (306 K), have further expanded the family of hydride-based superconducting materials. However, these materials require extremely high-pressure conditions for synthesis, which poses significant constraints on their practical applicability. Consequently, the discovery of high-temperature superconductors under ambient pressure has become particularly crucial. Notably, the experimental synthesis5 of (La,Pr)3Ni2O7 (45 K) under ambient pressure has garnered considerable attention as it exceeds the McMillan limit. Recent advancements in high-throughput computing and machine learning techniques have accelerated the discovery of superconducting materials, with Mg2XH6 (X = Rh, Ir, Pd, or Pt)33,34,35,36 exhibiting a Tc exceeding 80 K. These developments motivate our adoption of state-of-the-art AI technologies to further expedite the exploration of superconducting materials.
The conventional high-throughput screening approach, constrained by the limited exploration of chemical space, has shown fundamental limitations in discovering high-temperature superconductors. Our InvDesFlow-AL framework enables direct generation of materials with targeted properties, facilitating exploration in unbounded chemical space. To generate materials with ambient-pressure high-temperature superconductivity, we develop a multi-stage active learning strategy combining sequential model refinement and committee-based validation. The strategy initiates with a two-phase expected model change optimization. Phase I conducts domain adaptation—fine-tuning a pretrained model originally trained on diverse material types to better specialize in a target domain, such as conductive metals. By adapting the model with metal-specific data, it adjusts its internal representations to more accurately capture relevant structural and property features, enhancing its ability to generate materials with desired properties—through fine-tuning on metallic materials from the pre-training dataset, establishing essential electronic conductivity priors. Phase II implements superconducting specialization using recently discovered conventional superconductors33,34,35,36, progressively aligning the model’s generative space with superconducting characteristics. This sequential refinement ensures structural validity while enhancing target property awareness.
The optimized generator produces candidate superconductors that undergo dual-stage validation. We first introduce a superconducting graph neural network (SuperconGNN) to screen candidates by predicted critical temperature (Tc > 20 K threshold). Subsequently, we adopt DFT calculations to verify electronic structure features and superconducting stability. Validated materials enrich the training dataset through an active learning loop governed by a query-by-committee strategy. The iterative refinement process is driven by QBC-guided data selection, where a committee consisting of SuperconGNN and DFT evaluates candidate materials using a multi-objective scoring function. “Methods” section provides a detailed explanation of this multi-objective function.
As shown in Fig. 3a, through multiple iterative generation cycles, InvDesFlow-AL has identified superconducting materials spanning a wide temperature range, from low to high Tc. Notably, K2GaCuH6 and Na2GaCuH6 exhibit critical temperatures exceeding the McMillan limit (40 K), while Li2AuH6 and Na2LiAgH6 surpass the liquid nitrogen temperature threshold (77 K). Remarkably, Li2AuH6 reaches approximately 140 K, representing the highest Tc achieved by conventional superconductors to date.

a Comparison of previously reported high-temperature superconductors and newly discovered materials generated by InvDesFlow-AL. The superconducting transition temperatures of these newly discovered materials span a wide range, from the McMillan limit to the liquid nitrogen temperature region. The inset in (a) shows the crystal structure of Li2AuH6. b Phonon dispersion and phonon density of states of Li2AuH6, indicating its dynamical stability. (c) Electronic band structure and density of states of Li2AuH6.
Figure 3b presents the phonon dispersion relations and phonon density of states (DOS) of Li2AuH6, demonstrating its dynamical stability. Figure 3c presents the electronic structure of Li2AuH6, revealing its metallic character with multiple bands crossing the Fermi level. The atomic orbital-projected density of states (PDOS) analysis demonstrates that electronic states near the Fermi level are predominantly contributed by the Au-H octahedral coordination. A van Hove singularity is identified at the W point, generating a pronounced PDOS peak at the Fermi level, which is often associated with enhanced electronic correlations. The phonon dispersion of Li2AuH6 exhibits no imaginary frequencies at ambient pressure, confirming its dynamic stability. The spectrum consists of three distinct frequency regions separated by two energy gaps: the high-frequency region (>120 meV), dominated by hydrogen vibrations; the intermediate-frequency region (60–100 meV), primarily composed of H-related vibrations with partial Au contributions; and the low-frequency region (<50 meV), where mixed vibrations of Au and Li atoms are observed. The Eliashberg spectral function α2F(ω) and the cumulative electron-phonon coupling (EPC) constant λ(ω) are displayed in the inset, with the total EPC constant calculated to be λ = 2.84. The strong EPC predominantly arises from three key phonon modes: the Eg mode at Γ (140 meV), the A1g mode at X (20 meV), and the Eg mode at X (30 meV). First-principles analysis reveals that the robust electron-phonon interactions in Li2AuH6 originate primarily from the vibrational modes of the Au-H octahedral framework and Li atoms, which induce significant charge density modulations and drive strong Cooper pairing. This exceptionally large λ value places Li2AuH6 among the strongest electron-phonon coupled superconductors predicted to date, highlighting its potential as a promising high-temperature conventional superconductor. For a more detailed analysis of the Li2AuH6 crystalline material and its synthesis pathway, please refer to our another work37. A variety of additional superconducting materials were also discovered by InvDesFlow-AL, with their corresponding DFT calculation results presented in Supplementary Note A.
Stable structure prediction task
The crystal structure prediction task (CSP) is of great significance for accelerating the discovery of new materials, understanding the fundamental laws of matter, and guiding experimental synthesis. Earlier approaches combining machine-learned interatomic potentials, such as moment tensor potentials (MTP), with evolutionary algorithms like USPEX38 have significantly improved computational efficiency and prediction accuracy through active learning, successfully predicting complex material crystal structures. Moreover, the open-source software CrySPY39 integrates various search algorithms and machine learning-based screening strategies to automate and accelerate structure generation and energy evaluation, further advancing the field of crystal structure prediction. These works provide valuable foundations for our active learning-based crystal structure prediction approach. In recent years, AI methods such as CDVAE19, DiffCSP16, and CrystaLLM11 have achieved promising results in this task. However, these methods often require multiple samplings and do not provide a clear approach for selecting the best structure from the multiple predictions. As a result, the results from multiple samplings do not necessarily reflect the true prediction accuracy. We have significantly improved the accuracy of crystal structure prediction using an active learning-based approach, with the single-prediction accuracy surpassing the current best methods.
It is important to note that the functional material generation described in previous sections employs a de novo generative model, where the number of atoms in the unit cell serves as a conditional constraint. In contrast, the CSP task focuses on predicting crystal structures with fixed atomic compositions. For the CSP implementation, we first adopted a diversity sampling strategy to ensure the model learns structural information across diverse data distributions. This process utilized the Alex-MP-20 dataset released by MattergGen2 and crystal structures from GNoME1, with rigorous exclusion of test sets including Perov-5, MP-20, and MPTS-52 during data preprocessing. Using the expected model change strategy, we fine-tuned the model on the corresponding structure prediction training sets (Perov-5, MP-20, MPTS-52).
Additionally, we utilized a de novo generative model and fine-tuned it on the aforementioned training set. Based on the atomic number distribution, we generated 100,000 crystal structures. For the generated materials, structural relaxation was performed using the DPA2 potential function to filter out structurally stable materials. Subsequently, we employed the formation energy prediction model FormEGNN to identify crystal structures with the same chemical formula but lower structural energy, and then fine-tuned the model again. The feedback from DPA2 and FormEGNN is referred to as an active learning-based query-by-committee, where the committee assists in selecting the most valuable samples. A multi-objective scoring function is used here, introduced in the “Methods” section. To quantitatively evaluate the accuracy of predicted crystal structures, we adopt the root-mean-square error (RMSE) of atomic positions as a key metric. RMSE measures the average deviation between predicted and ground-truth atomic coordinates after optimal rigid-body alignment, and is defined as
where N is the number of atoms, and \({{\boldsymbol{X}}}_{i}^{{\rm{pred}}}\) and \({{\boldsymbol{X}}}_{i}^{{\rm{true}}}\) represent the predicted and true atomic positions, respectively. The Kabsch algorithm is applied to eliminate global translation and rotation. This metric enables a fair comparison with recent methods on widely used benchmark datasets. As shown in Table 1, MP-20 is a test set with a maximum of 20 atoms per unit cell, representing conventional crystalline materials in existing databases like the Materials Project27. On the MP-20 test set, InvDesFlow-AL achieved an RMSE of 0.0423 Å, surpassing not only the recently popular large language model-based crystal structure prediction algorithm CrystaLLM11 but also outperforming state-of-the-art methods such as DiffCSP16, CDVAE19, and EquiCSP40. MPTS-52, a dataset allowing up to 52 atoms per unit cell, further demonstrated the robustness of our approach. InvDesFlow-AL achieved an RMSE of 0.0725 Å, representing a 37% improvement over EquiCSP40.
To further evaluate the performance of InvDesFlow-AL, we conducted a systematic comparison against USPEX (A crystal structure prediction method based on evolutionary algorithms. It can predict stable or metastable crystal structures from scratch given only the chemical composition)38 on 15 representative compounds. The USPEX calculation results for these compounds were obtained from the supplementary materials of the DiffCSP study16, in which the authors performed these computations to benchmark the performance of DiffCSP against USPEX. In this work, we directly adopt their USPEX results as the baseline for comparison. In contrast, all InvDesFlow-AL results reported in Table 2 were independently computed by our team. The key observations are as follows. First, InvDesFlow-AL achieved an overall match rate of 73.33%, which is significantly higher than the 53.33% achieved by USPEX, indicating stronger robustness across diverse systems. Second, the average minimum root-mean-square deviation (RMSD) of candidate structures generated by InvDesFlow-AL was 0.0109 Å, lower than the 0.0159 Å obtained by USPEX, suggesting that our method produces atomic configurations closer to the reference structures. Third, in terms of efficiency, USPEX required approximately 12.5 h per compound on average, while InvDesFlow-AL generated candidate structures within about 10 s, corresponding to a speedup of over 4000 times and a substantial reduction in computational cost. Overall, InvDesFlow-AL outperforms the conventional USPEX method in success rate, structural accuracy, and computational efficiency, demonstrating its strong potential for large-scale and high-throughput materials discovery.
Generation of ultra-high temperature ceramics
Ultra-high temperature ceramics (UHTCs)41 constitute a category of advanced materials that preserve exceptional mechanical robustness and thermophysical stability under extreme conditions, including ultra-high temperatures (>2000 °C), aggressive oxidation, and corrosive atmospheres. These materials exhibit critical application value in aerospace, defense, and energy engineering. UHTCs are predominantly composed of refractory transition-metal borides, carbides, and nitrides, such as borides (XB2, X = Zr, Hf, Ta) and carbides (XC, X = Zr, Hf, Ta, Ti), all of which possess melting points exceeding 3000 °C. Owing to their superior integrated properties—encompassing oxidation/ablation resistance, high-temperature strength retention, and thermal shock resilience—UHTCs have attracted extensive scientific attention, particularly for applications in thermal protection systems of hypersonic vehicles and next-generation nuclear reactors.
Herein, we applied InvDesFlow-AL to generate UHTCs (see Fig. 4). This section aims to demonstrate the generalization capability of the pretrained model. We collected crystal structure data for 14 UHTCs, such as ZrB2, HfC, TiC. Based on the pre-trained crystal generation model, we first fine-tuned it on the 14 UHTC crystal materials. Using the fine-tuned generative model, we then generated 100 crystal structures, achieving 100% coverage of the 14 UHTC materials in the training set. Among the remaining generated materials, we checked whether their chemical formulas had been previously synthesized or calculated using DFT. Three materials (Fig. 4d–f), TaB2, ZrC, and HfN, were found to have been experimentally validated. TaB2 has a high melting point and excellent wear resistance, making it an ideal choice for ultra-high-temperature ceramic materials42. It can be synthesized using high-temperature synthesis43, chemical vapor deposition, and sol-gel techniques. ZrC has a NaCl-type structure with a melting point of 3540 °C and is used as a nuclear fuel cladding material44. HfN exhibits high infrared reflectance and can be applied as a thermal control coating for satellites45.

a–c Synthesized UHTCs (ZrB2, HfC, TiC) existing in the fine-tuning dataset, demonstrating the model’s capacity to reproduce known high-performance ceramics. d–f Novel UHTCs (TaB2, ZrC, HfN) are absent from the training data, whose exceptional properties-high-temperature resistance, oxidation resistance, and thermal shock resilience-have been validated through theoretical/experimental studies. This systematic validation underscores InvDesFlow-AL's capability to design unreported, high-stability ceramics beyond existing databases.
Discussion
From electronic devices and aerospace to quantum computing and energy transport and storage, the advancement of almost every frontier technology depends on breakthroughs in novel functional materials. We introduce InvDesFlow-AL, an active learning-based inverse materials design framework. Through InvDesFlow-AL, scientists can specify target properties—such as superconductivity, topology, and magnetism—to generate candidate crystal structures, thereby overcoming the limitations of empirical approaches. InvDesFlow-AL integrates generative AI, quantum mechanical methods, and thermodynamic and kinetic modeling to enable multiscale simulation for the discovery of new functional materials. In crystal-structure prediction, our model achieves an RMSE of 0.0423 Å, which is close to the typical systematic error observed in X-ray diffraction (XRD) measurements due to instrument calibration, sample preparation, and data processing. In the generation of low-formation-energy and low-Ehull materials, InvDesFlow-AL demonstrates the effectiveness of active learning through multiple iterations. Each round produces materials with progressively lower formation energies, ultimately generating 1,598,551 materials with Ehull < 50 meV. For high-temperature superconductor discovery, our framework identified Li2AuH6 as a candidate with an ultra-high superconducting transition temperature of 140 K. Additionally, we discovered a series of novel superconductors that span the range between the McMillan limit and the liquid nitrogen temperature regime. Outstanding materials of interest to humanity often lie outside existing crystal structure databases, limiting the performance of adapter-based or latent-space-controlled generative models when the training data is insufficient. InvDesFlow-AL is designed to overcome this constraint. By leveraging an active learning strategy, the framework uses data generated by the model, filters it using QBCs, and fine-tunes the generative model over multiple iterations. As a result, InvDesFlow-AL continuously evolves to generate increasingly superior functional materials.
However, whether the generative model within InvDesFlow-AL can successfully discover superior materials depends critically on the quality of the pretrain data, the architecture of the generative model, the design of the QBCs strategy, and the advancement of scientific theory. Different classes of functional materials exhibit varying degrees of data availability. For instance, high-temperature superconductors under ambient pressure are extremely scarce, whereas more than 20,000 superconductors with transition temperatures below 10 K have already been identified. Traditional high-throughput DFT calculations provide a broader dataset, and with the aid of machine learning, this field is expected to yield an increasing number of high-quality data points46. We are currently using an equivariant GNN architecture based on EGNN47 to implement a crystal generation model. However, AlphaFold 348 demonstrates that equivariance can be achieved through data augmentation (random rotations and translations), effectively eliminating the need for architectural equivariance, while achieving state-of-the-art performance in molecular docking. This inspires us to explore crystal representation architectures beyond the constraints of equivariant networks. We currently use a QBC framework composed of AI models and DFT calculations to effectively screen for high-quality data. However, the computational cost of DFT remains prohibitive. In the future, artificial intelligence models will accelerate density functional theory (DFT) calculations, making it possible to screen hundreds of millions of hypothetical crystal structures. Tools such as DeepH49. Furthermore, the development of scientific theory itself may impose limitations on new materials discovery. For example, recent studies50 have suggested that Li2AuH6 and Li2AgH6 represent the upper bound of the superconducting transition temperature for conventional BCS superconductors under ambient pressure. If this conclusion holds true, it would imply that the search for new materials within this system is nearly exhausted.
With the deep integration of AI into materials science, we anticipate that the generalized InvDesFlow-AL framework will continue to unleash its potential in several key materials domains in the future. In the domain of high-performance battery electrode materials, enhancing energy density fundamentally relies on the development of electrodes with both high capacity and high voltage51. The InvDesFlow-AL framework can construct multi-objective scoring function—based on theoretical capacity, voltage window, and electrochemical stability, thereby enabling the generation of novel anode and cathode candidates that simultaneously exhibit high theoretical capacity and high operating voltage52. In the context of the green energy transition, the development of lightweight, high-capacity hydrogen storage materials53 remains a critical bottleneck for the widespread adoption of hydrogen energy. Conventional approaches struggle to systematically identify materials that simultaneously meet key criteria such as low dehydrogenation temperature, high reversible capacity, and appropriate thermodynamic stability. The InvDesFlow-AL framework offers a natural advantage in optimizing hydrogen storage performance by constructing multi-objective scoring functions targeting hydrogen adsorption/desorption characteristics54. This enables the discovery of novel magnesium-based alloys, hydrides, and even composite materials with enhanced hydrogen storage capabilities. For next-generation bioinspired robotics, mechanical flexibility and stimuli responsiveness are essential for achieving closed-loop control encompassing perception, response, and regulation55. Looking ahead, the InvDesFlow-AL framework can be extended by constructing multi-objective scoring functions targeting properties such as flexibility, shape-memory behavior, and self-healing capabilities, thereby facilitating the rational design of novel intelligent materials for bioinspired applications. This approach has the potential to significantly accelerate the development of advanced humanoid robots. The ultimate goal of InvDesFlow-AL is to bridge computational discovery with experimental synthesis and industrial application, going beyond theoretical predictions. Leveraging its active learning mechanism, the candidate pool can be dynamically refined to prioritize the synthesis of high-value materials. Moreover, by integrating metrics such as cost evaluation and environmental sustainability, the framework offers robust decision-making support for real-world applications. By continuously expanding the functional boundaries and application depth of InvDesFlow-AL, we anticipate significant advancements in multiple strategic domains, driving transformative breakthroughs in next-generation energy, intelligent manufacturing, and bioengineering.
Methods
InvDesFlow-AL pretrained crystal generation model
Generating target functional materials is a crucial step in the inverse design of materials. However, prior to achieving desired attributes, generative models must ensure that synthesized materials inherently exhibit fundamental crystalline characteristics, including periodicity, symmetry, interatomic interactions, and chemically reasonable stoichiometry. To this end, we propose a pretrained crystal generation model. In the following, we will introduce the data representation, model architecture, and training method required for this model.
In crystalline materials, atoms arrange themselves in a periodic configuration where the fundamental building block is termed the unit cell, denoted as \({\mathcal{M}}=({\boldsymbol{A}},{\boldsymbol{F}},{\boldsymbol{L}})\). This structural unit consists of three primary components: \({\boldsymbol{A}}=[{{\boldsymbol{a}}}_{1},{{\boldsymbol{a}}}_{2},...,{{\boldsymbol{a}}}_{N}]\in {{\mathbb{R}}}^{h\times N}\) encodes the chemical species present in the unit cell, with h representing the dimensionality of atomic feature descriptors. The spatial arrangement of atoms is specified by \({\boldsymbol{F}}=[{{\boldsymbol{f}}}_{1},{{\boldsymbol{f}}}_{2},...,{{\boldsymbol{f}}}_{N}]\in {{\mathbb{R}}}^{3\times N}\), which records their three-dimensional fractional coordinates. The periodicity of the crystal framework is defined through the lattice matrix \({\boldsymbol{L}}=[{{\boldsymbol{l}}}_{1},{{\boldsymbol{l}}}_{2},{{\boldsymbol{l}}}_{3}]\in {{\mathbb{R}}}^{3\times 3}\), whose column vectors establish the basis vectors spanning the crystalline lattice system. This pretrained model adopts the equivariant graph neural network (EGNN) architecture16,47, which ensures translation equivariance by using relative coordinate differences and achieves rotation/reflection equivariance through squared relative distances. Additionally, EGNN maintains the permutation equivariance of graph neural networks, ensuring that permuted input atom orders yield correspondingly permuted outputs. It is worth noting that there are many frameworks for achieving equivariance, such as e3nn56 based on spherical harmonic representations, tensor field networks57, and other high-order tensor product networks. These network architectures can be easily integrated into our active learning-based training. The pretrained model is based on a diffusion generative model. During the generation process, the number of atoms remains unchanged, while atomic types and the lattice matrix undergo noise addition and denoising using the standard enoising diffusion probabilistic model14 framework. Fractional coordinates, on the other hand, are processed using a score-matching-based framework15. The loss function of the pretrained and fine-tuned crystal generation model is given by: \({{\mathcal{L}}}_{{\rm{total}}}={{\mathcal{L}}}_{{\rm{lattice}}}+{{\mathcal{L}}}_{{\rm{atom}}}+{{\mathcal{L}}}_{{\rm{coord}}}\). The detailed training and sampling processes are shown in Algorithm 1 and Algorithm 2. Training details and hyperparameter settings are provided in Supplementary Note B.
Algorithm 1
Training Procedure for InvDesFlow-AL Pretrained Crystal Generation Model 
Algorithm 2
Sampling Procedure for InvDesFlow-AL Pretrained Crystal Generation Model 
Active learning-based materials inverse design
Diversity sampling
The core of diversity sampling lies in constructing a pretraining dataset with broad structural diversity to ensure the generalizability of the generative model. Specifically, we systematically collected stable crystal materials spanning different space groups, chemical compositions, and functional categories (e.g., conductors, semiconductors, insulators, and magnetic materials) to build an inorganic training set across the periodic table (Alex-MP-202 and GNoME datasets1). This strategy explicitly enhances the model’s ability to explore diverse atomic types, lattice symmetries, and functional properties, enabling the unconditional generation of candidate materials with high stability, structural novelty, and chemical uniqueness.
Expected model change
The expected model change strategy enables functional adaptation of the generative model through targeted fine-tuning. Starting from a pretrained model, it first performs preliminary fine-tuning using known crystal structures of target functional materials (e.g., superconductors or low formation energy compounds) to establish a property-guided generative prior. An iterative generate-and-filter loop then follows, where high-value candidate structures—validated via query by committee—are progressively incorporated into the training set for multi-round fine-tuning. This progressive optimization drives the model toward the desired property space while maintaining structural validity, offering a scalable pathway for the inverse design of complex functional materials, such as ambient-pressure high-temperature superconductors.
Query-by-Committee
The query-by-committee strategy enables intelligent screening of generated samples by constructing a multi-model committee system. This approach integrates complementary evaluation models—such as structural relaxation potentials, property prediction neural networks, and first-principles calculation tools—to form a multi-objective decision-making committee. For each generated candidate material, committee members independently assess key metrics within their respective domains of expertise. In the generation of low formation energy materials, to select the most valuable data for iterative fine-tuning of the generative model in active learning, we designed a multi-objective scoring function to evaluate the newly generated crystal structures:
Similarly, in the generation of low Ehull materials, the multi-objective scoring function for selecting the most valuable data is:
in the above three equations, the symbol \({{\mathbb{I}}}_{{\rm{condition}}}\) denotes an indicator function, which takes the value 1 when the specified condition is satisfied, and 0 otherwise. For the generation process of high-temperature superconducting materials, we devised a multi-objective scoring function to assess the newly generated crystal structures to identify the most valuable data for iterative fine-tuning of the generative model during active learning:
The high-Tc priority term directs the model to generate structures within the high-temperature superconducting material space, prioritizing the inclusion of DFT-confirmed superconducting phases. Additionally, it utilizes SuperconGNN predictions of high-Tc superconducting materials as a reference for selection. The force convergence term ensures structural stability, while the synthesizability term balances synthesizability. These conditions together form the criteria for selecting the most valuable data. In the stable structure prediction task, the DPA-2 potential function optimizes the generated structures, and FormEGNN is used to select the crystals with the lowest formation energy. The multi-objective scoring function is given in Eq. (1). Regarding the scoring function design for UHTCs, it should be noted that due to the extremely limited number of available stable UHTC crystal structures (only 14), there is insufficient data to train a reliable property prediction model. Therefore, we did not construct a dedicated multi-objective scoring function.
The proposed QBC strategy in InvDesFlow-AL offers unique advantages compared to established multi-objective optimization techniques in materials discovery, such as Pareto optimization58, BLOX59, and probabilistic target range (PTR)60,61 methods. First, unlike traditional Pareto optimization, which requires explicit objective functions and comprehensive sampling of the Pareto front—often computationally prohibitive in high-dimensional material spaces—our QBC approach embeds domain-specific physical constraints directly into the generation process via scoring functions. For example, indicators such as atomic force convergence \({{\mathbb{I}}}_{{\rm{relaxation}}}\) and thermodynamic stability \({{\mathbb{I}}}_{{E}_{{\rm{hull}}} < 50{\rm{meV}}}\) are used to guide the model toward synthesizable candidates. This enables InvDesFlow-AL to discover over 1.6 million low-Ehull materials, including Li2AuH6 with a superconducting Tc above 140 K. Second, in contrast to BLOX, which emphasizes exploration by selecting out-of-distribution samples, our method integrates both exploration and exploitation. The diversity sampling phase ensures broad chemical space coverage, while the QBC-driven fine-tuning focuses on property-specific optimization. This hybrid mechanism achieves both novelty and functionality, overcoming BLOX’s limitations in goal-directed design. Third, compared to multi-objective PTR, which relies on the probability of satisfying predefined target ranges, QBC offers greater flexibility in handling conflicting or unranked objectives. Instead of static thresholds, InvDesFlow-AL applies a physically-informed, dynamically weighted scoring system (e.g., stability via − Eform and superconductivity via \({T}_{c}^{{\rm{DFT}}}\)), and leverages committee models (e.g., DFT, DPA-2, SuperconGNN) for real-time evaluation. This enables robust discovery under complex conditions, as demonstrated by the identification of Li2AuH6, which exhibits both low Ehull and ultrahigh Tc. Finally, InvDesFlow-AL can be extended by integrating ideas from Pareto and PTR methods to further enhance its committee mechanism. For example, Pareto-guided QBC can prioritize promising regions before fine-tuning, and probabilistic targets (e.g., \({{\mathbb{I}}}_{{T}_{c}\ > \ 40{\rm{K}}}\)) can act as auxiliary indicators in the scoring function. Such integration could improve efficiency in multi-property optimization, especially in data-scarce scenarios like ambient-pressure superconductors.
SuperconGNN is a superconducting transition temperature prediction model we developed based on an equivariant graph neural network architecture. As shown in Fig. 5a, the model consists of a crystal graph input layer, an embedding layer, an encoding layer, and a prediction layer.

a The input layer, Embedding layer, encoding layer, and prediction layer of the crystal graph in SuperconGNN. b Performance comparison of different models in predicting high-temperature superconducting materials. c Accuracy of AI predictions: whether AI can correctly classify materials with superconducting transition temperatures above 5 K, 10 K, ..., up to 60 K into their corresponding intervals.
The crystal structure is represented as a geometric graph \(({\mathcal{V}},{{\mathcal{E}}}_{{r}_{\max }})\), where nodes correspond to atoms within the unit cell. We employ pymatgen to construct a local environment graph by identifying bonding interactions between atoms. The crystallographic information file (CIF) of a structure is then converted into a graph representation that includes atomic types A, bonding connectivity \({\mathcal{E}}\), lattice parameters L, and periodic boundary information. In addition to the bonding-based edges obtained from the local environment analysis, we further construct a geometric graph based on atomic cartesian coordinates by connecting atoms within a cutoff distance of 5 Å. These distance-based connections are subsequently merged with the bonding-based edges to form the final geometric graph representation:
rab denotes the Euclidean distance between atoms a and b, fa represents the node scalar features, and fab corresponds to the edge scalar features of the bond (a, b) if it belongs to \({\mathcal{E}}\), and is 0 otherwise. The node features fa consist of a one-hot encoding of the atomic type combined with global lattice information (α, β, γ, a, b, c). The edge features include a 2D one-hot encoding indicating the presence or absence of a bond, along with the corresponding Gaussian-expanded interatomic distance.
The Encoding layers are based on tensor product layers56,62,63. At each layer, messages are generated for every node pair in the graph by applying tensor products between the current node features and the spherical harmonic representation of the corresponding normalized edge vector. The weights for these tensor products are computed as a function of the edge embeddings and the scalar features of the two connected nodes, where the scalar features of node a are denoted as \({{\bf{h}}}_{a}^{0}\). These messages are then aggregated at each node, and the resulting information is used to update its feature representation.
Here, \({{\mathcal{N}}}_{a}=\{b| (a,b)\in {{\mathcal{E}}}_{{r}_{\max }}\}\) denotes the set of neighboring atoms of atom a, where \({{\mathcal{E}}}_{{r}_{\max }}\) refers to the set of edges within a predefined cutoff radius. Y represents the spherical harmonics up to order ℓ = 2, and BN indicates an equivariant batch normalization layer. All learnable parameters are encapsulated in Ψ, which governs the weighting of tensor products. The resulting node features ha include both scalar and vector representations. Considering that the superconducting transition temperature of a crystal is invariant under SE(3) transformations, the final layer of our model employs an SE(3)-invariant linear layer from e3nn to map the node features of the crystal to an SE(3)-invariant representation. Additionally, since Tc is strictly greater than zero, we apply a ReLU activation function. Finally, the invariant representations of all atoms are aggregated to predict the superconducting transition temperature.
The discovery of high-temperature superconducting materials has long been a central goal in condensed matter physics. Accordingly, we focus on evaluating the model’s performance in the high-Tc regime. The yellow region in Fig. 5b illustrates the scatter plot of predicted values versus DFT-calculated values for materials with Tc > 20 K, as predicted by ALIGNN9, ALIGNN-H35, and our model SuperconGNN. A greater number of points deviating from the diagonal reference line indicates superior model performance. As shown, SuperconGNN successfully predicts all superconducting materials with Tc > 20 K within the highlighted region. The baseline model ALIGNN was trained on conventional superconductor datasets, whereas ALIGNN-H was trained specifically on hydride data. Due to variations in DFT computational settings, theoretical predictions of superconducting transition temperature (Tc) can exhibit discrepancies exceeding 10 K. Moreover, differing considerations of factors such as anisotropy among experts can lead to even larger deviations for the same material, with reported Tc values differing by more than 50 K—for example, Mg2IrH6 has been predicted to exhibit a Tc of both 160 K34 and 77 K33. Therefore, an excessive focus on reducing the AI prediction error (e.g., MAE below 10 K) is not meaningful in the context of discovering high-Tc superconductors. To address this, we propose a novel evaluation criterion, as illustrated in Fig. 5c. Specifically, a prediction is considered correct if the model successfully predicts Tc above a given threshold, which allows us to compute an accuracy score that better reflects the model’s capability in the high-Tc regime. Using this approach, we observe that SuperconGNN accurately predicts materials with Tc > 15 K and Tc > 20 K, significantly outperforming the baseline models. In the regime beyond the McMillan limit, SuperconGNN also achieves high predictive accuracy. Overall, SuperconGNN offers a novel, efficient, and accurate model for the discovery of high-Tc superconductors.
Regarding data partitioning, we follow the same strategy described in ref. 9, which involves splitting the 626 conventional superconductor data points into training, validation, and test sets with a ratio of 0.9:0.05:0.05. Since the specific data identifiers were not released by the authors, we performed a new split using the same proportions. Given that the majority of these data points correspond to materials with superconducting transition temperatures below 40 K, we further augmented the training set with 59 hydride crystal structures reported in ref. 35. We directly adopted the final checkpoint of our model as the screening model. To evaluate its screening capability, we added 12 superconducting candidates discovered by InvDesFlow-AL to the test set and used them to assess model performance in the corresponding figures.
We discuss the differences between InvDesFlow-AL and current reinforcement learning approaches. While reinforcement learning has been extensively applied in domains such as large language models and robotic control, achieving remarkable outcomes like GPT410 and DeepSeek64, methods like reinforcement learning from human feedback (RLHF)65 require proximal policy optimization (PPO)66 and additional reward model training, resulting in high computational complexity and training instability. Our active learning strategy resembles the direct preference optimization (DPO)67,68 in reinforcement learning by fine-tuning models directly with preference data, which offers lower computational overhead and eliminates the need for separate reward modeling. The distinction between InvDesFlow-AL and DPO lies in the former’s elimination of negative sampling, instead directly updating the model towards the distribution of preferred data generation.
The first-principles electronic calculation
As shown in Table 3, this includes the DFT Settings, EPC Calculation, and the corresponding BCS Theory for Li2AuH6. As shown in Fig. 3a previously, InvDesFlow-AL also identified many other superconducting materials, with the corresponding DFT calculation results provided in Supplementary Note A. As shown in Table 4, the detailed configuration parameters for the USPEX-based density functional theory (DFT) structure search are provided, including the evolutionary algorithm settings, relaxation methods, computational resources, and software versions used in the calculations.
Data availability
All the data, code~(https://github.com/xqh19970407/InvDesFlow-AL), and models have been open-sourced. We rely on PyTorch~(https://pytorch.org) for deep model training. We use specialized tools for the Vienna Abinitio Simulation Package~(https://www.vasp.at/).
Code availability
All the data, code (https://github.com/xqh19970407/InvDesFlow-AL), and models will be open-sourced after the paper is published. We rely on PyTorch (https://pytorch.org) for deep model training. We use specialized tools for the Vienna Abinitio Simulation Package (https://www.vasp.at/).
References
Merchant, A. et al. Scaling deep learning for materials discovery. Nature 624, 80–85 (2023).
Zeni, C. et al. A generative model for inorganic materials design. Nature 639, 624–632 (2025).
Xiao, X. et al. Aqueous-based recycling of perovskite photovoltaics. Nature 638, 670–675 (2025).
Sun, H. et al. Signatures of superconductivity near 80 K in a nickelate under high pressure. Nature 621, 493–498 (2023).
Zhou, G. et al. Ambient-pressure superconductivity onset above 40 K in (La,Pr)3Ni2O7 films. Nature 640, 641–646 (2025).
Gao, Z. et al. Shielding Pt/γ-Mo2N by inert nano-overlays enables stable H2 production. Nature 638, 690–696 (2025).
Jiang, H. et al. Two-dimensional Czochralski growth of single-crystal MoS2. Nat. Mater. 24, 188–196 (2025).
Jiao, Y. et al. Implantable batteries for bioelectronics. Acc. Mater. Res. 1, 1–6 (2025).
Choudhary, K. & Garrity, K. Designing high-TC superconductors with BCS-inspired screening, density functional theory, and deep-learning. npj Comput. Mater. 8, 244 (2022).
OpenAI, Achiam, J. & Adler, S. GPT-4 Technical Report. https://arxiv.org/abs/2303.08774 (2024).
Antunes, L. M., Butler, K. T. & Grau-Crespo, R. Crystal structure generation with autoregressive large language modeling. Nat. Commun. 15, 10570 (2024).
Han, X.-Q. et al. AI-driven inverse design of materials: past, present, and future. Chin. Phys. Lett. 42, 027403 (2025).
HAN, J. et al. A survey of geometric graph neural networks: data structures, models and applications. Front. Comput. Sci. https://journal.hep.com.cn/fcs/EN/abstract/article_52426.shtml (2025).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. In Proc. 34th International Conference on Neural Information Processing Systems, NIPS ’20 (Curran Associates Inc., 2020).
Song, Y. et al. Score-based generative modeling through stochastic differential equations. ICLR https://openreview.net/forum?id=PxTIG12RRHS (2021).
Jiao, R. et al. Crystal structure prediction by joint equivariant diffusion on lattices and fractional coordinates. In Proc. Workshop on ”Machine Learning for Materials” ICLR 2023 https://openreview.net/forum?id=VPByphdu24j (2023).
Ye, C.-Y., Weng, H.-M. & Wu, Q.-S. Con-CDVAE: a method for the conditional generation of crystal structures. Comput. Mater. Today 1, 100003 (2024).
Han, X.-Q. et al. InvDesFlow: an AI-driven materials inverse design workflow to explore possible high-temperature superconductors. Chin. Phys. Lett. 42, 047301 (2025).
Xie, T., Fu, X., Ganea, O.-E., Barzilay, R. & Jaakkola, T. S. Crystal diffusion variational autoencoder for periodic material generation. In International Conference on Learning Representations (2021).
Zhao, Y. et al. Physics guided deep learning for generative design of crystal materials with symmetry constraints. npj Comput. Mater. 9, 38 (2023).
Cao, Z.-D., Luo, X.-S., Lv, J. & Wang, L. Space group informed transformer for crystalline materials generation. Sci. Bull. 70, 3522−3533 (2025).
Ye, C. et al. Materials discovery acceleration by using condition generative methodology. https://arxiv.org/abs/2505.00076 (2025).
Li, Z., Liu, S., Ye, B., Srolovitz, D. J. & Wen, T. Active learning for conditional inverse design with crystal generation and foundation atomic models. https://arxiv.org/abs/2502.16984 (2025).
Hu, E. J. et al. LoRA: Low-rank adaptation of large language models. In Proc. International Conference on Learning Representations https://openreview.net/forum?id=nZeVKeeFYf9 (2022).
Houlsby, N. et al. Parameter-efficient transfer learning for NLP. In Chaudhuri, K. & Salakhutdinov, R. (eds.) Proc. 36th International Conference on Machine Learning (eds Chaudhuri, K. & Salakhutdinov, R.), vol. 97, 2790–2799. https://proceedings.mlr.press/v97/houlsby19a.html (PMLR, 2019).
Rebuffi, S.-A., Bilen, H. & Vedaldi, A. Learning multiple visual domains with residual adapters. In Proc. 31st International Conference on Neural Information Processing Systems, NIPS’17, 506–516 (Curran Associates Inc., 2017).
Jain, A. et al. Theory-Driven Data and Tools. In Handbook of Materials Modeling: Methods: Theory and Modeling, 1751–1784. https://doi.org/10.1007/978-3-319-44677-_60 (Springer, 2020).
Zhang, D. et al. DPA-2: a large atomic model as a multi-task learner. npj Comput. Mater. 10, 293 (2024).
Drozdov, A. P., Eremets, M. I., Troyan, I. A., Ksenofontov, V. & Shylin, S. I. Conventional superconductivity at 203 kelvin at high pressures in the sulfur hydride system. Nature 525, 73–76 (2015).
Drozdov, A. P. et al. Superconductivity at 250 K in lanthanum hydride under high pressures. Nature 569, 528–531 (2019).
Somayazulu, M. et al. Evidence for superconductivity above 260 K in lanthanum superhydride at megabar pressures. Phys. Rev. Lett. 122, 027001 (2019).
Zhong, X. et al. Prediction of above-room-temperature superconductivity in lanthanide/actinide extreme superhydrides. J. Am. Chem. Soc. 144, 13394–13400 (2022).
Sanna, A. et al. Prediction of ambient pressure conventional superconductivity above 80 K in hydride compounds. npj Comput. Mater. 10, 44 (2024).
Dolui, K. et al. Feasible route to high-temperature ambient-pressure hydride superconductivity. Phys. Rev. Lett. 132, 166001 (2024).
Cerqueira, T. F. T., Fang, Y.-W., Errea, I., Sanna, A. & Marques, M. A. L. Searching materials space for hydride superconductors at ambient pressure. Adv. Funct. Mater. 34, 2404043 (2024).
Zheng, F. et al. Prediction of ambient pressure superconductivity in cubic ternary hydrides with MH6 octahedra. Mater. Today Phys. 42, 101374 (2024).
Ouyang, Z. et al. High-temperature superconductivity in Li2AuH6 mediated by strong electron-phonon coupling under ambient pressure. Phys. Rev. B 111, L140501 (2025).
Podryabinkin, E. V., Tikhonov, E. V., Shapeev, A. V. & Oganov, A. R. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys. Rev. B 99, 064114 (2019).
Yamashita, T. et al. Cryspy: a crystal structure prediction tool accelerated by machine learning. Sci. Technol. Adv. Mater.: Methods 1, 87–97 (2021).
Lin, P. et al. Equivariant diffusion for crystal structure prediction. In Proc. 41st International Conference on Machine Learning (eds Salakhutdinov, R. et al.), vol. 235, 29890–29913. https://proceedings.mlr.press/v235/lin24b.html (PMLR, 2024).
Wyatt, B. C., Nemani, S. K., Hilmas, G. E., Opila, E. J. & Anasori, B. Ultra-high temperature ceramics for extreme environments. Nat. Rev. Mater. 9, 773–789 (2024).
Jaworska, L. Tantalum diboride obtained by reactive sintering-properties and wear resistance. DEStech Transactions on Environment, Energy and Earth Science (2016).
Zhang, X., Hilmas, G. & Fahrenholtz, W. Synthesis, densification, and mechanical properties of TaB2. Mater. Lett. 62, 4251–4253 (2008).
Katoh, Y., Vasudevamurthy, G., Nozawa, T. & Snead, L. L. Properties of zirconium carbide for nuclear fuel applications. J. Nucl. Mater. 441, 718–742 (2013).
Hu, C. et al. New design for highly durable infrared-reflective coatings. Light Sci. Appl. 7, 17175 (2018).
Schmidt, J. et al. Improving machine-learning models in materials science through large datasets. Mater. Today Phys. 48, 101560 (2024).
Satorras, V. G., Hoogeboom, E. & Welling, M. E(n) Equivariant Graph Neural Networks. In Proc. 38th International Conference on Machine Learning, vol. 139, 9323–9332. https://proceedings.mlr.press/v139/satorras21a.html (PMLR, 2021).
Abramson, J., Adler, J., Dunger, J. & Evans, R. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Li, H. et al. Deep-learning electronic-structure calculation of magnetic superstructures. Nat. Comput. Sci. 3, 321–327 (2023).
Gao, K. et al. The maximum Tc of conventional superconductors at ambient pressure. Nat. Commun. 16, 8253 (2025).
Wang, Y. Application-oriented design of machine learning paradigms for battery science. npj Comput. Mater. 11, 89 (2025).
Zhou, L. et al. Machine learning assisted prediction of cathode materials for zn-ion batteries. Adv. Theory Simul. 4, 2100196 (2021).
Zhou, P. et al. Machine learning in solid-state hydrogen storage materials: challenges and perspectives. Adv. Mater. 37, 2413430 (2025).
Zhang, X. et al. Atomic reconstruction for realizing stable solar-driven reversible hydrogen storage of magnesium hydride. Nat. Commun. 15, 2815 (2024).
Hao, Y. et al. A review of smart materials for the boost of soft actuators, soft sensors, and robotics applications. Chin. J. Mech. Eng. 35, 37 (2022).
Corso, G., Stärk, H., Jing, B., Barzilay, R. & Jaakkola, T. DiffDock: diffusion steps, twists, and turns for molecular docking. In Proc. International Conference on Learning Representations (ICLR) (2023).
Thomas, N. et al. Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds. https://arxiv.org/abs/1802.08219 (2018).
Motoyama, Y. et al. Bayesian optimization package: Physbo. Computer Phys. Commun. 278, 108405 (2022).
Terayama, K. et al. Pushing property limits in materials discovery via boundless objective-free exploration. Chem. Sci. 11, 5959–5968 (2020).
Takahashi, A., Kumagai, Y., Aoki, H., Tamura, R. & Oba, F. Adaptive sampling methods via machine learning for materials screening. Sci. Technol. Adv. Mater.: Methods 2, 55–66 (2022).
Takahashi, A., Terayama, K., Kumagai, Y., Tamura, R. & Oba, F. Fully autonomous materials screening methodology combining first-principles calculations, machine learning and high-performance computing system. Sci. Technol. Adv. Mater.: Methods 3, 2261834 (2023).
Geiger, M. & Smidt, T. e3nn: Euclidean neural networks. https://arxiv.org/abs/2207.09453 (2022).
Jing, B., Corso, G., Chang, J., Barzilay, R. & Jaakkola, T. Torsional diffusion for molecular conformer generation. In Advances in Neural Information Processing Systems (eds Koyejo, S. et al.), vol. 35, 24240–24253 https://proceedings.neurips.cc/paper_files/paper/2022/file/994545b2308bbbbc97e3e687ea9e464f-Paper-Conference.pdf (Curran Associates, Inc., 2022).
Guo, D. et al. DeepSeek-R1 incentivizes reasoning in LLMs through reinforcement learning. Nature 645, 633–638 (2025).
Ouyang, L. et al. Training language models to follow instructions with human feedback. In Proc. 36th International Conference on Neural Information Processing Systems, NIPS’22 (Curran Associates Inc., 2022).
Schulman, J., Wolski, F., Dhariwal, P., Radford, A. & Klimov, O. Proximal policy optimization algorithms. https://arxiv.org/abs/1707.06347 (2017).
Rafailov, R. et al. Direct preference optimization: your language model is secretly a reward model. In Advances in Neural Information Processing Systems (eds Oh, A. et al.), vol. 36, 53728–53741. https://proceedings.neurips.cc/paper_files/paper/2023/file/a85b405ed65c6477a4fe8302b5e06ce7-Paper-Conference.pdf (Curran Associates, Inc., 2023).
Cao, Z. & Wang, L. CrystalFormer-RL: Reinforcement Fine-Tuning for Materials Design. https://arxiv.org/abs/2504.02367 (2025).
Gebauer, N. W., Gastegger, M., Hessmann, S. S., Müller, K.-R. & Schütt, K. T. Inverse design of 3d molecular structures with conditional generative neural networks. Nat. Commun. 13, 1–11 (2022).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phy.: Condens. Matter 21, 395502 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hamann, D. R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2013).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Poncé, S., Margine, E., Verdi, C. & Giustino, F. EPW: electron-phonon coupling, transport and superconducting properties using maximally localized Wannier functions. Comp. Phys. Commun. 209, 116–133 (2016).
Pizzi, G. et al. Wannier90 as a community code: new features and applications. J. Phy. Condens. Matter 32, 165902 (2020).
Acknowledgements
This work was supported by the National Key R&D Program of China (Grants No. 2024YFA1408601) and the National Natural Science Foundation of China (Grant Nos. 12434009, 62476278,12204533). Computational resources were provided by the Physical Laboratory of High Performance Computing at Renmin University of China.
Author information
Authors and Affiliations
Contributions
X.Q.H. and Z.F.G. contributed to the ideation and design of the research and performed the research. X.Q.H., Z.F.G., and Z.Y.L. wrote the original paper. X.Q.H., Z.F.G., H.S., and Z.Y.L. edited the paper. All authors contributed to the research discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, XQ., Guo, PJ., Gao, ZF. et al. InvDesFlow-AL: active learning-based workflow for inverse design of functional materials. npj Comput Mater 11, 364 (2025). https://doi.org/10.1038/s41524-025-01830-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-025-01830-z
This article is cited by
-
Artificial intelligence-driven approaches for materials design and discovery
Nature Materials (2026)
