
Abstract
The representation of atomic configurations for machine learning models has led to numerous sets of descriptors. However, many descriptor sets are incomplete and/or functionally dependent. Incomplete sets cannot faithfully represent atomic environments. Yet complete constructions often suffer from a high degree of functional dependence, where some descriptors are functions of others. These redundant descriptors do not improve discrimination between atomic environments. We employ pattern recognition techniques to remove dependent descriptors to produce the smallest possible set that satisfies completeness. We apply this in two ways: First, we refine an existing description, the atomic cluster expansion. Second, we augment an incomplete construction, yielding a new message-passing neural network architecture that can recognize up to 5-body patterns. This architecture shows strong accuracy on state-of-the-art benchmarks while retaining low computational cost. Our results demonstrate the utility of this strategy to optimize descriptor sets across a range of descriptors and application datasets.
Similar content being viewed by others
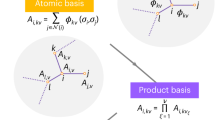
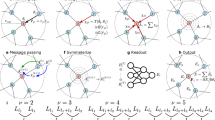
Data availability
The data used in this work is publicly accessible alongside previously published works, which have been referenced in this article accordingly. The ACEpotentials.jl code described in ref. 56 is available at the following URL: https://github.com/ACEsuit/ACEpotentials.jl. The code for HIP-HOP-NN has been incorporated into hippynn, an open-source library for atomistic machine learning, available at https://www.github.com/lanl/hippynn, along with examples training to the methane dataset and the ANI-1x dataset.
References
Botu, V., Batra, R., Chapman, J. & Ramprasad, R. Machine learning force fields: construction, validation, and outlook. J. Phys. Chem. C 121, 511–522 (2017).
Deringer, V. L., Caro, M. A. & Csányi, G. Machine learning interatomic potentials as emerging tools for materials science. Adv. Mater. 31, 1902765 (2019).
Fedik, N. et al. Extending machine learning beyond interatomic potentials for predicting molecular properties. Nat. Rev. Chem. 6, 653–672 (2022).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Kulichenko, M. et al. The rise of neural networks for materials and chemical dynamics. J. Phys. Chem. Lett. 12, 6227–6243 (2021).
Behler, J. Four generations of high-dimensional neural network potentials. Chem. Rev. 121, 10037–10072 (2021).
Musil, F. et al. Physics-inspired structural representations for molecules and materials. Chem. Rev. 121, 9759–9815 (2021).
No, K. T., Chang, B. H. & Kim, S. Y. Description of the potential energy surface of the water dimer with an artificial neural network. Chem. Phys. Lett. 271, 152 (1997).
Chmiela, S. et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 3, e1603015 (2017).
Chmiela, S., Sauceda, H. E., Müller, K.-R. & Tkatchenko, A. Towards exact molecular dynamics simulations with machine-learned force fields. Nat. Commun. 9, 3887 (2018).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Braams, B. J. & Bowman, J. M. Permutationally invariant potential energy surfaces in high dimensionality. Int. Rev. Phys. Chem. 28, 577–606 (2009).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Shapeev, A. V. Moment tensor potentials: a class of systematically improvable interatomic potentials. Multiscale Model. Simul. 14, 1153–1173 (2016).
Smith, J. S., Isayev, O. & Roitberg, A. E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 8, 3192–3203 (2017).
Schütt, K. T., Arbabzadah, F., Chmiela, S., Müller, K. R. & Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 8, 13890 (2017).
Schütt, K. T., Sauceda, H. E., Kindermans, P. J., Tkatchenko, A. & Müller, K. R. SchNet—a deep learning architecture for molecules and materials. J. Chem. Phys. 148, 241722 (2018).
Lubbers, N., Smith, J. S. & Barros, K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 148, 241715 (2018).
Unke, O. T. & Meuwly, M. Physnet: a neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 15, 3678–3693 (2019).
Zubatyuk, R., Smith, J. S., Leszczynski, J. & Isayev, O. Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network. Sci. Adv. 5, eaav6490 (2019).
Lindsey, R. K., Fried, L. E. & Goldman, N. Chimes: a force matched potential with explicit three-body interactions for molten carbon. J. Chem. Theory Comput. 13, 6222–6229 (2017).
Allen, A. E. A., Dusson, G., Ortner, C. & Csányi, G. Atomic permutationally invariant polynomials for fitting molecular force fields. Mach. Learn. Sci. Technol. 2, 025017 (2021).
Pozdnyakov, S. N. et al. Incompleteness of atomic structure representations. Phys. Rev. Lett. 125, 166001 (2020).
Thomas, N. et al. Tensor field networks: rotation- and translation-equivariant neural networks for 3D point clouds. Preprint at https://arxiv.org/abs/1802.08219 (2018).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 14, 579 (2023).
Gasteiger, J., Groß, J. & Günnemann, S. Directional message passing for molecular graphs. In Proc. International Conference on Learning Representations. https://openreview.net/forum?id=B1eWbxStPH (2020).
Wang, J. et al. E(n)-equivariant Cartesian tensor message passing interatomic potential. Nat. Commun. 15, 7607 (2024).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Chigaev, M. et al. Lightweight and effective tensor sensitivity for atomistic neural networks. J. Chem. Phys. 158, 184108 (2023).
Luo, S., Chen, T. & Krishnapriyan, A. S. Enabling efficient equivariant operations in the Fourier basis via Gaunt tensor products. In Proc. Twelfth International Conference on Learning Representations https://openreview.net/forum?id=mhyQXJ6JsK (2024).
Nigam, J., Pozdnyakov, S. & Ceriotti, M. Recursive evaluation and iterative contraction of N-body equivariant features. J. Chem. Phys. 153, 121101 (2020).
Kovács, D. P. et al. Linear atomic cluster expansion force fields for organic molecules: beyond RMSE. J. Chem. Theory Comput. 17, 7696–7711 (2021).
Lysogorskiy, Y. et al. Performant implementation of the atomic cluster expansion (pace) and application to copper and silicon. NPJ Comput. Mater. 7, 97 (2021).
Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 99, 014104 (2019).
Batatia, I. et al. The design space of E(3)-equivariant atom-centered interatomic potentials. Nat. Mach. Intell. 7, 56–67 (2025)
Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csányi, G. Mace: higher order equivariant message passing neural networks for fast and accurate force fields. Adv. Neural Inf. Process. Syst. 35, 11423–11436 (2022).
Frank, J. T., Unke, O. T., Müller, K.-R. & Chmiela, S. A Euclidean transformer for fast and stable machine learned force fields. Nat. Commun. 15, 6539 (2024).
Kabylda, A. et al. Molecular simulations with a pretrained neural network and universal pairwise force fields. J. Am. Chem. 147, 33723–33734 (2025).
Dusson, G. et al. Atomic cluster expansion: completeness, efficiency and stability. J. Comput. Phys. 454, 110946 (2022).
Cheng, B. Cartesian atomic cluster expansion for machine learning interatomic potentials. NPJ Comput. Mater. 10, 157 (2024).
Bochkarev, A., Lysogorskiy, Y. & Drautz, R. Graph atomic cluster expansion for semilocal interactions beyond equivariant message passing. Phys. Rev. X 14, 021036 (2024).
Nigam, J., Pozdnyakov, S. N., Huguenin-Dumittan, K. K. & Ceriotti, M. Completeness of atomic structure representations. APL Mach. Learn. 2, 016110 (2024).
Hornik, K., Stinchcombe, M. & White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 2, 359–366 (1989).
Hu, M.-K. Visual pattern recognition by moment invariants. IRE Trans. Inf. Theory 8, 179–187 (1962).
Dirilten, H. & Newman, T. G. Pattern matching under affine transformations. IEEE Trans. Comput. 26, 314–317 (1977).
Flusser, J. On the independence of rotation moment invariants. Pattern Recognit. 33, 1405–1410 (2000).
Langbein, M. & Hagen, H. A generalization of moment invariants on 2D vector fields to tensor fields of arbitrary order and dimension. In Proc. International Symposium on Visual Computing, 1151–1160 (Springer, 2009).
Flusser, J., Zitova, B. & Suk, T. 2D and 3D Image Analysis by Moments (Wiley, 2016).
Bujack, R., Zhang, X., Suk, T. & Rogers, D. Systematic generation of moment invariant bases for 2d and 3d tensor fields. Pattern Recognit. 123, 108313 (2022).
Jacobi, C. G. J. De determinantibus functionalibus. J. Reine Angew. Math. 1841, 319–359 (1841).
Bujack, R. & Hagen, H. Moment invariants for multi-dimensional data in Modeling, Analysis, and Visualization of Anisotropy, 43–64 (Springer, 2017).
Bujack, R., Shinkle, E., Allen, A., Suk, T. & Lubbers, N. Flexible moment-invariant bases from irreducible tensors. Preprint at https://arxiv.org/abs/2503.21939 (2025).
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Witt, W. C. et al. ACEpotentials.jl: a Julia implementation of the atomic cluster expansion. J. Chem. Phys. 159, 164101 (2023).
Pozdnyakov, S., Willatt, M. & Ceriotti, M. Randomly-displaced methane configurations. Materials Cloud Archive https://doi.org/10.24435/materialscloud:qy-dp (2020).
Montavon, G. et al. Machine learning of molecular electronic properties in chemical compound space. N. J. Phys. 15, 95003 (2013).
Ramakrishnan, R., Dral, P. O., Rupp, M. & von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 140022 (2014).
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018).
Smith, J. S. et al. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 7, 134 (2020).
Kovács, D. P., Batatia, I., Arany, E. S. & Csányi, G. Evaluation of the MACE force field architecture: from medicinal chemistry to materials science. J. Chem. Phys. 159, 044118 (2023).
Simeon, G. & de Fabritiis, G. TensorNet: Cartesian tensor representations for efficient learning of molecular potentials. In Proc. Advances in Neural Information Processing Systems. 36, 37334–37353 (2023).
Kovács, D. P. et al. MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules. J. Am. Chem. Soc. 147, 17598–17611 (2025).
Reiss, T. The revised fundamental theorem of moment invariants. IEEE Trans. Pattern Anal. Mach. Intell. 13, 830–834 (1991).
Uhrin, M. Through the eyes of a descriptor: constructing complete, invertible descriptions of atomic environments. Phys. Rev. B 104, 144110 (2021).
Lo, C. & Don, H. 3-D moment forms: their construction and application to object identification and positioning. IEEE Trans. Pattern Anal. Mach. Intell. 11, 1053–1064 (1989).
Coope, J. A. R., Snider, R. F. & McCourt, F. R. Irreducible Cartesian tensors. J. Chem. Phys. 43, 2269–2275 (1965).
Taylor, J. K. An introduction to graphical tensor notation for mechanistic interpretability. Preprint at https://arxiv.org/abs/2402.01790 (2024).
Suk, T. & Flusser, J. Graph method for generating affine moment invariants. In Proc. 17th International Conference on Pattern Recognition (ICPR 2004), Vol. 2, 192–195 (IEEE, 2004).
Darby, J. P. et al. Tensor-reduced atomic density representations. Phys. Rev. Lett. 131, 028001 (2023).
Wu, Y. & He, K. Group normalization. In Proc. European Conference on Computer Vision (ECCV), 3–19 (2018).
Hagberg, A. A., Schult, D. A. & Swart, P. J. Exploring network structure, dynamics, and function using NetworkX. In Proc. 7th Python in Science Conference (eds Varoquaux, G. et al.) 11–15 (2008).
Ansel, J. et. al. PyTorch 2: Faster Machine Learning Through Dynamic Python Bytecode Transformation and Graph Compilation. In 29th ACM International Conference on Architectural Support for Programming Languages and Operating Systems. 2, 929–947 (2024)
Smith, S. L., Kindermans, P.-J. & Le, Q. V. Don’t decay the learning rate, increase the batch size. In Proc. International Conference on Learning Representations. https://openreview.net/forum?id=B1Yy1BxCZ (2018).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In International Conference on Learning Representations. https://arxiv.org/abs/1412.6980 (2015).
Bergstra, J., Bardenet, R., Bengio, Y. & Kégl, B. Algorithms for hyper-parameter optimization. In Proc. Advances in Neural Information Processing Systems. 24, 2546–2554 (2011).
Liaw, R. et al. Tune: a research platform for distributed model selection and training. In The ICML 2018 AutoML Workshop. https://arxiv.org/abs/1807.05118 (2018).
Bergstra, J., Yamins, D. & Cox, D. Making a science of model search: hyperparameter optimization in hundreds of dimensions for vision architectures. PMLR 28, 115–123 (2013).
Lubbers, N. et al. hippynn. GitHub https://github.com/lanl/hippynn (2025).
Schütt, K., Unke, O. & Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra. In Proc. International Conference on Machine Learning, 9377–9388 (PMLR, 2021).
Thölke, P. & Fabritiis, G. D. Equivariant transformers for neural network based molecular potentials. In Proc. International Conference on Learning Representations. https://openreview.net/forum?id=zNHzqZ9wrRB (2022).
Gasteiger, J., Giri, S., Margraf, J. T. & Günnemann, S. Fast and uncertainty-aware directional message passing for non-equilibrium molecules. In Machine Learning for Molecules Workshop at NeurIPS 2020. https://arxiv.org/abs/2011.14115 (2020).
Jo, J., Kwak, B., Lee, B. & Yoon, S. Flexible dual-branched message-passing neural network for a molecular property prediction. ACS Omega 7, 4234–4244 (2022).
Zaverkin, V., Holzmüller, D., Bonfirraro, L. & Kästner, J. Transfer learning for chemically accurate interatomic neural network potentials. Phys. Chem. Chem. Phys. 25, 5383–5396 (2023).
Haghighatlari, M. et al. Newtonnet: a newtonian message passing network for deep learning of interatomic potentials and forces. Digit. Discov. 1, 333–343 (2022).
Acknowledgements
The authors are thankful for productive conversations with Kipton Barros, Cristina Garcia Cardona, Ying Wai Li, Yen Ting Lin, Sakib Matin, and Pieter Swart. We would like to thank Kei Davis for his feedback on this manuscript. We gratefully acknowledge the support of the U.S. Department of Energy through the LANL Laboratory Directed Research and Development Program under project numbers 20250145ER, 20230293ER for this work. This research used resources provided by the LANL Institutional Computing (IC) Program and the CCS-7 Darwin cluster at LANL. LANL is operated by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy (Contract No. 89233218NCA000001).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.E.A.A., E.S., R.B., N.L.; Formal analysis: A.E.A.A., E.S., N.L.; Investigation: A.E.A.A., E.S., N.L.; Methodology: A.E.A.A., E.S., R.B., N.L.; Software: A.E.A.A., E.S., R.B., N.L.; Visualization: A.E.A.A., E.S., N.L.; Writing—original draft: A.E.A.A., E.S., R.B., N.L.; Writing—review and editing: A.E.A.A., E.S., R.B., N.L.; Supervision: R.B., N.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Allen, A.E.A., Shinkle, E., Bujack, R. et al. Optimal invariant sets for atomistic machine learning. npj Comput Mater (2026). https://doi.org/10.1038/s41524-025-01948-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-025-01948-0
