
Abstract
Ferroelectric perovskites with giant spontaneous polarization have extensive applications in electronic devices, energy conversion, sensor and so on. However, the rapid discovery of new perovskites with giant polarization remains an open challenge especially when thousands of candidates are treated. Here, combining machine learning (ML) and first-principles calculations, we successfully predict 8 perovskites with giant polarization from 2021 different possible compounds, among which seven candidates have never been reported before. These perovskites have large c/a ratio and giant polarization compared to the reported ferroelectric perovskites, and room temperature stability. Among them, the polarization of SnFeO3 with G-AFM magnetic ordering is as high as 138.63 µC/cm2. The non-magnetic SrPbO3 and magnetic EuSnO3 not only exhibit giant polarization, but also possess band gaps close to the ideal value for photovoltaic applications, showing great potential in the field of ferroelectric photovoltaics. Besides, polarity and metallicity coexist in SnFeO3 and CaTaO3, which are suggested to have potential applications in fields such as spintronics and superconductivity. This work thus provides an effective strategy for discovering new functional materials.
Similar content being viewed by others
Introduction
In recent years, functional materials have been particularly important in industrial development. Ferroelectric materials have gained extensive attention in the scientific community due to their spontaneous polarization characteristic, which exhibit nonzero electric dipole moment even without an external electric field1,2,3,4,5,6. The most common ferroelectric materials currently include perovskite, layered bismuth oxide, and boracite7. Tetragonal perovskite materials with a high c/a ratio often have giant spontaneous polarization, which is referred to as supertetragonal (ST) perovskites8. Supertetragonal perovskites, such as BiFeO3, PbTiO3, BiCoO3, and PbVO3, show great potential in the fields of photodetectors, ferroelectric nanocapacitors, and piezoelectric oscillators due to their spontaneous polarization8,9,10,11,12,13,14. However, these compounds are rare and generally require extreme synthesis conditions. For example, supertetragonal lead titanate (PbTiO3) with an out–of–plane–to–in–plane lattice parameter ratio (c/a) of 1.238 and giant polarization of 236.3 µC/cm2 is obtained in epitaxial composite thin films of tetragonal PbTiO3 by Zhang and Chen via interphase strain8. Stress is also a key factor in regulating the c/a ratio and polarization intensity14,15,16,17,18,19. Perovskites such as BiScO3, BiMnO3, BiFeO3 and BiCoO3 exhibit an increase in the c/a ratio with increasing negative pressure20. Conversely, under positive pressure, the c/a ratio gradually decreases as the pressure increases. Ion implantation can produce defect-driven true supertetragonal phase with single-domain-state in ferroelectric BiFeO3 thin films with c/a values of ~1.316. Therefore, it is necessary to explore novel supertetragonal perovskites that can exist stably under ambient conditions and exhibit giant polarization for applications in ferroelectric devices.
Due to the complexity and diversity of perovskites structures, Density Functional Theory-based high-throughput calculations are too expensive and time-consuming, not to mention experiments. Fortunately, the launch of the Materials Genome Initiative (MGI) has brought exciting prospects for addressing this dilemma, which aims to accelerate the research and application process of new materials21,22,23,24,25,26,27. In recent years, with the maturity of material synthesis and characterization, as well as data storage technology, the field of materials has gradually entered the big data era28. In the past, the MGI has given rise to a multitude of high-throughput computational platforms and databases such as the Materials Project (MP)29, Open Quantum Materials Database30, Harvard Clean Energy Project31, Electronic Structure Project (ESP)32, Computational Materials Repository33, and Novel Materials Discovery34. Information like atomic positions, space groups, lattice constants, and symmetries can be found in these databases, providing material engineers with ready access to the properties of known materials. With the rapid development of artificial intelligence technology, the machine learning (ML) technique has made significant advancements in materials design. For example, using ML, 15 promising lithium-ion battery solid electrolyte materials were screened out by Jalem et al.35. Kaufmann et al. successfully predict the entropy-forming ability of 70 new compositions by ML model, and several of these predictions were validated by density functional theory calculations (DFT) and experimental synthesis36. Combining ML and DFT, six orthorhombic lead-free hybrid organic-inorganic perovskites (HOIPs) with proper bandgap for solar cells and room temperature thermal stability were screened out by Lu et al. from 5158 unexplored HOIPs37. By bypassing complex quantum mechanics, ML technology can not only greatly accelerate materials design with high accuracy, but also reveal the structure-property relationships within materials from large material datasets.
Here, we develop a target-driven method to discover supertetragonal perovskites ABX3 (A = monovalent or divalent cation; B = divalent or tetravalent cation; X = oxygen or halide anion) based on ML technique and DFT calculations. We first train our ML model from 95 reported perovskites, and predict the lattice parameter ratio c/a of 2021 different possible perovskites. A close structure-property relationship mapping the c/a of perovskites is concurrently excavated from ML data, in which the ranges of tolerance factor (t), octahedral factor (µ), electronegativity of A-site element (EA), electronegativity of X-site element (Ex), first ionization of A-site element (FA), and ionic radius of X-site element (RIX) are defined for supertetragonal perovskites. After further screening, eight stable supertetragonal perovskites are selected as promising ferroelectric materials with giant polarization. These newly screened supertetragonal perovskites provide a valuable theoretical reference for related experimental investigation.
Results
Design framework
Our material screening process consists of 6 parts, i.e., Input dataset, Feature engineering, ML model construction, Prediction dataset, Materials screening, and DFT validation, which are schematically illustrated in Fig. 1. As the foundation of ML, the first step is to prepare a train-test dataset. The train-test dataset consists of input data and output data, which are the descriptor and the target performance, respectively. To remove redundant features and improve the accuracy of the ML model, feature engineering is performed to retain the most important features for the target data. Then, the ML modeling is constructed by using classification algorithms. Next, the trained ML model is applied to a prediction dataset, where the predicted material is screened based on certain constraints. Finally, DFT calculations and Ab initio molecular dynamics (AIMD) simulations are used to study the electronic properties and stability of the candidate perovskites selected from the ML simulation.

The entire workflow consists of six parts: input dataset preparation, feature engineering, ML model construction, prediction, materials screening and DFT validation.
Dataset
The input data for this study, containing 95 perovskites, is obtained from Materials Project (MP)29. The selected 95 perovskite compounds belong to the family of perovskites of chemical formula ABX3. Generally, it is considered that if the lattice constant ratio of c/a is greater than 1.2, then the perovskite can be defined as a supertetragonal perovskite material8. Accordingly, the value of 1.2 is used as a threshold to divide the c/a values in the dataset. Figure 2a visualizes the division of the dataset, where blue and green dots represent the c/a values less than and greater than 1.2, respectively. For the classification modeling, the c/a values less than 1.2 are considered as 0, and greater or equal to 1.2 as 1, to conduct a binary classification. As shown in Fig. 2b, the numbers 0 and 1 are 76 and 19, respectively. Data analysis and visualization can also reveal the hidden trends and periodicity in the perovskite dataset. The impact on the c/a ratio is evaluated based on several important descriptors, as shown in Fig. 2c–f. It can be observed from the figures that with the increase of Ex, the value of c/a tends to increase; it tends to exhibit a larger c/a when t is between 0.7 and 1.0; the lower the boiling point of the monatomic element at the A-site, the larger the c/a value tends to be; the smaller the ionic radius of the element at the X-site, the larger the c/a value tends to be.

a Visualization of train/test dataset, where green and blue dots represent c/a less and greater than 1.2, respectively; b Number of samples in binary classification, where 0 and 1 represent c/a less and greater than 1.2, respectively; c The relationship between c/a and electronegativity of X atom; d The relationship between and c/a and tolerance factor; e The relationship between c/a and boiling point of single substance for B-site element; f The relationship between c/a and Ion radius of X-site element. The dotted box represents the most appropriate range for each feature in the train/test dataset.
Indeed, there are many choices for the A-, B-, and X-site elements in perovskites. As illustrated in Fig. 3a, when X is F, Cl, Br, or I, the A should be a monovalent cation, and B is a divalent cation; when X is O, the A should be a divalent cation, and B is a tetravalent cation. Consequently, for X = F, Cl, Br, or I, we consider 5 monovalent cations and 43 divalent cations from the periodic table for the A-site and B-site, respectively. In the case of X = O, we consider 43 divalent cations for the A-site and 27 tetravalent cations for the B-site. Thus, we obtain 2021 different possible perovskite compounds, which are the potential candidates we will explore in this study.

Both train/test and prediction datasets incorporate the materials modified via the element mixing strategy and the structure-property relationship between c/a values and features in prediction datasets a Virtual space for ABX3 perovskites, b Electronegativity of X atom Ex, c Tolerance factor t, d A Boiling point of single substance BPA and e Ion radius of X-site element RIX. The green and blue dots represent c/a greater and less than 1.2.
Feature engineering
For any ML method directed at predefining and specifying material properties, it typically relies on a certain number of features (descriptors)24,37. These features not only uniquely define each material in the input dataset, but also are related to the physical and chemical properties that are expected. While there might be many factors influencing the target properties of the material, the number of features must be reasonable. The best strategy is to choose features capable of completely representing the material properties, and the number of such features should be far less than the number of materials in the input dataset, to avoid the curse of dimensionality38. In this work, 41 initial features are selected (see Supplementary Table S1 for a complete list of features), including ionic radius, tolerance factor, and electronegativity to collectively describe the chemical space of the perovskites. Pearson correlation coefficients are calculated in the feature engineering step to eliminate the redundant features, which are depicted in the heat map for each feature pair (see Fig. S1). The heat map demonstrates that a significant number of raw features are highly correlated with each other, and some of them should be eliminated. The features with the Pearson correlation coefficients >0.9 or <−0.9 are removed, such that only the appropriate one in the feature pair remains for the next round. Consequently, 10 features are selected from the raw features based on the Pearson correlation coefficients (Fig. 4a). The new feature set contains structural features (tolerance factor t, octahedral factor µ) as well as the elemental properties of A, B and X site ions, including electronegativity of A-site element EA, first ionization energy of A-site element FIA, density of the simple substance corresponding to A-site element DA, boiling point of the simple substance corresponding to the A-site element BPA, boiling point of the simple substance corresponding to the B-site element BPB, electronegativity of the X-site element EX, ionic radius of the A-site element RIA and ionic radius of the X-site element RIX. It turns out that the descriptors most correlated with c/a are EX, t, BPA, and RIX, as indicated by the feature importance ranking shown in Fig. 4b.

a The heat map of the Pearson correlation coefficient matrix among the 10 selected features and target c/a values. b The 10 selected features are ranked according to their Pearson correlations with the target c/a values. c Training model for a random forest classifier. d Test model for random forest classifier. e Training model for Extra Trees classifier and f Test model for Extra Trees classifier.
Machine learning models
An appropriate ML algorithm is important for ML modeling. In this work, two ML algorithms, i.e., Random Forest classifier and Extra Trees classifier, are compared, with 70% (66 samples) of the original dataset used as the training set and 30% (29 samples) as the test set. Figures 4c, d represent training and test set models established by using a Random Forest classifier, respectively. As shown in Table S2, this classification algorithm achieves a model accuracy of 0.924 on the training set and 0.897 on the test set. However, it is noted that this model still predicts 5 samples originally labeled 1 as type 0 in the training set and predicts 3 samples that should have been type 1 as type 0 in the test set. Due to the limited number of type 1 samples in the dataset, although the predictions of the model for type 0 samples are relatively accurate and the overall accuracy is acceptable, there are still significant errors in predicting type 1 samples, resulting in a recall of 0.643 in the training set and only 0.40 in the test set. These results show that the Random Forest classifier model is not suitable for our prediction goals to some extent. Therefore, another ML model is established by using the Extra Trees classifier. The obtained training and test set models are presented in Fig. 4e, f, respectively. It can be observed that this classification algorithm achieves an accuracy of 1 in both the training set and test set, where the real type and predicted type are the same, suggesting that all predictions are correct. Moreover, the consistently high accuracy observed in cross-validation (as shown in Fig. S2) further mitigates concerns that the enhanced performance of the model may arise from overfitting. Therefore, the Extra Trees classifier model can be used as a predictive model for further analysis.
Materials screening and first-principles evaluation
We predict the c/a ratio of perovskite materials in the predicted space of 2021 candidates via the trained ML model. As shown in Fig. 5, to screen out structurally stable supertetragonal perovskite materials, more screening is required. First, 130 perovskites are screened out from the total 2021 perovskites with ML predicted c/a ratio as type 1. Based on the stability of the structure37, 109 samples with a tolerance factor between 0.78 and 1 are selected, among which 67 samples with an octahedral factor between 0.4 and 0.7 are further selected. As compared with the 95 input samples from the previous training and test sets included in the 2021 total samples, there are 11 duplicates of perovskite types with c/a ratios greater than 1.2 in the output samples. Consequently, 56 new perovskite materials are selected in total, all of which possess ideal c/a ratios and stable structures. Considering that oxides are relatively rich in earth, as well as cheap and stable32,39,40, perovskite oxides are retained for further investigations. It should be noted that the reason of including halides in the dataset lies in their contribution to enhancing the diversity of the training and test sets, which in turn facilitates the construction of a more accurate and robust ML model.

The screening is based on four criteria: whether c/a exceeds 1.2, whether the structure is theoretically stable, whether it is new and the species of X-site element.
Ground state properties of selected supertetragonal perovskites
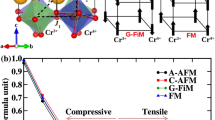
After further first-principles calculations validation, there are 11 perovskites that stand out first with ordered structure, including three non-magnetic perovskites, i.e., CaSnO3, SrPbO3, CaWO3, and eight magnetic perovskites, i.e., EuPbO3, EuSnO3, BiVO3, SnFeO3, SrFeO3, CaTaO3, SrTbO3, and CaTbO3. Since magnetic perovskites may exhibit different magnetic ordering states, it is necessary to perform first-principles calculations on different magnetic ordering states for one material to find the lowest energy state (See Table S3 for details). Figure 6b illustrates four different magnetic orderings, i.e., ferromagnetic (FM), A-type antiferromagnetic (A-AFM), C-type antiferromagnetic (C-AFM), and G-type antiferromagnetic (G-AFM), where red and black arrows represent spin up and spin down ordering of magnetic atoms, respectively. Figure 6c illustrates the energy differences between various AFM magnetic states and FM states, as well as the c/a ratios for eight magnetic perovskites. According to Fig. 6c, for magnetic perovskites SrFeO3, SrTbO3, and CaTbO3, the c/a ratios are all less than 1.2. These three perovskites, thus, are excluded from further consideration. Besides, for G-AFM and A-AFM EuPbO3, FM and A-AFM EuSnO3, FM and A-AFM BiVO3, as well as C-AFM and G-AFM SnFeO3, they not only exhibit the lowest energy but also have c/a ratios greater than 1.2. Hence, they are considered as ideal candidates for supertetragonal perovskites. Although the CaTaO3 with FM and A-AFM magnetic orderings do not possess the lowest energy, but their c/a ratios are greater than 1.2, indicative of its potential as supertetragonal perovskites. Obviously, the magnetic orderings of these perovskites have nonnegligible impacts on the lattice parameters and ground state energy of the material, highlighting the importance of magnetism as a factor that researchers cannot ignore. To the end, 8 perovskites are finally screened out, including three non-magnetic perovskites, i.e., CaSnO3, SrPbO3, CaWO3, and five magnetic perovskites, i.e., G-AFM and A-AFM EuPbO3, FM and A-AFM EuSnO3, G-AFM and C-AFM SnFeO3, FM and A-AFM CaTaO3, and FM and A-AFM BiVO3 (see Fig. 6a for detail structures).

a optimized geometrical structures of 8 selected supertetragonal perovskites, b illustration of different magnetic orderings where red and black arrows represent spin up and spin down, c energy differences between various magnetic orderings and the FM ordering, and c/a values for magnetic perovskites with different magnetic orderings. The value inside the parentheses is the c/a ratio.
Thermal and chemical stabilities of eight selected supertetragonal perovskites
Based on the optimized geometrical structures, AIMD simulations are further carried out to evaluate the thermal stability of selected supertetragonal perovskites. As shown in Fig. 7, although the energies of SrPbO3, CaWO3, and EuSnO3 exhibit relatively large fluctuations, all the screened supertetragonal materials keep an ordered crystal structure within a 5000 fs process at a temperature of 300 K. To clarify the origin of these fluctuations, we present the geometrical structures of all screened materials at their minimum and maximum energy in Fig. S3. For SrPbO3, CaWO3, and EuSnO3, while the temporary structural fluctuations observed at the maxima may suggest local instabilities, such deviations are expected under finite-temperature molecular dynamics simulations and do not necessarily undermine the overall thermodynamic stability of the phase. The recovery to the supertetragonal configuration at the energy minima further indicates that the polar distortion is robust against thermal perturbations, supporting the feasibility of stabilizing these phases experimentally under ambient conditions. The other five materials exhibit only small energy fluctuations throughout the entire AIMD simulation process and retain a well-preserved supertetragonal phase at both the maximum and minimum energy states. Therefore, it can be concluded that the supertetragonal materials screened in this work can remain stable at room temperature.

a CaSnO3, b SrPbO3, c CaWO3, d EuPbO3, e EuSnO3 f SnFeO3 g CaTaO3 and h BiVO3. The AIMD simulations are performed at room temperature (300 K) by using the Nosé-Hoover method. Geometrical structures of each compound before and after AIMD simulations are presented. Min and max represent the minimum and maximum energy points, respectively.
In addition to the thermal stability analysis, we have also calculated the formation energy to evaluate the chemical stability of screened materials. Table S4 and Table S5 present the chemical potentials of constituent elements and formation energies of the eight screened supertetragonal materials, respectively. It is shown that all formation energies are negative, ranging from −1.2 to −2.7 eV/atom. These results are close to the formation energies of CaTiO3 and BaTiO3 (~ −3.5 eV/atom) that have been reported experimentally41, suggesting that the screened materials are chemically stable and have the potential to be synthesized experimentally.
Ferroelectric properties of eight selected supertetragonal perovskites
Subsequently, ferroelectric properties of these perovskites are investigated. Considering that the ability to switch polarization under an electric field is the key to define a ferroelectric material42,43, here we perform nudged elastic band (NEB) calculations to evaluate the polarization reversal pathways and energy barriers of the screened materials (see Fig. S4). The calculated energy barriers for the eight compounds lie in the range of 0.175–0.720 eV/f.u., comparable with the values of ferroelectric CuCrS2 (0.23 eV/f.u.)44 and BiFeO3 (0.43 eV/f.u.)45. Although the supertetragonal phases of CaWO3, EuSnO3, and CaTaO3 are predicted to be metastable, they still exhibit inversion-symmetry breaking and maintain distinct polar distortions. Obviously, the screened materials possess reversible polarization, indicative of their potential to be ferroelectrics. In addition, the exact values of polarization are calculated and summarized in Table 1. For non-magnetic CaSnO3, SrPbO3, and CaWO3, the polarizations are predicted to be 103.248, 99.86, and 117.438 μC/cm2, respectively. As for magnetic perovskites, it is found that magnetic ordering has different influences in different compounds. For EuPbO3 and EuSnO3, small differences exist in the polarization between different magnetic states, since the polarizations of 105.186 and 95.255 μC/cm2 for their A-AFM state are only 7.091 and 4.259 μC/cm2 larger than those of G-AFM and FM states, respectively. As for SnFeO3, the influence of magnetic ordering on the polarization is larger than the cases of EuPbO3 and EuSnO3, as indicated by the difference of 12.614 μC/cm2 between G-AFM and C-AFM states. In the case of CaTaO3 and BiVO3, the magnetic ordering has almost no effect on polarization, which only shows the difference of 1.53 and 0.467 μC/cm2 between FM and A-AFM states, respectively. Obviously, all the perovskites exhibit giant polarization, varying from 90.996 to 138.627 µC/cm2. As compared with the traditional ferroelectric material BiFeO3 with spontaneous polarization of 150 µC/cm2 driven by epitaxial strain46 and defect engineering47, the PbTiO3 with polarization of 75 μC/cm2 with high Curie temperature and high thermoelectric coefficient48,49, the BiCoO3 with polarization of 126 µC/cm2 predicted by DFT within the local spin-density approximation50 and the PbVO3 with polarization above 100 µC/cm2 under ambient and high pressure14, the supertetragonal perovskites that are screened out in this work are stable at room temperature and can be obtained without any external conditions, suggesting that they are all potential excellent ferroelectric materials. Notably, the polarization of BiVO3 has been predicted to be 128 µC/cm2 by Abbassi et al. employing DFT calculation with the generalized gradient approximation (GGA) and modified Becke Johnson approximation51, which agrees well with our calculated value of ~125 µC/cm2, further validating the robust predictive performance of our ML model and the reproducibility of our polarization results.
The intrinsic spontaneous polarization, which is widely recognized as the most important feature of a ferroelectric perovskite, depends on its structural information, such as axial ratio c/a or off-center ion displacements52,53,54. To explore the origin of the different polarization of eight perovskite oxides, we further analyze the relationship between polarization and structural parameters of the predicted materials (see Table S6 for details). Figure 8a presents the relationship between ΔB-O and the c/a ratio. It can be seen that the ΔB-O is linearly correlated with c/a to a certain degree, since the A-site atom has more space for displacement than the B-site atom due to the fact that A is located between oxygen octahedra, while B is inside the octahedra54. Consequently, as the ΔB-O increases, the A-site atom will displace much more than the B-site atom to keep the structure stable, resulting in an increase in c/a. Figures 8b, c illustrate the relationships between the ΔB-O and polarization as well as between the c/a and polarization, respectively. Obviously, there is no strong correlation between ΔB-O and polarization, as well as between c/a ratio and polarization in different supertetragonal materials. This is different from the case of PbTiO354, for which polarization shows a positive correlation with c/a ratio. The presented results suggest that this mechanism apply only to the same material, but not to different kinds of materials. Furthermore, we conduct a detailed analysis of the born effective charges and volumes of eight supertetragonal perovskites. As shown in Fig. 8d, the polarization shows a strong linear negative correlation with the volume, i.e., the smaller the volume, the larger the polarization. This suggests that the polarization of the system is affected by its size, which is mainly because a reduced unit cell volume enhances the dipole density per unit area, thus giving rise to the relatively large polarization. Theoretically, Zhang et al. tuned the structure of supertetragonal PbTiO3 via strain to modulate the polarization strength; Tariq et al. changed the unit cell volume by varying the film thickness of ATiO3 (A = Ba and Pb) and applying strain engineering, and found that the polarization strength is affected54,55. These investigations demonstrate that the change in volume has a significant impact on the polarization strength, agreeing well with our calculations. Besides, it is found that there is no relationship between polarization and born effective charge of the B-site atom (Z*B) as well as the O atom (Z*O), as shown in Fig. S10. However, Fig. 8e, f shows that the polarization is almost linearly correlated with the Born effective charge of the A-site atom (Z*A) and O1 atom (Z*O1). This finding agrees well with the results of feature engineering shown in Fig. 4b, suggesting that the type of A-site element has a significant impact on the polarization of supertetragonal perovskites.

The relationships between a ΔB-O and c/a ratio; b ΔB-O and polarization; c c/a ratio and polarization; d volume of unit cell and polarization; e born effective charge of A-site atom (Z*A) and polarization; f born effective charge of O1 (Z*O1) and polarization of 8 screened supertetragonal perovskites. In (a), dO and dB represent the displacement of the B-site atom and O atom along the c axis and ΔB-O = |dB-dO | , respectively. Since O has two different sites, the O at the top of the octahedra is defined as O1.
To gain insight into the mechanisms underlying the giant c/a and polarization of these supertetragonal perovskites, the electron localization function (ELF)56 on the (100) plane of representative perovskites is further calculated and shown in Fig. 9. The ELF values can be ranged from 0 to 1. If it is close to 0, it means that the cation-anion bond is of ideal ionic character; if it is close to 1, it represents that the cation-anion bond is close to ideal covalent nature57. The ELF for A-AFM EuPbO3 and FM BiVO3, which are characterized by the largest and smallest c/a ratios among eight screened supertetragonal perovskites, is illustrated in Fig. 9a, b, respectively. It is evident that the charge density of the A-site atom Eu is significantly higher than that of Bi, which results in a stronger interaction between Eu and O compared to the interaction between Bi and O58. Thus, the displacement of O towards Eu is larger than that of Bi, leading to the fact that ΔPb-O is larger than ΔV-O. Since the c/a ratio has a strong positive correlation with ΔB-O, as shown in Fig. 8b, the c/a ratio of A-AFM EuPbO3 is larger. For the C-AFM SnFeO3 and FM EuSnO3 with the largest and smallest polarization in eight screened supertetragonal perovskites, the ELF is illustrated in Fig. 9c, d, respectively. One can see from the figures that the density of electron gas surrounding the A-site atom Sn is significantly lower than that of Eu. For cations, fewer localized electrons typically exhibit greater positive oxidation states, which corresponds to a larger Born effective charge59. This analysis, in conjunction with the insights from Fig. 8d–e, accounts for the observed larger polarization in C-AFM SnFeO3.

a, b ELF and 2D image for A-AFM EuPbO3 and FM BiVO3 with the biggest and smallest c/a ratio, and c, d for C-AFM SnFeO3 and FM EuSnO3 with the biggest and smallest polarization in 8 screened supertetragonal perovskites, respectively.
Electronic structures of eight selected supertetragonal perovskites
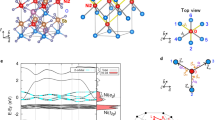
Furthermore, the electronic structures of eight selected supertetragonal perovskites are studied (see Figs. S6 and S7 for more details). The PBE functional under GGA approximation is employed first. As shown in Figs. S8 and S9, only CaSnO3 has a bandgap value of 1.21 eV and the other seven perovskites show metallic characteristics. Considering that the GGA/PBE functional generally underestimates the band gap of materials, the Heyd–Scuseria–Ernzerhof (HSE) screened hybrid functional is further employed, which is known to be more successful than standard DFT in predicting the bandgaps of a wide range of materials60,61,62. The specific values of the bandgaps are presented in Table 1. As illustrated in Fig. 10a–c, three non-magnetic supertetragonal perovskites exhibit insulating or semiconducting behavior. The band structures of CaSnO3 and SrPbO3 show indirect character with bandgap values of 2.99 eV and 1.14 eV, respectively, while CaWO3 is of a direct bandgap about 0.44 eV. Among five magnetic supertetragonal perovskites, the SnFeO3, CaTaO3, and FM BiVO3 exhibit no bandgap, indicative of metallic characteristics, while the EuPbO3, EuSnO3, and A-AFM BiVO3 are of semiconducting character (Fig. 10d–h). It is noted that the non-magnetic SrPbO3 and magnetic EuSnO3 not only exhibit giant polarization, but also possess band gaps close to the ideal value of 1.2 eV for photovoltaic applications, showing great potential in the field of ferroelectric photovoltaics63,64,65. Besides, in SnFeO3 and CaTaO3, polarity and metallicity coexist in the same phase, i.e., SnFeO3 and CaTaO3 are polar metals in nature. Therefore, these two compounds may have wide implications in the fields of topology, ferroelectricity, magnetoelectricity, spintronics, and superconductivity66,67,68,69.

a CaSnO3, b SrPbO3, c CaWO3, d EuPbO3 with G-AFM magnetic ordering, e EuSnO3 with FM magnetic ordering, f SnFeO3 with C-AFM magnetic ordering, g CaTaO3 with FM magnetic ordering and h BiVO3 with FM magnetic ordering. The red and black lines represent spin-up and spin-down states, respectively.
As shown in Fig. 11, for non-magnetic supertetragonal perovskites (Fig. 11a–c), the valence band maxima (VBM) are partly contributed by the O 2p orbitals and some orbitals from the metal atoms at the B site, while the conduction band minima (CBM) are mainly contributed by the orbitals of the B site atom. For EuPbO3 with G-AFM magnetic ordering and EuSnO3 with FM magnetic ordering (Fig. 11d, e), the VBM is partly contributed by the Eu 4 f orbital, and the CBM is mainly contributed by O 2p orbitals. For SnFeO3 with C-AFM magnetic ordering (Fig. 11f), CaTaO3 with FM magnetic ordering (Fig. 11g), and BiVO3 with FM ordering (Fig. 11h), they have no bandgap, and the electrons distributed on the Fermi level are primarily contributed by the d orbitals of metal atoms at the B site.

a CaSnO3, b SrPbO3, c CaWO3, d EuPbO3 with G-AFM magnetic ordering, e EuSnO3 with FM magnetic ordering, f SnFeO3 with C-AFM magnetic ordering, g CaTaO3 with FM magnetic ordering and h BiVO3 with FM magnetic ordering. The solid and dash lines represent spin-up and spin-down states, respectively.
Physical insights between descriptors and structures
To further understand the structure-property relationships in the eight predicted materials, we analyze descriptors with physical significance. It is easy to find that the descriptor of electronegativity is important for the c/a ratio from Fig. 4b. Electronegativity describes the ability of an atom or ion to attract electrons in a chemical bond70. Here, the electronegativity of the A-site element is denoted as EA, which ranges from 0.79 to 2.54 (see source data). Figure 4b shows that EA is negatively correlated with c/a, i.e., a smaller EA corresponds to a larger c/a. As shown in Table 2, the screened eight perovskites have EA values close to 1, with the highest being 2.02, which are within the lower end of the range. The electronegativity of element X (EX) is positively correlated with c/a and has a value range of 2.66–3.98 (see source data). The EX of oxygen is 3.44, which lies towards the higher end of the range. One can see from Fig. 4b that the value of c/a is positively correlated with the electronegativity of the X-site element and negatively correlated with the electronegativity of the A-site element. This is because X-site ions tend to gain electrons, while A-site ions tend to lose electrons. The stronger the electronegativity of the X-site element, the easier it is to attract the A-site elements. Conversely, the lower the electronegativity of the A-site element, the easier it is to be attracted. This leads to a structural offset, resulting in an increasing trend in the c/a ratio.
FA represents the first ionization energy of the A-site element, and it shows a negative correlation with the c/a ratio (see Fig. 4b). The predicted FA ranges from 3.89 to 10.49 KJ/mol (see source data). Most of the selected eight perovskites have FA values around 5–6, which are towards the lower end of the range. The first ionization energy is the amount of energy required to remove the outermost electron from an atom71. An atom with a lower first ionization energy is more easily attracted by an ion with a negative charge. Therefore, the smaller the first ionization energy of the A element, the more easily it is attracted by the X-site element, leading to structural deviation and an increasing trend in c/a ratio.
RIA refers to the radius of the A-site ion, with a predicted spatial value range of 43–167 pm (see source data). As shown in Fig. 4b, there is a positive correlation between RIA and the c/a ratio. The majority of the eight selected perovskites have RIA around 110 pm, which is on the larger side of the range. On the other hand, the Ionic radius of element X (RIX) is negatively correlated with the c/a value (see Fig. 4b), with a predicted spatial range of 133–220 pm (see source data). The RIX of oxygen is 140 pm, which is on the smaller side of the range. The ionic radius of an element is determined by the number of charges contained in the ion and the changes in the electron configuration72. The smaller the nuclear charge of an A-site ion, the larger its radius will be, and the smaller the nuclear charge, the more easily the ion can be attracted by X-site ions. The same analysis applies to X-site ions as well, leading to a structural shift and an increasing trend in c/a ratio.
The structure-property relationships between c/a values and features in prediction datasets are shown in Fig. 3b–e. When EX and t are larger, c/a tends to be greater than 1.2; whereas when BPA and RIX are smaller, c/a tends to be less than 1.2. The descriptor values and their importance rankings corresponding to the selected perovskites are aligned, from which we can get several guiding suggestions for subsequent predictions of supertetragonal perovskites with large c/a ratios, i.e., the elements on the A-site should have a smaller electronegativity, a lower first ionization energy, a lower density, a lower boiling point, and a larger ionic radius. Besides, the boiling point of the B-site atom should be high, the ionic radius associated with the X-site element should be small, and its electronegativity should be large. This provides guidance for accelerating the discovery of supertetragonal materials with giant polarization, as well as for other functional materials.
Discussions
Combining ML technology and DFT calculations, we have developed a highly efficient method for discovering supertetragonal perovskites with giant polarization. Through screening 2021 perovskite materials, we successfully identify eight stable supertetragonal perovskites, i.e., CaSnO3, SrPbO3, CaWO3, EuPbO3, EuSnO3, SnFeO3, CaTaO3, and BiVO3, among which seven candidates have not been reported before. The eight supertetragonal perovskites selected by ML exhibit significantly giant polarization, varying from 90.996 to 138.627 μC/cm2. The further NEB calculations show that these materials possess switchable polarization under an external field and are promising ferroelectrics. Unlike other normal ferroelectric perovskites, which need extreme synthesis conditions, they are thermally stable under ambient condition and no external conditions are needed, thereby leading to a substantial reduction in material development costs. Furthermore, these selected supertetragonal perovskites demonstrate superior theoretical stability and hold promise for practical production applications. Among them, SrPbO3 and EuSnO3 possess an ideal bandgap of around 1.2 eV and giant polarization, which are thus proposed to be potential ferroelectric photovoltaic candidates. Meanwhile, SnFeO3 and CaTaO3 are predicted to be polar metals, which have potential applications in the fields of topology, spintronics, and superconductivity. These results underscore the potential of our research approach in facilitating the design of novel functional materials for practical application. More importantly, we have established a clear structure-property relationship that offers valuable insight for the discovery of supertetragonal materials with giant polarization. Our finding highlights the crucial impact of various factors on the c/a ratio of tetragonal perovskites, such as the electronegativity of the X and A-site elements, tolerance factor, ionic radius of the X-site element, and boiling point of the single substance for the B-site element, providing guidance for the design of perovskites with enhanced polarization.
Methods
Density functional theory calculation
All the DFT calculations are performed using the Vienna Ab Initio Simulation Package73,74,75. The interaction between ions and electrons is described using the projector augmented wave76 method, and the exchange-correlation effects between electrons are treated using the functional of Perdew–Burke–Ernzerhof (PBE)77 under the generalized gradient approximation (GGA)78. The kinetic-energy cutoff is set to be 600 eV and a 9 × 9 × 7 Gamma k-point mesh is employed. The convergence criterion for total energies is 1 × 10−5 eV. The Gaussian smearing (ISMEAR = 0) and SIGMA = 0.05 (corresponding to a smearing temperature of approximately 580 K) are applied. For magnetic perovskites, it is necessary to consider different magnetic orderings, and the calculations are performed with a 2 × 2 × 2 supercell consisting of 40 atoms and 4 × 4 × 3 Gamma k-point mesh. The electronic structures are calculated by hybrid DFT in the framework of HSE60 based on the relaxed structures. AIMD simulations are performed at room temperature by using the Nosé-Hoover method79,80. The polarization switching pathway is determined using the NEB method81. The Born effective charges are calculated using density functional perturbation theory82. It should be noted that the Born effective charges are calculated under the adiabatic approximation. In particular, the polar metals investigated in our work are fundamentally different from traditional metals (see Fig. S11 for details). In polar metals, the electrons around atoms are localized, which leads to a large electron linear response under perturbation, resulting in finite Born effective charges. For the investigated systems in this work, there are several atoms with large atomic numbers, such as Eu or Pb. To investigate if the spin-orbit coupling (SOC) effect is necessary, we select EuPbO3 as a representative candidate and calculate its geometrical structure and band structure, considering the SOC effect. Table S7 compares the lattice parameters, and Fig. S5 compares the band structure of EuPbO3 obtained by the PBE and PBE + SOC methods. It can be seen that consideration of the SOC effect has a slight effect on the geometrical structure and electronic structure. Therefore, the SOC effect is not considered in this work.
Machine learning model construction
The Random Forest classifier algorithm83 is adopted to build the ML models, and the hyperparameters are: max depth = 4, random state = 0, n jobs = 2. Besides, the Extra Trees classifier algorithm84 is also adopted to build the ML models, and the hyperparameters are: n estimators = 10, max depth = None, min samples split = 2, random state = 0.
In this work, we handle a binary classification task, so the confusion matrix is a 2×2 matrix that reports the number of true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) as follows85,86:
The performance indicator can be used to compare different classifiers. We usually take the model with the best performance and the common indicators are as follows:
Formation energy
The formation energy of ABO3 perovskites, (\({E}_{f}^{AB{O}_{3}}\)) is calculated according to Equation:
where \({E}_{t}^{AB{O}_{3}}\) is the total energy of the perovskites, μA/B is the chemical potential of element A/B in its bulk phase, and μO is 1/2 chemical potential in oxygen gas phase87,88.
Spontaneous polarization
An excellent ferroelectric material should have a large spontaneous polarization. The polarization can be estimated by:
where \(\Delta {\mu }_{j\beta }\) is the displacement of ions j in the Cartesian direction β, \({Z}_{j\alpha \beta }^{\ast }\) is the Born effective charge tensor, e is the charge of an electron, and \(\varOmega\) is the cell volume89,90.
Data availability
The source data and codes used in this study are available at the https://github.com/Winger0203/ML_ABX3_ST_perovskites.
References
Xu, Y. Ferroelectric materials and their applications. (Elsevier, 2013).
Martin, L. W. & Rappe, A. M. Thin-film ferroelectric materials and their applications. Nat. Rev. Mater. 2, 1–14 (2016).
Mikolajick, T. et al. Next generation ferroelectric materials for semiconductor process integration and their applications. J. Appl. Phys. 129, 100901 (2021).
Yang, Y. et al. An ultrathin ferroelectric perovskite oxide layer for high-performance hole transport material free carbon based halide perovskite solar cells. Adv. Funct. Mater. 29, 1806506 (2019).
Zhang, C. C. et al. Polarized ferroelectric polymers for high-performance perovskite solar cells. Adv. Mater. 31, 1902222 (2019).
Han, X., Ji, Y. & Yang, Y. Ferroelectric photovoltaic materials and devices. Adv. Funct. Mater. 32, 2109625 (2022).
Rabe, K. M., Ahn, C. H. & Triscone, J.-M. Physics of ferroelectrics: a modern perspective. Vol. 105 (Springer Science & Business Media, 2007).
Zhang, L. et al. Giant polarization in super-tetragonal thin films through interphase strain. Science 361, 494–497 (2018).
Zhang, L. et al. Controllable ferromagnetism in super-tetragonal PbTiO3 through strain engineering. Nano Lett. 20, 881–886 (2019).
Li, M. et al. Microstructure-dependent thermal stability of super-tetragonal nanocomposite films through in situ TEM/EELS study. ACS Appl. Mater. Interfaces 14, 52316–52323 (2022).
Lachheb, M. et al. Surface and bulk ferroelectric phase transition in super-tetragonal BiFeO3 thin films. Phys. Rev. Mater. 5, 024410 (2021).
Fan, Z. et al. Stable ferroelectric perovskite structure with giant axial ratio and polarization in epitaxial BiFe0.6Ga0.4O3 thin films. ACS Appl. Mater. Interfaces 7, 2648–2653 (2015).
Shpanchenko, R. V. et al. Synthesis, structure, and properties of new perovskite PbVO3. Chem. Mat. 16, 3267–3273 (2004).
Belik, A. A., Azuma, M., Saito, T., Shimakawa, Y. & Takano, M. Crystallographic features and tetragonal phase stability of PbVO3, a new member of PbTiO3 family. Chem. Mat. 17, 269–273 (2005).
Belik, A. A. et al. Neutron powder diffraction study on the crystal and magnetic structures of BiCoO3. Chem. Mat. 18, 798–803 (2006).
Tinte, S., Rabe, K. M. & Vanderbilt, D. Anomalous enhancement of tetragonality in PbTiO3 induced by negative pressure. Phys. Rev. B 68, 144105 (2003).
Damodaran, A. R., Breckenfeld, E., Chen, Z., Lee, S. & Martin, L. W. Enhancement of ferroelectric Curie temperature in BaTiO3 films via strain-induced defect dipole alignment. Adv. Mater. 26, 6341–6347 (2014).
Wang, J. et al. Negative-pressure-induced enhancement in a freestanding ferroelectric. Nat. Mater. 14, 985–990 (2015).
Kvasov, A. et al. Piezoelectric enhancement under negative pressure. Nat. Commun. 7, 12136 (2016).
Singh, A., Singh, V. N., Canadell, E., Íñiguez, J. & Diéguez, O. Polymorphism in Bi-based perovskite oxides: a first-principles study. Phys. Rev. Mater. 2, 104417 (2018).
Le, T., Epa, V. C., Burden, F. R. & Winkler, D. A. Quantitative structure–property relationship modeling of diverse materials properties. Chem. Rev. 112, 2889–2919 (2012).
Tabor, D. P. et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat. Rev. Mater. 3, 5–20 (2018).
Balachandran, P. V., Kowalski, B., Sehirlioglu, A. & Lookman, T. Experimental search for high-temperature ferroelectric perovskites guided by two-step machine learning. Nat. Commun. 9, 1668 (2018).
Hu, W., Zhang, L. & Pan, Z. Designing two-dimensional halide perovskites based on high-throughput calculations and machine learning. ACS Appl. Mater. Interfaces 14, 21596–21604 (2022).
Xu, G. et al. Machine learning-accelerated discovery and design of electrode materials and electrolytes for lithium ion batteries. Energy Storage Mater. 72, 103710 (2024).
Chen, J. et al. Accelerating discovery of next-generation power electronics materials via high-throughput ab initio screening. npj Comput. Mater. 11, 249 (2025).
Yin, P. et al. Machine-learning-accelerated design of high-performance platinum intermetallic nanoparticle fuel cell catalysts. Nat. Commun. 15, 415 (2024).
Korolev, V., Mitrofanov, A., Eliseev, A. & Tkachenko, V. Machine-learning-assisted search for functional materials over extended chemical space. Mater. Horiz. 7, 2710–2718 (2020).
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65, 1501–1509 (2013).
Curtarolo, S. et al. AFLOWLIB. ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Ortiz, C., Eriksson, O. & Klintenberg, M. Data mining and accelerated electronic structure theory as a tool in the search for new functional materials. Comput. Mater. Sci. 44, 1042–1049 (2009).
Landis, D. D. et al. The computational materials repository. Comput. Sci. Eng. 14, 51–57 (2012).
Draxl, C. & Scheffler, M. NOMAD: the FAIR concept for big data-driven materials science. MRS Bull. 43, 676–682 (2018).
Jalem, R., Nakayama, M. & Kasuga, T. An efficient rule-based screening approach for discovering fast lithium ion conductors using density functional theory and artificial neural networks. J. Mater. Chem. A 2, 720–734 (2014).
Kaufmann, K. et al. Discovery of high-entropy ceramics via machine learning. npj Comput. Mater. 6, 42 (2020).
Lu, S. et al. Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning. Nat. Commun. 9, 3405 (2018).
Nasrabadi, N. M. Pattern recognition and machine learning. J. Electron. Imaging 16, 049901 (2007).
Moure, C. & Peña, O. Recent advances in perovskites: processing and properties. Prog. Solid State Chem. 43, 123–148 (2015).
Hwang, J. et al. Perovskites in catalysis and electrocatalysis. Science 358, 751–756 (2017).
Horton, M. K. et al. Accelerated data-driven materials science with the Materials Project. Nat. Mater. 24, 1–11 (2025).
Mu, X. et al. Polarization switching of HfO2 ferroelectric in bulk and electrode/ferroelectric/electrode heterostructure. npj Comput. Mater. 11, 126 (2025).
Xu, R. et al. Ferroelectric polarization reversal via successive ferroelastic transitions. Nat. Mater. 14, 79–86 (2015).
Zhong, T., Li, X., Wu, M. & Liu, J.-M. Room-temperature multiferroicity and diversified magnetoelectric couplings in 2D materials. Natl. Sci. Rev. 7, 373–380 (2020).
Ravindran, P., Vidya, R., Kjekshus, A., Fjellvåg, H. & Eriksson, O. Theoretical investigation of magnetoelectric behavior in BiFeO3. Phys. Rev. B 74, 224412 (2006).
Zhang, J. et al. Microscopic origin of the giant ferroelectric polarization in tetragonal-like BiFeO3. Phys. Rev. Lett. 107, 147602 (2011).
Chen, C. et al. Controllable defect driven symmetry change and domain structure evolution in BiFeO3 with enhanced tetragonality. Nanoscale 11, 8110–8118 (2019).
Tuttle, B., Payne, D. & Mukherjee, J. Ferroelectric materials for dielectric power conversion. Ferroelectrics 27, 219–222 (1980).
Ali, H. E. H., Ricote, J., Calzada, M., Bretos, I. & Jiménez, R. The influence of the crystallization temperature on the reliability of PbTiO3 thin films prepared by chemical solution deposition. J. Eur. Ceram. Soc. 37, 1449–1458 (2017).
Uratani, Y., Shishidou, T., Ishii, F. & Oguchi, T. First-principles predictions of giant electric polarization. Jpn. J. Appl. Phys. 44, 7130 (2005).
Abbassi, A., Zaari, H., Azahaf, C., Ez-Zahraouy, H. & Benyoussef, A. Spontaneous polarization and magnetic investigation of BiXO3 (X= Co, Mn, Fe, V, Zn): first-principle study. J. Supercond. Nov. Magn. 29, 487–491 (2016).
Ederer, C. & Spaldin, N. A. Effect of epitaxial strain on the spontaneous polarization of thin film ferroelectrics. Phys. Rev. Lett. 95, 257601 (2005).
Qi, T., Grinberg, I. & Rappe, A. M. Correlations between tetragonality, polarization, and ionic displacement in PbTiO3-derived ferroelectric perovskite solid solutions. Phys. Rev. B 82, 134113 (2010).
Zhang, F. et al. Correlations between polarization and structural information of supertetragonal PbTiO3. Phys. Rev. B 105, 024106 (2022).
Tariq, A. & Nazir, S. Tunable ferroelectric polarization of the bulk and free standing ATiO3 (A = Ba and Pb) thin films via unit-cell thicknesses and strain engineering. AIP Adv. 7, https://doi.org/10.1063/1.5009038 (2017).
Savin, A., Nesper, R., Wengert, S. & Fässler, T. F. ELF: the electron localization function. Angew. Chem. Int. Ed. 36, 1808–1832 (1997).
Liu, J. et al. Electronic structure and anion engineering for perovskite oxysulfide BaTi (O, S)3. J. Vac. Sci. Technol. A 40, 012801 (2022).
Li, M. et al. Supertetragonal BaZrS3: a promising perovskite sulphide with giant ferroelectricity and low band gap. Sci. China-Phys. Mech. Astron. 67, 1–8 (2024).
Xie, L. & Zhu, J. The electronic structures, born effective charges, and interatomic force constants in BaMO3 (M= Ti, Zr, Hf, Sn): a comparative first-principles study. J. Am. Ceram. Soc. 95, 3597–3604 (2012).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Deák, P., Aradi, B., Frauenheim, T., Janzén, E. & Gali, A. Accurate defect levels obtained from the HSE06 range-separated hybrid functional. Phys. Rev. B 81, 153203 (2010).
Deák, P., Aradi, B. & Frauenheim, T. Polaronic effects in TiO2 calculated by the HSE06 hybrid functional: dopant passivation by carrier self-trapping. Phys. Rev. B 83, 155207 (2011).
Matsuo, H., Noguchi, Y. & Miyayama, M. Gap-state engineering of visible-light-active ferroelectrics for photovoltaic applications. Nat. Commun. 8, 207 (2017).
Yang, S. et al. Above-bandgap voltages from ferroelectric photovoltaic devices. Nat. Nanotechnol. 5, 143–147 (2010).
Paillard, C. et al. Photovoltaics with ferroelectrics: current status and beyond. Adv. Mater. 28, 5153–5168 (2016).
Bhowal, S. & Spaldin, N. A. Polar metals: principles and prospects. Ann. Rev. Mater. Res. 53, 53–79 (2023).
Cao, Y. et al. Artificial two-dimensional polar metal at room temperature. Nat. Commun. 9, 1547 (2018).
Zhang, J. et al. A correlated ferromagnetic polar metal by design. Nat. Mater. 23, 1–8 (2024).
Oh, Y. S., Wang, L., Lee, H., Choi, W. S. & Kim, T. H. Polar perturbations in functional oxide heterostructures. Adv. Funct. Mater. 33, 2302261 (2023).
Iczkowski, R. P. & Margrave, J. L. Electronegativity. J. Am. Chem. Soc. 83, 3547–3551 (1961).
Lang, P. F. & Smith, B. C. Ionization energies of atoms and atomic ions. J. Chem. Educ. 80, 938 (2003).
Rahm, M., Hoffmann, R. & Ashcroft, N. Atomic and ionic radii of elements 1–96. Chem. Eur. J. 22, 14625–14632 (2016).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
White, J. & Bird, D. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations. Phys. Rev. B 50, 4954 (1994).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695 (1985).
Sheppard, D., Xiao, P., Chemelewski, W., Johnson, D. D. & Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 136, 074103 (2012).
Baroni, S., De Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515 (2001).
Pal, M. Random forest classifier for remote sensing classification. Int. J. Remote Sens. 26, 217–222 (2005).
Sharaff, A. & Gupta, H. in Advances in Computer Communication and Computational Sciences: Proc. IC4S. 189–197 (Springer, 2017).
Caelen, O. A Bayesian interpretation of the confusion matrix. Ann. Math. Artif. Intell. 81, 429–450 (2017).
Deng, X., Liu, Q., Deng, Y. & Mahadevan, S. An improved method to construct basic probability assignment based on the confusion matrix for classification problem. Inf. Sci. 340, 250–261 (2016).
Emery, A. A. & Wolverton, C. High-throughput DFT calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci. Data 4, 1–10 (2017).
Guo, X. et al. Tackling the activity and selectivity challenges of electrocatalysts toward the nitrogen reduction reaction via atomically dispersed biatom catalysts. J. Am. Chem. Soc. 142, 5709–5721 (2020).
Neaton, J., Ederer, C., Waghmare, U., Spaldin, N. & Rabe, K. First-principles study of spontaneous polarization in multiferroic BiFeO3. Phys. Rev. B 71, 014113 (2005).
Zhang, S. et al. Band-gap reduction in (BiCrO3)m/(BiFeO3)n superlattices: designing low-band-gap ferroelectrics. Phys. Rev. Appl. 10, 044004 (2018).
Acknowledgements
H.Y. Xiao was supported by the Joint Funds of the National Natural Science Foundation of China (Grant No. U1930120). L. Qiao acknowledges the support by National Natural Science Foundation of China (Grant No. 52072059). The theoretical calculations are performed using the supercomputer resources at TianHe-1 located at National Supercomputer Center in Tianjin.
Author information
Authors and Affiliations
Contributions
H.X. conceptualized the project. H.X. and L.Q. supervised the project. W.H. conducted the DFT calculations, data set preparation, and training of the machine learning model. W.H., Z.W., M.L., S.F., H.Q., X.L., and X.Z. analyzed the data and interpreted the results. W.H. drafted the manuscript. All authors reviewed and edited the manuscript and have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, W., Wu, Z., Li, M. et al. Accelerated discovery of supertetragonal perovskites with giant polarization via machine learning. npj Comput Mater 12, 103 (2026). https://doi.org/10.1038/s41524-026-01970-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-026-01970-w
