
Abstract
The high prevalence of comorbid mental disorders (CMDs) in patients with inflammatory bowel disease (IBD) is well-documented. This study delves into the intricate CMD-IBD relationship through comprehensive analyses using human variants, gut microbiome, and anxiety/depression estimates from a cohort of 507 IBD patients and 75 controls. Notably, patients with IBD, especially those with CMD, exhibited lower diversity than controls. We identified 106 differentially abundant taxa (DATs) in IBD patients compared to controls and 21 DATs distinguishing CMD-affected from CMD-free IBD patients. Microbial IBD-risk scores, reflecting an individual’s microbial burden for IBD, revealed a significant enrichment of IBD-risk signatures in CMD-affected patients compared to CMD-free patients. Additionally, there was an IBD-risk variant potentially regulating the abundance of an IBD/CMD-associated DAT, suggesting an interplay between IBD-risk variants and dysbiosis in CMD. Our investigation underscores the pivotal role of IBD-associated gut dysbiosis in predisposing IBD patients to CMD, partially through genetic variant-mediated mechanisms.
Similar content being viewed by others


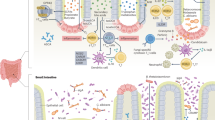
Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), causes relapsing and remitting inflammation of the gastrointestinal tract, which has a major impact on the quality of life1,2. Although the etiology of IBD is largely unknown, several risk factors have been identified including genetic variants, immunological cytokines, gut dysbiosis, and environmental triggers2,3,4,5.
Several epidemiological studies have reported higher frequencies of comorbid mental disorders (CMDs) in patients with IBD. The pooled prevalence of anxiety and depression in patients with inactive IBD has been estimated to be 31.1% and 25.2%, respectively6. These prevalence estimates were much higher than those in the general population (3.8% for anxiety and 3.4% for depression)7.
Previous reports have provided evidence supporting the association between mental disorders and IBD8. For example, IBD patients with high disease activity exhibited a higher prevalence of anxiety and depression (57.6% and 38.9%, respectively)6. Similarly, IBD patients with CMD showed worse prognosis and a high risk of bowel surgery especially in patients with CD9,10. A population-based analysis revealed that depression prior to IBD diagnosis was associated with an increased likelihood of IBD development, especially if they experienced gastrointestinal symptoms before the diagnosis of depression11. In addition, treatment with antidepressants was reported to reduce risk of IBD12. These results indicate that mental disorders do not occur simply due to IBD-related confounding effects on mood (e.g., disease activity, fatigue, flare, frustration, high medical expense).
The accumulated evidence of molecular signatures in both IBD and mental disorders suggested the presence of shared biomarkers in both diseases, suggesting common pathological pathways potentially involved in a bidirectional gut-brain axis13,14,15. A gut microbiome study that compared the microbial composition between IBD patients with CMD and those without CMD emphasized abundance changes in a large number of individual taxa according to CMD16. Our recent observations indicate that fecal microbiota transplantation from patients suffering from both IBD and depression induces severe IBD-like colitis and depression-like behaviors in mice17. Considering the gut dysbiosis in patients with IBD and the reported gut-brain axis in mental disorders18, it could be worth investigating the correlation of IBD-specific and CMD-specific changes of microbial abundance in a single cohort.
With respect to human genetic factors, a weak but significant genetic correlation was observed between depression and IBD in terms of disease-specific effect sizes of genome-wide variants19. Some of the IBD-risk variants in stress regulator genes were enriched in open chromatin regions interacted with promoters in a brain tissue19. However, the individual genetic variants that explain the pleiotropic mechanism involved in two diseases remain unclear.
In this study, we investigated human genetic variants, gut microbiota, and their interplay that confer risk of CMD in patients with IBD, by profiling the microbial diversity, abundance level, polygenic risk scores (PRS), and microbial quantitative trait loci (mbQTL) in a single-center cohort with a homogenous genetic background.
Results
Summary of study design
This study aimed to comprehensively understand the IBD-risk human genetic factors and gut microbial dysbiosis associated with the frequent development of comorbid anxiety and depression in patients with IBD. To this end, we recruited 507 patients with IBD (UC, n = 290; CD, n = 217) and 75 healthy controls to generate genome-wide variant data from peripheral blood, 16S rRNA amplicon sequencing data from fecal samples, and/or HADS data (Fig. 1). There were no significant differences in age, body mass index (BMI), cigarette smoking, and alcohol consumption between IBD cases and healthy controls, except sex ratio (Table 1). HADS information was obtained from a subset of patients with IBD (n = 225; 95 UC + 130 CD). A total of 29 patients with IBD (12.9%; n = 29; 13 UC + 16 CD) had significant anxiety and/or depression with HADS ≥ 11. Non-psychological characteristics were not significantly different between CMD-free and CMD-affected patients with IBD (Table 2).

The schematic diagram illustrates the sample groups in the Kyung Hee University Hospital cohort according to data availability and analysis methods.
To dissect the potential causal relationships between IBD-risk factors and mental disorders (depicted in Fig. 2), we first tested whether IBD-risk microbial signatures, including microbial diversity and taxon-level abundance, were more predominant in IBD patients with CMD than in those without CMD. Second, we tested whether the effects of genome-wide variants on IBD were correlated with those on mental disorders in the general population and whether PRSs for IBD or mental disorders were higher in IBD patients with CMD than in those without CMD. Finally, we tested whether IBD-risk genetic variants mediate changes in the abundance of microbiota that confer risk of CMD.

The figure shows a simplified representation of the research scope and strategies to investigate the contributions of IBD-risk factors to the development of CMD using a large Korean case-control cohort with gut microbial and human genetic data and clinical outcomes.
Enhanced IBD-specific gut dysbiosis in IBD patients with CMD
Fecal microbiota composition was examined using 16S rRNA amplicon sequencing for both IBD patients and healthy controls (Fig. 3). We estimated gut microbial diversity and genus-level abundance in Korean patients with IBD and healthy controls. Shannon alpha diversity index was significantly lower in patients with IBD (mean alpha diversity \(\bar{\alpha }\) = 3.98) than healthy controls (\(\bar{\alpha }\) = 4.85) at the genus level (P = 3.93 ×10−15; Fig. 4a), showing similar differences in alpha diversity between each IBD subtype and the control group (P = 2.90 ×10−11 for UC and P = 6.26 ×10−19 for CD, respectively; Supplementary Figure 1). The stratification of patients with IBD according to CMD status revealed that IBD patients with CMD had even less alpha diversity (\(\bar{\alpha }\) = 3.59) in the gut microbiota compared to controls (P = 6.69 ×10−9) or CMD-free IBD patients (\(\bar{\alpha }\) = 3.93, P = 0.047; Fig. 4a). Alpha diversity in the anxiety group (\(\bar{\alpha }\) = 3.39) was relatively lower than that in the depression group (\(\bar{\alpha }\) = 3.75); however, the difference was not statistically significant (P = 0.189).

Each stacked bar indicates the relative abundance of microbial families in each sample from (a) UC, (b) CD, and (c) heathy control groups. Families with a median relative abundance of less than 1% are depicted in gray. Sample groups are divided based on the availability of HADS data and the affected status of CMD.

Alpha diversity was assessed based on the Shannon index (a), Simpson index (b), richness (c), and Faith’s PD index (d) at the genus level. P-values for statistical differences in alpha diversity were calculated using t-test between healthy controls and patients with IBD and between CMD-free and CMD-affected IBD patients. e Beta diversity was measured based on the Bray–Curtis dissimilarity of individuals’ genus abundance and visualized in a PCoA plot according to the top two principal components (PCs). The centroid of each cluster is marked by a diamond. The top PC that explained the largest proportion (20.22%) of variance in microbial composition in the study subjects is shown in a boxplot according to the affected status. The P-value from a Jonckheere-Terpstra test (PJT) was calculated to test for a significant trend toward higher PC1 values in the order of healthy controls, CMD-free IBD patients, and CMD-affected IBD patients.
Consistently, other diversity indices were significantly lower in patients with IBD than healthy controls (P = 4.32 ×10−13 for Simpson index, P = 9.82 ×10−13 for richness, P = 1.06 ×10−11 for Faith’ phylogenetic diversity (PD) index), especially in CMD-affected IBD patients than CMD-free IBD patients (P = 0.0604 for Simpson index, P = 7.77 ×10−3 for richness, P = 0.0104 for Faith’s PD index; Fig. 4). In the same analysis for each subtype (UC and CD) of IBD, we observed a similar trend of decreased alpha diversity in patients with CMD but this did not achieve statistical significance, probably due to the decreased sample sizes in a stratification analysis (Supplementary Fig. 1).
We then evaluated beta diversity using the individual-to-individual Bray–Curtis dissimilarity from the gut microbiome composition at the genus level. There was a significant dissimilarity of the gut microbiome composition between patients with IBD and healthy controls (P ≤ 0.001; Fig. 4e; see also Supplementary Fig. 2). In a principal coordinate analysis (PCoA) for the Bray–Curtis dissimilarity, the top principal component (PC1) explained 20.22% of the variance in the dissimilarity of gut microbiome composition among individuals and were significantly higher in patients with IBD than healthy controls (P = 8.07 ×10−20; Fig. 4e). In addition, there was a significant trend toward higher PC1 values in the order of healthy controls, CMD-free IBD patients, and CMD-affected IBD patients (P = 2.24 ×10−14 in a Jonckheere-Terpstra test among the three groups, P = 0.0345 between CMD-free and CMD-affected IBD patient groups). Thus, it is tempting to suggest that a part of the microbiota that largely contributes to the top PCs may explain the risk of both CMD and IBD.
Given the alpha and beta diversity analysis results, we hypothesized that a part of the IBD-associated gut microbiota is further involved in the development of CMD, which may explain the high prevalence of depression and anxiety in patients with IBD. In this context, we aimed to investigate the taxa whose relative abundance changes were associated with the risk of both IBD and CMD with the same direction of abundance changes in these two diseases.
We identified 106 differentially abundant taxa (DATs), including nine phyla, 10 classes, 15 orders, 28 families, and 44 genera, between patients with IBD and healthy controls at a false discovery rate threshold of 5% (Supplementary Data 1; Fig. 5a). Of the IBD-associated DATs with complete taxonomic classification information (n = 75), a total of 20 identified taxa (26.7%) have already been reported to exhibit differential abundance in previous IBD gut microbial studies at various taxonomic levels20,21, showing the consistent directions of abundance changes in patients with IBD; they included the phyla Firmicutes, Proteobacteria, and Tenericutes; the classes Gammaproteobacteria and Mollicutes; the order Clostridiales; the families Christensenellaceae, Enterococcaceae, and Mogibacteriaceae; the genera Actinomyces, Barnesiella, Bifidobacterium, Bilophila, Butyricimonas, Campylobacter, Coprococcus, Eggerthella, Enterococcus, Paraprevotella, and Pediococcus. In addition, a large number of IBD-associated DATs were newly identified by taking advantage of using a large-scale IBD cohort with an underrepresented ethnicity (Supplementary Data 1). Considering the ancestral heterogeneity and the significant number of replicated associations of known IBD-associated taxa, novel IBD-associated DATs are likely to be useful biomarkers and therapeutic targets, especially in East Asian patients with IBD.

Log2-fold changes (LFCs) in microbial abundance were estimated at the various taxonomic levels in a differential abundance analysis between healthy controls and patients with IBD (a), and between CMD-free and CMD-affected IBD patients (b). c The LFCs were then plotted in volcano plots based on their PFDR values. The scatter plot represents phenotype-specific LFCs for genera associated with either IBD or CMD (PFDR < 0.05). Data points highlighted in red with error bars of 95% confidence intervals indicate the genera whose abundance changes were significant in both phenotypes. Their names were provided with the initial of the lowest taxonomic rank that was classified. N.S. not significant.
In parallel, we identified the significantly differential abundance of 21 taxa between CMD-free and CMD-affected patients with IBD (Supplementary Data 1; PFDR < 0.05; Fig. 5b). Of the identified DATs with complete taxonomic classification information (n = 18), 10 taxa (55.6%) have been associated with anxiety or depression in previous studies16,22,23,24,25. Intriguingly, 13 CMD-associated taxa were also IBD-associated taxa in our cohort, with 11 showing the same directions of the abundance changes in both CMD and IBD, which was significantly more consistent than expected by chance (P = 6.05 ×10−4 in a binomial test; Fig. 5c; see also Supplementary Data 1). This result indicated that patients with CMD-affected IBD demonstrated a significantly greater deviation in the abundance levels of these taxa from those observed in healthy controls, in comparison to the degree of deviation exhibited by patients with CMD-free IBD from controls. This trend is consistent with the distribution of PC1 values among the three phenotypic groups in the PCoA results. All genera associated with both IBD and CMD showed significant correlations with PC1 values (Supplementary Data 2). We also noted that 14 IBD-associated genera, even without statistical evidence of CMD associations, showed consistent directions of abundance changes in IBD and CMD (Supplementary Fig. 3).
To assess whether the CMD-specific microbial change enriches the microbial risk factors for IBD, we newly developed a single statistic to indicate the IBD-specific microbial burden, referred to as microbial risk score (MRS), which is a linear summation of the normalized abundance levels of CMD-associated genera in an individual weighted by the pre-estimated IBD-specific effect sizes (see the Methods section). We found that a higher MRS for IBD is associated with an increased risk of CMD in patients with IBD (P = 7.33 ×10−3). When the study subjects were dichotomized into two groups at the median MRS, the high-MRS group had a large odds ratio of 5.0 (95% confidence interval = 1.7–15.4) for CMD development in a multivariate logistic regression (Table 3).
No evidence of IBD-risk genetic variants on the susceptibility to CMD in patients with IBD
Genome-wide association studies have identified many risk loci for IBD26,27 and mental disorders28,29. A positive correlation between the disease-risk effects of genome-wide genetic variants on IBD and depression has been previously reported using linkage disequilibrium score regression (LDSC)19. Thus, it is likely that a high genetic burden from the reported IBD-risk variants increases the susceptibility to CMD in patients with IBD.
To test this hypothesis, we calculated individual disease-specific PRSs based on the reported disease effect sizes of genetically independent risk variants (n = 136 for IBD, 87 for UC, 119 for CD, 70 for anxiety, or 49 for depression) from previous genome-wide association studies (GWASs) in multiple ancestral populations26,27,28,29, as described in the Methods section. We note that the previously reported significance of the LDSC-based genetic correlation between IBD and mental disorders was replicated in the public GWAS results used in our PRS analysis (Supplementary Table 2).
In a combined analysis with additional out-of-study controls, the patients with IBD showed significantly higher disease-specific PRSs for IBD, CD, UC, and depression than those of the controls (8.17 ×10−13 ≤ P ≤ 0.0182; Supplementary Fig. 4) except for anxiety. However, there was no evidence of the involvement of the genetic burden of IBD-risk variants in the development of CMD (P = 0.641). Moreover, patients with CMD did not show significantly higher PRSs for anxiety and depression than those without CMD (P ≥ 0.321). This result indicates a relatively weak genetic effect of mental disorder-risk variants on the development of CMD in patients with IBD or suggests that the development of CMD in IBD may have a different etiology from the general mental disorders that develop in the general population.
Regulatory effects of IBD-risk variants on CMD-risk microbial abundance levels
We searched for mbQTLs, which represent human genetic loci associated with relative microbial abundance, for each CMD-risk taxon through a genus-wide case-only GWAS using both microbial abundance and genetic data from patients with IBD, followed by a meta-analysis with a previous mbQTL from a larger-scale study30 involving multiple cohorts. We evaluated the genetic overlap of mbQTLs for CMD-risk taxa with known risk variants for IBD and mental disorders.
Among the 11 taxa associated with both IBD and CMD with consistent directions of abundance changes, we identified a single locus where the T allele of rs35866622 in the FUT2-FUT1 locus was associated with the lower relative abundance of the genus Ruminococcus at the genome-wide significance threshold of 5 × 10−8 (torques group; P = 2.51 × 10−8, β = −0.061, 95% confidence interval= −0.082 to −0.040; Supplementary Figure 5). The genus Ruminococcus is a well-known IBD-associated taxon31 and our study showed a significantly low level in its relative abundance in both patients with IBD (compared to controls; P = 6.83 × 10−3) and patients with CMD-affected IBD (compared to patients with CMD-free IBD; P = 4.13 × 10−2). In addition, the identified human mbQTL variant rs35866622 for the genus Ruminococcus has been reported as a lead disease-risk variant of IBD and CD, and its T allele conferred risk of the diseases26,27. Therefore, it is plausible to suspect that the IBD-risk allele in the FUT2-FUT1 locus has been associated with lower relative abundance of the genus Ruminococcus, thus potentially increasing the genetic liability to both IBD and CMD.
The locus around rs35866622 contains a number of proxy variants of the lead variant in a high linkage disequilibrium (LD), including a nonsense variant rs601338 in FUT2 (r2 = 1.00 and 0.80 in 1KGP East Asians and 1KGP Europeans, respectively). FUT2 encodes α-(1, 2)-fucosyltransferase enzymes responsible for the secretion of H antigens in body fluids, including the gut mucosa. The absence of H antigens in the gut mucosal fluid, caused by two copies of nonfunctional FUT2 alleles with the nonsense allele of rs601338, has been reported to have a large impact on the abundance levels of several taxa30,32, some of which have been previously associated with IBD-associated dysbiosis30,32,33.
Discussion
To our knowledge, this study represents the inaugural investigation of both host genetic and gut microbial factors in IBD patients afflicted with CMD. Our integrative analysis of the human genome and gut microbiome revealed that IBD-risk dysbiosis was significantly enhanced in patients with CMD, whose gut microbiota was characterized by less diversity and more dysregulated abundance levels of IBD-associated microbial taxa than those without CMD. An MRS analysis showed that an enrichment of microbial IBD-risk factors dramatically increases susceptibility to CMD. Moreover, an IBD-risk mbQTL allele was associated with a lower relative abundance of the genus Ruminococcus, which exhibited protective associations against both IBD and CMD. Our results strongly suggest that the gut-brain axis plays a crucial role in the development of CMD in patients with IBD, with IBD-specific gut dysbiosis primarily contributing to CMD risk and a host-gut microbiota interaction partially mediating this relationship.
We identified five genera associated with the risk of both IBD and CMD, with the same direction of relative abundance changes in both conditions (Fig. 5c). This indicates that extremely severe alteration in the abundance of IBD-associated taxa in patients with CMD-affected IBD. For instance, the genus Coprococcus is 5.7-fold higher in patients with IBD than in healthy controls and 2.8-fold in patients with CMD compared to those without CMD. Indeed, the same genus was 8.6-fold lower in CMD-affected IBD patients than in healthy controls (Supplementary Data 1).
The genus Coprococcus is a butyrate-producing microbe involved in the hypothalamic-pituitary-adrenal axis and is known to be depleted in individuals with depression and stress18,34. The reduced production of short-chain fatty acids, including butyrate, facilitates inflammation or barrier dysfunction in the gastrointestinal tract35,36. In addition, a recent study suggested a possible role of butyrate-unrelated mechanisms in Coprococcus in association with quality of life18.
Another important observation was that the IBD-risk variant rs35866622 was an mbQTL for CMD-associated bacteria (Supplementary Fig. 5). We examined the genetic effects on the compositional changes in the microbiome to understand the mediating effect between IBD-risk variants and CMDs through alterations in the microbiome. The IBD-risk mbQTL is suggested to regulate the IBD/CMD-associated genus Ruminococcus by regulating the secretion of H antigens in the gut mucosa32,37,38. This finding suggests that the genetic burden of IBD partially contributes to the frequent occurrence of mental disorders in patients with IBD, suggesting that FUT2 proteins and H antigens might be potential therapeutic targets for IBD and CMD. Ruminococcus spp., butyrate-producing bacteria that are abundant in the gut microbial community39,40, were also lower in patients with IBD in our study, especially in patients with CMD. A similar lower level of Ruminococcus torques was previously reported in patients with CD41. A discussion of the other IBD/CMD-associated genera is provided in Supplementary Note 1.
Notably, CMD-associated taxa without significant IBD associations need to be further investigated to understand whether they are IBD-independent mental disorder-risk factors or secondary outcomes from mental disorder-mediated effects on microbiome composition. For example, we found a CMD association of the orders Coriobacteriales and Pasteurellales, the family Pasteurellaceae, and the genus Collinsella, which were previously reported to be associated with anxiety or depression in the same direction as abundance changes in affected individuals (Supplementary Data 1).
Although a significant but weak genetic correlation between mental disorder in the general population and IBD was observed in an analysis using large-scale public GWAS results (Supplementary Table 2), the PRS-level genetic burden for IBD or mental disorders was not significantly different between CMD-affected and CMD-free IBD patients in our study cohort (Supplementary Fig. 4). Given the small sample size for the genetic association analysis, our results indicate that pleiotropic variants for the risk of IBD and mental disorders have little or much weaker effects on the development of CMD in patients with IBD than the effects of gut dysbiosis on CMD.
A case-only study in Switzerland was previously conducted to identify DATs in the gut correlated with Hospital Anxiety and Depression Scale (HADS; HADS-A for anxiety; HADS-D for depression) scores in patients with IBD in remission16. None of the reported CMD-associated genera with complete classification information (n = 14; eight genera tested in our study) were replicated in our analyses (Supplementary Data 1). The inconsistent results may indicate ancestry-specific heterogeneity in microbial effects on CMDs between European and East Asian populations, possibly due to the population-specific gut microbial community and the population-specific abundance levels of CMD-associated taxa that could be associated with genetic background, diet, and lifestyle, as well as the degree of statistical power in a differential microbial abundance analysis. In addition, the Swiss sample size (n = 204) was moderate and was further stratified in the differential abundance analysis according to the type of IBD or CMD. Additionally, the statistical models used in the two studies differed (linear regression vs. generalized linear regression using a negative binomial distribution).
This study provided unique opportunities to examine the gut microbiome and human genome-wide single nucleotide polymorphism (SNP) data simultaneously in a single center cohort, which allowed us to investigate gut dysbiosis and human genetic data separately, as well as to explore their interconnections. In addition, conducting both case-control and case-only microbial analyses within the same cohort could offer more reliable identification of shared risk factors between IBD and CMD, as compared to conducting separate analyses using two independent cohorts with different conditions such as genetic background, diet, and disease activities.
Nevertheless, this study has five major limitations. First, the number of CMD-affected patients was relatively small, although our sample size was relatively large among the published microbial studies on IBD. Large effects of gut dysbiosis were detected with strong statistical confidence in our study. However, it is likely that very small changes in microbial abundance and PRS in patients with CMD would not be detectable in our sample size. Second, the analyses of PRS and mbQTL were largely dependent on public resources generated from European populations. There is uncertainty regarding the cross-ancestry transferability of IBD PRS variants and the different genetic architectures of European and Korean populations in terms of allelic frequency and LD. Better analysis will be ensured by public resources generated from future East Asian-specific genome-wide association studies to identify mbQTLs and IBD susceptibility loci. Third, our analyses rely on stool samples, which may not fully represent the gut microbiome composition. Forth, our analyses assess differences in relative abundances rather than absolute abundances. Therefore, conclusions regarding abundance changes, such as that observed with Ruminococcus, signify differences in abundance relative to other taxa rather than absolute abundance difference. Five, the HADS information was not available for the control group. Although we rigorously reviewed clinical records to exclude individuals with a history of or current mental health conditions, we cannot rule out the possibility of including controls with high HADS values.
In summary, this study demonstrated how IBD-risk factors in the gut microbiota and human genetic variants are associated with prevalent comorbid depression and anxiety in patients with IBD through a series of comprehensive analyses in a large single-center prospective IBD cohort. The identified gut bacteria and genetic markers may serve as predictive biomarkers and therapeutic targets for mental disorders, thus providing new insights and opportunities to explore the underlying mechanisms of CMD development in IBD.
Methods
Study cohort
All patients and healthy controls were enrolled at the IBD Center of Kyung Hee University Hospital (Seoul, Republic of Korea) between April 2018 and October 2022, after providing written informed consent. This study was approved by the Institutional Review Board of Kyung Hee University Hospital (KHUH 2018-03-006) and compiled with all relevant ethical regulations including the Declaration of Helsinki. The patients met the diagnostic criteria of CD or UC based on clinical, biochemical, stool, and endoscopic findings using histopathological and cross-sectional imaging methods42. Clinical disease activity was measured using the Harvey–Bradshaw index (HBI) for CD and the Mayo score for UC. Clinical remission was defined as HBI ≤ 4 and Mayo score ≤243.
We obtained genomic and fecal microbial DNAs from genetically unrelated individuals over 17 years of age and collected basic demographic and clinical data from all participants (Table 1). We carefully selected healthy controls from a pool of randomly recruited participants who met stringent criteria to ensure a rigorous comparison with IBD patients collected during the same period. Exclusion criteria comprised individuals who had consistently consumed probiotics for over a week in the past three months, those who had used medications affecting the gastrointestinal system (such as painkillers and anti-inflammatory drugs) in the last week, participants who had taken antibiotics in the previous month, and those who had recently experienced acute gastrointestinal issues such as diarrhea and stomach pain. After applying these exclusion criteria, the remaining healthy controls had no serious illnesses, including gastrointestinal inflammation, and had no family history of IBD or mental disorders at enrollment. Additionally, healthy controls currently undergoing treatment for mental disorders, and similar conditions were strictly excluded during the screening process. The fecal samples were collected using the Stool Nucleic Acid Collection and Preservation Tube (NORGEN BioTek) and promptly stored at −80 °C until DNA extraction44.
Psychometric evaluation
The HADS45, a standardized psychological scale containing anxiety and depression subscales named HADS-A and HADS-D, respectively, was used for the psychometric testing of 225 patients with IBD. Significant anxiety and depression were defined as HADS-A ≥ 11 and HADS-D ≥ 11, respectively, as previously described46.
16S rRNA amplicon sequencing and microbial abundance analysis
The V3 to V4 regions of the 16S rRNA genes in fecal DNA were amplified using Agilent Herculase II fusion DNA Polymerase and Illumina Nextera XT library preparation kit v2 for paired-end sequencing on the MiSeq platform. The sequences of the targeted regions of the 16S rRNA genes were reconstructed by merging high-quality paired-end reads after trimming adapter bases and low-quality bases using Trimmomatic47 and QIIME248. We employed an amplicon sequence variant (ASV) method49 to denoise sequence errors and remove chimeric sequences. Taxonomic classification and phylogenetic annotation were performed using reference sequences from Greengenes v13.8 database50. ASVs were collapsed into taxonomic levels from phylum to genus for downstream analysis.
For diversity analyses, we employed the ‘diversity core-metrics-phylogenetic’ function in QIIME248, subsampling reads to the minimum sampling depth across samples using rarefaction51. We assessed the Shannon index, species richness by observed features, Simpson index, and Faith’s PD index to quantify the alpha diversity of the gut microbial community using library size-corrected sequencing data52,53,54. The beta diversity of the gut microbial composition among individual samples was examined using a PCoA based on the Bray–Curtis dissimilarity estimated from normalized microbial abundance levels using DESeq255 and vegan56 packages. Differential beta diversity between sample groups of interest was evaluated by distance-based permutational multivariate analysis of variance using the adonis package56. Analysis of the differential abundance of the gut microbiota was performed using DESeq2 to estimate the log2-fold changes in abundance between sample groups for only taxa present in at least 10% of samples from any phenotypic group used in the comparison. Sex, age, BMI, alcohol consumption, and current smoking status were adjusted in the case-control analysis. In the case-only differential abundance analysis, we additionally included IBD subtypes disease activity scores, and disease duration as covariates.
IBD MRS estimation
To understand the difference in the IBD-specific microbial burden between IBD patients with CMD and without CMD, the MRS for IBD was newly developed based on the abundance of CMD-associated genera, which were identified in differential abundance analysis, in each individual and the pre-estimated IBD-specific effect sizes of the same genera. The equation for the IBD MRS of an individual \(k\) (\({{MRS}}_{k}\)) is
where \(n\) is the number of CMD-associated genera, \({z}_{i}\) is the normal transformed DESeq-normalized read count for the \(i\)-th genus in the individual \(k\), and \(\sqrt{\left|{\beta }_{i}\right|}\) is the scaling factor to make \({z}_{i}\) have a variance as much as the absolute value of \({\beta }_{i}\) that is the pre-estimated log2-fold change in the abundance of the \(i\)-th genus in patients with IBD compared to healthy controls in the study. \(\mathrm{sgn}\left({\beta }_{i}\right)\) is the sign function that returns +1 or −1 according to the sign of \({\beta }_{i}\).
Genotyping and whole genome imputation
Genome-wide data of SNPs in a subset of patients with IBD were generated using the Korea Biobank Array57 to estimate PRSs for IBD, depression, and anxiety in patients with or without CMD and to explore human mbQTLs for the bacteria of interest. After a general quality control procedure for the genotyping data (Supplementary Table 1), 541,449 high-quality variants with call rates ≥97%, minor allele frequencies ≥1%, and P values for Hardy–Weinberg equilibrium ≥1 ×10−7 were retained in 225 unrelated patients who showed a homogeneous genetic background and per-sample genotyping call rates ≥0.95. Whole genome imputation was performed using Minimac358 with the reference panel derived from the 1000 Genomes Project phase 3 (1KGP3) data, after phasing the genotyping data into haplotypes using SHAPEIT software (v2.r904)59. All variants with imputation quality scores (R2) > 0.4 (n = 10,689,279) were subjected to subsequent analyses.
Genetic correlation
We estimated the between-trait genetic correlations of effect sizes of genome-wide variants on IBD, depression, and anxiety. We used the large-scale European GWAS summary statistics for IBD27, depression, and anxiety28,29, but not Asian GWAS statistics because our meta-analysis results for mbQTLs were mostly derived from European populations and there were no publicly available Asian GWAS summary statistics for the three traits. Genetic correlations were calculated using LDSC60 with pre-estimated scores of LD in the 1KGP3 European population.
PRS calculation
We calculated individual-level PRSs for IBD and mental disorders in IBD patients with HADS information based on the reported effect sizes of 136 variants for IBD (87 for UC, 119 for CD)26,27, 49 for depression, and 70 for anxiety28,29. The variants used for the PRS calculation were genetically and physically independent of each other (population-specific r2 < 0.2 and distance >1 Mb). They exceeded the GWAS significance threshold for disease risk in previous studies and are listed in our genetic data. Disease-specific PRS distributions in Korean individuals were obtained from the same variant sets in the whole genome imputation data of the Korean National Institute of Health cohort (n = 72,179)61.
MbQTL analysis for microbial taxa associated with both IBD and CMD
Genome-wide mbQTL analysis was performed to test for genetic associations with the abundance of the taxa of interest. The microbial abundance level of each taxon was normalized across samples using the rank-based inverse normal transformation. A multivariable linear regression model between microbial abundance and the allelic dosage of each genetic variant was examined using RVTESTS62, with adjustment for potential confounders such as sex, age, BMI, alcohol consumption, current smoking, IBD subtype, disease activity score, mental disorder status, and the top five genotypic principal components. A genome-wide mbQTL meta-analysis was performed using our result and a previous large-scale mbQTL analysis result30 (from 18,340 individuals; Europeans = 78.0%, non-Europeans = 22.0%) by METAL63 using the inverse variance-weighted fixed-effects model.
Data availability
All data relevant to the study are included in the article or uploaded as supplementary information. The 16S rRNA amplicon sequencing data generated in this study is available in the NCBI repository under accession number SRA: PRJNA1101759. The other data are available from the corresponding authors upon reasonable request.
Code availability
The software packages used in this study are open access. The analytic codes utilized in this study for microbiome analysis and mbQTL analysis via the link below: https://github.com/KimLabKHU/16S-rRNA-microbiome/tree/main.
References
Conway, G. et al. The impact of co-existing immune-mediated diseases on phenotype and outcomes in inflammatory bowel diseases. Aliment Pharm. Ther. 45, 814–823 (2017).
Lee, M. & Chang, E. B. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology 160, 524–537 (2021).
Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 7247238 (2019).
Levine, A., Sigall Boneh, R. & Wine, E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut 67, 1726–1738 (2018).
Yilmaz, B. et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 25, 323–336 (2019).
Barberio, B., Zamani, M., Black, C. J., Savarino, E. V. & Ford, A. C. Prevalence of symptoms of anxiety and depression in patients with inflammatory bowel disease: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 6, 359–370 (2021).
Saloni Dattani, H. R. A. M. R. Mental Health, https://ourworldindata.org/mental-health (2021).
Hu, S., Chen, Y., Chen, Y. & Wang, C. Depression and Anxiety Disorders in Patients With Inflammatory Bowel Disease. Front Psychiatry 12, 714057 (2021).
Fairbrass, K. M. et al. Bidirectional brain-gut axis effects influence mood and prognosis in IBD: a systematic review and meta-analysis. Gut 71, 1773–1780 (2021).
Ananthakrishnan, A. N. et al. Psychiatric co-morbidity is associated with increased risk of surgery in Crohn’s disease. Aliment Pharm. Ther. 37, 445–454 (2013).
Blackwell, J. et al. Depression in individuals who subsequently develop inflammatory bowel disease: a population-based nested case-control study. Gut 70, 1642–1648 (2021).
Frolkis, A. D. et al. Depression increases the risk of inflammatory bowel disease, which may be mitigated by the use of antidepressants in the treatment of depression. Gut 68, 1606–1612 (2019).
Iordache, M. M. et al. Intestinal Permeability and Depression in Patients with Inflammatory Bowel Disease. J. Clin. Med. 11, 5121 (2022).
Haroon, E. et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol. Psychiatry 21, 1351–1357 (2016).
Cui, W. et al. Crosstalk Between Inflammation and Glutamate System in Depression: Signaling Pathway and Molecular Biomarkers for Ketamine’s Antidepressant Effect. Mol. Neurobiol. 56, 3484–3500 (2019).
Humbel, F. et al. Association of Alterations in Intestinal Microbiota With Impaired Psychological Function in Patients With Inflammatory Bowel Diseases in Remission. Clin. Gastroenterol. Hepatol. 18, 2019–2029 e2011 (2020).
Jang, H. M. et al. Transplantation of fecal microbiota from patients with inflammatory bowel disease and depression alters immune response and behavior in recipient mice. Sci. Rep. 11, 20406 (2021).
Valles-Colomer, M. et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 4, 623–632 (2019).
Lasconi, C. et al. Variant-to-Gene-Mapping Analyses Reveal a Role for the Hypothalamus in Genetic Susceptibility to Inflammatory Bowel Disease. Cell Mol. Gastroenterol. Hepatol. 11, 667–682 (2021).
Imhann, F. et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 67, 108–119 (2018).
Abdel-Rahman, L. I. H. & Morgan, X. C. Searching for a Consensus Among Inflammatory Bowel Disease Studies: A Systematic Meta-Analysis. Inflamm. Bowel Dis. 29, 125–139 (2023).
Cheung, S. G. et al. Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 10, 34 (2019).
Janssens, Y. et al. Disbiome database: linking the microbiome to disease. BMC Microbiol 18, 50 (2018).
Simpson, C. A. et al. The gut microbiota in anxiety and depression - A systematic review. Clin. Psychol. Rev. 83, 101943 (2021).
Radjabzadeh, D. et al. Gut microbiome-wide association study of depressive symptoms. Nat. Commun. 13, 7128 (2022).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Thorp, J. G. et al. Symptom-level modelling unravels the shared genetic architecture of anxiety and depression. Nat. Hum. Behav. 5, 1432–1442 (2021).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Kurilshikov, A. et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165 (2021).
Schirmer, M., Garner, A., Vlamakis, H. & Xavier, R. J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 17, 497–511 (2019).
Ruhlemann, M. C. et al. Genome-wide association study in 8,956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 53, 147–155 (2021).
Cheng, S. et al. Altered gut microbiome in FUT2 loss-of-function mutants in support of personalized medicine for inflammatory bowel diseases. J. Genet. Genomics 48, 771–780 (2021).
Zhao, L. et al. Bidirectional gut-brain-microbiota axis as a potential link between inflammatory bowel disease and ischemic stroke. J. Neuroinflam. 15, 339 (2018).
Alipour, M. et al. Mucosal Barrier Depletion and Loss of Bacterial Diversity are Primary Abnormalities in Paediatric Ulcerative Colitis. J. Crohns Colitis 10, 462–471 (2016).
Wang, H. et al. Aberrant Gut Microbiome Contributes to Intestinal Oxidative Stress, Barrier Dysfunction, Inflammation and Systemic Autoimmune Responses in MRL/lpr Mice. Front. Immunol. 12, 651191 (2021).
Rausch, P. et al. Multigenerational Influences of the Fut2 Gene on the Dynamics of the Gut Microbiota in Mice. Front Microbiol 8, 991 (2017).
Giampaoli, O., Conta, G., Calvani, R. & Miccheli, A. Can the FUT2 Non-secretor Phenotype Associated With Gut Microbiota Increase the Children Susceptibility for Type 1 Diabetes? A Mini Review. Front. Nutr. 7, 606171 (2020).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
Zhu, L. B., Zhang, Y. C., Huang, H. H. & Lin, J. Prospects for clinical applications of butyrate-producing bacteria. World J. Clin. Pediatr. 10, 84–92 (2021).
Takahashi, K. et al. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 93, 59–65 (2016).
Maaser, C. et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis 13, 144–164 (2019).
Sturm, A. et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 2: IBD scores and general principles and technical aspects. J. Crohns Colitis 13, 273–284 (2019).
Carroll, I. M., Ringel-Kulka, T., Siddle, J. P., Klaenhammer, T. R. & Ringel, Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PLoS One 7, e46953 (2012).
Hinz, A. et al. Anxiety and depression in the general population in Colombia: reference values of the Hospital Anxiety and Depression Scale (HADS). Soc. Psychiatry Psychiatr. Epidemiol. 49, 41–49 (2014).
Mondolo, F. et al. The validity of the hospital anxiety and depression scale and the geriatric depression scale in Parkinson’s disease. Behav. Neurol. 17, 109–115 (2006).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Amir, A. et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2. https://doi.org/10.1128/mSystems.00191-16 (2017)
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ. Microbiol 72, 5069–5072 (2006).
Sanders, H. L. Marine benthic diversity: a comparative study. Am. Naturalist 102, 243–282 (1968).
Shannon, C. E. The mathematical theory of communication. 1963. MD Comput 14, 306–317 (1997).
Simpson, E. H. Measurement of Diversity. Nature 163, 688–688 (1949).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
vegan: Community Ecology Package. R package version 2.5-7 (2020).
Moon, S. et al. The Korea Biobank Array: Design and Identification of Coding Variants Associated with Blood Biochemical Traits. Sci. Rep. 9, 1382 (2019).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287 (2016).
O’Connell, J. et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet 10, e1004234 (2014).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet 47, 1236–1241 (2015).
Kim, Y. J. et al. The contribution of common and rare genetic variants to variation in metabolic traits in 288,137 East Asians. Nat. Commun. 13, 6642 (2022).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI23C0661) and the Medical Research Center Program through the National Research Foundation (NRF) of Korea, funded by the Ministry of Science and ICT (NRF-2017R1A5A2014768).
Author information
Authors and Affiliations
Contributions
C.K.L. and K.K. designed the research. C.K.L., H.J.K., G.Y.S. and S.J.O. collected the data. J.L., S.J.O., K.K. and C.K.L. analyzed the data, with assistance from E.H. and G.Y.S. J.L., S.J.O., K.K. and C.K.L. wrote the manuscript. All authors contributed to the revision and approval of paper for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, J., Oh, S.J., Ha, E. et al. Gut microbial and human genetic signatures of inflammatory bowel disease increase risk of comorbid mental disorders. npj Genom. Med. 9, 52 (2024). https://doi.org/10.1038/s41525-024-00440-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41525-024-00440-w
This article is cited by
-
The relationships between depression, inflammation and self-reported disease activity in IBD and their impact on healthcare usage
BMC Gastroenterology (2025)
-
Cross-Trait Cross-Genome Cross-Organ Analysis of Gastrointestinal Disorders and Depression
Digestive Diseases and Sciences (2025)
