
Abstract
Advances in genomic technologies have revolutionized the diagnosis of rare genetic diseases, leading to the emergence of precision therapies. However, there remains significant effort ahead to ensure the promise of precision medicine translates to improved outcomes. Here, we discuss the challenges in advancing precision child health and highlight how international collaborations such as the International Precision Child Health Partnership, which embed research into clinical care, can maximize benefits for children globally.
Similar content being viewed by others

Background
The burden of rare disease
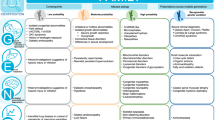
Rare disease, defined variably across the world, refers to conditions that affect fewer than 1 in 2000 individuals1. While individually rare, they in aggregate become common, with recent estimates suggesting that there are more than 10,000 rare diseases affecting up to 10% of the population (Fig. 1)2,3. While rare diseases are associated with morbidity and mortality across the lifespan, the majority have onset in childhood4. These conditions are often severe, chronic, and complex, and are frequently progressive, degenerative, and life-threatening; approximately one in three children with a rare disease will not live to see their fifth birthday5,6,7. Rare diseases also adversely impact their families, with caregivers facing psychosocial and economic burdens that contribute to reduced quality of life8,9,10. At a societal level, rare diseases, which can also be contributors to ‘common disease,’ have a high economic impact on healthcare systems and societies11,12,13. In the USA, the cost of rare diseases is estimated to be more than $1 trillion annually, including direct medical costs as well as indirect and non-medical costs, such as lost productivity14. Rare disease diagnosis and treatment therefore represent an urgent unmet global need.

Epidemiologic and genetic data highlight the urgent unmet need for improving the diagnosis and treatment of rare disease, with a coordinated and comprehensive approach to advancing discovery and translation in precision child health given the high genetic and phenotypic heterogeneity. In total, 12% of disease-associated genes have established interventions that may modify outcomes, which highlights both the imperative for prompt diagnosis, but also that most rare diseases lack a precision treatment currently2,4,21,22,129,130,131,132,133. Created with Biorender.com.
Challenges and opportunities for diagnosis of rare disease
Currently, over 70% of rare diseases have a known or presumed genetic cause4. Identifying a molecular genetic diagnosis for a child with a rare disease is the first step in delivering precision medicine to improve outcomes. Advances in genomic technologies, notably exome sequencing (ES) and more recently genome sequencing (GS), and initial integration of these genomic tests into clinical settings in some high-income countries, have considerably improved diagnostic yields for rare diseases15,16,17,18,19,20.
A genetic diagnosis ends the diagnostic odyssey for the child and their family and often informs clinical management, treatment, prognosis, and recurrence risk counseling, access to resources and support, and eligibility for natural history studies and clinical trials of novel precision therapies. Even with precision child health still in its relative infancy, over 600 rare disease genes that cause severe childhood-onset disorders—12% of all disease genes—have an established intervention that can modify outcomes, which highlights the urgency for, and potential immediate benefits of, improving diagnosis21,22.
Not surprisingly, access to genomic testing varies widely across and within countries23. In resource-poor countries, access to genomic testing is limited24. Even within well-resourced countries, barriers to equitable access remain substantial, and include inadequate payor coverage and inequities in reaching patients from medically underserved and minority populations25,26,27. Broad implementation and equity of access is also dependent on workforce education, availability of clinical, laboratory and bioinformatic personnel, and accredited laboratory facilities28.
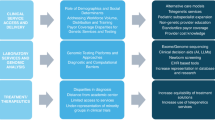
However, even with access to timely genomic tests, fewer than 50% of children with suspected rare genetic diseases receive an accurate genetic diagnosis17,29,30. There are numerous factors limiting diagnostic yield, and therefore strategies to improve this will need to be multipronged, including ways to resolve variants of uncertain significance (VUS), identification and validation of novel disease genes, advances in informatic strategies, new diagnostic techniques and disease models, and overcoming tissue-specific challenges for diagnosis (Fig. 2)31,32,33.

Strategic advancements and methodological innovations are essential for enhancing diagnostic accuracy and overcoming current bottlenecks. Key priority areas include improved ability to rapidly resolve variants of unknown significance (VUS), identification of novel disease genes, advancing informatic capabilities, and overcoming diagnostic challenges with emerging technologies134,135. Created with Biorender.com.
Despite great advances in knowledge, the consequences of much of the rare genetic variation found in any human genome remains uncertain. Resolution of VUS is a priority that needs improved in silico methods to predict pathogenicity and in vitro methods to demonstrate their functional consequences. Advances in understanding gene–disease associations, variable expressivity, and incomplete penetrance are critical, with over 30% of disease-associated genes demonstrating phenotypic pleiotropy34,35. An area of great urgency is one of population equity, defined in this case as the under-representation of populations with ancestry from outside of Europe in reference databases, which may limit the capacity for global variant interpretation36. Numerous national and international efforts are underway to address this issue, including the latest iteration of the gnomAD population database (v4.0). GnomAD now includes ~138,000 individuals of non-European ancestry, although participants of European ancestry still comprise 77% of the individuals in this database, highlighting that further work to increase diversity is required37,38,39.
New diagnostic techniques, including long-read sequencing and ‘multi-omic’ technologies, such as transcriptomics, methylation studies, proteomics, and metabolomics, can be utilized as an adjunct to, or aid in overcoming limitations of clinical ES and GS40,41,42,43. Recent work supplementing trio GS with additional bioinformatic analyses and transcriptome sequencing reported increased diagnostic yield from 47% to 54% in a cohort of critically ill infants and children30. Novel technologies hold considerable promise for increasing diagnostic yield, although variant interpretation remains difficult due to insufficient population data. The utility and role of new technologies within diagnostic pathways are currently unclear and likely to differ between conditions, depending on their genetic landscape and affected tissues.
Coordinated international efforts are needed to address these knowledge gaps, and it is imperative that their focus extends to building infrastructure to overcome barriers that limit advances. Priorities include the development of frameworks and systems for responsible sharing and use of data and samples, and addressing the silo-ing of clinically acquired genomic data, much of which is not available for use in research. Breaking down research-clinical barriers, with appropriate privacy safeguards in place, would enable access to clinical data for research, benefit individual patients through further analysis of their own data, and advance science through making large datasets available for interrogation.
Challenges and opportunities for rare disease treatment
Despite recent progress in the development of targeted therapies (Table 1), the promise of precision medicine remains out of reach for most children with a rare disease, which have historically been less attractive to commercial development given the small number of affected individuals44,45,46. Treatments modifying the underlying disease mechanism are more likely to be transformative than less specific, symptom-based treatments, given their greater likelihood of improving many, if not all, features of a condition. Such therapies could be targeted to the individual disease gene or could be pathway-based for related disorders (e.g., mTORopathies, RASopathies) (Fig. 3)47,48. Precision nutritional and vitamin or trace element-based therapies, and some enzyme replacement therapies, have established roles as treatment of some rare diseases, particularly genetic metabolic disorders49. More recently, pioneering clinical trials have demonstrated proof-of-concept for developing safe and effective precision therapies acting at the DNA, RNA, or protein level for rare diseases50,51. Some of these, such as gene-based therapies for spinal muscular atrophy and sickle cell disease, and protein-based treatments for cystic fibrosis, have transformed patient outcomes and received regulatory approval for use in clinical practice52,53,54. Researchers have also reported progress toward precision therapies customized to patient-specific DNA variants for ultra-rare diseases, such as ‘n-of-1’ antisense oligonucleotide therapy, gene therapy, and the promising and versatile prime editing technique, which can insert, delete, or change selected base pairs55,56,57.

Level of intervention, e.g., at a DNA/RNA/protein/pathway or network level, appears on the left of the figure. Examples of methods of novel therapeutic approaches appear on the right. Text in green indicates examples of previous or current clinical trials136,137,138,139, text in orange indicates evidence from pre-clinical models140,141. In early-onset severe SCN2A-related epilepsy, variants result in a GOF and thus seizures are responsive to sodium channel blocking antiseizure medication, as are neonatal and infantile epilepsies due to LOF variants in KCNQ2, likely due to co-localization with sodium channels66,142. l-serine acts as an alternate agonist at NMDA receptors to improve both seizure and non-seizure outcomes in LOF GRIN neurodevelopmental disorders143. Pathway-focused approaches are possible in metabolic epilepsies such as GLUT1-related epilepsy, disorders of the Vitamin B6 pathway, and biotinidase deficiency, but also as a broad approach in focal epilepsies related to mTOR pathway dysfunction including tuberous sclerosis complex, germline and somatic MTOR variants, and GATOR1 complex (DEPDC5, NPRL2, NPRL3) epilepsies144,145,146,147,148,149. Network-based approaches remain limited, but there is emerging evidence of targeted stimulation strategies in severe epilepsies such as Lennox Gastaut syndrome, and development of activity-based gene therapies which may allow targeted interruption of epileptic networks by targeting overactive neurons150,151,152,153. Created with Biorender.com. AAV adeno-associated virus, EKC engineered potassium channel, CRISPRa CRISPR activation, CRISPRi CRISPR inhibition, ASO antisense oligonucleotide, siRNA small interfering RNA, ssRNA single strand RNA, GOF gain-of-function, LOF loss-of-function, MTOR mammalian target of rapamycin, DBS deep brain stimulation, RNS responsive neurostimulation.
Barriers to accelerating the development and translation of promising novel therapies are multiple58. First, genetic diagnosis is a key precursor to implementation of precision therapies. Beyond simply achieving a diagnosis, it is imperative that diagnostic testing is not delayed, to enable prompt implementation of precision therapies where available early in the disease course when they are likely to have the greatest impact and reduce the development of comorbidities. A precise genetic diagnosis, with understanding of the type of functional impact, is required for entry into the limited but growing number of targeted trials of novel therapies.
Second, multidisciplinary collaborations are required, to connect clinician–researchers (also known as physician–scientists) with fundamental scientists to advance understanding of disease mechanisms, and forge partnerships with industry, disease foundations, and patient advocacy groups to identify opportunities for the development of novel therapies and rapid translation to the clinic.
Third, the development of precision treatments requires understanding of underlying pathological mechanisms, phenotypes, and natural history of rare disease, knowledge of which is often limited by small patient numbers and limited opportunities for data collection59. The development of standards for reporting phenotypic data in publications on rare disease genes is critical. These are vital to support studies of rare disease, particularly those based on large, federated datasets, to ensure consistent reporting of clinical and outcome data, including ethnicity and medications which are recorded differently in different countries60,61. Further, improvements to clinical disease coding are necessary to ensure precise documentation of rare genetic disorders, many of which are not precisely coded with current systems, as a diagnosis in an individual’s electronic medical record62. International data-sharing models can streamline and standardize the collection of phenomic and genomic real-world data to compile clinical information over time, and establish genotype–phenotype-disease mechanism correlations. These are required to delineate disease subgroups and patient trajectories, informing patient selection, trial designs, and outcome measures for clinical trials. Progress related to genetic epilepsies due to ion channel disorders (e.g., SCN1A, SCN2A) provides important examples of how critical these elements are, as each gene is now associated with a phenotypic spectrum related to specific disease mechanisms (Fig. 4)63,64,65,66,67. Both gain- and loss-of-function phenotypes are recognized, and disease subgroups will require different molecularly based treatments, both for effect and to avoid exacerbating disease if the ‘wrong’ treatment is given. Phenotypes of variable severity have also been identified both between and within these disease subgroups, not all of which will require novel therapies, as some patients will have favorable outcomes with existing treatments. While optimizing processes for the collection of clinical data, the burden on families must remain manageable. Multidisciplinary clinics with research infrastructure, which collect comprehensive, standardized data as part of a clinical assessment, are one means of advancing knowledge of phenotypes and natural history at the same time as addressing clinical needs; such clinics are valued by both families and referring clinicians68,69.

The gene encoding the Nav1.1 sodium channel, SCN1A, provides an example of one gene causing more than one disease. This gene is associated with both gain- and loss-of-function disease mechanisms, and phenotypes of variable severity, only some of which are associated with poor outcomes63,154,155. Clinical trials of novel therapies for Dravet syndrome (SCN1A loss-of-function phenotype), including AAV9 gene therapy and an antisense oligonucleotide, are underway136,156. Much work has been done to understand phenotypic variability, natural history, and phenotype–disease mechanism correlations, which has been critical for therapeutic development, clinical trial design and candidate selection, and outcome measurements64,86,157,158,159. The more recently identified gain-of-function SCN1A phenotypes will require different treatments63,160. Created with Biorender.com.
It is notable that the pathway to novel therapies has to date been a long one, with the median time between genetic target discovery and regulatory drug approval being 25 years70. As the number of clinical genetic diagnoses increases, issues of efficiency and cost-effectiveness in bringing novel therapies to clinical practice must be managed.
Once a potential precision treatment is identified, the small number of patients with individual genetic disorders worldwide makes translation of novel therapies extremely challenging. Standard funding and regulatory pathways may not be suitable and risk failure for this vulnerable group of patients for whom access to treatment is often time-critical. While well-established ‘best practice’ trial design—randomized controlled trials—may not always be appropriate or feasible, cautionary tales highlight that steps to carefully evaluate promising therapies should not be missed. A good example is the use of quinidine in KCNT1-related epilepsies, epilepsy of infancy with migrating focal seizures (EIMFS), and autosomal dominant sleep-related hypermotor epilepsy (ADSHE)71,72,73,74. Rigorous in vitro evaluation using Xenopus oocytes demonstrated pathogenic variants result in gain-of-function which could be reversed using the cardiac drug, quinidine75. Initial case reports suggested benefit in children with EIMFS, although subsequent reports had mixed findings (including seizure worsening) and not uncommonly reported prolongation of the QT interval, raising concerns that toxicity may limit effectiveness76,77,78. A subsequent order-randomized, blinded, placebo-controlled cross-over study of six patients with ADSHE, adequately powered due to large seizure burden, did not demonstrate improvement in seizures, and confirmed anecdotal evidence of toxicity limiting dose escalation79. Such examples highlight that appropriately designed and well-conducted clinical trials are essential to confirm benefit in patients using novel therapies studied in pre-clinical models.
Innovative trial designs using ‘n-of-1’ approaches, adaptive designs or considering within-patient change may enable rigorous evaluation with a relatively small number of patients80,81,82. Where randomized controlled trials are not ethical or practical due to patient numbers or disease course, natural history data can be used as control group data, further underscoring the importance of careful, longitudinal phenotypic study. Such ‘historical control’ data have been integral to demonstrating the benefit of novel treatments for spinal muscular atrophy and neuronal ceroid lipofuscinosis type 2 disease83,84. For conditions like these, where fatality is inevitable without treatment, comparative improvement from an intervention may be easier to determine than less severe conditions or those with greater phenotypic heterogeneity. This aside, natural history studies are being undertaken in other rare diseases, such as the severe epilepsy SCN1A-Dravet syndrome, against which the effect of novel therapies can be compared85,86. Overall, the assessment of new treatments requires novel approaches around both trial design and outcome measures. While some approaches may be bespoke, determining commonalities across diseases may enable economies of scale.
With increasing submissions for regulatory approval for orphan drugs, regulatory bodies have had to adapt processes and requirements that lack suitability for rare disease, to ensure that treatments which may transform patient outcomes are not stymied or delayed by pathways that are not fit for purpose. Programs such as the Food and Drug Administration’s Rare Disease Endpoint Advancement Pilot Program and the Medicines and Healthcare Products Regulatory Agency’s Innovative Licensing and Access Pathway represent responses of regulatory bodies to adapt to the unique challenges of rare disease clinical trials and treatment development, with the aim of accelerating time to market87,88. Further efficiencies, requiring input from all stakeholders will surely follow, as the number of rare disease treatments increase. A number of rare disease research networks, such as the Rare Diseases Clinical Research Network in the United States, and the Maternal Infant Child and Youth Research Network and the RareKids-CAN: Pediatric Rare Disease Clinical Trials and Treatment Network in Canada, have been recently established89,90,91. Cross-border networks enhance and expand upon national efforts, providing the infrastructure, expertise, and patient numbers needed to scale up and streamline rare disease trials and long-term follow-up using standardized tools. The development of processes and platforms upon which to evaluate and, ultimately, implement novel therapies, as has been successfully executed in the oncology setting, will be key for efficiency and to avoid ‘reinventing the wheel’ with each rare disease. A shift to considering clinical trials to be part of standard clinical care will be important for the successful implementation and assessment of benefit of new treatments92.
The need for collaboration
The promise of potential treatments is driving a worldwide impetus to implement precision child health, but in the traditional silos of a single clinical center, state, or even country, progress in addressing gaps and unmet needs is slow and inefficient. For maximum impact, solutions require coordinated international efforts by experts to accelerate accurate diagnosis and effective management of rare disorders and to implement evidence-based practice changes feasibly, efficiently, and equitably.
A coordinated approach on a global scale can energize collaboration and maximize the output of research efforts by building upon the diverse expertise and existing efforts underway across the world, reducing duplication of efforts. Consortium approaches support access to a much greater number of patients and generate more data than could be achieved in a single jurisdiction, thereby enabling researchers to ask novel questions and identify previously unknown patterns and gene–disease relationships.
The International Precision Child Health Partnership
As many rare diseases have pediatric onset, with evolution and often progression of phenotype over time, it is logical to assume that the optimal time for intervention is as early as possible in childhood to maximize benefits to the individual, family, and health system4. The pediatric setting is therefore the ideal place in which to tackle the massive challenges of precision medicine for rare disease.
The International Precision Child Health Partnership (IPCHiP) is a multinational collaborative formed in 2020 to advance precision child health (Box 1). Bringing together four leading paediatric institutes -Boston Children’s Hospital; UCL Great Ormond Street Institute for Child Health and Great Ormond Street Hospital (London); the Murdoch Children’s Research Institute with The Royal Children’s Hospital and the University of Melbourne Department of Paediatrics (Melbourne Children’s Campus) (Melbourne); and The Hospital for Sick Children (SickKids) (Toronto), IPCHiP is inspired by a common vision to accelerate genomic discovery and translation to clinical care. Leveraging medical and scientific expertise at each site, and expanding upon local initiatives, IPCHiP is building bridges between clinical practice, and clinical and fundamental research, effectively ‘bringing science to patients’ in real time17,93. By embedding research in clinical practice, IPCHiP is generating the evidence needed to advance precision child health and provide a model for implementing change more widely.
IPCHiP aims to facilitate collaboration on the scientific investigation of rare diseases, create innovative diagnostic and therapeutic solutions, accelerate progress through the development of methods for acquiring, sharing and analyzing genomic and phenotypic data across institutions, and ultimately, improve access to genomic diagnoses and novel treatments in order to optimize outcomes for children with rare diseases. IPCHiP is developing and adopting global best practices for precision child health, such as establishing foundations and standards for federated data sharing. To this end, IPCHiP has recently been designated a ‘Driver Project’ of the Global Alliance for Genomics and Health (GA4GH) [https://www.ga4gh.org/news_item/2023-driver-projects/]94.
IPCHiP is considered a priority initiative at each of our world-leading institutes. This shared mindset means that the partnership is prioritized at each of our institutes to ensure studies are supported. It is also highly valuable external to our institutes, in that a productive international partnership can improve grant funding success, and be an important voice in advocating for the development of standards. Ongoing prioritization of this partnership will ensure that the IPCHiP sites can collectively be greater than the sum of their parts in advancing precision child health.
At the core of IPCHiP is the design, institutional support, and launch of multi-site cohort studies. Each study will seek to address one or more of the following goals: (1) improve diagnosis, (2) improve treatment, and (3) improve access to precision health strategies for people with rare disease. Its initial flagship project, Gene-STEPS (Shortening Time of Evaluation in Paediatric Epilepsy Services), has demonstrated the value of this multicenter approach17.
IPCHiP project: Gene-STEPS
The Gene-STEPS study commenced in Australia, Canada, England, and the United States in late 2021, aiming to determine the yield and utility of rapid trio genome sequencing (rGS) in infants with new-onset epilepsy and to demonstrate the impact of rapid genetic diagnosis. Numerous studies have highlighted the high yield and cost-effectiveness of (non-rapid) genomic testing in infants with epilepsy16,95,96. Infantile-onset epilepsies frequently have a monogenic basis, with over 1000 monogenic epilepsy genes now identified97. An increasing number have treatment implications, including some that are based on specific genetic mechanisms and their downstream effects17. However, management changes are often initiated late in the course of disease given long turn-around-times for clinical genomic testing. During this delay, the infant accrues the negative impacts of months of uncontrolled seizures, compounded by an ongoing epileptic encephalopathy, and exposure to many antiseizure medications, on cognitive and motor development98,99,100. In Gene-STEPS, infants presenting to an IPCHiP center undergo rGS with a turn-around time of 3 weeks or less for results from an accredited laboratory, ensuring immediate action with prompt initiation of optimal therapy and cessation of contraindicated drugs17. Findings from its pilot year, indicated high diagnostic yield, with 43% of infants with epilepsy receiving a genetic diagnosis. The results of rGS informed clinical care in 98% of infants with a diagnostic result, including guiding choice of treatments in 56%, and informing overall prognosis in 86%. Importantly, a ‘negative’ (non-diagnostic for the primary indication) result influenced management in 23% of infants in whom no etiological diagnosis for epilepsy was identified. For example, specific genetic diagnoses might dissuade a clinical team from pursuing epilepsy surgery for focal epilepsy, and in the absence of these genetic diagnoses, surgery could be considered. Ongoing Gene-STEPS work aims to understand the long-term impact of early etiologic diagnosis on seizure and developmental outcomes, and to determine the cost-effectiveness and acceptability to families of rGS. Collectively, data from this study will provide evidence to guide the implementation of rGS into routine clinical practice for the benefit of children globally.
To our knowledge, Gene-STEPS is the first prospective study of rGS primarily outside of an intensive care unit (ICU) setting, with over 80% of infants recruited from outpatient or non-ICU inpatient settings, or conducted in a disease-specific cohort. The processes used provide a blueprint for future studies of the utility of rapid genomic testing in time-critical or potentially treatment-changing scenarios managed predominantly outside ICU, such as infantile hypotonia and renal disease.
Gene-STEPS has now recruited over 400 infants. Rapid GS is now well-established as standard practice at our sites, with most (>90%) parents of eligible infants consenting to rGS via the study. The success of Gene-STEPS required extensive system changes to embed research into clinical practice (Table 2 and Fig. 5). This complex process relied heavily on group synergy and enthusiasm, aided substantially by existing collaborative relationships of the lead investigators. A community of clinician–scientists, laboratory scientists, and other researchers was built, to establish each of the processes required, and to interface with and engage the many clinicians caring for infants with epilepsy. The complexity of readying each part of the study varied between sites: for example, rGS was available as a clinical test at some centers (albeit used in very select settings) but needed to be established in a short time frame at others. Mentoring and sponsorship from the IPCHiP Executive Committee, and the support of each IPCHiP institute in providing funding for Gene-STEPS, were critical. Institutional backing included support for sequencing and bioinformatic research structures that could seamlessly interface with clinical laboratory medicine systems, coordinator time to support enrollment into Gene-STEPS, support for physicians and genetic counselors to provide pre-testing counseling, ongoing data analysis, and return of results, and support for social scientists to explore impact on quality of life. Without institutional support, a study such as Gene-STEPS would be extremely difficult to conduct, given the paucity of funding opportunities for international studies. However, such investment yields returns that support future work and increase local capabilities: Gene-STEPS investigators have received 11 competitive grants to continue this work, which support numerous employees and training positions, and the IPCHiP collaboration has also brought new philanthropic funders to the table across the four partner countries. Pleasingly, some of the awarded grants supported the multidisciplinary team science required for studies such as Gene-STEPS. However, none provided funding to more than one site, highlighting a significant barrier to international collaboration, which will be important to address.

BCH = Boston Children’s Hospital, GOS ICH = Great Ormond Street Institute for Child Health, MCC = Melbourne Children’s Campus (Murdoch Children’s Research Institute and the Royal Children’s Hospital), SickKids = The Hospital for Sick Children. Color of orange squares denotes the availability of each resource required for the study in clinical practice at each site prior to the commencement of Gene-STEPS. Solid orange = clinically available and routine for this patient group, orange diagonal lines = clinically available but not routine, orange grid lines = was not available clinically and needed to be established, research = white. *Other assessments include measures of adaptive function, quality of life, and clinical impact of genomic testing. **Advanced-omic testing includes RNA sequencing, long-read genome sequencing.
IPCHiP initiatives: toward federated data sharing
While each IPCHiP institute has generated robust and comprehensive datasets through Gene-STEPS, the sharing of clinical and genetic information across sites and jurisdictional borders remains a challenge. Biomedical data are sensitive, as they involve the private data of individuals and families, and can trigger legal restrictions on disclosure and international data transfer. One of the top priorities of IPCHiP is to explore innovative approaches to overcome challenges in the way data are currently shared, accessed, and used internationally (Table 3).
One such potential approach is federation, a data-sharing model where requesting parties bring their analysis software to the data (‘data visiting’). Federation offers an alternative to traditional sharing models of copying data locally; an approach not feasible for IPCHiP institutes bound by jurisdictional policy and regulation. In federated models, the data remain within the host institution, without exposing individual-level personal information, facilitating compliance with legislation like the General Data Protection Regulation (GDPR)94,101. We plan to use data generated from Gene-STEPS to pilot the establishment of a federated data network and set the stage to jointly analyze clinical and genomic data generated in collaborative projects for any pediatric rare disease. We envision that access to the federated ecosystem would be scalable and adhere to the FAIR (Findable, Accessible, Interoperable, and Reusable) principles102.
Contributors to IPCHiP are already working with international partners (e.g., Undiagnosed Disease Network International (UDNI), Matchmaker Exchange (MME), National Initiatives Forum at GA4GH), to develop the governance and infrastructure needed to establish a federated network. IPCHiP investigators recognize that the challenges their sites face in data sharing are also challenges for many other countries and institutions, and see great value in being able to share resources and experiences to advance precision child health.
IPCHiP imperatives: accessibility and equity in precision child health
Ensuring access to precision diagnosis and treatments at scale in a sustainable and equitable way is daunting but essential. Broadly, there needs to be consideration of what success may look like, such as an increase in tests sent and diagnoses made, percentage of diagnoses that lead to changes in management or precision treatment, equity of access to diagnosis and treatment, and development of systems for efficient and cost-effective deployment of novel therapies. IPCHiP provides an opportunity to generate evidence to guide access and use of diagnostic technologies and precision therapies in clinical practice, although we acknowledge that requirements for, and barriers to, ‘real-world’ implementation of precision child health, will vary across different health systems, given differences in resourcing and funding models across countries. Bringing hospitals and researchers, and the governments and payors that fund them, to the same table is a priority, as advances will be limited if health and medical research systems work in isolation.
It is critical that approaches to improving precision medicine involve all stakeholders, including patient and public involvement. This has already been demonstrated to be an efficient approach, influencing speed of translation into clinical practice and acceptability to target populations. Work in prenatal diagnosis and the use of rapid genomics in ICU systems have gone beyond studying diagnostic yield and clinical impact, to understanding economic impacts, family experience and requirements for health system resourcing, efficiently providing comprehensive bodies of evidence to support implementation and funding of testing in clinical practice103,104,105,106,107,108,109. Linking researchers with clinicians also increases the speed of translation and provides the opportunity to utilize implementation-effectiveness frameworks to test run the role of diagnostics and interventions in the setting in which they will ultimately be used110.
Priorities for wider implementation should include the development of frameworks for the rigorous evaluation of diagnostic technologies and new therapies in a standardized way, so centers can all achieve the same goal and contribute data constructively. The IPCHiP consortium is ideally placed to do this given the expertise and developing infrastructure, already piloted through Gene-STEPS17. Work should be undertaken with stakeholders, to address key needs, models of care (e.g., remote consent), training requirements, and financial viability, to ensure scalability and equity, looking toward efficiency of systems utilizing new ways of working and upskilling of staff. This said, there must be strong consideration given to the fact that solutions for large well-resourced clinical academic institutions, or in particular countries or cultures, may not be applicable universally111,112,113,114.
The way forward
Through collaboration across continents, health systems, academic institutions, medical and scientific disciplines, IPCHiP’s pilot efforts focused on infants with epilepsy have demonstrated the potential for success in advancing precision diagnosis for children with rare diseases; we encourage others to take a similar approach to studies in rare disease. We have seen a rapid paradigm and culture shift in the approach our four institutions’ clinicians take to the diagnosis of an infant with unexplained epilepsy, with Gene-STEPS eligibility considered on day 1 of presentation in both outpatient and inpatient settings. IPCHiP will expand its work on rapid genomic testing to other genetically heterogeneous conditions that would potentially benefit from prompt diagnosis and institution of optimal treatment. Its next cohort, GEMStone, will utilize this approach in neonates and infants with hypotonia.
For several decades, genetic and genomic research studies have enrolled participants with a given condition, taken data and samples, and yielded ground-breaking advances that eventually improve care for subsequent generations of patients with that condition. By embedding research into clinical practice, and compressing timeframes from presentation to diagnosis, studies like Gene-STEPS accelerate discovery and ensure benefit in this generation of patients.
While studies such as Gene-STEPS can promptly advance precision child health, it is important to acknowledge that the current systems and models of healthcare and medical research are not set up to enable such work. Efficiently improving outcomes for all people with rare disease will take a truly visionary outlook. Whole-of-system changes are needed to bridge structures that are typically siloed. We strongly advocate for further funding mechanisms to encourage team science and team medicine efforts, across institutes and across countries.
Genomic sequencing in childhood will set the scene for life-long application of genomic data, including the possibility of tackling major population health issues (e.g., risk identification, risk reduction, and treatment for common non-monogenic disorders such as heart disease), and large-scale application of genomic data in carrier screening for people in their reproductive years115,116. Accelerating precision child health is therefore an investment beyond the setting of pediatric rare disease, holding great promise for improving health and quality of life of all the world’s population.
References
Richter, T. et al. Rare disease terminology and definitions-a systematic global review: report of the ISPOR rare disease special interest group. Value Health 18, 906–914 (2015).
Haendel, M. et al. How many rare diseases are there? Nat. Rev. Drug Discov. 19, 77–78 (2020).
Ferreira, C. R. The burden of rare diseases. Am. J. Med. Genet. A 179, 885–892 (2019).
Nguengang Wakap, S. et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 28, 165–173 (2020).
Dodge, J. A. et al. The importance of rare diseases: from the gene to society. Arch. Dis. Child. 96, 791–792 (2011).
Wojcik, M. H. et al. The unrecognized mortality burden of genetic disorders in infancy. Am. J. Public Health 111, S156–S162 (2021).
Kingsmore, S. F. et al. Measurement of genetic diseases as a cause of mortality in infants receiving whole genome sequencing. npj Genom. Med. 5, 49 (2020).
Campbell, J. D. et al. Assessing the impact of caring for a child with Dravet syndrome: results of a caregiver survey. Epilepsy Behav. 80, 152–156 (2018).
Landfeldt, E. et al. Caregiver burden of spinal muscular atrophy: a systematic review. Pharmacoeconomics 41, 275–293 (2023).
Valcarcel-Nazco, C. et al. Health-related quality of life and perceived burden of informal caregivers of patients with rare diseases in selected European countries. Int. J. Environ. Res. Public Health 19, 8208 (2022).
Chung, C. C. Y. et al. Socio-economic costs of rare diseases and the risk of financial hardship: a cross-sectional study. Lancet Reg. Health West. Pac. 34, 100711 (2023).
Navarrete-Opazo, A. A., Singh, M., Tisdale, A., Cutillo, C. M. & Garrison, S. R. Can you hear us now? The impact of health-care utilization by rare disease patients in the United States. Genet. Med. 23, 2194–2201 (2021).
Walker, C. E. et al. The collective impact of rare diseases in Western Australia: an estimate using a population-based cohort. Genet. Med. 19, 546–552 (2017).
Yang, G. et al. The national economic burden of rare disease in the United States in 2019. Orphanet J. Rare Dis. 17, 163 (2022).
Wojcik, M. H. et al. Genome sequencing for diagnosing rare diseases. N. Engl. J. Med. 390, 1985–1997 (2024).
Sheidley, B. R. et al. Genetic testing for the epilepsies: a systematic review. Epilepsia 63, 375–387 (2022).
D’Gama, A. M. et al. Evaluation of the feasibility, diagnostic yield, and clinical utility of rapid genome sequencing in infantile epilepsy (Gene-STEPS): an international, multicentre, pilot cohort study. Lancet Neurol. 22, 812–825 (2023).
Splinter, K. et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 379, 2131–2139 (2018).
Tan, T. Y. et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 171, 855–862 (2017).
Costain, G., Cohn, R. D., Scherer, S. W. & Marshall, C. R. Genome sequencing as a diagnostic test. CMAJ 193, E1626–E1629 (2021).
Downie, L. et al. Gene selection for genomic newborn screening: moving toward consensus? Genet. Med. 26, 101077 (2024).
Lunke, S. et al. Prospective cohort study of genomic newborn screening: BabyScreen+ pilot study protocol. BMJ Open 14, e081426 (2024).
Phillips, K. A., Douglas, M. P., Wordsworth, S., Buchanan, J. & Marshall, D. A. Availability and funding of clinical genomic sequencing globally. BMJ Glob. Health 6, e004415 (2021).
Helmy, M., Awad, M. & Mosa, K. A. Limited resources of genome sequencing in developing countries: challenges and solutions. Appl. Transl. Genom. 9, 15–19 (2016).
Luke, J. et al. Investigating disparity in access to Australian clinical genetic health services for Aboriginal and Torres Strait Islander people. Nat. Commun. 13, 4966 (2022).
Mordaunt, D. A., Dalziel, K., Goranitis, I. & Stark, Z. Uptake of funded genomic testing for syndromic and non-syndromic intellectual disability in Australia. Eur. J. Hum. Genet. 31, 977–979 (2023).
Lee, S. S., Appelbaum, P. S. & Chung, W. K. Challenges and potential solutions to health disparities in genomic medicine. Cell 185, 2007–2010 (2022).
Stark, Z. et al. Australian genomics: outcomes of a 5-year national program to accelerate the integration of genomics in healthcare. Am. J. Hum. Genet. 110, 419–426 (2023).
Wright, C. F. et al. Genomic diagnosis of rare pediatric disease in the United Kingdom and Ireland. N. Engl. J. Med. 388, 1559–1571 (2023).
Lunke, S. et al. Integrated multi-omics for rapid rare disease diagnosis on a national scale. Nat. Med. 29, 1681–1691 (2023).
Ye, Z. et al. Cerebrospinal fluid liquid biopsy for detecting somatic mosaicism in brain. Brain Commun. 3, fcaa235 (2021).
Kernohan, K. D. & Boycott, K. M. The expanding diagnostic toolbox for rare genetic diseases. Nat. Rev. Genet. 25, 401–415 (2024).
Marwaha, S., Knowles, J. W. & Ashley, E. A. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med. 14, 23 (2022).
Posey, J. E. et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 21, 798–812 (2019).
Online Mendelian Inheritance in Man (McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD) www.omim.org (2024).
Jooma, S., Hahn, M. J., Hindorff, L. A. & Bonham, V. L. Defining and achieving health equity in genomic medicine. Ethn. Dis. 29, 173–178 (2019).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Wong, E. et al. The Singapore National Precision Medicine Strategy. Nat. Genet. 55, 178–186 (2023).
Ju, D., Hui, D., Hammond, D. A., Wonkam, A. & Tishkoff, S. A. Importance of including non-European populations in large human genetic studies to enhance precision medicine. Annu. Rev. Biomed. Data Sci. 5, 321–339 (2022).
Ketkar, S., Burrage, L. C. & Lee, B. RNA sequencing as a diagnostic tool. JAMA 329, 85–86 (2023).
Sanford Kobayashi, E. et al. Approaches to long-read sequencing in a clinical setting to improve diagnostic rate. Sci. Rep. 12, 16945 (2022).
Kerkhof, J. et al. Diagnostic utility and reporting recommendations for clinical DNA methylation episignature testing in genetically undiagnosed rare diseases. Genet. Med. 26, 101075 (2024).
Chantada-Vazquez, M. D. P., Bravo, S. B., Barbosa-Gouveia, S., Alvarez, J. V. & Couce, M. L. Proteomics in inherited metabolic disorders. Int. J. Mol. Sci. 23, 14744 (2022).
Tesi, B. et al. Precision medicine in rare diseases: What is next? J. Intern. Med. 294, 397–412 (2023).
Groft, S. C., Posada, M. & Taruscio, D. Progress, challenges and global approaches to rare diseases. Acta Paediatr. 110, 2711–2716 (2021).
Haque, B. et al. Contemporary aetiologies of medical complexity in children: a cohort study. Arch. Dis. Child. 108, 147–149 (2023).
Balestrini, S., Mei, D., Sisodiya, S. M. & Guerrini, R. Steps to improve precision medicine in epilepsy. Mol. Diagn. Ther. 27, 661–672 (2023).
Saint-Laurent, C., Mazeyrie, L., Yart, A. & Edouard, T. Novel therapeutic perspectives in Noonan syndrome and RASopathies. Eur. J. Pediatr. 183, 1011–1019 (2024).
Hoytema van Konijnenburg, E. M. M. et al. Treatable inherited metabolic disorders causing intellectual disability: 2021 review and digital app. Orphanet J. Rare Dis. 16, 170 (2021).
Lauffer, M. C., van Roon-Mom, W., Aartsma-Rus, A. & Collaborative, N. Possibilities and limitations of antisense oligonucleotide therapies for the treatment of monogenic disorders. Commun. Med. 4, 6 (2024).
Dunbar, C. E. et al. Gene therapy comes of age. Science 359, eaan4672 (2018).
Sutharsan, S. et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: a 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 10, 267–277 (2022).
Chaytow, H., Faller, K. M. E., Huang, Y. T. & Gillingwater, T. H. Spinal muscular atrophy: from approved therapies to future therapeutic targets for personalized medicine. Cell Rep. Med. 2, 100346 (2021).
Frangoul, H. et al. Exagamglogene autotemcel for severe sickle cell disease. N. Engl. J. Med. 390, 1649–1662 (2024).
Kim, J. et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 381, 1644–1652 (2019).
Dowling, J. J. et al. AAV gene therapy for hereditary spastic paraplegia type 50: a phase 1 trial in a single patient. Nat. Med. 30, 1882–1887 (2024).
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177 (2023).
O’Shea, R., Ma, A. S., Jamieson, R. V. & Rankin, N. M. Precision medicine in Australia: now is the time to get it right. Med. J. Aust. 217, 559–563 (2022).
Palmer, E. E., Howell, K. & Scheffer, I. E. Natural history studies and clinical trial readiness for genetic developmental and epileptic encephalopathies. Neurotherapeutics 18, 1432–1444 (2021).
Routen, A. et al. Strategies to record and use ethnicity information in routine health data. Nat. Med. 28, 1338–1342 (2022).
Kotecha, D. et al. CODE-EHR best practice framework for the use of structured electronic healthcare records in clinical research. BMJ 378, e069048 (2022).
AlMail, A. et al. Consensus reporting guidelines to address gaps in descriptions of ultra-rare genetic conditions. npj Genom. Med. 9, 27 (2024).
Brunklaus, A. et al. The gain of function SCN1A disorder spectrum: novel epilepsy phenotypes and therapeutic implications. Brain 145, 3816–3831 (2022).
Brunklaus, A. et al. Development and validation of a prediction model for early diagnosis of SCN1A-related epilepsies. Neurology 98, e1163–e1174 (2022).
Howell, K. B. et al. SCN2A encephalopathy: a major cause of epilepsy of infancy with migrating focal seizures. Neurology 85, 958–966 (2015).
Wolff, M. et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336 (2017).
Berecki, G. et al. Functional correlates of clinical phenotype and severity in recurrent SCN2A variants. Commun. Biol. 5, 515 (2022).
Summers, J. et al. An integrated clinical approach to children at genetic risk for neurodevelopmental and psychiatric conditions: interdisciplinary collaboration and research infrastructure. J. Neurodev. Disord. 16, 37 (2024).
Smith, L. A. et al. A model program for translational medicine in epilepsy genetics. J. Child Neurol. 32, 429–436 (2017).
Trajanoska, K. et al. From target discovery to clinical drug development with human genetics. Nature 620, 737–745 (2023).
Zuberi, S. M. et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 63, 1349–1397 (2022).
Barcia, G. et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259 (2012).
Heron, S. E. et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44, 1188–1190 (2012).
McTague, A. et al. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology 90, e55–e66 (2018).
Milligan, C. J. et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 75, 581–590 (2014).
Bearden, D. et al. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol. 76, 457–461 (2014).
Xu, D. et al. Precision therapy with quinidine of KCNT1-related epileptic disorders: a systematic review. Br. J. Clin. Pharmacol. 88, 5096–5112 (2022).
Mikati, M. A. et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann. Neurol. 78, 995–999 (2015).
Mullen, S. A. et al. Precision therapy for epilepsy due to KCNT1 mutations: a randomized trial of oral quinidine. Neurology 90, e67–e72 (2018).
Geoerger, B. et al. Precision cancer medicine platform trials: concepts and design of AcSe-ESMART. Eur. J. Cancer 208, 114201 (2024).
Subbiah, V. The next generation of evidence-based medicine. Nat. Med .29, 49–58 (2023).
Duan, X. P. et al. New clinical trial design in precision medicine: discovery, development and direction. Signal Transduct. Target. Ther. 9, 57 (2024).
Schulz, A. et al. Study of intraventricular cerliponase alfa for CLN2 disease. N. Engl. J. Med. 378, 1898–1907 (2018).
Finkel, R. S. et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2016).
Sullivan, J. et al. Adaptive functioning and neurodevelopment in patients with Dravet syndrome: 12-month interim analysis of the BUTTERFLY observational study. Epilepsy Behav. 151, 109604 (2024).
Perry, M. S. et al. Severe communication delays are independent of seizure burden and persist despite contemporary treatments in SCN1A+ Dravet syndrome: insights from the ENVISION natural history study. Epilepsia 65, 322–337 (2024).
FDA. Rare Disease Endpoint Advancement Pilot Program (US Food and Drug Administration, 2024).
MHRA. Innovative Licensing and Access Pathway (Medicines and Healthcare products Regulatory Agency, 2021).
Rubinstein, Y. R. et al. The case for open science: rare diseases. JAMIA Open 3, 472–486 (2020).
Lumsden, J. M. & Urv, T. K. The rare diseases clinical research network: a model for clinical trial readiness. Ther. Adv. Rare Dis. 4, 26330040231219272 (2023).
Attar, S. et al. Harmonizing quality improvement metrics across global trial networks to advance paediatric clinical trials delivery. Ther. Innov. Regul. Sci. 58, 953–964 (2024).
Lorentzos, M. S. et al. Providing Australian children and adolescents with equitable access to new and emerging therapies through clinical trials: a call to action. Med. J. Aust. 220, 121–125 (2024).
Rockowitz, S. et al. Children’s rare disease cohorts: an integrative research and clinical genomics initiative. npj Genom. Med. 5, 29 (2020).
Rehm, H. L. et al. GA4GH: international policies and standards for data sharing across genomic research and healthcare. Cell Genom. 1, 100029 (2021).
Howell, K. B. et al. A population-based cost-effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia 59, 1177–1187 (2018).
Smith, L. et al. Genetic testing and counseling for the unexplained epilepsies: an evidence-based practice guideline of the National Society of Genetic Counselors. J. Genet. Couns. 32, 266–280 (2023).
Oliver, K. L. et al. Genes4Epilepsy: an epilepsy gene resource. Epilepsia 64, 1368–1375 (2023).
Eisermann, M. M. et al. Infantile spasms in Down syndrome-effects of delayed anticonvulsive treatment. Epilepsy Res. 55, 21–27 (2003).
Berg, A. T., Loddenkemper, T. & Baca, C. B. Diagnostic delays in children with early onset epilepsy: impact, reasons, and opportunities to improve care. Epilepsia 55, 123–132 (2014).
Loddenkemper, T. et al. Developmental outcome after epilepsy surgery in infancy. Pediatrics 119, 930–935 (2007).
Thorogood, A. et al. International Federation of Genomic Medicine Databases using GA4GH standards. Cell Genom. 1, 100032 (2021).
Wilkinson, M. D. et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Mellis, R., Chandler, N. & Chitty, L. S. Next-generation sequencing and the impact on prenatal diagnosis. Expert Rev. Mol. Diagn. 18, 689–699 (2018).
French, C. E. et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 45, 627–636 (2019).
Australian Genomics Health Alliance Acute Care Flagship, et al.Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. JAMA 323, 2503–2511 (2020).
McInnes-Dean, H. et al. ‘Something that helped the whole picture’: experiences of parents offered rapid prenatal exome sequencing in routine clinical care in the English National Health Service. Prenat. Diagn. 44, 465–479 (2024).
Bowman-Smart, H. et al. ‘Diagnostic shock’: the impact of results from ultrarapid genomic sequencing of critically unwell children on aspects of family functioning. Eur. J. Hum. Genet. 30, 1036–1043 (2022).
Goranitis, I. et al. Is faster better? An economic evaluation of rapid and ultra-rapid genomic testing in critically ill infants and children. Genet. Med. 24, 1037–1044 (2022).
Lynch, F., Nisselle, A., Stark, Z., Gaff, C. L. & McClaren, B. Genetics follow up after rapid genomic sequencing in intensive care: current practices and recommendations for service delivery. Eur. J. Hum. Genet. 30, 1276–1282 (2022).
Brown, H. L., Sherburn, I. A., Gaff, C., Taylor, N. & Best, S. Structured approaches to implementation of clinical genomics: a scoping review. Genet. Med. 24, 1415–1424 (2022).
Wilmshurst, J. M. et al. Access to pediatric neurology training and services worldwide: a survey by the International Child Neurology Association. Neurology 101, 798–808 (2023).
Esterhuizen, A. I. et al. Precision medicine for developmental and epileptic encephalopathies in Africa-strategies for a resource-limited setting. Genet. Med. 25, 100333 (2023).
Best, S., Vidic, N., An, K., Collins, F. & White, S. M. A systematic review of geographical inequities for accessing clinical genomic and genetic services for non-cancer related rare disease. Eur. J. Hum. Genet. 30, 645–652 (2022).
Baynam, G. et al. Global health for rare diseases through primary care. Lancet Glob. Health 12, e1192–e1199 (2024).
Chung, B. H. Y., Chau, J. F. T. & Wong, G. K. Rare versus common diseases: a false dichotomy in precision medicine. npj Genom. Med. 6, 19 (2021).
Archibald, A. D. et al. The Australian Reproductive Genetic Carrier Screening Project (Mackenzie’s Mission): design and implementation. J. Pers. Med. 12, 1781 (2022).
Heneghan, M., Southern, K. W., Murphy, J., Sinha, I. P. & Nevitt, S. J. Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del). Cochrane Database Syst. Rev. 11, CD010966 (2023).
Hisert, K. B. et al. Understanding and addressing the needs of people with cystic fibrosis in the era of CFTR modulator therapy. Lancet Respir. Med. 11, 916–931 (2023).
Lopez, A., Daly, C., Vega-Hernandez, G., MacGregor, G. & Rubin, J. L. Elexacaftor/tezacaftor/ivacaftor projected survival and long-term health outcomes in people with cystic fibrosis homozygous for F508del. J. Cyst. Fibros. 22, 607–614 (2023).
Ong, T. & Ramsey, B. W. Cystic fibrosis: a review. JAMA 329, 1859–1871 (2023).
Guide, S. V. et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N. Engl. J. Med. 387, 2211–2219 (2022).
Maguire, A. M. et al. Durability of voretigene neparvovec for biallelic RPE65-mediated inherited retinal disease: phase 3 results at 3 and 4 years. Ophthalmology 128, 1460–1468 (2021).
Pierrache, L. H. M. et al. Longitudinal study of Rpe65-associated inherited retinal degenerations. Retina 40, 1812–1828 (2020).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
Bender, M. A. & Carlberg, K Sickle Cell Disease. In: GeneReviews® [Internet]. (eds Adam, M. P. et al.). (Seattle (WA): University of Washington, Seattle; 1993–2025). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1377/
Payne, A. B. et al. Trends in sickle cell disease-related mortality in the United States, 1979 to 2017. Ann. Emerg. Med. 76, S28–S36 (2020).
Prior, T. W., Leach, M. E. & Finanger, E. Spinal muscular atrophy. In GeneReviews® (eds Adam, M. P. et al.) (Seattle, WA). University of Washington, Seattle; 1993–2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352/
GA4GH. GA4GH Driver Project - International Precision Child Health Partnership. https://www.ga4gh.org/driver_project/international-precision-child-health-partnership-ipchip/ (2023).
Amaral, P. et al. The status of the human gene catalogue. Nature 622, 41–47 (2023).
GENCODEGENES.org/human. GENCODE.
Bick, D. et al. An online compendium of treatable genetic disorders. Am. J. Med. Genet. C Semin. Med. Genet. 187, 48–54 (2021).
www.rx-genes.com. Rx-Genes.
data.unicef.org. UNICEF.
Liao, W. W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Vollger, M. R. et al. Increased mutation and gene conversion within human segmental duplications. Nature 617, 325–334 (2023).
Tanenhaus, A. et al. Cell-selective adeno-associated virus-mediated SCN1A gene regulation therapy rescues mortality and seizure phenotypes in a Dravet syndrome mouse model and is well tolerated in nonhuman primates. Hum. Gene Ther. 33, 579–597 (2022).
Snowball, A. et al. Epilepsy gene therapy using an engineered potassium channel. J. Neurosci. 39, 3159–3169 (2019).
Almacellas Barbanoj, A. et al. Anti-seizure gene therapy for focal cortical dysplasia. Brain 147, 542–553 (2024).
Wengert, E. R. et al. Targeted augmentation of nuclear gene output (TANGO) of Scn1a rescues parvalbumin interneuron excitability and reduces seizures in a mouse model of Dravet Syndrome. Brain Res. 1775, 147743 (2022).
Colasante, G. et al. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol. Ther. 28, 235–253 (2020).
Colasante, G. et al. In vivo CRISPRa decreases seizures and rescues cognitive deficits in a rodent model of epilepsy. Brain 143, 891–905 (2020).
Pisano, T. et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 56, 685–691 (2015).
Julia-Palacios, N. et al. l-serine treatment in patients with GRIN-related encephalopathy: a phase 2A, non-randomized study. Brain 147, 1653–1666 (2024).
Wang, D., Pascual, J. M. & De Vivo, D. Glucose transporter type 1 deficiency syndrome. In GeneReviews® [Internet]. (eds Adam, M. P. et al.) (Seattle, WA): University of Washington, Seattle; 1993-2025). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1430/
Wilson, M. P., Plecko, B., Mills, P. B. & Clayton, P. T. Disorders affecting vitamin B(6) metabolism. J. Inherit. Metab. Dis. 42, 629–646 (2019).
Wolf, B. Biotinidase deficiency. In GeneReviews® [Internet]. (eds Adam, M. P. et al.) (Seattle, WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1322/
French, J. A. et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388, 2153–2163 (2016).
Moloney, P. B. et al. Everolimus precision therapy for the GATOR1-related epilepsies: a case series. Eur. J. Neurol. 30, 3341–3346 (2023).
Xu, Q. et al. mTOR inhibitors as a new therapeutic strategy in treatment resistant epilepsy in hemimegalencephaly: a case report. J. Child Neurol. 34, 132–138 (2019).
Dalic, L. J. et al. DBS of thalamic centromedian nucleus for Lennox-Gastaut syndrome (ESTEL Trial). Ann. Neurol. 91, 253–267 (2022).
Warren, A. E. L. et al. Targeting thalamocortical circuits for closed-loop stimulation in Lennox-Gastaut syndrome. Brain Commun. 6, fcae161 (2024).
Lieb, A. et al. Biochemical autoregulatory gene therapy for focal epilepsy. Nat. Med. 24, 1324–1329 (2018).
Qiu, Y. et al. On-demand cell-autonomous gene therapy for brain circuit disorders. Science 378, 523–532 (2022).
Harkin, L. A. et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 130, 843–852 (2007).
Scheffer, I. E. & Nabbout, R. SCN1A-related phenotypes: epilepsy and beyond. Epilepsia 60, S17–S24 (2019).
Han, Z. et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 12, eaaz6100 (2020).
Brunklaus, A., Ellis, R., Reavey, E., Forbes, G. H. & Zuberi, S. M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 135, 2329–2336 (2012).
Gallagher, D. et al. Genotype-phenotype associations in 1018 individuals with SCN1A-related epilepsies. Epilepsia 65, 1046–1059 (2024).
Li, W., Schneider, A. L. & Scheffer, I. E. Defining Dravet syndrome: an essential pre-requisite for precision medicine trials. Epilepsia 62, 2205–2217 (2021).
Sadleir, L. G. et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology 89, 1035–1042 (2017).
Costain, G., Cohn, R. D. & Malkin, D. Precision child health: an emerging paradigm for paediatric quality and safety. Curr. Treat. Options Pediatr. 6, 317–324 (2020).
Chancellor, D., Barrett, D., Nguyen-Jatkoe, L., Millington, S. & Eckhardt, F. The state of cell and gene therapy in 2023. Mol. Ther. 31, 3376–3388 (2023).
Papaioannou, I., Owen, J. S. & Yanez-Munoz, R. J. Clinical applications of gene therapy for rare diseases: a review. Int. J. Exp. Pathol. 104, 154–176 (2023).
Acknowledgements
We thank the patients, families, referring clinicians and the teams working on the Gene-STEPS study, and other IPCHiP projects. We are grateful for funding and infrastructure support provided by our institutes, as well as grant and philanthropic support. The research conducted at the MCC was supported by the Victorian Government’s Operational Infrastructure Support Program, and received funding from the Murdoch Children’s Research Institute, The Royal Children’s Hospital Foundation (RCHF), the Australian Government’s Medical Research Futures Fund (MRFF), and UCB Australia. The Chair in Genomic Medicine awarded to J.C. is generously supported by RCHF. K.B.H. is supported by a clinician-scientist fellowship from the MCC. The research conducted at SickKids is supported by the University of Toronto McLaughlin Centre, the SickKids Foundation, and Patsy and Jamie Anderson. S.W.S. holds the Northbridge Chair in Pediatric Research at the Hospital for Sick Children and University of Toronto. The research conducted at UCL Great Ormond Street Institute for Child Health and Great Ormond Street Hospital is supported by the National Institute of Health Research (NIHR) Biomedical Research Centre at Great Ormond Street Hospital (GOSH). J.H.C. holds an endowed chair at UCL Great Ormond Street Institute of Child Health, and grants from NIHR, EPSRC, GOSH Charity, ERUK, and the Waterloo Foundation. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. The research conducted at BCH is supported by Boston Children’s Hospital Children’s Rare Disease Cohorts Initiative, the Boston Children’s Hospital IDDRC Molecular Genetics Core Laboratory funded by P50HD105351 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the NIH, the One8 Foundation, the Robinson Fund for Transformative Research in Epilepsy, the American Academy of Pediatrics Marshall Klaus Neonatal-Perinatal Research Award, and the Diamond Blackfan Chair in Neuroscience Research. A.M.D. was supported by T32HD098061 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of NIH.
Author information
Authors and Affiliations
Contributions
All authors (K.B.H., S.M.W., A.Mc.T., A.M.D., G.C., A.P., I.E.S., V.C., L.D.S., S.E.M.S., M.W., A.D., N.S., P.S., A.H.B., L.S.C., R.D.C., C.R.M., N.C.A., K.N.N., J.H.C., J.C., and S.W.S.) contributed to the conceptualization and design of the paper, and reviewed and edited the manuscript. The original draft was written by K.B.H., S.M.W., A.M.D., A.Mc.T., G.C., A.P., L.D.S., C.R.M., J.H.C., J.C., and S.W.S. The figures were designed by K.B.H., A.Mc.T., and S.E.M.S. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
K.B.H. has received research funding from UCB Australia, Praxis Precision Medicines and RogCon Biosciences, Inc., has served on an advisory board for UCB Australia, and is a member of the Scientific and Medical Board for SCN2A Asia-Pacific. N.C.A. is on the Boards of Directors of Novartis, Charles River Laboratories and Maze Therapeutics, and the Scientific Advisory Board of Dyne Therapeutics. A.H.B. has received consulting fees from Astellas Gene Therapies, GLG Inc., Guidepoint Global, and F. Hoffman-La Roche, is on the Scientific Advisory Board of Kate Therapeutics, and holds equity in Kate Therapeutics and Kinea Bio. J.H.C. has acted as an investigator for studies with GW Pharma/Jazz Pharmaceuticals, Zogenix/UCB Pharma, Vitaflo, Stoke Therapeutics, and Ultragenyx. She has been a speaker and on advisory boards for Jazz Pharmaceuticals, UCB, Biocodex, and Nutricia; all remuneration has been paid to her department. J.C. is a member of the Drug Monitoring and Safety Board for Anavex Life Sciences Corp, the Endpoint Adjudication Committee for Taysha Gene Therapies, the Scientific and Medical Advisory Committees of the Childhood Dementia Initiative and the Rett Syndrome Association of Australia, and is the director of the Mito Foundation of Australia. S.W.S. served on the scientific advisory committee of Population Bio, Deep Genomics and intellectual property from his research held at the Hospital for Sick Children and licensed to Athena Diagnostics and Population Bio. S.W.S., R.D.C., and C.R.M. are Editors or Editorial Board Members of npj Genomic Medicine; they were not part of the peer review or decision-making processes for this manuscript. S.M.W., A.McT., A.M.D., G.C., A.P., I.E.S., V.C., L.D.S., S.E.M.S., M.W., A.D., N.S., P.S., L.S.C., and K.N.N. report no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Howell, K.B., White, S.M., McTague, A. et al. International Precision Child Health Partnership (IPCHiP): an initiative to accelerate discovery and improve outcomes in rare pediatric disease. npj Genom. Med. 10, 13 (2025). https://doi.org/10.1038/s41525-025-00474-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41525-025-00474-8
