
Abstract
Parkinson’s Disease (PD) is driven by pathological aggregates of alpha-synuclein (αSyn), whose formation is facilitated by impaired glycosphingolipid metabolism via acidic glucocerebrosidase (GCase). We investigated glucosylceramide (GlcCer) accumulation in human, mouse, and cellular PD models. Lipidomic analyses revealed elevated plasma GlcCer, especially GlcCer24:1, and a shift in phosphatidylcholine (PC) species in PD patients. PD patient skin fibroblasts accumulated more GlcCer under lysosomal stress. GlcCer and sulfatides (SHexCer) were increased in Pink1−/−SNCAA53T PD mouse brains, and HT22 neurons exposed to preformed αSyn fibrils accumulated GlcCer and ceramides. GlcCer24:1 enhanced fibril toxicity, but had no direct or indirect effect on G-protein coupled receptors. RNAseq of GlcCer24:1-treated dorsal root ganglion neurons showed upregulation of glycolipid response genes, similar to pathogen-related signaling. These data indicate extracellular GlcCer is elevated in PD and triggers innate immune responses in sensory neurons.
Similar content being viewed by others


Introduction
The pathology of PD is caused by deposits of oligomeric forms of alpha-synuclein (gene SNCA, αSyn)1,2. Formation of αSyn fibrils is precipitated by glucosylceramides or their glucosylsphingosine metabolites3,4,5, which per se can form twisted ribbon, amyloid-like structures6. Oligomers and fibrils of αSyn spread from cell to cell via extracellular vesicles7 or directly via tunneling nanotubes8 or prion-like cell-to-cell transfer9. The primary and probably only pathway for removal is via autophagolysosomal degradation10,11,12,13. If it fails and lysosomes are overloaded with αSyn14, or if the lysosomal pH and calcium gradients decline under the load of accumulated lipids, lysosomal membranes become leaky15. The resulting release of proteases into the cytoplasm leads to autoproteolysis if it is not prevented by lysophagy, i.e. autophagy of damaged lysosomes15,16. Lysophagy can prevent cell death to some extent but eventually leads into a vicious cycle unless overloaded or damaged lysosomes are expelled as exosomes for temporary relief17, but it results in further spreading of αSyn18,19. Accumulation of αSyn in lysosomes interferes with the catalytic activity of lysosomal enzymes, in particular glucocerebrosidase 1 (gene GBA1, GCase), which is required for degradation of glucosylceramides (GlcCer). In turn, GCase malfunctioning exacerbates deficits of αSyn-degradation3,20,21,22. Accordingly, mutations in genes involved in glycosphingolipid transport such as ATP10B23 or degradation such as VPS3524,25 increase the risk and severity of PD. Particularly mutant GBA1 accelerates αSyn pathology26,27. It is still not completely understood how heterozygous PD-associated GBA1 mutations lead to a strong increase of the risk for developing PD28,29 and the severity of PD, in particular PD-associated dementia. Homozygous “loss-of-function” GBA1 mutations are causative for Gaucher disease30. Such GBA1 mutations, but not the most frequent PD-associated E326K mutation, lead to protein misfolding and hence early degradation, or failure of transport and delivery to the lysosomes31, which depends on the shuttle protein, lysosome integrated membrane protein 2 (LIMP2)32,33. However, misfolding or substantial loss of GCase protein expression or loss of catalytic activity is mostly not seen in heterozygous carriers (as in PD), and mechanistically, the bidirectional pathology of dysfunctional GCase with αSyn is only partially understood34. It was hypothesized that mutant GCase directly precipitates αSyn oligomerization, that accumulation of GCase substrates, mostly GlcCer and other glycosphingolipids, drive αSyn oligomerization, that GCase deficiency results in a loss of lysosomal function and capacity for clearance and turnover of αSyn, that increased oligomeric αSyn in the lysosomes impairs membrane targeting and hence, activity of GCase, and that GCase substrates in turn shield αSyn oligomers from degradation21,35, all finally leading to lysosomal leakage and impairment of mitochondria36. High levels of mutant misfolded GCase protein may also evoke an excessive unfolded-protein-response in the endoplasmic reticulum37 or saturation of the ubiquitin–proteasome pathway38, and defective generation of extracellular vesicles39. It is likely that multiple mechanisms are involved in the bidirectional GCase-αSyn pathology in PD. Dopaminergic neurons of the substantia nigra are particularly vulnerable to αSyn oligomers, and their death manifests as typical PD motor function deficits. Even earlier, but often unrecognized, sensory and autonomic neurons succumb to αSyn and lipid overload, manifesting in sensory loss, chronic muscular, neuropathic and visceral pain and headache40,41. We have shown that sporadic PD patients have increased plasma concentrations of glucosylceramides, which was associated with a loss of thermal perception, mechanical hypersensitivity in Quantitative Sensory Tests, and high pain ratings42. The results suggested that accumulation of glucosylceramides may be common phenomenon in PD, not restricted to the about 5–15% PD patients carrying a pathogenic GBA1 mutation28.
To obtain further insight into the pattern of glycosphingolipids and their impact on other lipids we used a back-translational approach and assessed lipidomic patterns in human PD plasma versus controls, patient derived primary fibroblasts, brain tissue of PD mice carrying a double PD-causative Pink1 deletion plus SNCA mutation (SNCA A53T) and in HT22 hippocampal neurons stimulated with preformed αSyn fibrils, and then we investigated effects of GlcCer in vitro on i) G-protein coupled receptors (GPCRs), ii) handling of preformed aSyn fibrils and iii) transcriptomic changes in vulnerable primary neurons.
GlcCer or HexCer were increased in all PD species/sites that were examined but GlcCer did not affect the uptake of αSyn fibrils or function of GPCRs, the latter suggested to be a target of glucosylsphingosines (GlcSph). Instead, exposure of primary sensory neurons to GlcCer 24:1 resulted in a transcriptomic switch reminiscent of cell responses to pathogen-derived glycolipids and suggests that extracellular GlcCer may mimic pathogenic threats. Since sensory neurons are a site of early premotor manifestations of PD and a source of αSyn spreading the observed mechanism may be relevant to the progression of the disease and potentially susceptible to therapeutic intervention.
Results
Increased plasma glucosylceramides in PD patients
In previous studies, we42 and others43,44 have observed increased plasma concentrations of ceramides and glucosylceramides or glucosylsphingosines in patients with idiopathic PD who were not carrying GBA1 mutations that are associated with mal-folding or dysfunction of the GBA1 gene product, lysosomal glucocerebrosidase alpha (GCase). To gain further insight into PD-associated ceramide pathology we explored and compared lipidomic patterns in plasma (Fig. 1) and primary fibroblasts of PD patients (Fig. 2), in brain tissue of PD-mice (Fig. 3), and in HT22 mouse hippocampal neurons loaded with αSyn preformed fibrils (Figs. 4 and 5).

Patients (n = 16 f, 34 m) and controls (n = 25 f, 25 m) were age-matched <60-70+ years old at the time of blood sampling. Demographic details are shown in Supplementary Table 1. A Volcano plots show the Log2(fold change) on the x-axis versus the negative Log10 of the t-test P-value on the Y-axis. Increased lipids in PD are in red, reduced in blue. Lipids with statistically significant P-value but below the threshold for fold change are in orange. The Volcano plots reveals changes in FA, HexCer and PC which are detailed in (B–D). Please note that hexosylceramides (HexCer) represent GlcCer and GalCer. Hex2Cer mostly LacCer. B Fatty acids (FA) female and male PD patients and controls. C Hexosylceramides obtained by targeted and untargeted (TOF) lipidomic analyses. D Phospahtidylcholines (PC). The line is the mean, the whiskers show the SD. Each scatter is a subject. Data were submitted to 2-way ANOVA and subsequent posthoc analysis for each lipid species using a adjustment of alpha according to Sidak. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. CAR carnitines, CER ceramides, CE cholesterol ester, DG diglycerides, FA fatty acids, HexCer hexosylceramides, LPC lysophosphatidylcholines, LPE lysophosphatidylethanolamines, LPG lysophosphatidylglycerols, LPI lysophosphatidylinositols, PC phosphatidylcholines, PE phosphatidylethanolamines, PD phosphatidylglycerols, PI phosphatidylinositols, SM sphingomyelins, ST sterols, TG triglycerides, UbiQ ubiquitin, –O ether bound.

Primary fibroblast cultures were obtained from 3 mm skin biopsies of the lower leg from n = 12 (9 f, 3 m) HC and n = 13 (4 f, 9 m) PD patients. Sphingolipids were analyzed by targeted LC-MS/MS from 2.5 ×105 primary human fibroblasts per sample, and concentrations in ng/ml were auto-scaled to have an common mean and variance of 1 (Z-score). Sub-confluent cultures were stimulated with 12.5 µM pimozide (PIM 12.5 µM = 1/5th of EC50) or vehicle (1:10000 DMSO) and harvested at 24 h. A Box/scatter plots of sphingolipid z-scores. Pimozide treatment raised Cer 24:1 and GlcCer’s and reduced sphingosine (SPH d18:1) predominantly in PD-PHF. The box shows the interquartile range, the line is the median, whiskers show minimum to maximum, scatters are individual subjects. Statistics: 2-way ANOVA and subsequent t-tests with adjustment of alpha according to Sidak for the between subject factor (4 groups) *P < 0.05, **P < 0.01, ***P < 0.001. B Score plots of a canonical discrimination analyses using sphingolipid concentrations as input. Clusters of vehicle treated PHF are overlapping, but pimozide treated PD-PHF differ from HC-PHF. C Paired analysis of for the most abundant GlcCer 16:0 and GlcCer24:1. PD fibroblasts show a stronger PIM evoked increase. D Violin plots reveal stronger variance of sphingolipid levels in PHF of PD patients than controls. Data as in (A).

A Ceramides and hexosylceramides in brain tissue (cortex, subcortex, midbrain) in Pink1−/−SNCAA53T and Sv129FVB wildtype control mice (age 50–60 weeks) analyzed by targeted LC-MS/MS analysis. Each scatter is one mouse. Data were submitted to 2-way ANOVA using the between subject factor “genotype” and the within subject factor “ceramide”, followed by posthoc t-tests with adjustment of alpha according to Sidak for genotype. *P < 0.05, ****P < 0.0001. B Ceramides and hexosylceramides in brain tissue (cortex, subcortex, midbrain) in Pink1−/−SNCAA53T and Sv129FVB wildtype control mice (age 50–60 weeks) analyzed by untargeted UHPLC-MS/MS lipidomic screen. Data were analyzed using 2-way ANOVA with the between subject factor “genotype” and the within subject factor “ceramide”, followed by posthoc analysis for each lipid using the false discovery rate for adjustment of alpha. Heatmaps of top 50 regulated lipids are presented in Supplementary Fig S3.

A Ceramide and hexosylceramide concentrations in picograms per 100,000 HT22 mouse hippocampal neurons as assessed by targeted LC-MS/MS analysis. Cells were seeded in the absence or presence of PFF at 2 µg/ml or 6 µg/ml in culture flasks and grown for 48 h. Control cells were treated with the respective volume of DMSO or were left untreated (naïve HT22). Each group consisted in 8 replicates. Data were submitted to 2-way ANOVA and subsequent posthoc t-test using an adjustment of alpha according to Sidak for the between subject factor “treatment” (i.e. 4 groups). *P < 0.05, **P < 0.01. High PFF increased all ceramides. B Heatmap of top 50 regulated lipids in HT22 mouse hippocampal neurons as assessed by UHPLC-MS/MS lipidomic analysis. Cells were treated as explained in (A). AUC/IS values were square root transformed and auto-scaled to have a common mean and variance of 1 (z-scores). Lipids were clustered according to Euclidean distance metrics using the Ward method. C Principal Component Analysis of HT22 lipidomic data showing a 3D-score scatter plot of the first three PC. Each scatter shows one sample. The circles are 90% confidence elipsoids.

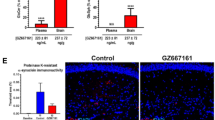
Immunofluorescent studies show the uptake of AF555-labeled αSyn preformed fibrils in HT22 mouse hippocampal neurons. Pretreatment with GlcCer24:1 (1 µM) had no significant impact on the PFF-AF555 fluorescence uptake in HT22 cells (pink). Wheat germ agglutin WGA-AF488 (yellow) is used as counterstain of plasma membranes (early time point) and endolysosomes (late time point). Hoechst-33342 is used as nuclear stain. Additional zoom-in images are shown in Supplementary Fig. S5.
Plasma lipidomic studies of 50 PD patients versus 50 age-matched healthy controls (HC) revealed increased hexosylceramides in PD, particularly GlcCer24:1 (Fig. 1A, Supplementary Fig. S1), stronger in male than female PD patients (Fig. 1C), whereas a number of mono- and poly-unsaturated fatty acids were reduced without differences between sexes (Fig. 1B). In addition, phosphatidylcholine species (PC) shifted from long chain poly-unsaturated PC species to shorter mono-unsaturated PCs (Fig. 1D) without overall change of summed PCs, which agrees with a recent study where a set of serum lipids including PC species allowed for a better prediction of PD progression/outcome than age, sex and risk gene LRRK2 mutation status45. PCs are connected with GlcCer via the phospholipid translocase ATP10 (ATP10A, 10B and 10D), which flips PC in exchange with GlcCer in the plasma membrane, and contributes thereby to membrane dynamics23,46,47, and is considered as PD risk gene23. A recent study shows that ATP10 rather than GCase accounts for increased GlcCer in PD48.
Plasma lipidomic data and metadata are available at https://www.ebi.ac.uk/biostudies with the accession number S-BSST1880. https://www.ebi.ac.uk/biostudies/studies/S-BSST1880?key=a0dc6f53-0365-410a-96e3-d83e51a04282.
Increased glucosylceramides in PD primary fibroblasts upon stimulation with pimozide
GlcCer are generated in the ER, from there transported and inserted into the plasma membrane and membranes of organelles, and they are degraded in the lysosomes49. Considering the autophagolysosomal pathophysiology of PD the most likely mechanism underlying increased GlcCer in plasma is mal-degradation in the lysosomes12,14,50,51. To study this aspect we used primary patient derived fibroblasts that were stimulated with pimozide52.
Patient derived primary fibroblasts are considered a viable model for studying neurodegenerative changes due to their metabolic and biochemical relationships with neurons53. In particular, the phenotype of skin fibroblasts of late-onset sporadic PD subjects recapitulated features of iPSC derived midbrain dopamine neurons from the same patient, including PD-associated changes in morphology, mitochondrial function, and autophagy54. Fibroblasts of PD patients express about 5-10 fold more αSyn55 and are used for early diagnosis of prodromal symptoms56. Further, sphingolipid changes found in PD fibroblasts precipitated the aggregation of αSyn57. Primary patient derived fibroblasts were therefore chosen to further study GlcCer alterations.
To mimic PD-like lysosomal dysfunctionality, primary human fibroblasts (PHF) of 13 PD patients and 12 HC were stimulated with pimozide, which is a lysosome-tropic, antipsychotic drug. As a weak base (pKa 8.36) pimozide is trapped in lysosomes but does not cause major lysosomal alkalinization and therefore does not cause complete disruption of lysosomal functions like chloroquine52. At high concentrations, its accumulation in lysosomes can cause lysosomal membrane permeabilization and lysophagy15 but at lower concentrations it mainly interferes with lysosomal (sphingo)-lipid metabolism that manifests in an increase of ceramides and GlcCer in cell lysates15,52. It was therefore an ideal stimulus to assess subtle differences of lysosomal functions in PD versus control fibroblasts.
The IC50 in primary human fibroblasts (PHF) of pimozide-evoked cell death was 90-100 µM (Supplementary Fig. S2). To assess effects on lipid homeostasis, PHF were stimulated for 24 h with 1/8th of the IC50 (i.e. 12.5 µM). At baseline, there was no difference between PD and HC fibroblasts in any of the analyzed sphingolipids (Fig. 2). Pimozide increased ceramides and reduced sphingoid base sphingolipids (sphingosine, S1P) in both PD and HC fibroblasts in line with previous observations in tumor cells15, but the pimozide-evoked increase of glucosylceramides was stronger in PD fibroblasts (Fig. 2A, C). Canonical discrimination analyses using all sphingolipids as input was able to distinguish PD-PIM versus HC-PIM fibroblasts (Fig. 2B). Further, violin plots of Z-transformed sphingolipids revealed higher variability of PD fibroblasts as compared with HC fibroblasts (Fig. 2D). The results suggest higher vulnerability of PD-fibroblasts towards a pimozide-evoked raise of lysosomal pH possibly because PD fibroblasts have a higher lysosomal burden of αSyn55 and therefore lower capacity to adjust lysosomal hydrolase activity.
Fibroblast sphingolipid raw data and metadata are available at https://www.ebi.ac.uk/biostudies with the accession number S-BSST1881. https://www.ebi.ac.uk/biostudies/studies/S-BSST1881?key=4e8ae53f-17f0-4179-80ff-40392e418270.
Increased ceramides and hexosylceramides in PD mouse brain
Plasma hexosylceramides are maintained and replenished from peripheral and central sources. The relative contribution is unknown. Elevated plasma levels in PD patients suggested disease-associated metabolic changes, but plasma levels represent the extracellular compartment and may not necessarily directly reflect brain tissue. Therefore, we used PD-mice to get direct insight into brain ceramides. As explained in the description of the PD mouse model, we opted for Pink1−/−SNCAA53T mice because they combine features of human PD including a premotor disease with sensory symptoms and development of spontaneous motor deficits.
Targeted (Fig. 3A) and untargeted (Fig. 3B) lipidomic studies revealed increased ceramides in the brain of double mutant Pink1−/−SNCAA53T mice compared with wildtype Sv129-FVB control mice, in agreement with previous ceramide studies in Pink1−/− mouse brain58. Importantly, Pink1−/−SNCAA53T mice had no clinical symptoms of PD motor disease at the time of tissue sampling at ~12 months of age. Ceramides, hexosylceramides (GlcCer or GalCer) and sulfatides (SHexCer) were increased in Pink1−/−SNCAA53T brains whereas acetyl-HexCer (AHexCer) were decreased (Fig. 3B). Lipidomic analyses further revealed increased diacylglycerols (DG) and lysophosphatidylethanolamines (LPE) in Pink1−/−SNCAA53T brains (Supplementary Fig. 3). DGs are normally low in the brain, and high brain DGs have been suggested to indicate CNS disease59. LPEs are contained in Lewy bodies and likely precipitate αSyn aggregation, depending on C-chain length and saturation43. Lipidomic heat maps of candidate lipids also show a switch of PC species not equal but corresponding to the patterns in human plasma, mainly a decrease of long-chain PC-O and increase of shorter PCs, mostly with low saturation.
Brain sphingolipid and lipidomic and metabolomic raw data and metadata are available at available at https://www.ebi.ac.uk/biostudies with the accession number S-BSST1888. https://www.ebi.ac.uk/biostudies/studies/S-BSST1888?key=6230dc37-d998-4eca-bf0f-3502f7164655.
Increased ceramides in HT22 neurons upon ingestion of preformed αSyn fibrils
Mutant GBA1 and αSyn mutually increase neuronal damage likely converging on lysosomal collapse22,50 and mitochondrial toxicity36. To mimic the αSyn part, HT22 mouse hippocampal neurons were exposed to active preformed αSyn fibrils, which were supplied via the culture medium. Using confocal live imaging microscopy it was confirmed that HT22 cells ingested PFF, thus creating PD-like HT22. HT22 cell were chosen as a model because of their well-established sensitivity to PD-inducing pro-oxidative agents, expression of dopamine receptors, clathrin endocytosis machinery and the observed reliable uptake of αSyn-PFF.
Lipidomic analyses of such PD-like HT22 showed that the exposure and ingestion of αSyn fibrils increased ceramides and hexosylceramides. The higher dose of 6 μg/ml caused stronger changes than the low dose of 2 µg/ml (Fig. 4A). αSyn fibrils alone had no effect on cell viability as assessed by WST-1 assays (Supplementary Fig. 4A, B), nor did it consistently affect polar metabolites. At high concentration, αSyn-PFF treatment resulted in an increase of LDH activity in supernatants in some cultures (Supplementary Fig. S4C), suggesting damage of the cell membrane (cytosolic LDH) or lysosomal exocytosis (lysosomal LDH). Lipidomic analyses (Fig. 4B, C) revealed that ceramides were among the top upregulated lipids exposed to αSyn PFF. In agreement with mouse brain and human plasma, PFF ingestion was associated with an increase of short-chain PC but decrease of long-chain PC (Supplementary Fig. S4C). In addition, heatmap analyses showed a strong increase of phosphatidylinositol (PI) species which had not been observed in mouse brain or human plasma. Score plots of a principal component analysis showed a distinction of PFF-treated versus not-treated cells, but no PFF dose-dependent difference.
HT22 lipidomic and metabolomic data and metadata are available at https://www.ebi.ac.uk/biostudies with the accession number S-BSST1897. https://www.ebi.ac.uk/biostudies/studies/S-BSST1897?key=d2a20a65-a88f-44a4-9654-672a72f8a9ac.
No significant increase of PFF ingestion in the presence of GlcCer24:1
It has been suggested that GBA1 deficiency exaggerates the exosomal spreading of αSyn60,61 likely because the release of exosomes which are enriched in GlcCer62 is a compensatory mechanisms to get rid of excess GlcCer. Spreading also requires subsequent uptake in recipient cells. Therefore we studied in a series of confocal live imaging experiments if extracellular GlcCer 24:1 enhanced the uptake or distribution of αSyn PFF in HT22 hippocampal neurons. The plasma membrane was visualized with fluorescent wheat germ agglutinin (WGA-AF488). During the observation time, WGA was taken up and moved to the lysosomes (Supplementary Fig. S5 WGA co-staining with LysoID®). Hence, in the first images, WGA is localized mostly at the outer membrane (left panel in Fig. 5), whereas it co-segregates with αSyn PFF in the lysosomes at late images (right panel, Supplementary Fig. S5 co-staining with LysoID®). The middle panel is a time point in between. GlcCer 24:1 treatment had no overt effect on the quantity of ingested αSyn PFF but the cell size was more variable in GlcCer 24:1 plus PFF-treated cell cultures and some cells had thin cell-to-cell contacts (Fig. 5 bottom row left image, Supplementary Fig. S5 Zoom-in) and WST-1 assays showed that the combination of GlcCer 24:1 plus αSyn PFF for 48 h increased the percentage of non-viable cells (Supplementary Fig. S4).
No activation of G-protein coupled receptors by GlcCer 24:1 and 18:1
High levels of GlcCer in extracellular fluids (plasma) suggest that GlcCer get into contact with the outer membrane, membrane receptors, transporters or channels. We have previously observed that GlcCer in culture medium increased stimulated calcium fluxes in a fraction of neurons, but not per se without additional stimulus63. GlcCer are important components of membrane microdomains and hence involved in the regulation of membrane receptor insertion and function64,65. Considering the importance of dopamine receptors in PD and previous reports showing activation of serotonin receptors with GlcSph we assessed putative direct effects of the top PD-associated GlcCer candidates in human plasma, GlcCer18:1 and GlcCer24:1, on G-protein coupled receptors via beta-arrestin screening and dynamic mass redistribution (DMR) assays. Compared with the positive control (carbachol, muscarinic GPCR), GlcCer18:1 and GlcCer24:1 had no effect on any of the screened GPCRs (Fig. 6A, B; Supplementary Fig. S6 showing replicates) at a reasonable concentration range up to 10 µM. At 10 µM, GlcCer24:1 increased beta-arrestin binding up to 3-fold in HTLA cells with heterologous expression of OPRL1 (opioid related receptor, nociceptin/orphanin FQ). The result was reproducible in quadruple replicates (Supplementary Fig. S6) but still weak compared with the positive controls.

A Beta arrestin-based screening of GPCR activity in a heterologous expression model in COS cells upon stimulation with GlcCer 18:1 versus vehicle at 1, 5 and 10 µM. The heatmap shows the mean of four replicates for candidate GPCR selected from an initial screen (330 GPCRs, 8 neg and 4 × 2 pos controls). Details in Supplementary Fig. 1. B As in A but stimulation with GlcCer 24:1 C: Dynamic Mass Redistribution (DMR) analysis of candidate GPCRs stimulated with GlcCer 18:1 or GlcCer 24:1 at 1 µM. DMR measures a ligand-induced shift in resonant wavelength in picometers. Three candidate GPCR were tested with GlcCer 18:1 (upper left) and 14 with GlcCer 24:1. Carbachol activation of muscarinic receptor CHRM1 was used as positive control (right Y-axis). MCHR1 melanin-concentrating hormone receptor 1, FFA3 free fatty acid receptor 3 (GPR41), CXCR7 CXC-type chemokine receptor 7, GPR4 G-protein coupled receptor 4, pH-sensing, GPRC5A orphan GPCR, alias: retinoic acid-induced protein 3 (RAI3), GPR77 C5a anaphylatoxin chemotactic receptor, GPR82 and GPR85 both orphan, P2RY8 P2Y purinoceptor, NPSR1 neuropeptide S receptor 1, GRM5 metabotropic glutamate receptor, GPPR gastrin releasing peptide receptor, GPR123 adhesion GPCR, GPR143 ocular albinism type 1 (OA1), receptor for tyrosine, L-dopa and dopamine, BDKRB2 bradykinin receptor B2, AVPR2 arginine vasopressin receptor 2, ADRA1B alpha-1B adrenoreceptor.
GlcCer 24:1 causes inflammatory transcriptional response in primary mouse neurons
The loss of motor functions in PD is often preceded with non-motor symptoms originating from manifestations of the disease in the peripheral somatosensory and autonomous nervous systems66,67. The premotor phase may last for years and causes (among others) PD-associated sensory neuropathy68 with sensory loss and pain69,70,71, and restless leg syndrome72. In “body-first” PD, αSyn spreading originates from the peripheral nervous system73,74,75. Somatosensory neurons are therefore in many cases the site of the disease onset where the progression may still be responsive to disease-modifying intervention. We have shown previously that sensory loss and pain is associated with high GlcCer in plasma (PD patients) or DRGs and sciatic nerve tissue (mice) and that treatment of dorsal root ganglia (DRG) neurons with GlcCer 24:1 leads to an exaggerated calcium influx upon stimulation42,63. Therefore, we used adult DRG neurons to gain further insight into the effects of GlcCer using mRNA sequencing to reveal transcriptomic changes that occur when DRG neurons are exposed to GlcCer 24:1 (Fig. 7).

A Volcano plots of regulation of mRNA expression assessed by RNAseq in primary sensory neurons of the dorsal root ganglia from adult mice treated with 1 µM GlcCer24:1 versus vehicle. The x-axis shows the Log2(Fold change) of TMM-normalized counts. The y-axis shows the negative logarithm of the t-test P-value. The data are from n = 4 cultures per condition. Upregulated genes are shown in red, downregulated in blue. B Candidate genes were selected according to the FDR-adjusted P-value and are sorted according to abundance and presented as TMM-normalized reads (trimmed mean of m values). C Candidate genes were submitted to GO and pathway enrichment analysis using DAVID, STRING and Panther consistently showing an enrichment of genes involved in “response to glycolipid”.
RNAseq revealed an upregulation of a number of membrane-associated genes (GO CC: membrane) and overrepresentation of genes involved in the “response to glycolipids” (GO BP or similar terms such as “response to LPS”, “response to pathogens”). The results suggest that extracellular GlcCer can induce inflammatory responses similar to those induced by pathogens. The results agree with a recent study showing that glucosylceramide accumulation due to GCase deficiency caused macrophage activation and neuroinflammation76. Candidate genes, gene descriptions and GO studies are presented in a Supplementary Excel file. RNAseq data have been deposited as GEO dataset with the provisional accession number GSE262573.
To review GEO accession GSE262573.
Enter token clytuucuxjupzmj into the box.
Discussion
Lewy bodies are primarily composed of α-synuclein fibrils intertwined with lipids. Mutations in genes responsible for lipid degradation or transport, such as GBA1, ATP13a2 and ATP10b, have been identified as factors that increase the risk of PD23,77. Although mechanistically complex, lipidomic research suggests that an accumulation of glycosylated sphingolipids may enhance the pathogenicity of αSyn3,4,78 by precipitating the formation of αSyn fibrils6. In the present study, we show that glucosylceramides are increased in the plasma of PD patients, in the brain of PD mice, in primary human fibroblasts of PD patients treated with pimozide and in HT22 hippocampal neurons after ingestion of preformed αSyn fibrils, together strongly suggesting that αSyn interferes with metabolic homeostasis and/or release of hexosylceramides. The predominant candidate was GlcCer 24:1. Except AHexCer in the mouse brain, all GlcCer (HexCer) species were increased in human PD, or PD-models. However, GlcCer 24:1 (and GlcCer 18:1 in some experiments) had little effect on cell viability, morphology, ingestion of αSyn, or GPCR activation which agrees with a recent study that shows high plasma GlcCer in association with PD but not in a causative manner. The authors concluded that GlcCer per se was not pathogenic48 but changes of GlcCer need to be considered in the context of more complex changes of lipids that are connected via metabolic and transport pathways. In particular, we and several previous studies observed changes in a number of phosphatidylcholine (PC) species43,79,80,81,82 whose specific functions are presently mostly elusive, but determine the secondary structure of αSyn oligomers83,84 and are intricately linked to endolysosomal transfer and membrane insertion of GlcCer via flippases77,79,85. Nonetheless, alterations of HexCer are the most robust finding in PD and models of PD and our RNAseq studies have shown that GlcCer 24:1 treatment of primary neurons elicited a pro-inflammatory response. In support, a recent study in a PD fly model showed that GlcCer caused cellular immune activation76.
Considering the close association of glucosylceramides with the risk and the course of PD we asked how elevated glucosylceramides might contribute to an unfavorable course of PD. We considered three putative mechanisms: (i) by activation or inhibition of GPCR signaling (based on studies with hexosylsphingosines86,87), (ii) by facilitating αSyn uptake and (iii) by causing a glycolipid-like immune response. We found no evidence for the first two hypotheses but some support for the third.
In β-arrestin-based GPCR screening assays, GlcCer 24:1 induced a dose-dependent increase of β-arrestin binding in OPRL1 positive clones (nociception/orphanin-FQ) up to 3-fold. However, the effect was weak compared with the positive control carbachol, which increased the β-arrestin signal up to 50-fold upon binding to muscarinic choline receptors (CHRM1). In DMR-based GPCR screening, GlcCer had no effect. We infer that GlcCer 24:1 (and 18:1) do not activate or inhibit GPCRs at concentrations which might be reached in extracellular fluids. Importantly, GlcCer 24:1 or 18:1 had no direct effect on dopaminergic receptors which could have been a putative disease-relevant site of action. It is of note that glucosylsphingosines (GlcSph), which are the deacylated (lyso-) form of glucosylceramides, were found to activate serotonin receptors88 and that glucosylsphingolipids may indirectly affect GPCR by changes of membrane composition and lipid rafts89.
Both, GlcCer and GlcSph accumulate in brain and peripheral organs in Gaucher disease90, but GlcSph are preferred as “biomarkers” in Gaucher disease91 because GBA1 deficient macrophages (Gaucher cells) were reported to accumulate predominantly the lyso-glycosphingolipid92, which however does not confer more or less toxicity to the acylated or deacylated version for neurons in the context of neuronal type Gaucher disease and PD, but might explain why GlcSph has gained more attention93,94,95 and is analyzed for newborn Gaucher screening96,97. But GlcCer and gangliosides rather than GlcSph were found to precipitate αSyn oligomerization in acidic environments in vitro98.
αSyn monomers normally bind to the surface of synaptic vesicles99,100. Their oligomerization properties enable vesicle tethering but αSyn is not essential for synaptic vesicle cycling and release101. However, excess or mutant or phosphorylated αSyn adopts beta sheet rich conformations which are prone to oligomerization, further aggregation and fibril formation and spread like prion-like proteins9,102,103. αSyn uses several means of cell-to-cell spreading104,105 including transsynaptic transfer from neuron to neuron, traveling through tunneling nanotubes8,51,106, packaging into and release of extracellular vesicles which can reach far distant target cells and are taken up endocytosis7,107. Further, αSyn can be expelled via lysosomal exocytosis likely together with GlcCer thereby creating a hostile microenvironment108. GlcCer are also components of exosomal membranes109 and may assist in packaging of αSyn into exosomes110, and define the target cells111. Our experiment created a neuronal microenvironment with excess free non-exosome-packed αSyn fibrils (plus/minus GlcCer24:1) and high extracellular GlcCer, a situation that occurs when αSyn fibrils or aggregates are expelled from cells via exocytosis or lysosomal exocytosis. Because of the excess of PFF, all neurons ingested the fibrils, and our experiment did not allow for live-observation of αSyn-PFF spreading from one population of neurons to another. In extracellular fluids, GlcCer likely bind to albumin or other proteins, but the relative contribution of exosomal, albumin/protein-bound and free GlcCer is unknown. Under the experimental conditions of this study, GlcCer did neither increase the uptake of αSyn-PFF nor the accumulation in lysosomes. However, results may differ in mixed cultures that include immune cells and glia, and/or in compartmentalized cultures with localized preformed fibril (PFF) seeds.
In myeloid immune cells, GlcCer have been described as ligands of Mincle (Clec4e)112,113, which is an inducible C-type lectin and innate immune receptor of macrophages, neutrophils and dendritic cells that senses glycolipids of the mycobacterial cell wall (trehalose 6,6’-dimycolate, known as cord factor)114,115 and other glycolipid species derived from pathogens or damaged and dead cells116,117,118. Mincle (Clec4e) was weakly expressed in our primary sensory neuron cultures (RNAseq studies). For comparison, Clec4e total read counts ranged from 11 to 70, whereas total read counts of marker genes for DRG neurons were in the range of 100–4000, e.g. TRPA1 ̴100, TRPV1 ̴300, Scn10a (Nav1.8) ̴∼800, TAC1 (Substance P) ̴4000, and relevant lipid-sensitive GPCRs were e.g. LPAR5 ̴∼15, PTGER4 (EP4) ̴∼50, CNR1 (CB1) ̴∼300, S1PR1 ̴∼70 and S1PR3 ̴∼600. Hence, Clec4e expression was low but still in a range where its activation may result in measurable effects. In line with this reasoning we found that treatment of primary sensory neurons with GlcCer 24:1 resulted in a transcriptional response similar to that observed by stimulation of cells with bacterial glycolipids. Gene ontology analyses of upregulated genes showed an enrichment of terms like “toll-like receptor signaling”, “positive regulation of innate immune response”, “pattern recognition receptor signaling”. The data suggest that high extracellular GlcCer may induce an innate immune response, which is adequate for the defence against mycobacterium but may contribute to neuroinflammation if it were to occur in the brain. The latter is supported by a study where GlcCer stimulated microglia phagocytosed living neurons119. We used primary sensory neurons because they are a site of early PD manifestations and sensitive to GlcCer but it needs to be assessed in future studies if GlcCer elicits similar innate immune responses in neurons of the CNS.
C-type lectin activation leads to spleen tyrosine kinase (Syk) mediated downstream signaling involving phospholipase C, protein kinase C, MAP kinases and nuclear factor kappa B (NFκB). Considering that the most frequent loss-of-function mutation, GBA1 N370S, that causes Gaucher disease confers resistance to tuberculosis in a zebrafish model120 it is tempting to speculate that GlcCer-mediated Mincle/Clec4e activation is protective against pathogens but possibly harmful if it sustains a microglial attack of neurons. It has been shown that Syk inhibitors reduce neuroinflammation, but so far only in models of stroke or vascular brain injury121, not yet for Gaucher disease or GBA1-associated Parkinson’s disease. It is of note that a recent study suggested to add a Mincle activating adjuvant to αSyn-peptides to enhance the efficacy of active PD vaccination122. The vaccine consisted in a glucan-αSyn conjugate, i.e. not a lipid-conjugate but the strategy reduced fibril propagation in an in vivo synuclein-seeding-spreading model. Hence, although the molecular target of GlcCer remains unidentified its immune-stimulating effect might be beneficial if it could be harnessed to enhance fibril elimination.
The present study has limitations. It focused on GlcCer in non-GBA1-associated PD, but it would have been valuable to include some confirmed GBA1 mutant patients and GBA1-mutant mice for comparison as “positive” control, particularly because we did not find quantifiable glucosylsphingosine species in our lipidomic analysis, which are supposed to be biomarkers for Gaucher disease. Although the frequency of PD-associated GBA1 mutations is low in Germany, it is a limitation that our patients were not genotyped for GBA1 or other PD-associated genes that affect lipid metabolism or transport. Hippocampal mouse HT22 neurons and primary mouse DRG neurons are valuable models for PD research but human CNS neurons may respond differently to PFF or GlcCer which needs to be assessed in human neurons or organoids in future studies.
In summary, we show that extracellular GlcCer is increased in PD and in PD models likely originating from defective lysosomal functions and the data suggest that extracellular GlcCer may induce an innate immune response in peripheral sensory neurons. Such immune activation may sustain a proinflammatory state but may also attract glia and immune cells to remove extracellular αSyn.
Methods
PD patients versus age-matched healthy controls
PD patients, diagnosed using ICD10 diagnostic criteria for Parkinson’s Disease, were consecutively recruited from the Movement Disorders Department of Neurology of the University Hospital in Frankfurt am Main, Germany as described42. Aged healthy control subjects were consecutively recruited from spouse and friends of PD patients and from the outpatient stroke unit of the University Hospital in Frankfurt am Main. Demographic data of patients and controls have been shown in detail in ref. 42. PD disease severity was assessed according to the Hoehn Yahr rating scale in “ON” periods for patients who experienced substantial disease fluctuations (16 out of 50 patients). Patients were not routinely genotyped for PD-associated GBA1 mutations because the frequency in Germany is low (5-10%). Informed written consent was obtained from all subjects. The study was approved by the Institutional Ethics Committee of the University Hospital of Frankfurt (Approval #458/16, date 13.01.2017, amendment 01.10.2018). Data acquisition and blood sample collection adhered to the Declaration of Helsinki. Demographic data are presented in Supplementary Table 1.
Skin biopsy and primary fibroblast culture of PD patients and controls
After local anesthesia by intradermal injection of lidocaine a 3 mm skin punch biopsy was obtained from the skin of the lower leg 10–15 cm above the ankle. The biopsy was placed in ice-cold DMEM medium for transport. Culture dishes (6-well plates) were prepared by coating the surfaces with FBS and dried under the clean bench. The biopsy was placed in a culture dish within 1 h and cautiously covered with fibroblast medium (low glucose DMEM (100 mg/l), 10% FBS, 1x MEM Non-Essential Amino Acids, 2 mM glutamine, 1% PenStrep) and fibroblasts were allowed to grow for 3–4 days. Fibroblasts were harvested with trypsin and sub-cultured in T75 flasks in fibroblast medium or in DMEM high glucose (5 g/l), 10% FBS, 1% PenStrep. Targeted lipidomic analyses were obtained from naïve cultures and upon stimulation with pimozide versus vehicle. WST-1 assays and immunofluorescence analyses were used to assess EC50 values of pimozide in PHF.
For lipidomic analyses, primary human fibroblast (PHF) cultures were stimulated with 1/8th of the EC50 of pimozide (i.e. 12.5 µM; Merck, #573110-100MG) or vehicle (DMSO 1:10000) for 24 h, in two rounds, each consisting in 25 PHF cultures (13 PD, 12 HC cultures) of matched HC and PD patients. Sub-confluent cultures were split and plated on 10 cm culture dishes, each two for DMSO (Control) and Pimozide. Cells were allowed to attach and grow for 24 h, before treatments were added and cells grown for another 24 h. Cells were washed 3 × in 1× PBS, harvested by trypsinization, pelleted, washed, and adjusted to 250,000 cells per cell pellet which was frozen at −80° until lipid analyses.
Demographic data of PFB donors are presented in Supplementary Table 2.
Pimozide IC50 in primary human fibroblasts
WST-1 and sulforhodamine B (SRB) cell viability/cell death assays were used to define stimulating but non-toxic pimozide concentrations and conditions. PHF were seeded in 100 µl medium/well of a 96 well plate, 5000 per well and allowed to grow for 24 h. The medium was replaced with full medium containing Pimozide at concentrations ranging from 1 to 256 µM (log2 scale 1–8 µM) or 1:10000 DMSO (=vehicle). The next day, 10 µl WST-1 reagent (Merck #11644807001) was added to each well and cells were incubated for 60 min at 37 °C. Formation of the formazan dye, which relies on the metabolic activity was then measured via its absorbance in a multi-well spectrophotometer (TECAN Infinite F200 Pro, RRID: SCR_020543) at 405 nm and 620 nm (reference). Subsequently, the reaction was stopped by adding 20 µl 50% trichloroacetic acid, TCA (w/v) to a final concentration of 10% TCA for 1 h at 4 °C. Wells were washed with distilled water 7-times and were dried. To quantify cell mass, fixed cells were stained by adding 100 µl 0.4% (w/v) sulforhodamine B (SRB) in 1% acetic acid for 30 min at room temperature under shaking. Wells were washed with 1% acetic acid 5-times and cells were lysed by adding 250 µl 10 mM Tris/HCl pH 10.5 for 10 min at room temperature. Absorbance of SRB at 540 nm was measured with TECAN plate reader. The percentage of viable cells compared with DMSO was plotted versus the log2 concentration and fitted with a inhibitory sigmoidal Emax model in GraphPad Prism 9 or 10 (RRID:SCR_002798).
The survival of HT22 cells in the presence of GlcCer24:1 or vehicle and AF555-labeled αSyn-PFF was tested using the WST-1 reagent (Merck #11644807001) as described above. HT22 cells were seeded in 96 well plates (4000 cells/well) and treated with 1 µM GlcCer24:1, with or without αSyn-PFF. After 48 h of incubation, 10 µl of the WST-1 reagent was added to the cells and after a 60 min incubation at 37 °C the absorbance was measured at 450 nm and 620 nm (reference) in a multi-well spectrophotometer as above. Subsequently, the SRB assay was performed.
Double mutant Pink1 −/− SNCA A53T Parkinson-model mice
Homozygous Pink1−/− plus SNCAA53T double mutant mice and Pink1−/− single mutant mice were generated as described in refs. 63,123 by crossing Pink1−/− mice (MGI:3850370) with PrPmtA mice expressing human mutant SNCA A53T (mtA) under the prion promoter (PrP) (MGI:3723258) and then, interbreeding the littermates. Mice are available from Jackson lab as cryopreserved sperm (FVB;129-Pink1tm1Aub X Tg(Prnp-SNCA*A53T)AAub/J; Strain #:017678). Double mutant mice are referred to as Pink1−/−SNCAA53T or in short, Pink1SNCA.
We used Pink1−/−SNCAA53T double mutant mice because unlike other PD models, they develop spontaneous PD-like motor symptoms with an onset at >15 months of age and a frequency of about 20-30 percent123 and they combine the complex PD pathophysiology of pro-oxidative and dysfunctional autophagolysosomal pathways12,124,125,126. Further, in contrast to single mutant mice they develop sensory premotor deficits reminiscent of the human premotor phase that manifests with a sensory loss and pain127 and is associated with increased plasma GlcCer species42. The onset of sensory deficits in Pink1−/−SNCAA53T double mutant is at around 6 months of age and is progressive127. Using 1-year-old mice ensured the presence of PD-like pathology (mitochondrial morphology127, respiratory dysfunction127, synuclein aggregates123, ceramide accumulation127) but avoided overt serious motor deficits and suffering of the animals. Overall, Pink1−/−SNCAA53T double mutant mice show many features of human PD.
Mice had free access to water and food, and they were maintained in climate-controlled rooms with a 12 h light-dark cycle. The maintenance of the breeding colony of Pink1−/−SNCAA53T and observation up to onset of PD symptoms were approved by the local Ethics Committee for animal research (Darmstadt, Germany; FK1032 (12.02.205-11.02.2020) and FK1131 (17.02.2020-31.12.2023). The studies adhered to the guidelines of the Society of Laboratory Animals (GV-SOLAS) and were in line with the European and German regulations for animal research. Mice were observed for ~12 months and were euthanized to obtain tissue and plasma samples for lipid analyses before onset of overt clinical symptoms which occur in about 20 percent of mice >18 months63,123.
Mice tissue collection: brain and plasma
Mice were euthanized with carbon dioxide and blood withdrawal by cardiac puncture, whereby blood was collected into K3+ EDTA tubes, centrifugated at 1300 × g for 5 min, and plasma was transferred to a fresh tube and snap frozen on dry ice or in liquid nitrogen. The brain was dissected for lipidomic and proteomic analyses. Cerebellum and olfactory bulb were removed, and the brain was cut sagittal. Left and right halves were weighed with precision scales and snap frozen on dry ice. Samples were stored at −80 °C until analysis.
HT22 cell culture, αSyn-PFF loading, and immunofluorescence analysis
HT22 is an immortalized mouse hippocampal neuronal cell line which are derived from hippocampal neuronal HT4 cells and are widely used as an in vitro model of PD because of their high sensitivity to glutamate excitotoxicity and PD-inducing toxicants like MPP+ (1-methyl-4-phenylpyridinium) and rotenone128,129. They express dopamine D4 receptors130.
HT22 mouse immortalized hippocampal neurons (RRID:CVCL_0321) were grown in High Glucose Dulbecco’s modified eagle medium (DMEM, Gibco) containing 10% fetal bovine serum (FBS, Gibco), 2 mM L-glutamine (Gibco) and 1x penicillin/streptomycin (Sigma) at 37 °C and in 5% CO2 atmosphere in a humidified cell culture incubator.
For stimulation of HT22 neurons with pre-formed active aSyn fibrils (aSyn-PFF) (A53T Mutant Alpha Synuclein Active Protein, MyBioSource), sub-confluent cells were trypsinized, pelleted, and resuspended in full medium containing αSyn-PFF and seeded in T75 flasks (Greiner) to achieve a final concentration of 2 µg/ml PFF or 6 µg/ml PFF and cell density of 0.4 ×106 cells/flask. Control cells were plated in full medium without PFF.
For lipid analyses cells were incubated for 72 h and harvested by trypsinization. Cells were washed 3-times with 1× PBS, resuspended in 1 ml 1× PBS, counted and adjusted to 250,000 cells per sample, finally pelleted. The supernatant was discarded and pellets stored at −80 °C until analysis. For lipid analysis, 8 replicates were generated for each treatment.
For analysis of cell viability upon exposure with αSyn-PFF, WST-1 and LDH activity assays were used. The WST-1 assay is based on the cleavage of the water-soluble tetrazolium salt by cellular enzymes to a formazan dye, which is quantified by measuring absorbance and correlates with the number of metabolically active viable cells. Cells were seed in 96-well plates (3500 cells/well) and were cultured for 24 h or 48 h with/without 2 µg/ml αSyn-PFF. The medium was then replaced with 100 μl of fresh medium (per well), and 10 µl WST-1 solution were added to each well. After 90 min in the incubator, the absorbance was read on a multimode microplate reader (SpectraMax i3X, RRID:SCR_026346) at 450 nm, and the reference wavelength of 620 nm.
For analysis of LDH activity, 5 µl cell culture supernatant and NADH standards (0–12.5 nmol/well) were pipetted into the wells of a 96 well plate. The master reaction mix consisting of assay buffer and substrate mix was added to the wells. The absorbance was measured on a microplate reader at 450 nm every 5 min until the value of the most active sample exceeded the value of the highest standard. The absorbance was corrected with the blank, and LDH activity was calculated as described in the manufacturer’s protocol (Sigma).
For live cell immunofluorescence analyses of αSyn-PFF uptake, HT22 cells were seeded in 8-well cover glass bottom culture slides in full medium in the presence of 1 µM GlcCer24:1 (AvantiPolar Lipids #860549) or vehicle (2:1 mixture of chloroform and methanol, final dilution 1:10000) for 48 h. The chambers were coated with Poly-L-lysine (0.01%, Sigma) before seeding of 5000 cells per well. Then 2 µg/ml αSyn-PFF labeled with a fluorophore (Alexa Fluor 555 Microscale Protein Labeling Kit, Invitrogen) were added to the medium and incubated for 24 h. Live culture images were obtained on a Leica Stellaris 8 fluorescent confocal microscope (RRID:SCR_024660). To visualize the plasma membrane and the nucleus, cultures were incubated 15 min before imaging with wheat germ agglutinin-Alexa Fluor 488 reagent (WGA, Invitrogen). Hoechst 33342 was used as nuclear stain (Thermo Scientific). For analysis in FIJI ImageJ, mono-channel images were converted to 8-bit images, stacked and arranged in montages. Using the threshold default-algorithm, images were converted to binary masks which were then submitted to the particle counter for each image, and the immunofluorescent (IF) area was used for further quantitative analysis. The red-PFF IF area was normalized by the respective Hoechst IF area representing the nuclei, and the area ratio was submitted to statistical t-test comparisons.
Beta-arrestin screening of G-protein activation by GlcCer24:1
PRESTO-Tango beta-arrestin assay131 was used to screen for the activation profile of heterologously expressed G-protein coupled receptors.
The assay uses HTLA cells which are HEK293T cells stably expressing a luciferase reporter gene and stably expressing a human β-arrestin-2 fused to Tobacco Etch Virus protease. 5000 HTLA cells were seeded in a white, transparent and poly-L-Lysin-coated 384-well plate from PerkinElmer. The cells were co-transfected after 6 h with a plasmid from the PRESTO-Tango kit (Addgene #1000000068). We used a mixture of 10 ng plasmid and 0.04 µl Lipofectamine 2000 per well performing the transfection using the protocol described by Kroeze et al.131. We used GFP as a transfection control and 100 µM carbachol as an assay control at the muscarinic M5 receptor. After 24 h, the medium was aspirated and replaced by 45 µl of serum-free medium. The ligand (5 µl) was then added at a final concentration of 1, 5 and 10 µM for ~24 h. Afterwards, the medium was aspirated, and the cells lysed using 50 µl of bright-Glo reagent (Promega) diluted 10X with 1x PBS. After 15 min of incubation with the lysis buffer, the luminescence (endpoint, 1500 ms integration time) was measured using a flexstation 3 device.
Dynamic Mass Redistribution Assay (DMR)
Label-free dynamic mass redistribution enables real-time detection of refractive index alterations on biosensor-coated microplates that originate from stimulus-induced changes in the total biomass in proximity to the sensor surface. Dynamic mass redistribution monitoring was performed with a Corning EPIC BT system. 5000 HEK293 cells were seeded in a transparent and poly-L-Lysin precoated 384-well plate from Corning. The transfection was done as described for the beta arrestin screen. After 48 h, the culture medium was removed. Assay buffer was added and cells incubated in assay buffer for 2 before a 1 min baseline was recorded. Diluted lipids (final concentration 1 µM) or positive control were then added and DMR signals were acquired every 3 s for a period of 60 min. The readout is the shift of resonance wavelength over time ∆λ(t) shown as response curves. Carbachol effects on muscarinic receptors were used as positive control.
Primary DRG neuron culture
To assess gene regulations under treatment with GlcCer24:1, primary sensory neurons were prepared from naïve adult C57BL6 mice which had free access to food and water, were kept in climate-controlled rooms with a 12 h light-dark cycle. Primary adult neuron-enriched cultures of dorsal root ganglia (DRG) of each four mice per group were prepared by dissecting DRGs of adult mice into Hank’s balanced salt solution (HBSS, Merck), followed by digestion with 5 mg/ml collagenase A (Millipore) and 1 mg/ml dispase II (Roche Diagnostics, Germany) before treatment with DNase (Sigma, 250 U per sample). Triturated cells were centrifuged through a 15% fat-free bovine serum albumin (BSA) solution. Primary sensory neurons were seeded on poly-L-lysine and laminin-coated cover slips (9–12 cover slips per mouse). Cells were cultured in serum-free Neurobasal medium (Gibco) containing 1x B27 supplement, 1x Pen/Strep, 200 ng/ml nerve growth factor and 2 mM L-glutamine at 37 °C and 5% CO2 and 95% humidity. Neurons were cultured in the presence of 1 µM GlcCer24:1 or vehicle (2:1 mixture of chloroform and methanol, final dilution 1:10000) and submitted to RNAseq.
RNA sequencing in DRG neurons upon stimulation with GlcCer24:1
The day after seeding of primary DRG neurons, GlcCer24:1 was added to the medium at a final concentration of 1 µM. Control cells received the same volume of vehicle. Cells were collected two days after adding treatments. Total RNA was isolated using a single-cell RNA extraction kit (PicoPure, Thermo Fisher). Quantity of total RNA was determined using Invitrogen’s Qubit HS assay and quality was checked on an Agilent 2100 Bioanalyzer Instrument (RRID:SCR_018043). First-strand cDNA was prepared and amplified from 1 ng total RNA using SMART Seq v4 Ultra Low-Input RNA Kit for sequencing (TaKaRa) according to the manual compatible with Illumina NGS. Barcoded sequencing libraries were subsequently prepared using the low-input library preparation kit (Sv4 PLUS kit, TaKaRa). The Sv4 PLUS kit includes the first stand cDNA synthesis using SMART technology and library preparation that incorporates enzymatic fragmentation and stem-loop adapters to construct high-quality, Illumina-compatible libraries. Concentrations were determined using Invitrogen’s Qubit HS assay and fragment size was analyzed on Agilent’s 2100 Bioanalyzer on a HS DNA chip. RNA sequencing was performed of 4 biological replicates per group using an Illumina NextGen 2000 system (RRID:SCR_023614).
The sequence alignment was done with Qiagen’s CLC genomic workbench (v. 22, RRID:SCR_011853). Sequence reads were trimmed for adapter sequences and low-quality sequences using CLC genomic workbench standard settings of RNA sequencing (quality limit 0.05). Sequence reads were aligned to the reference genome mm10 provided from UCSC (GRCm38) as template, using CLC’s default setting for RNAseq. Read counts extraction and normalization (TMM) were done using CLC genomic workbench. The expression value unit is the trimmed mean of M-values, TMM (EdgeR algorithm). TMM reads were Log2 transformed.
Differential gene expression was assessed using t-tests and fold change using CLC genomic workbench. Genes were filtered for at least 4 valid values out of 8 samples to exclude low-expression genes. The P value was set at 0.05 and adjusted according to the False Discovery Rate (FDR). Hierarchical clustering with Euclidean distance metrics was used to assess gene expression patterns. Genes were ranked according to P and q value, fold change and abundance. Ranked genes were submitted to gene ontology enrichment analysis using DAVID (Database for Annotation, Visualization and Integrated Discovery132; RRID:SCR_001881) and ExpressAnalyst (https://www.expressanalyst.ca/) (RRID:SCR_025651)133. The RNAseq data have been deposited via the Gene Expression Omnibus (RRID:SCR_005012) as GEO dataset with the provisional accession number GSE262573.
Lipidomic and metabolomic analyses (plasma, cells, supernatant)
Human blood was collected in 9 ml K3 + EDTA tubes, mouse blood in 500 µl K + EDTA tubes, centrifuged at 1300 × g or 1500 × g and plasma was transferred into 1.5 ml tubes and stored at −80 °C until further use. Cells were collected by trypsinization, washed, pelleted, resuspended in 100 µl counted and adjusted to yield 1.5x 10exp6 cells per pellet (experiment-1) or 2.5x 10exp5 cells (experiment-2). Finally, the cells were pelleted at high speed and supernatant removed with 100 µl and subsequently 20 µl pipet. The “dry” pellet was frozen at −80 °C until lipid/metabolite extraction. Fibroblasts were homogenized as explained below (targeted analyses). Lipidomic and metabolomic analysis were conducted applying the same procedure as previously described134. Lipidomic analysis of the human plasma samples used a slightly different procedure135. Targeted sphingolipid analyses were done in plasma, cell and tissue extracts essentially as described previously42 using liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS), according to procedures described in detail in ref. 136. Further protocol details for extraction and analyses of different matrices (cells, tissue, plasma) are described in the supplementary methods (Excel file).
Statistics
Group data are presented as mean ± SD or median ± IQR as specified in the respective figure legends. Data were analyzed with SPSS 29 (RRID:SCR_016479) and GraphPad Prism 9 or 10 (RRID:SCR_002798), Origin Pro 2024 (RRID:SCR_014212), and MetaboAnalyst 5.0 (RRID:SCR_016723) (https://www.metaboanalyst.ca)137. Bioinformatic analysis of “omic” data (RNAseq, lipidomic, metabolomic) is explained in the respective paragraphs. Area under the curve (AUC) data of lipidomic and metabolomic analyses transformed to square root values to adjust skewed distributions. For testing the null-hypothesis that groups were identical, two groups were compared with 2-sided, unpaired Student’s t tests. Lipidomic and metabolomic data were submitted to 2-way analysis of variance (ANOVA) using e.g., the factors “feature” (e.g. lipid, metabolite) and ‘group’ (e.g. PD versus HC with/without stimulation). In case of significant differences, groups were mutually compared using post hoc t-tests according to Šidák or false discovery rate (FDR). The meaning of asterisks in figures is explained in the legends. ANOVA-simultaneous component analysis (ASCA)138 was used for analysis of multiple covariates on lipidomic data from patients and fibroblasts. ASCA is a combination of ANOVA and PCA plus feature extraction method for multivariate data to model two major components and their interaction. The meta-data for patients included sex, age, BMI, telomere length, and sensory loss. For multivariate analyses, data were normalized to have a common mean and variance of 1 (Z-scores = (x − x̄)/SD). Volcano plots were used to assess fold differences of lipids versus the negative logarithm (Log10) of the t-test P value according to standard procedures. Partial Least Square Discriminant analysis (PLS-DA) or canonical discrimination analysis were used to assess group membership and variable importance.
Data availability
All data that were analyzed for the study are presented within the manuscript or supplementary files. The source datasets supporting the conclusions of this article are available in the following repositories: RNAseq data have been deposited to NCBI's GEO repository with the accession number GSE262573. Processed lipidomic data have been deposited BioStudies (https://www.ebi.ac.uk/biostudies). The accession numbers are given below the respective results. The datasets are:Plasma lipidomic data of PD patients and healthy controls: S-BSST1880, https://www.ebi.ac.uk/biostudies/studies/S-BSST1880?key=a0dc6f53-0365-410a-96e3-d83e51a04282. Targeted sphingolipids in cells extracts of primary PD and HC fibroblasts with/without pimozide: S-BSST1881. https://www.ebi.ac.uk/biostudies/studies/S-BSST1881?key=4e8ae53f-17f0-4179-80ff-40392e418270. Untargeted lipidomics and targeted sphingolipids of brain tissue of Pink1SNCA versus wildtype mice: S-BSST1888, https://www.ebi.ac.uk/biostudies/studies/S-BSST1888?key=6230dc37-d998-4eca-bf0f-3502f7164655. Targeted and untargeted lipidomic and metabolomic analyses in HT22 cells extracts with/without preformed fibrils: S-BSST1897, https://www.ebi.ac.uk/biostudies/studies/S-BSST1897?key=d2a20a65-a88f-44a4-9654-672a72f8a9ac.
References
McCann, H., Cartwright, H. & Halliday, G. M. Neuropathology of α-synuclein propagation and braak hypothesis. Mov. Disord. 31, 152–160 (2016).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Mazzulli, J. R. et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 (2011).
Zunke, F. et al. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron 97, 92–107.e10 (2018).
Taguchi, Y. V. et al. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 37, 9617–9631 (2017).
Paul, A. et al. Glucosylceramide associated with gaucher disease forms amyloid-like twisted ribbon fibrils that induce α-synuclein aggregation. ACS Nano 15, 11854–11868 (2021).
Danzer, K. M. et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7, 42 (2012).
Chakraborty, R., Nonaka, T., Hasegawa, M. & Zurzolo, C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of α-Synuclein and mitochondria. Cell Death Dis. 14, 329 (2023).
Bernis, M. E. et al. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol. Commun. 3, 75 (2015).
Yang, F. et al. Crosstalk between the proteasome system and autophagy in the clearance of alpha-synuclein. Acta Pharm. Sin. 34, 674–680 (2013).
Xilouri, M., Vogiatzi, T., Vekrellis, K. & Stefanis, L. alpha-synuclein degradation by autophagic pathways: a potential key to Parkinson’s disease pathogenesis. Autophagy 4, 917–919 (2008).
Pan, T., Kondo, S., Le, W. & Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 131, 1969–1978 (2008).
Xilouri, M. et al. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 136, 2130–2146 (2013).
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004).
Meyer, N. et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy 17, 3424–3443 (2021).
Tegeder, I. & Kögel, D. When lipid homeostasis runs havoc: Lipotoxicity links lysosomal dysfunction to autophagy. Matrix Biol. 100-101, 99–117 (2021).
Tsunemi, T. et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca(2+) Channel Agonists Protects Human Dopaminergic Neurons from α-Synuclein Toxicity. J. Neurosci. 39, 5760–5772 (2019).
Xie, Y. X. et al. Lysosomal exocytosis releases pathogenic α-synuclein species from neurons in synucleinopathy models. Nat. Commun. 13, 4918 (2022).
Bae, E. J. et al. TNF-α promotes α-synuclein propagation through stimulation of senescence-associated lysosomal exocytosis. Exp. Mol. Med. 54, 788–800 (2022).
Du, T. T. et al. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 11, 1803–1820 (2015).
Muñoz, S. S., Petersen, D., Marlet, F. R., Kücükköse, E. & Galvagnion, C. The interplay between Glucocerebrosidase, α-synuclein and lipids in human models of Parkinson’s disease. Biophys. Chem. 273, 106534 (2021).
Murphy, K. E. et al. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848 (2014).
Martin, S. et al. Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol 139, 1001–1024 (2020).
Miura, E. et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 71, 1–13 (2014).
Tang, F. L. et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35, 10613–10628 (2015).
Migdalska-Richards, A. et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 140, 2706–2721 (2017).
Neumann, J. et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794 (2009).
den Heijer, J. M. et al. A large-scale full GBA1 gene screening in Parkinson’s disease in the Netherlands. Mov. Disord. 35, 1667–1674 (2020).
Duran, R. et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 28, 232–236 (2013).
Horowitz, M., Braunstein, H., Zimran, A., Revel-Vilk, S. & Goker-Alpan, O. Lysosomal functions and dysfunctions: molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv. Drug Deliv. Rev. 187, 114402 (2022).
Pietrafesa, D., et al. Investigating the impact of the Parkinson’s-associated GBA1 E326K mutation on β-glucocerebrosidase dimerization and interactome dynamics through an in silico approach. Int. J. Mol. Sci. 25, 11443 (2024).
Reczek, D. et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131, 770–783 (2007).
Rothaug, M. et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc. Natl Acad. Sci. USA 111, 15573–15578 (2014).
Aflaki, E., Westbroek, W. & Sidransky, E. The complicated relationship between Gaucher disease and Parkinsonism: insights from a rare disease. Neuron 93, 737–746 (2017).
Do, J., McKinney, C., Sharma, P. & Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 14, 36 (2019).
Rubilar, J. C., Outeiro, T. F. & Klein, A. D. The lysosomal β-glucocerebrosidase strikes mitochondria: implications for Parkinson’s therapeutics. Brain 147, 2610–2620 (2024).
Garcia-Sanz, P. et al. N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 32, 1409–1422 (2017).
Osellame, L. D. & Duchen, M. R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 9, 1633–1635 (2013).
Thomas, R. E. et al. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLoS Genet. 14, e1007694 (2018).
Wu, Y. H., Lee, W. J., Chen, Y. H., Chang, M. H. & Lin, C. H. Premotor symptoms as predictors of outcome in Parkinsons disease: a case-control study. PLoS One 11, e0161271 (2016).
Pont-Sunyer, C. et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov. Disord. 30, 229–237 (2015).
Klatt-Schreiner, K. et al. High glucosylceramides and low anandamide contribute to sensory loss and pain in Parkinson’s disease. Mov. Disord. 35, 1822–1833 (2020).
Galper, J. et al. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain 145, 3472–3487 (2022).
Leyns, C. E. G. et al. Glucocerebrosidase activity and lipid levels are related to protein pathologies in Parkinson’s disease. NPJ Parkinsons Dis. 9, 74 (2023).
Galper, J. et al. Prediction of motor and non-motor Parkinson’s disease symptoms using serum lipidomics and machine learning: a 2-year study. NPJ Parkinsons Dis. 10, 123 (2024).
Kita, N. et al. Glucosylceramide flippases contribute to cellular glucosylceramide homeostasis. J. Lipid Res. 65, 100508 (2024).
Norris, A. C. et al. Deficiency of the lipid flippase ATP10A causes diet-induced dyslipidemia in female mice. Sci. Rep. 14, 343 (2024).
Somerville, E. N. et al. Plasma glucosylceramide levels are regulated by ATP10D and are not involved in Parkinson’s disease pathogenesis. Ann. Neurol. 97, 873–878 (2025).
Ishibashi, Y., Kohyama-Koganeya, A. & Hirabayashi, Y. New insights on glucosylated lipids: metabolism and functions. Biochim Biophys. Acta 1831, 1475–1485 (2013).
Mazzulli, J. R. et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 (2011).
Abounit, S. et al. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138 (2016).
Zhong, Y. et al. Combinatorial targeting of glutamine metabolism and lysosomal-based lipid metabolism effectively suppresses glioblastoma. Cell Rep. Med. 5, 101706 (2024).
Olesen, M. A., Villavicencio-Tejo, F. & Quintanilla, R. A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegener.11, 36 (2022).
Corenblum, M. J. et al. Parallel neurodegenerative phenotypes in sporadic Parkinson’s disease fibroblasts and midbrain dopamine neurons. Prog. Neurobiol. 229, 102501 (2023).
Hoepken, H. H. et al. Parkinson patient fibroblasts show increased alpha-synuclein expression. Exp. Neurol. 212, 307–313 (2008).
Doppler, K. et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol. 133, 535–545 (2017).
Galvagnion, C. et al. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. Brain 145, 1038–1051 (2022).
Torres-Odio, S. et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflam.14, 154 (2017).
Li, Y., Wu, Q., Hu, E., Wang, Y. & Lu, H. Quantitative mass spectrometry imaging of metabolomes and lipidomes for tracking changes and therapeutic response in traumatic brain injury surrounding injured area at chronic phase. ACS Chem. Neurosci. 12, 1363–1375 (2021).
Kurzawa-Akanbi, M. et al. Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders. Acta Neuropathol. 142, 961–984 (2021).
Papadopoulos, V. E. et al. Modulation of beta-glucocerebrosidase increases alpha-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum. Mol. Genet. 27, 1696–1710 (2018).
Yuyama, K. & Igarashi, Y. Linking glycosphingolipids to Alzheimer’s amyloid-ß: extracellular vesicles and functional plant materials. Glycoconj. J. 39, 613–618 (2022).
Valek, L. et al. Prodromal sensory neuropathy in Pink1(−/−) SNCA(A53T) double mutant Parkinson mice. Neuropathol. Appl Neurobiol. 47, 1060–1079 (2021).
Yokoyama, N. et al. Multiplicity of glycosphingolipid-enriched microdomain-driven immune signaling. Int. J. Mol. Sci. 22, 9565 (2021).
Patel, H. H., Murray, F. & Insel, P. A. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb. Exp. Pharm. 186, 167–184 (2008).
Leite Silva, A. B. R. et al. Premotor, nonmotor and motor symptoms of Parkinson’s disease: a new clinical state of the art. Ageing Res. Rev. 84, 101834 (2023).
Mahlknecht, P., Seppi, K. & Poewe, W. The concept of prodromal Parkinson’s disease. J. Parkinsons Dis. 5, 681–697 (2015).
Vacchi, E. et al. Alpha-synuclein oligomers and small nerve fiber pathology in skin are potential biomarkers of Parkinson’s disease. NPJ Parkinsons Dis. 7, 119 (2021).
Schrag, A., Horsfall, L., Walters, K., Noyce, A. & Petersen, I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 14, 57–64 (2015).
O’Sullivan, S. S. et al. Nonmotor symptoms as presenting complaints in Parkinson’s disease: a clinicopathological study. Mov. Disord. 23, 101–106 (2008).
Baig, F. et al. Delineating nonmotor symptoms in early Parkinson’s disease and first-degree relatives. Mov. Disord. 30, 1759–1766 (2015).
Calzetti, S. et al. Non-length-dependent somatosensory small fiber pathology presenting with restless legs syndrome in pre-motor Parkinson’s disease. Evidence from skin biopsy in four patients. J. Clin. Neurosci. 69, 139–142 (2019).
Horsager, J. et al. Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain 143, 3077–3088 (2020).
Passaretti, M. et al. Clinical progression and genetic pathways in body-first and brain-first Parkinson’s disease. Mol. Neurodegener. 20, 74 (2025).
Ferreira, N. et al. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 9, 31 (2021).
Vincow, E. S. et al. Glucocerebrosidase deficiency leads to neuropathology via cellular immune activation. PLoS Genet. 20, e1011105 (2024).
Wouters, R. et al. The lipid flippase ATP10B enables cellular lipid uptake under stress conditions. Biochim. Biophys. Acta Mol. Cell Res. 1871, 119652 (2024).
Suzuki, M. et al. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant alpha-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 24, 6675–6686 (2015).
Martin, S. et al. Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 139, 1001–1024 (2020).
López de Frutos L. et al. Serum phospholipid profile changes in Gaucher disease and Parkinson’s disease. Int. J. Mol. Sci. 23, 10387 (2022).
Carrillo, F. et al. Multiomics approach identifies dysregulated lipidomic and proteomic networks in Parkinson’s disease patients mutated in TMEM175. NPJ Parkinsons Dis. 11, 23 (2025).
Kalecký, K. & Bottiglieri, T. Targeted metabolomic analysis in Parkinson’s disease brain frontal cortex and putamen with relation to cognitive impairment. NPJ Parkinsons Dis. 9, 84 (2023).
Dou, T. & Kurouski, D. Phosphatidylcholine and phosphatidylserine uniquely modify the secondary structure of α-synuclein oligomers formed in their presence at the early stages of protein aggregation. ACS Chem. Neurosci. 13, 2380–2385 (2022).
Dou, T., Matveyenka, M. & Kurouski, D. Elucidation of secondary structure and toxicity of α-synuclein oligomers and fibrils grown in the presence of phosphatidylcholine and phosphatidylserine. ACS Chem. Neurosci. 14, 3183–3191 (2023).
Naito, T. et al. Phospholipid flippase ATP10A translocates phosphatidylcholine and is involved in plasma membrane dynamics. J. Biol. Chem. 290, 15004–15017 (2015).
Tomura, H., Mogi, C., Sato, K. & Okajima, F. Proton-sensing and lysolipid-sensitive G-protein-coupled receptors: a novel type of multi-functional receptors. Cell Sig. 17, 1466–1476 (2005).
Seuwen, K., Ludwig, M. G. & Wolf, R. M. Receptors for protons or lipid messengers or both?. J. Recept Sig. Transduct. Res 26, 599–610 (2006).
Afzal, R. & Shim, W. S. Glucosylsphingosine activates serotonin receptor 2a and 2b: implication of a novel itch signaling pathway. Biomol. Ther. 25, 497–503 (2017).
Garcia-Ruiz, C., Morales, A. & Fernández-Checa, J. C. Glycosphingolipids and cell death: one aim, many ways. Apoptosis 20, 607–620 (2015).
Nilsson, O. & Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 39, 709–718 (1982).
Marano, M. et al. Increased glucosylsphingosine levels and Gaucher disease in GBA1-associated Parkinson’s disease. Parkinsonism Relat. Disord. 124, 107023 (2024).
Dekker, N. et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood 118, e118–e127 (2011).
Mahoney-Crane, C. L. et al. Neuronopathic GBA1L444P mutation accelerates glucosylsphingosine levels and formation of hippocampal alpha-synuclein inclusions. J. Neurosci. 43, 501–521 (2023).
Elstein, D. et al. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naive and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 122, 113–120 (2017).
Peterschmitt, M. J., Foster, M. C., Ji, A. J., Zajdel, M. B. & Cox, G. F. Plasma glucosylsphingosine correlations with baseline disease burden and response to eliglustat in two clinical trials of previously untreated adults with Gaucher disease type 1. Mol. Genet. Metab. 138, 107527 (2023).
Li, Y. T. et al. Selective extraction and effective separation of galactosylsphingosine (psychosine) and glucosylsphingosine from other glycosphingolipids in pathological tissue samples. Neurochem Res. 36, 1612–1622 (2011).
Rolfs, A. et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a Non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS One 8, e79732 (2013).
Gaspar, R., Pallbo, J., Weininger, U., Linse, S. & Sparr, E. Ganglioside lipids accelerate alpha-synuclein amyloid formation. Biochim. Biophys. Acta Proteins Proteom. 1866, 1062–1072 (2018).
Bodner, C. R., Maltsev, A. S., Dobson, C. M. & Bax, A. Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 49, 862–871 (2010).
Das, T. et al. The role of membrane affinity and binding modes in alpha-synuclein regulation of vesicle release and trafficking. Biomolecules 12, 1816 (2022).
Lotharius, J. & Brundin, P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 3, 932–942 (2002).
Peelaerts, W. et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 (2015).
Masuda-Suzukake, M. et al. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2, 88 (2014).
Uchihara, T. & Giasson, B. I. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 131, 49–73 (2016).
Prymaczok, N. C. et al. Cell-to-cell transmitted alpha-synuclein recapitulates experimental Parkinson’s disease. NPJ Parkinsons Dis. 10, 10 (2024).
Scheiblich, H. et al. Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron 112, 3106–25.e8 (2024).
Guo, M. et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 143, 1476–1497 (2020).
Poehler, A. M. et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy 10, 2171–2192 (2014).
Scesa, G., Moyano, A. L., Bongarzone, E. R. & Givogri, M. I. Port-to-port delivery: Mobilization of toxic sphingolipids via extracellular vesicles. J. Neurosci. Res. 94, 1333–1340 (2016).
Stykel, M. G. et al. α-Synuclein mutation impairs processing of endomembrane compartments and promotes exocytosis and seeding of α-synuclein pathology. Cell Rep. 35, 109099 (2021).
Verderio, C., Gabrielli, M. & Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 59, 1325–1340 (2018).
Nagata, M. et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl Acad. Sci. USA 114, E3285–e94 (2017).
Sharma, A. et al. Glycolipid Metabolite β-Glucosylceramide Is a Neutrophil Extracellular Trap-Inducing Ligand of Mincle Released during Bacterial Infection and Inflammation. J. Immunol. 209, 391–400 (2022).
Behler, F. et al. Macrophage-inducible C-type lectin Mincle-expressing dendritic cells contribute to control of splenic Mycobacterium bovis BCG infection in mice. Infect. Immun. 83, 184–196 (2015).
Ishikawa, E. et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888 (2009).
Kostarnoy, A. V. et al. Receptor Mincle promotes skin allergies and is capable of recognizing cholesterol sulfate. Proc. Natl Acad. Sci. USA 114, E2758–e65 (2017).
Patin, E. C., Orr, S. J. & Schaible, U. E. Macrophage inducible C-type lectin as a multifunctional player in immunity. Front. Immunol. 8, 861 (2017).
Williams, S. J. Sensing lipids with mincle: structure and function. Front. Immunol. 8, 1662 (2017).
Shimizu, T. et al. Direct activation of microglia by β-glucosylceramide causes phagocytosis of neurons that exacerbates Gaucher disease. Immunity 56, 307–319.e8 (2023).
Fan, J. et al. Gaucher disease protects against tuberculosis. Proc. Natl Acad. Sci. USA 120, e2217673120 (2023).
Xie, Y. et al. Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav. Immun. 60, 346–360 (2017).
Schmidhuber, S. et al. A novel C-type lectin receptor-targeted α-synuclein-based parkinson vaccine induces potent immune responses and therapeutic efficacy in mice. Vaccines 10, 1432 (2022).
Gispert, S. et al. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 24, 1061–1076 (2015).
Abou-Sleiman, P. M., Muqit, M. M. & Wood, N. W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7, 207–219 (2006).
Maiti, P., Manna, J. & Dunbar, G. L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 6, 28 (2017).
Plotegher, N. & Duchen, M. R. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front Cell Dev. Biol. 5, 110 (2017).
Valek, L., Auburger, G. & Tegeder, I. Sensory neuropathy and nociception in rodent models of Parkinson’s disease. Dis. Model. Mech. 12, dmm039396 (2019).
Bastian, P. et al. Regulation of mitochondrial dynamics in Parkinson’s disease-is 2-methoxyestradiol a missing piece? Antioxidants 10, 248 (2021).
Sheng, Y. C. et al. TIGAR plays neuroprotective roles in MPP(+)/MPTP-induced Parkinson’s disease by alleviating ferroptosis. Eur. J. Pharm. 995, 177430 (2025).
Shimada, S. et al. Activation of dopamine D4 receptors is protective against hypoxia/reoxygenation-induced cell death in HT22 cells. J. Pharm. Sci. 114, 217–224 (2010).
Kroeze, W. K. et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015).
Dennis, G. Jr. et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3 (2003).
Liu, P. et al. ExpressAnalyst: a unified platform for RNA-sequencing analysis in non-model species. Nat. Commun. 14, 2995 (2023).
Sens, A. et al. Pre-analytical sample handling standardization for reliable measurement of metabolites and lipids in LC-MS-based clinical research. J. Mass Spectrom. Adv. Clin. Lab 28, 35–46 (2023).
Hahnefeld, L. et al. Implementation of lipidomics in clinical routine: can fluoride/citrate blood sampling tubes improve preanalytical stability?. Talanta 209, 120593 (2020).
Brunkhorst-Kanaan, N. et al. Targeted lipidomics reveal derangement of ceramides in major depression and bipolar disorder. Metabolism 95, 65–76 (2019).
Pang, Z. et al. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 17, 1735–1761 (2022).
Smilde, A. K. et al. ANOVA-simultaneous component analysis (ASCA): a new tool for analyzing designed metabolomics data. Bioinformatics 21, 3043–3048 (2005).
Acknowledgements
The study was supported by the Deutsche Forschungsgemeinschaft (CRC1039 A03 to I.T.; CRC1039 A04 to SO; CRC1039 Z01 and 445757098 to G.G.; TE 322/11-1 to I.T.). The funders had no role in the study design, data analysis, interpretation or decision to publish. The authors would like to thank Yannick Schreiber, Carlo Angioni, Alena Sens and Pia Viktoria Wagner for technical support in performing the LC-HRMS and LC-MS/MS experiments, and Yannick Jäger for technical support with beta-arrestin G-protein screening studies.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.F. and L.V. created primary cultures and performed experiments in primary neurons and fibroblasts, K.K.S. recruited and examined patients, A.W.S. conducted RNAseq studies, L.H. established, performed and supervised untargeted lipidomic analyses, S.T. analyzed sphingolipids, D.T. and R.G. supervised lipid analyses, G.G. acquired funding and resources, W.A. and S.O. performed and analyzed beta-arrestin G-protein and DMR screens, I.T. conceived and designed the study, acquired funding and ethical approval, performed experiments with primary cells, obtained skin biopsies, compiled and analyzed data, made the figures and tables and wrote the paper (draft and editing). All authors contributed to writing or editing parts of the manuscript and agree to the latest version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Franck, L., Hahnefeld, L., Valek, L. et al. Elevated hexosylceramides in Parkinson’s disease cause gene upregulations in neurons mimicking responses to pathogens. npj Parkinsons Dis. 11, 268 (2025). https://doi.org/10.1038/s41531-025-01114-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01114-9
This article is cited by
-
Selective neuronal restoration of progranulin does not prevent the frontotemporal dementia like-phenotype of progranulin knockout mice
Journal of Neuroinflammation (2026)
