
Abstract
Genome-wide association study of Parkinson’s disease (PD) identified common variants associated with lysosomal mechanism, including TMEM175, SCARB2, and CTSB. We investigated the association between common and rare variants across populations using cohorts from the Global Parkinson’s Genetics Program (GP2) (33,733 cases and 18,703 controls from ten ancestries). In the European cohort, we confirmed significant associations with PD risk for all known genetic risk variants across the three genes and TMEM175 p. Met393Thr as an independent genome-wide significant signal. Additionally, a novel independent signal, SCARB2 rs11547135, was detected. The burden analysis linked PD to SCARB2 in African American, Ashkenazi Jewish and East Asian cohorts. Single variants-based tests identified rare missense variants in SCARB2 in several populations. Our study reinforces the association of lysosomal genetic variants with PD risk, revealing genetic heterogeneity across populations.
Similar content being viewed by others

Introduction
Glucocerebrosidase (GCase), encoded by GBA1, is a lysosomal enzyme critical for maintaining lysosomal protein and lipid homeostasis. GCase deficiency causes accumulation of alpha-synuclein(α-syn)1 and variants in GBA1 are recognized as a common genetic risk factor for PD2. The latest genome-wide association study (GWAS) in European derived populations identified common genetic variants associated with lysosomal dysfunction that contribute to Parkinson’s disease (PD) risk, with GBA1 (2025 PD GWAS, p.E365K (rs2230288, odds ratio [OR] = 1.66)) emerging as a key locus3,4,5. Genetic variants in other lysosome-related genes (including transmembrane protein 175 [TMEM175], Scavenger Receptor Class B Member 2 [SCARB2] and Cathepsin B [CTSB]) are also associated with PD risk and age at onset6,7,8. These candidate genes were prioritized in this study based on (1) GBA1’s role as a major lysosomal driver of PD pathogenesis; (2) functional convergence of TMEM175, SCARB2, and CTSB with GCase activity and lysosomal function—either through direct interaction (SCARB2)9, pH regulation (TMEM175)10,11, or cathepsin B(CTSB) in mediating prosaposin cleavage to form saposin C, the lysosomal coactivator of GCase12,13. In addition to their mechanistic relevance, these three genes were identified as genome-wide significant loci in large-scale GWAS of PD3, further supporting their contribution to disease risk. Together, these genes interact to provide a cohesive framework to dissect lysosomal mechanisms in PD. Their distinct roles are detailed below.
The TMEM175 gene encodes a proton-selective ion channel located on lysosomal membranes. It mediates the lysosomal H+ leak that balances vacuolar-type H + -ATPase (V-ATPase) activity which helps maintain lysosomal pH homeostasis11. Two common coding variants in the TMEM175 gene, p. Met393Thr (rs34311866) and p. Gln65Pro (rs34884217), show opposite effects on PD susceptibility in several populations7,14,15. The GWAS identified p. Met393Thr associated with increased risk (2025 PD GWAS, OR = 1.19)3,5 and earlier age of onset of PD14,16.
SCARB2, known as lysosomal integral membrane protein-2 (LIMP-2), is an intracellular receptor which shuffles GCase from the endoplasmic reticulum to the lysosome. Variants in this gene may lead to functional and structural lysosomal dysfunction9. Two common intronic variants, rs6812193 and rs6825004 (2025 PD GWAS, rs6825004, OR = 0.96; rs6812193, OR = 0.93), have been associated with the risk of PD in several genetic studies3,5,9,17,18,19.
CTSB is a cysteine protease, which plays an essential role in lysosomal degradation of α-syn12. The CTSB locus harbors an intronic variant (rs1293298) which is a common genetic risk factor for PD (2025 PD GWAS, OR = 0.93)3,5. Interestingly, this variant also modifies PD risk in GBA1 carriers, suggesting an interaction between GBA1 and CTSB6. This variant lowers the penetrance of GBA1 mutations, reducing the risk of PD in carriers.
To overcome limitations of prior studies, including insufficient sample sizes for robust statistical inference, ancestry heterogeneity or Eurocentric cohort bias, and fragmented assessments of lysosomal genes contributions, we leveraged large-scale cohorts of genotyping data from GP2 (Global Parkinson’s Genetics Program) and investigated association of common and rare variants in lysosomal related genes (TMEM175, SCARB2, CTSB and GBA1) with PD across ten populations.
Results
Association analysis of known common risk variants in TMEM175, SCARB2, and CTSB across ten populations
To validate previously reported GWAS hits, we tested the association of the following variants with PD risk in the GP2-EUR cohort: TMEM175 (p. Met393Thr and p. Gln65Pro), SCARB2 (rs6812193 and rs6825004), and CTSB (rs1293298). We replicated previous findings that both TMEM175 variants and CTSB variant were significant in the GP2-EUR cohort (Fig. 1a, b, e, Supplementary Table 1a and 1c). However, both variants in SCARB2, rs6812193 and rs6825004 did not remain significant after correction for multiple testing (Fig. 1c, d, Supplementary Table 1b).

a TMEM175 p.M393T (rs34311866), b TMEM175 p.Q65P (rs34884217), c SCARB2 rs6812193, d SCARB2 rs6825004, and e CTSB rs1293298. P values were corrected for multiple testing across ten ancestries and five genetic variants using the Benjamini–Hochberg method to control the false discovery rate. Abbreviations: AAC African American, AFR African, AJ Ashkenazi Jewish, AMR Latino and indigenous Americas, CAS Central Asian, EAS East Asian, EUR European, MDE Middle Eastern, SAS South Asian, CAH Complex Admixture History.
The known risk factor in TMEM175, p. Met393Thr was found to be significantly associated with PD in the GP2-SAS cohort [corrected P value = 0.010, OR (95%CI) = 1.83(1.28–2.61)], and GP2-AJ cohort [corrected P value = 0.023, OR (95%CI) = 1.43 (1.13–1.80)]. The remaining nominally significant associations did not surpass multiple testing correction (Fig. 1 and Supplementary Table 1a–c). Notably, the TMEM175 p. Met393Thr had a MAF below 0.01 in the African ancestry and p. Gln65Pro variant in the African and East Asian ancestry and were therefore not analysed.
Identification of independent signals with conditional analysis across ten populations
The GWAS signal in the TMEM175 gene, p. Met393Thr was validated as an independent signal in the GP2-EUR cohort (pJ = 6.65e-16) (Table 1; Supplementary Fig. 1a). In the GP2-AJ cohort, rs6599388 (pJ = 2.88e-06) was identified as an additional distinct signal (Table 1; Supplementary Fig. 1b). We found rs6599388 was correlated with the previously reported variant p. Met393Thr (D’ = 0.87, R2 = 0.39, P < 0.0001) in the GP2-AJ cohort. In the GP2-EAS cohort, another signal rs3755956 was identified as a novel independent signal, showing no correlation with p. Met393Thr (D’ = 0.12, R2 = 0.0003, P = 0.59) (Table 1; Supplementary Fig. 1c).
We identified one independent signal, rs11547135(pJ = 1.01e-05), in SCARB2 in the GP2-EUR cohort (Table 1; Supplementary Fig. 2a). rs11547135 was not correlated with previously reported variants rs6812193(D’ = 0.43, R2 = 0.056, P < 0.0001) and rs6825004(D’ = 0.27, R2 = 0.073, P < 0.0001). Additionally, colocalization analysis showed that rs11547135 was strongly associated with SCARB2 expression (posterior probability for a shared causal variant, H4 > 0.99; Supplementary Table 2). Consistent findings were observed across the eQTLGen and GTEx datasets, indicating that the effect allele T was associated with upregulated gene expression in both blood and brain tissues. One additional novel independent genetic variants in SCARB2, rs530111925(pJ = 3.22e-05) was identified in the GP2-AJ cohort(Table 1; Supplementary Fig. 2b) and this variant was not correlated with rs6812193(D’ = 0.31, R2 = 0.0027, P < 0.0001) and rs6825004(D’ = 0.14, R2 = 0.00035, P = 0.12). In the GP2-AMR cohort, one additional distinct signal, rs73828719 (pJ = 3.71e-05) was correlated with rs6812193 (D’ = 0.87, R2 = 0.26, P < 0.0001) but not correlated with rs6825004 (D’ = 0.19, R2 = 0.0028, P < 0.0001)(Table 1; Supplementary Fig. 2c).
We identified one conditionally distinct association signal in CTSB, rs1293289(pJ = 1.14e-06), in the GP2-EUR cohort, but rs1293289 was correlated with the known PD GWAS hit rs1293298 (D’ = 0.97, R2 = 0.80, P < 0.0001) (Table 1; Supplementary Fig. 3a). In the GP2-AMR cohort, one independent novel genetic variants in CTSB, rs73551266(pJ = 1.56e-04) was identified (Table 1; Supplementary Fig. 3b) and rs73551266 was independent of rs1293298(D’ = 0.0038, R2 = 0.00, P = 0.96).
Burden analysis with GP2 neurobooster array
To test if an aggregate burden of rare variants in TMEM175, SCARB2 and CTSB contributes to the risk of PD, SKAT-O analysis was conducted within the GP2 Neurobooster array data across ten populations (Table 2).
No significant associations were detected for rare exonic and non-synonymous and LoF variants in TMEM175 and CTSB across ten GP2 cohorts. In addition, we conducted SKAT-O analyses including the known GWAS hits TMEM175 p. Met393Thr and p. Gln65Pro as covariates, and the results were summarized in Supplementary Table 3a.
Similarly, a SKAT-O analysis for SCARB2 was run to detect a potential genetic burden in PD cases versus controls (Table 2). Subsequently, a SKAT test was performed to further validate SKAT-O results according to the rho value20 (Supplementary Table 3b). However, no associations survived multiple-testing correction (Table 2). To determine which single variants may be driving the observed association with PD risk, a single-variant-based test was conducted. Nominal associations were detected in the GP2-AAC, GP2-AJ, and GP2-EAS cohorts; however, none remained significant after multiple-testing correction (Table 3).
Discussion
We show that previous GWAS hits of three lysosomal related genes (TMEM175, SCARB2 and CTSB) were successfully replicated using large scale genotyping array data. All previously detected variants were significant in the cohort of European ancestry and in some additional ancestry cohorts. The GCTA-COJO analysis was performed across all ten cohorts to identify novel independent signals in these three lysosome-related genes. Significant associations were observed in GP2-EUR, GP2-AJ, GP2-EAS, and GP2-AMR cohorts. Furthermore, SKAT-O analysis revealed genetic burdens for SCARB2, showing nominal significant associations in distinct cohorts. Collapsed burden tests and SKAT tests were further used to validate these findings. Moreover, we detected single rare variants contributing to PD risk through single-variant-based tests.
The missense variant p. Met393Thr in the TMEM175 gene was the most strongly associated signal in the region. The association reached genome-wide significance and TMEM175 p. Met393Thr was identified as a single independent risk factor of PD in the European population, aligning with previous reports3. In addition, it was associated with PD in Ashkenazi Jewish and South Asian populations, which could be possibly due to a haplotype block including p. Met393Thr variant in these other populations. Another common variant rs34884217 p. Gln65Pro, as a secondary signal in the same locus, was found to be associated with reduced PD risk in a previous study21. In this study, the frequency of the protective allele was also significantly higher in the controls than in cases in European population. Notably, due to the MAF threshold of 0.01 for common variants, TMEM175 p. Met393Thr and p. Gln65Pro in the African, and TMEM175 p. Gln65Pro in the East Asian populations, were not included in the analysis in these populations.
We went further to identify independent signals in TMEM175 using GCTA-COJO analysis in other populations. Genotypes at rs6599388 in the TMEM175 gene were detected as an additional distinct intronic variant for PD risk with GCTA-COJO in the Ashkenazi Jewish population. Of note, TMEM175 rs6599388 was correlated with p. Met393Thr. It was also previously reported to be associated with PD in GWA studies in the European population (2011 PD GWAS, rs6599388, odds ratio [OR] = 1.16)22. In the East Asian population, conditional analysis identified rs3755956 as a novel, independent association signal at the TMEM175 locus.
No statistically significant associations of rare variants in the TMEM175 gene were found for any ancestral populations. A previous study did not show significant results in 2657 patients and 3647 controls in a TMEM175 burden analysis23. Our finding was consistent with this previous study, supported by sufficient statistical power, suggesting that a cumulative burden of rare variants in the TMEM175 gene does not contribute significantly to PD risk.
Prior studies in European-ancestry cohorts reported associations at rs6812193 and rs6825004 in SCARB2 with PD risk3,9,18,19. However, after correcting for multiple testing across variants and ancestries, none of the associations remained significant, neither in Europeans nor in any other population.
Besides this, we identified one novel independent non-coding common genetic variant (rs11547135) in the European population. Colocalization analysis showed rs11547135 was strongly associated with SCARB2 expression in blood (eQTLGen) and brain (GTEx) tissue, with consistent positive effect directions. This concordance suggested a shared regulatory mechanism influencing gene expression across tissues, supporting its role as a potential functional variant contributing to PD risk. Further functional studies are warranted to validate this regulatory mechanism and to elucidate its contribution to the molecular pathways in PD pathogenesis. In the Ashkenazi Jewish population, rs530111925 at the SCARB2 locus was identified as a novel, independent variant. Excessive genetic burden of SCARB2 rare variants was not replicated in the European population. Previous studies had shown a possible association between SCARB2 rare variants and PD risk with SKAT-O test8,23. This may be attributed to genetic and demographic differences among populations from different regions or lack of statistical power. Interestingly, SCARB2 rare variants in exonic regions showed nominal associations with PD risk in three other populations: the African American, the Ashkenazi Jewish, and the East Asian cohorts. We found one novel single genetic variant, p. Val149Met, was associated with increased PD risk in the African American population. In the Ashkenazi Jewish cohort, single-variant-based tests indicated that p. Met159Val might have a protective effect on PD. In the East Asian population, we identified two nominally significant missense variants, p. Asp194Asn and p. Ile144Leu, which exhibited discordant effects on PD. None of these significant single genetic variants has previously been reported in the context of PD risk. Furthermore, rare variant analysis of SCARB2 revealed mixed effect directions, with a balanced effect magnitude across the two directions. In such scenarios, the aggregate effect may appear null, despite the presence of biologically meaningful associations. The bidirectional effects have also been observed in genes like TMEM175 and CTSB, where opposing variant effects can obscure the cumulative genetic burden on disease risk. These findings underscore a key limitation of conventional burden analyses that rely on the assumption of effect directionality. On the other hand, understanding such discordance was crucial as it suggested that targeting a single specific pathogenic variant—rather than the gene as a whole—may be a more effective strategy in clinical interventions.
We found CTSB rs1293298 as a significant risk factor in the European population. Of note, CTSB rs1293298 in East Asians was excluded from the common variant analyses due to MAF < 0.01. In the European cohort, we identified a conditionally distinct common intronic variant (rs1293289), linked to the known GWAS hit CTSB rs1293298. In addition, CTSB rs73551266 was detected as a novel independent variant in the Latino and indigenous Americas population.
A recent study observed a nominal association between CTSB rare variants and PD risk in a single Ashkenazi Jewish cohort24. However, we did not replicate this result in large European cohort (24,208 cases, 9662 controls) and any other populations. In contrast to the previous study, we excluded carriers of GBA1 variants from all cohorts to minimize potential bias, which may partly explain the discrepancy. Our findings further support the limitation of rare variant burden analysis, particularly in genes like TMEM175 and CTSB, where both risk and protective variants may co-exist. Besides, future studies should focus on larger sample size in other populations to increase robustness.
The associations of known GWAS hits with PD in other populations were observed although not reaching genome wide significance. This discrepancy may reflect distinct LD patterns, differences in haplotype structures among populations or limited statistical power due to smaller cohort size. Future haplotype-resolved analyses (e.g., phased whole-genome sequencing or ancestry-stratified fine-mapping) could clarify whether these associations arise from shared causal variants present on divergent haplotypes or population-specific functional variants embedded within risk haplotypes. On the other hand, our study illustrates heterogeneity in genetic associations across populations in PD. These combined analyses emphasize these three lysosomal related genes (TMEM175, SCARB2 and CTSB) driving PD risk via both common variants and rare variants. Future research should prioritize detailed functional characterization of these novel variants and their potential patho-mechanism in diverse populations to uncover their roles, facilitating improved understanding and potential development of targeted interventions for PD.
The strength of this study is the analysis of a large-scale case-control cohort from the GP2 Neurobooster array with a population of European ancestry and nine additional populations. The observed replication may, in part, be influenced by partial sample overlap with the 2019 PD GWAS cohort. Notably, 7 out of the 126 cohorts included in our study were included in the previous Nalls’s PD GWAS. It is not possible to identify exactly which samples overlap, because the genotyping arrays are so different that kinship analysis does not identify related/duplicated samples. In addition, as GBA1 lies adjacent to its highly homologous pseudogene GBAP1, accurate variant calling in short-read data such as the Neurobooster array can be challenging. Moreover, the array does not capture all rare variants in GBA1 gene, which may result in some carriers not being detected. These factors should be taken into account when interpreting the findings. Although conditional analysis identified independent signals in distinct populations, the strong linkage disequilibrium (LD) between variants and the lack of replication in non-European cohorts indicated potential population specificity. Therefore, these associations should be interpreted with caution, and further large-scale studies across diverse ancestries are essential to confirm their robustness and generalizability. In the single-variant analyses, the very small number of carriers for ultra-rare variants can yield unstable effect-size estimates and spuriously small p-values under the score test framework. These signals should be viewed cautiously until replicated in larger cohorts, and further methodological development for ultra-rare variants will be warranted.
Importantly, our study reinforces the association between lysosomal genes and PD risk in the European population. The number of non-European individuals will need to be increased in future studies, being clearly smaller compared to the European cohort. PD is a complex disorder with many genetic risk factors at multiple loci contributing to disease risk. Therefore, the novel variants reported in this study should be further investigated in terms of functional effects on PD pathogenesis in the future.
Methods
Subjects
To validate the association between common variants in lysosomal genes from the reported GWA study of PD3, large-scale Neurobooster genotyping imputed data obtained from GP2 release 10 (https://gp2.org/the-components-of-gp2s-10th-data-release/) was analysed, which contains 33,733 cases and 18,703 controls in ten ancestries: GP2-African American (GP2-AAC: 369 cases, 756 controls), GP2-African (GP2-AFR: 1147 cases, 2169 controls), GP2-Ashkenazi Jewish (GP2-AJ: 1285 cases, 408 controls), GP2-Latino and indigenous Americas (GP2-AMR: 1917 cases, 1402 controls), GP2-Central Asian (GP2-CAS: 734 cases, 578 controls), GP2-East Asian (GP2-EAS: 2377 cases, 2598 controls), GP2-European (GP2-EUR: 24,208 cases, 9662 controls), GP2-Middle Eastern (GP2-MDE: 724 cases, 551 controls), GP2-South Asian (GP2-SAS: 309 cases, 261 controls), GP2-Complex Admixture History (GP2-CAH: 663 cases, 318 controls). Populations with less than 100 individuals (i.e. Finnish) were excluded. Given the strong influence of GBA1 on lysosomal biology and its close interactions with other lysosomal related genes, individuals carrying established GBA1 risk-associated variants were removed across all ancestries to avoid inflating the apparent contribution of other lysosomal related genes and to assess their independent effects. This approach referred to a previous study25 and minimize potential bias from co-occurrence with GBA1 variants. The number of individuals removed was provided in Supplementary Table 4. Quality control analyses at a sample and variant level for this GP2 dataset were described elsewhere (https://github.com/GP2code/GenoTools)26. At the sample level, quality control included call rate outliers(--mind, 0.05), biologic sex mismatches (--check-sex, default cutoffs: 0.25 ≤ F ≤ 0.75), relatedness check (--grm-cutoff, in GCTA; Usually set at 0.125 to remove first cousins or more related individuals), and heterozygosity rate outliers (--het, default range: −0.15 ≤ F ≤ 0.15). At the variant level, quality control included missingness by case control (--test-missing, using P > 1e-04), missing by haplotype (--test-mishap, using P > 1e-04), Hardy–Weinberg equilibrium (--filter-controls --hwe, using P > 1e-04) and variant missingness (--geno, 0.05). Cases and controls with missing covariates (including gender and age) were removed.
Gene region extraction and variants annotation
Regions of each gene were extracted with PLINK 2.0 based on the gene locations according to the Ensembl genome browser (https://www.ensembl.org/Homo_sapiens/), including 50 kb downstream and upstream of the genes. Variants were annotated using ANNOVAR27, and variants were defined as (1) exonic, (2) non-synonymous and loss of function (LoF). LoF included stopgain, stoploss and frameshift variants.
Statistical analysis
The number of principal components was chosen by the scree plot of explained variance (Supplementary Fig. 4), which showed a clear elbow after PC5; accordingly, PC1–PC5 were included as covariates.
Association analyses were performed with PLINK 2.028, adjusted by gender, age and the first five genetic principal components (PCs) in each ancestry.
Conditional and joint (COJO) analysis was conducted to determine if there were independent signals per ancestry with genome-wide complex trait analysis (GCTA) software version 1.94.129,30. To identify gene-wide significant signals in the absence of genome-wide significance (P < 5 × 10⁻⁸), we established gene-specific significant thresholds through linkage disequilibrium (LD) pruning followed by Bonferroni correction. LD pruning was performed to define a p-value threshold for the COJO analysis in GP2 NeuroBooster array (parameters: --indep-pairwise 50 50 0.5). We identified 107 independent variants in TMEM175, 141 variants in SCARB2 and 116 variants in CTSB. Therefore, the thresholds for P values which were used in conditional analysis were: 4.67e-4 (0.05/107, TMEM175), 3.55e-4 (0.05/141, SCARB2), 4.31e-4 (0.05/116, CTSB). In addition, LDpair tool (https://ldlink.nih.gov/?tab=ldpair) was used to validate if these signals were independent of known GWAS hits. Regional plots were generated with the online tool LocusZoom.js (https://statgen.github.io/localzoom/)31.
Colocalization analysis was performed to assess whether the genetic variant colocalized with expression quantitative trait locus(eQTL) signals using eQTLGen public dataset(https://www.eqtlgen.org/cis-eqtls.html), following previously described pipelines32(https://rhreynolds.github.io/RBD-GWAS-analysis/, https://github.com/manuelatan/PD-survival-GWAS/). The eQTLGen consortium provides eQTL data representing gene expression in whole blood. To further assess tissue specificity and consistency of effect direction, the GTEx public dataset (https://www.gtexportal.org/home/) was additionally examined to validate the association in brain tissue.
GP2 NeuroBooster array dataset was used to assess whether TMEM175, SCARB2, and CTSB have an excessive genetic burden on PD. Gene-based burden tests were performed using RVTESTS33 to assess the cumulative effects of rare variants on the risk for PD. We defined rare variants as MAF ≤ 1% (--freqUpper 0.01) and applied this same threshold to both gene-based and single-variant-based tests. We used the default Beta (1,25) kernel weights, which up-weight rare variants; and all other parameters were set by default. Analyses were performed twice for each gene, once for all exonic variants and once for the stricter subset of non-synonymous +LoF variants. Furthermore, single-variant-based tests were conducted with RVTESTS (https://zhanxw.github.io/rvtests/#single-variant-tests-1) using a score test under a logistic regression model (--single score) for the case–control phenotype to find single statistically significant variants which have an impact on PD risk33. All burden analyses were adjusted for age, gender and PC1-PC5.
To account for multiple testing, raw p-values were adjusted using the Benjamini–Hochberg (BH) method to control false discovery rate (FDR). For the association analysis of known common genetic variant, corrections were applied across ten ancestries and five genetic variants. For the SKAT-O tests, p-values were adjusted across ten ancestries, three genes, and two variant categories. For the single-variant analyses, corrections were applied to all single-variant tests across ten ancestries using the same method.
All analyses were performed using R statistical language, Python and Linux programming language on Terra (https://terra.bio/) and Verily workbench (https://workbench.verily.com/).
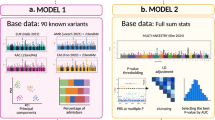
Our overall analytic approach is illustrated in Fig. 2.

General overview of analysis.
Ethics and consent
Written informed consent was obtained from all participants (or their parent/legal guardian in the case of individuals under 16 years of age). For multi-centre studies, each collaborating site secured approval from their local ethics committee, and consent procedures were reviewed by the Operations and Compliance Working Group (OCWG) of GP2 to ensure compliance with international standards and local data-sharing regulations. A list of all participating sites can be found on the GP2 website (https://gp2.org/cohort-dashboard-advanced/). Detailed information on ethics approvals and consent procedures is available on the GP2 website (https://gp2.org/resources/consent-guidelines/).
Data availability
Data used in the preparation of this article were obtained from the Global Parkinson’s Genetics Program (GP2; https://gp2.org). Specifically, we used Tier 2 data from GP2 release 10 (https://doi.org/10.5281/zenodo.15748014). GP2 data are available on AMP PD (https://amp-pd.org).
Code availability
All code generated for this article, and the identifiers for all software programs and packages used, are available on GitHub (https://github.com/GP2code/TMEM175_SCARB2_CTSB_PDrisk) and were given a persistent identifier via Zenodo (https://doi.org/10.5281/zenodo.15799510).
Change history
13 April 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41531-026-01351-6
References
Mazzulli, J. R. et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 (2011).
Lerche, S. et al. The mutation matters: CSF profiles of GCase, sphingolipids, alpha-synuclein in PD(GBA). Mov. Disord. 36, 1216–1228 (2021).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Navarro-Romero, A., Montpeyo, M. & Martinez-Vicente, M. The emerging role of the lysosome in Parkinson’s Disease. Cells 9, 2399 (2020).
Leonard, H. L. & Global Parkinson’s Genetics, P. Novel Parkinson’s disease genetic risk factors within and across european populations. medRxiv (2025).
Blauwendraat, C. et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143, 234–248 (2020).
Jinn, S. et al. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum. Mol. Genet 28, 3244–3254 (2019).
Robak, L. A. et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203 (2017).
Alcalay, R. N. et al. SCARB2 variants and glucocerebrosidase activity in Parkinson’s disease. NPJ Parkinsons Dis. 2, 16004 (2016).
Jinn, S. et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 114, 2389–2394 (2017).
Hu, M. et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell 185, 2292–2308 e20 (2022).
McGlinchey, R. P. & Lee, J. C. Cysteine cathepsins are essential in lysosomal degradation of alpha-synuclein. Proc. Natl. Acad. Sci. USA 112, 9322–9327 (2015).
Kim, M. J., Jeong, H. & Krainc, D. Lysosomal ceramides regulate cathepsin B-mediated processing of saposin C and glucocerebrosidase activity. Hum. Mol. Genet 31, 2424–2437 (2022).
Iwaki, H. et al. Genetic risk of Parkinson disease and progression:: an analysis of 13 longitudinal cohorts. Neurol. Genet 5, e348 (2019).
Grover, S. et al. Genome-wide association and meta-analysis of age at onset in Parkinson disease: evidence from the COURAGE-PD consortium. Neurology 99, e698–e710 (2022).
Blauwendraat, C. et al. Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov. Disord. 34, 866–875 (2019).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516 (2017).
Do, C. B. et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 7, e1002141 (2011).
Hopfner, F. et al. The role of SCARB2 as susceptibility factor in Parkinson’s disease. Mov. Disord. 28, 538–540 (2013).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Krohn, L. et al. Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann. Neurol. 87, 139–153 (2020).
Nalls, M. A. et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649 (2011).
Rudakou, U. et al. Targeted sequencing of Parkinson’s disease loci genes highlights SYT11, FGF20 and other associations. Brain 144, 462–472 (2021).
Jones-Tabah, J. et al. The Parkinson’s disease risk gene cathepsin B promotes fibrillar alpha-synuclein clearance, lysosomal function and glucocerebrosidase activity in dopaminergic neurons. Mol. Neurodegener. 19, 88 (2024).
Lange, L. M. et al. The global landscape of genetic variation in Parkinson’s disease: multi-ancestry insights into established disease genes and their translational relevance. medRxiv (2025).
Vitale, D. et al. GenoTools: an open-source Python package for efficient genotype data quality control and analysis. G3 15, jkae268 (2025).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Hill, A. et al. Stepwise distributed open innovation contests for software development: acceleration of genome-wide association analysis. Gigascience 6, 1–10 (2017).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet 44, 369–375 (2012).
Boughton, A. P. et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics 37, 3017–3018 (2021).
Krohn, L. et al. Genome-wide association study of REM sleep behavior disorder identifies polygenic risk and brain expression effects. Nat. Commun. 13, 7496 (2022).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016).
Acknowledgements
This project was supported by the Global Parkinson’s Genetics Program (GP2; https://gp2.org). GP2 is funded by the Aligning Science Across Parkinson’s (ASAP) initiative and implemented by The Michael J. Fox Foundation for Parkinson’s Research (MJFF). For a complete list of GP2 members, see https://doi.org/10.5281/zenodo.7904831. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. W.S. acknowledges the support of the China Scholarship Council program (Project ID: 202207040033).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
W.S. contributed to the design of the study, M.T. supervised the data analysis, W.S. contributed to detailed analytical workflows, data visualization and drafted the manuscript, C.S., M.T., T.G. contributed to the guidance of the whole process and manuscript check. All listed authors reviewed the manuscript and provided comments and revisions prior to submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A full list of members and their affiliations appears in the Supplementary Information.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, W., Schulte, C., Gasser, T. et al. TMEM175, SCARB2 and CTSB associations with Parkinson’s disease risk across populations. npj Parkinsons Dis. 11, 348 (2025). https://doi.org/10.1038/s41531-025-01180-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01180-z
This article is cited by
-
DNA nanodevices detect an acidic nanolayer on the lysosomal surface
Nature Cell Biology (2026)
