
Abstract
Cellular processes evolve dynamically across time and space. Single-cell and spatial omics technologies have provided high-resolution snapshots of gene expression, greatly expanding the capability to characterize cellular states. This review summarizes recent modeling strategies for time-series and spatiotemporal transcriptomic data, emphasizing links between dynamical systems, generative modeling, and biological insight. These approaches illustrate how computational tools can deepen our understanding of the dynamic nature of single cells.
Similar content being viewed by others

Introduction
Biological processes are inherently dynamic. From lineage bifurcations in embryogenesis to the gradual remodeling of tissues in regeneration and disease, cells continuously evolve across both time and space. Understanding these transitions requires not only high-resolution molecular measurements but also computational methods that can connect static observations into continuous trajectories1,2,3,4,5,6,7,8,9. In recent years, the development of single-cell and spatial omics technologies has greatly expanded our ability to measure cellular states. Single-cell RNA sequencing (scRNA-seq)10,11,12 has revealed the diversity of cell types and states, while temporally resolved scRNA-seq4,13,14 enables sampling across multiple time points. Spatial transcriptomics (ST)15,16,17,18,19,20,21,22 has introduced spatial context into these measurements, and the emergence of time series ST23 offers the opportunity to investigate how cellular organization changes simultaneously across time and space. Together, these technologies mark an important step from static cellular atlases toward dynamic, spatiotemporal reconstructions of biological processes, and constitute the foundation of the recent efforts to build the AI-empowered virtual cells24,25,26.
Despite these progresses, the majority of omics-based data remain fragmented. For instance, snapshot-based scRNA-seq provides only a single-time-point measurement of each cell. Methods such as pseudotime inference27,28,29 and RNA velocity30,31,32,33,34 have therefore been developed to infer dynamics from these static data, ordering cells along latent trajectories or inferring velocity directions of change based on splicing kinetics. These approaches, while highly valuable, are inherently designed for single-time-point snapshot data. Meanwhile, time-series data, such as temporally resolved scRNA-seq or time series ST, directly capture population-level changes across multiple time points and hold the potential to reconstruct faithful cellular trajectories and regulatory mechanisms. Using these richer datasets requires dedicated mathematical and computational frameworks that extend beyond single snapshot-based tools.
A wide range of dynamical modeling strategies have then been proposed, including formulations based on dynamical systems (e.g., ordinary differential equations, ODEs; stochastic differential equations; and partial differential equations) as well as generative frameworks such as optimal transport14,35,36,37,38, flow matching39,40,41,42,43,44, unbalanced transport45,46,47,48, Schrödinger bridges (SB)49,50,51, and mean-field approaches52,53,54,55. These methods provide powerful tools to couple distributions across time and to account for proliferation, death, noise, and cell-cell interactions. Each framework offers distinct advantages and is well-suited to the analysis of multi-time-point or spatiotemporal data.
Several reviews have provided comprehensive summaries of this rapidly developing field. For instance, recent surveys have reviewed or benchmarked pseudotime inference methods56,57, summarized RNA velocity approaches31,56,58,59, and highlighted optimal transport theory in single-cell or spatial omics4,5,14,60. In37, it systematically discussed how concepts from dynamical systems and generative modeling can be applied to spatiotemporal single-cell data. Other reviews have highlighted complementary perspectives, including developmental landscapes and energy-based formulations1,3,9.
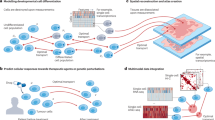
This review takes a distinct perspective. We concentrate specifically on methods designed to analyze time-series single-cell and ST data, including temporally resolved scRNA-seq and spatiotemporal ST. We aim to provide a unified, biologically accessible framework: (i) introducing the mathematical concepts and foundations in intuitive terms, (ii) reviewing key algorithms with a focus on their data requirements, design principles and practical applications, and (iii) offering practical guidelines for selecting, applying, and interpreting these methods in real biological studies. By clarifying the connections between data types, modeling choices, and biological insights, this review will serve as a bridge between theoretical advances and experimental practice, empowering the community to more effectively integrate dynamical modeling into their research. To this end, we introduce the idea of spatiot emporal Dynamical Generative Model (stDGM), a framework that conceptually encompasses the methods discussed (Fig. 1) and implemented in our accompanying software package CytoBridge.

A The analysis starts with raw data from single-cell sequencing, which is converted into a time-resolved, unpaired gene expression and then projected into a low-dimensional embedding. B stDGM model the evolution of the initial cell distribution to a target distribution over time. This approach accounts for key biological processes, including cell differentiation, growth/ death, stochastic effects, and cell-cell interactions. C The trained model enables a rich suite of downstream analyses, such as (a) visualizing velocity streamlines; (b) generating cell states and trajectories at unobserved time points; (c) identifying regions of high cellular proliferation; (d) mapping cell fate stability via a Waddington-like potential landscape; (e) inferring fate probabilities; and (f) constructing gene regulatory networks and predicting the effects of perturbations. The Figure was created in BioRender. Zhang, Z. (2025) under the license https://BioRender.com/f6etpat.
This paper is organized as follows. Section mathematical foundation introduces the mathematical principles of dynamical modeling. Section Algorithms Implementation reviews key algorithmic approaches for temporally resolved single-cell data and spatiotemporal data. Section Practical Guidelines provides practical guidelines for method selection and interpretation. Finally, we summarize the insights and conclude with future perspectives and open challenges in Section Conclusion and Future Directions.
Mathematical foundation
In this section, we summarize the mathematical foundations that underpin trajectory inference and spatiotemporal data integration in single-cell biology (Table 1). The core idea is to treat observed cell populations as samples from distributions that evolve over time and to transport one distribution into another in a way that respects biological constraints in reality. These theories form a hierarchical toolbox, ranging from deterministic to stochastic, from mass-conserving to unbalanced, and from individual to interacting populations. Together, they offer principled ways to decode developmental trajectories and tissue organization from spatiotemporal single-cell data. The historical lineage of key mathematical theories of stDGM is summarized in (Fig. 2A).

A Timeline traces the conceptual lineage from 18th-century static optimal transport to the recent unbalanced mean-field Schrödinger bridges, incrementally relaxing assumptions to match biological complexity. B The contemporary algorithmic zoo is compactly charted along three axes: data assumption, modeling strategy, and training method. The dense grid of named tools demonstrates a fast-evolving ecosystem where theoretical advances have already been packaged into practical, user-ready implementations. The algorithms are still in rapid expansion, and due to the limitation of the authors' scope, some relevant algorithms might not be included.
Static optimal transport
Static optimal transport (static OT) provides a principled way to relate two unpaired cellular populations sampled at distinct time points61. Formally, let \({\bf{X}}\in {{\mathbb{R}}}^{N\times G}\) and \({\bf{Y}}\in {{\mathbb{R}}}^{M\times G}\) be the gene expression matrices collected at two distinct time points t1 and t2, where each row represents a cell embedded in the G-dimensional transcriptomic space. Then, one can define two marginal distributions ν0 ∈ CN and ν1 ∈ CM at t1 and t2, respectively, on the probability simplex \({C}_{N}=\{{\bf{a}}\in {{\mathbb{R}}}^{N}| \sum {a}_{i}=1,{\bf{a}}\ge 0\}\). The well-known Kantorovich formulation62 of the static OT task is to find the nonnegative coupling \({\boldsymbol{\pi }}\in {{\mathbb{R}}}_{\ge 0}^{N\times M}\) that minimizes the total transportation cost:
where the transport plan \({\boldsymbol{\pi }}\in {{\mathbb{R}}}^{N\times M}\) must satisfy the marginal constraints π1M = ν0 and π⊤1N = ν1. Each entry cij: = c(xi, yj) of the cost matrix c = [cij] quantifies the dissimilarity between the transcriptomic profiles of cell i at time t1 and cell j at time t2, typically chosen as the squared Euclidean distance. The resulting optimal coupling π assigns each cell from ν0 to ν1 in the least-cost fashion where πij denotes the mass transported from cell i to j.
Intuitively, the OT model assumes that similar cell states in gene expression space are more likely to be coupled in the cell-fate decision process across time points. Using such a method63, identifies the heterogeneous EMT responses in a scRNAseq time course data of MCF10A cells treated by TGF-beta.
Dynamical optimal transport
Developmental biology seeks to understand how one cell population continuously reshapes itself into another. To generate continuous trajectories from cells at any time point and with different expressions rather than merely assigning pseudotime to all cells in static snapshots, dynamical optimal transport presents a continuous mechanistic modeling framework. By treating cell-state transitions as a smooth, mass-preserving flow, this approach recasts lineage progression in the language of continuum fluid dynamics. In the Benamou-Brenier framework64, the single-cell trajectories satisfy the ODE dXt = b(Xt, t)dt, where b(Xt, t) describes the nonlinear gene regulatory dynamics that drive the cell-state transitions, analogous to the concept of RNA velocity30,31,34. Suppose the gene expression matrices are sampled from a smooth and time-dependent probability density function ρ(x, t), the spatiotemporal dynamics of the density are governed by the continuity equation
where \({\bf{b}}({\bf{x}},t)\in {{\mathbb{R}}}^{G}\) here naturally could be reinterpreted as the velocity field of the density movement. The transport map from the initial to final conditions is not unique; OT resolves this ambiguity by choosing the one that minimizes the total kinetic energy, resulting in the Wasserstein distance between two probability distributions. This is expressed as a minimization with the cost:
and the continuity equation constraint. One important feature of dynamical OT type methods is that they serve as natural generative models. After the vector field in cell state space is learned, the processes of single cells at unobserved temporal points could be simulated through the inferred model. Theoretically, it has be shown that this dynamical OT is equivalent to the static OT when the cost cij = ||xi− yj||264.
Combining dynamical OT with other biological priors, such as RNA velocity or cell growth, TrajectoryNet generates continuous, nonlinear trajectories in both simulated and real biological systems, uncovering cell differentiation paths in human embryoid-body data that align with previously reported biological findings65.
Unbalanced dynamical optimal transport
To faithfully model biological systems in which cells proliferate (mass creation) and undergo apoptosis (mass destruction), one needs to relax the classical assumption of strict mass conservation, that is, the number of cell is permitted to change over time. Such biological constraints have motivated the introduction of unbalanced optimal transport, which is increasingly popular for connecting a time series of densities with different masses. To explicitly and continuously encode cell growth and death, unbalanced dynamical optimal transport introduces a spatiotemporal variable growth rate function \(g({\bf{x}},t):{{\mathbb{R}}}^{G}\times [0,1]\to {\mathbb{R}}\), which acts as a source-sink term in the continuity equation45,46:
and the initial and final conditions: ρ( ⋅ , 0) = ν0, ρ( ⋅ , 1) = ν1. In this setup, Wasserstein and Fisher-Rao (WFR) distance45,66 has been used to optimize transport dynamics with respect to both velocity and growth energy. It minimizes the combined WFR metrics defined as:
subject to the continuity Eq. (3).
Powered by dynamical unbalanced optimal transport, TIGON67 reconstructs cell-state transition dynamics during EMT and detects the cellular proliferation peak at the intermediate stage, consistent with the biology that intermediate-state cells transiently reacquire stem-like potency68,69,70,71,72.
Schrödinger bridge problem (SB)
To capture the prevalent stochasticity of single-cell trajectories during the cell-fate decision process73, the SB framework explicitly models random fluctuations rather than relying on purely deterministic transport. It seeks to determine the most probable evolution between a specified initial distribution ν0 and a terminal distribution ν1 relative to a prescribed reference stochastic process. Formally, the problem is described as an optimal control problem whose objective is to minimize the Kullback-Leibler (KL) divergence DKL, an idea that traces back to Schrödinger (1932) and subsequent stochastic control treatments74,75:
where \({\mu }_{[0,1]}^{{\bf{X}}}\) is the probability measure on \({\mathcal{C}}([0,1],{{\mathbb{R}}}^{G})\) induced by the stochastic process \({\{{{\bf{X}}}_{t}\}}_{0\le t\le 1}\). At each time t, the one-time marginal of measure \({\mu }_{[0,1]}^{{\bf{X}}}\) is denoted \({\mu }_{t}^{{\bf{X}}}\) and possesses the density ρ(x, t). Concretely, each cell’s gene expression state \({{\bf{X}}}_{t}\in {{\mathbb{R}}}^{G}\) can be assumed to evolve as dXt = b(Xt, t) dt + σ(Xt, t) dWt, where \({\{{{\bf{W}}}_{t}\}}_{t\ge 0}\) is a standard multidimensional Brownian motion (with dimension G) and \(\sigma :{{\mathbb{R}}}^{G}\times [0,1]\to {{\mathbb{R}}}^{G\times G}\) denotes the diffusion coefficient. The reference measure \({\mu }_{[0,1]}^{{\bf{Y}}}\) is generated by the uncontrolled diffusion dYt = σ(Yt, t) dWt. Simply put, it aims to identify the cell-state transition dynamics from purely stochastic motion to dynamics driven by both clear gene regulation forces and stochastic components either intrinsically from gene expression process or from the fluctuating environment. In this formulation, the problem can be equivalently transformed to minimizing the cost50,51,75,76:
where a(x, t) = σ(x, t)σ⊤(x, t) and the optimization is taken over all pairs of functions ρ satisfying ρ( ⋅ , 0) = ν0, ρ( ⋅ , 1) = ν1. Additionally, the pair (ρ, b) needs to satisfy the Fokker-Planck Equation:
where \({\nabla }_{{\bf{x}}}^{2}:({\bf{a}}\rho )={\sum }_{ij}{\partial }_{ij}({{\bf{a}}}_{ij}\rho )\), coupled with asymptotic vanishing boundary condition: \({\lim }_{| {\bf{x}}| \to \infty }\rho ({\bf{x}},t)=0\).
Inspired by SB, SF2M reconstructed high-dimensional trajectories of differentiating mouse embryonic stem cells from five unpaired scRNA-seq time-points and accurately predicted an unseen day-6 population77. Its built-in Brownian-bridge noise term captured the probabilistic bifurcation of pluripotent cells into mesoderm and ectoderm lineages.
Regularized Unbalanced Optimal Transport (RUOT)
In the study of cellular dynamics, where both random changes and processes such as cell growth and death occur, the RUOT framework offers a natural advancement of traditional optimal transport models51,78,79. Specifically, when the diffusion coefficient is isotropic, i.e., a(x, t) = σ2(t)I, the density evolution follows the Fokker–Planck equation with a source term:
where g(x, t) denotes the net growth rate and the boundary condition \({\lim }_{| x| \to \infty }\rho ({\bf{x}},t)=0\) ensures integrability. The corresponding optimization problem seeks to minimize an action functional that balances kinetic energy against a growth penalty:
subject to the dynamics (7) and marginal constraints ρ( ⋅ , 0) = ν0, ρ( ⋅ , 1) = ν1. Here \(\Psi :{\mathbb{R}}\to [0,+\infty ]\) is a convex penalty that controls deviations from mass conservation. Note that in the definition if \(\Psi \left(g\right)=+\infty\) unless g = 0 and \(\,\Psi \left(0\right)=0\), then it implies g(x, t) = 0 and the RUOT problem is equivalent to the special case of the SB Problem, characterized by a(x, t) = σ2(t) I. If σ(t) → 0 and \(\Psi \left({\bf{x}},t\right)=| g({\bf{x}},t){| }^{2}\), this degenerates to the unbalanced dynamic optimal transport with WFR metrics.
Based on such framework, DeepRUOT51 also traces the unbalanced continuous epithelial-intermediate-mesenchymal path with greater accuracy, illustrating how the method merges RNA-velocity drift, genuine growth/death, and stochastic effects to achieve smooth temporal cell population interpolation in real scRNA-seq data.
Hamilton-Jacobi-Bellman (HJB) Equation and Optimal Transport
The Hamilton-Jacobi-Bellman (HJB)80,81 equation is a fundamental tool in the field of stochastic optimal control, providing an effective framework for solving optimization problems involving stochastic processes. In the context of optimal transport, the HJB equation plays a crucial role in characterizing the optimal control strategies that minimize the cost of transporting one probability distribution to another. When solving optimal transport and its variants, the Fokker-Planck equation serves as a constraint that can be incorporated into the optimization objective via Lagrange multipliers. Taking the RUOT problem as an example51, the augmented objective function is given by:
Then the problem can be treated as an unconstrained optimization problem, with variational derivatives with respect to ρ, b and g yielding three optimality conditions. In particular, the optimality condition obtained from the variation with respect to ρ provides the evolution equation for the Lagrange multiplier over time, which is the HJB equation. By deriving the optimality condition, one can show that only one scalar field λ(x, t) needs to be trained48, sparing us from learning the RNA velocity drift b, the growth rate g and the density ρ (or the associated energy landscape, the score function) separately, yielding the faster and more stable optimization. The optimality condition and HJB equation for the RUOT problem is:
When g = 0, this equation reduces to the HJB equation for the SB problem. When σ = 0 with \(\Psi (g)=\frac{1}{2}{g}^{2}\), it reduces to the HJB equation for the Dynamical Unbalanced OT problem that employs the WFR metric.
In mouse blood hematopoiesis, Var-RUOT48 reaches the smallest action, trains faster and shows lower variance, all benefitted from learning one scalar field that simultaneously produces the straighter lineage-splitting trajectory and the accurate upstream-to-downstream decay of cellular growth rate, which is consistent with the knowledge of proliferating stem cells in biology.
Mean-Field Schrödinger Bridge (MFSB)
The classical SB problem reconstructs the most-probable trajectory between two observed distributions under the assumption that the underlying cells (i.e., particles) are independent. In comparison, biological reality is shaped by persistent cell-cell communications that couple individual fates into a collective process. To capture this coupling, the bridge problem can be extended to a mean-field setting82,83. Consider N particles evolving under the influence of a kernel of symmetric interaction K. The discretized McKean-Vlasov stochastic dynamics84 has the form
where the first term captures the interactions between particles quantified by the interacting kernel K, and the second term involves \({{\bf{W}}}_{t}^{i}\), which are independent standard Brownian motions for i = 1, …, N. The empirical measures at t = 0 and t = 1 are observed to be close to the prescribed probability measures ν0 and ν1, respectively. The discrete system has a mean field limit with density ρ(x, t) satisfies the McKean-Vlasov PDE \({\partial }_{t}\rho +{\nabla }_{{\bf{x}}}\cdot \left(\rho \,{\mathbb{K}}\rho \right)=\frac{1}{2}{\sigma }^{2}(t){\Delta }_{{\bf{x}}}\rho\) with \({\mathbb{K}}\rho ({\bf{x}},t):={\int}_{{{\mathbb{R}}}^{G}}{\bf{K}}({\bf{x}},{\bf{y}})\rho ({\bf{y}},t){\rm{d}}{\bf{y}}\).
In the context of the mean-field SB, one also seeks for a velocity field b(x, t) that drives the continuity equation
between the prescribed marginals ν0 and ν1. Here one chooses the reference process as the mean-field Mckean-Vlasov dynamics instead of the Brownian motion in Section. By incorporating a prescribed interaction kernel k(x, y) which modulates how strongly position y influences position x (e.g., nearest neighbor kernel or Gaussian kernel) in (11), one can approximate the interaction kernel K by taking the ansatz K(x, y) = − k(x, y) ∇xV(x − y), where V is a scalar interaction potential to be learned. Among all velocity fields b, one that minimizes the action functional, which is exactly Eq. (2) in the dynamical OT, is selected.
Optimizers of this MFSB problem characterize the most probable collective evolution of an interacting cellular population that is consistent with the observed initial and final statistics.
Unbalanced Mean-Field Schrödinger Bridge (UMFSB)
To simultaneously account for (i) collective cell-cell interactions, (ii) stochastic single-cell dynamics, and (iii) unbalanced mass changes driven by proliferation and death, the Mean-Field SB is unified with the regularized unbalanced optimal transport framework, resulting in the UMFSB model54. This model seeks the most-probable collective evolution point clouds of dynamically interacting cells between observed snapshots whose total mass may differ. Formally, UMFSB is a variational problem.
subject to the Fokker-Planck equation of McKean-Vlasov process
where \({\mathbb{K}}\rho ({\bf{x}}):={\int}_{{{\mathbb{R}}}^{G}}{\bf{K}}({\bf{x}},{\bf{y}})\rho ({\bf{y}},t){\rm{d}}{\bf{y}}\), \(\Psi :{\mathbb{R}}\to [0,\infty ]\) is a convex penalty, typically Ψ(g) = g2 that regulates mass deviations, and α > 0 balances transport energy against growth cost. In the limit σ → 0 with Ψ(g) = g2 UMFSB reduces to unbalanced dynamic optimal transport; with g ≡ 0 and Ψ(g) = + ∞ unless g = 0 it becomes the Mean-Field SB; and when both σ → 0 and g ≡ 0 classical optimal transport with interaction is recovered.
Using the UMFSB framework,54 reveals the interaction force that draws transcriptionally similar cells closer together in mouse hematopoiesis data. Especially compared the single-cell drift b with the interaction force \({\mathbb{K}}\rho\), the correlation shows a clear time-dependence: early in the time-course the attraction nudges progenitors toward differentiation, whereas later it restrains them from completing terminal stage. These results show both the necessity of embedding cell-cell interactions in dynamic models and the model’s capacity to learn those interactions directly from data.
Gromov-Wasserstein Optimal Transport (GWOT)
GWOT provides a principled way to compare or align two populations of cells, even when they are measured in entirely different feature spaces or different biological samples across time points36,85,86,87. In essence, GW transport asks how one cellular population could be “morphed” into another while preserving the internal relationships between cells, rather than depending on shared coordinates or matched features. This flexibility makes GW particularly appealing for single-cell biology, where distinct experimental conditions, temporal points, modalities, or technologies often produce data embedded in incompatible measurement spaces. By contrast, classical optimal transport (OT) assumes that both datasets lie in the same coordinate system and directly penalizes the cost of moving cellular mass from one point to another in that shared space.
In the GWOT formulation one posits two discrete metric-measure spaces \({{\mathcal{X}}}_{0}=({{\bf{X}}}_{{\bf{0}}},{d}_{{{\bf{x}}}_{{\bf{0}}}},{\nu }_{0})\) and \({{\mathcal{X}}}_{1}=({{\bf{X}}}_{{\bf{1}}},{d}_{{{\bf{x}}}_{{\bf{1}}}},{\nu }_{1})\). Here \({{\mathcal{X}}}_{0}\) and \({{\mathcal{X}}}_{1}\) are finite sets of cells, \({d}_{{{\bf{x}}}_{{\bf{0}}}}\) and \({d}_{{{\bf{x}}}_{{\bf{1}}}}\) are intrinsic distance matrices (they might be Euclidean, diffusion, or correlation distances), and ν0, ν1 are probability vectors that weight each cell, possibly reflecting sequencing depth or prior knowledge. The GW problem seeks a coupling matrix \({\boldsymbol{\pi }}\in {{\mathbb{R}}}_{\ge 0}^{N\times M}\) whose marginals recover the prescribed masses and that minimizes the total structural distortion
Intuitively, whenever cells \({{\bf{x}}}_{i}^{0}\) and \({{\bf{x}}}_{k}^{0}\) are far apart in their own geometry, the cells \({{\bf{x}}}_{j}^{1}\) and \({{\bf{x}}}_{\ell }^{1}\) to which they are coupled should be far apart in the second geometry, and vice-versa. The exponent p (usually p = 2) controls the sensitivity to large mismatches. The resulting matrix π can be read as a soft many-to-many assignment, producing lineage-like correspondences across experiments without any gene-wise alignment.
Using a fused GWOT framework, MOSCOT36 reconstructed spatiotemporal organogenesis trajectories in mouse embryogenesis especially for the heart and brain region, mapping how cells migrate, differentiate, and reorganize across both position and developmental stages.
Rigid Body Transformation Invariant Optimal Transport (RBTI-OT) and Spatiotemporal Dynamics Learning (stVCR)
In contrast to the GW framework, which preserves relational structures without imposing explicit alignment of the underlying spaces, rigid body transformation invariant optimal transport (RBTI-OT)88 explicitly models the deformation between the two measure spaces as a rigid body transformation, comprising only rotations and translations, to unify them into a common coordinate system. This approach is particularly suited to scenarios like aligning ST data across time points, where differences in the measurement spaces of the data are assumed to be alignable solely through rigid body transformations, enabling direct comparison in a shared Euclidean space rather than relying on intrinsic distances alone89. Formally, given two discrete spaces \({{\mathcal{X}}}_{0}=({{\bf{Z}}}_{{\bf{0}}},{\nu }_{0})\) and \({{\mathcal{X}}}_{1}=({{\bf{Z}}}_{{\bf{1}}},{\nu }_{1})\), where Z0 and Z1 are sets of points in potentially misaligned coordinate systems (Z is the coordinate variable in ST), RBTI-OT seeks a coupling matrix \({\boldsymbol{\pi }}\in {{\mathbb{R}}}_{\ge 0}^{N\times M}\) with marginals matching ν0 and ν1, along with an optimal transformation G = (R, r) from the set \({\mathcal{G}}\) of rotations R and translations r, minimizing the total transportation cost
where G(zj): = Rzj + r. Both RBTI-OT and GW are designed for data in distinct measurement spaces, dispensing with the need for shared features or coordinates in classical OT; however, RBTI-OT’s explicit parameterization of \({\mathcal{G}}\) as rigid motions allows for recovery of the transformation itself, whereas GW achieves flexibility through relational invariance but at a higher computational cost due to its quartic objective and lack of coordinate unification. The problem is solved via alternating minimization: first updating π as a static OT problem with fixed G, then solving for G as a weighted Procrustes alignment90 given π. Extensions to affine transformations or other constrained deformations are straightforward, and entropic regularization can be similarly applied to smooth the optimization. Finally, since RBTI-OT unifies the distributions into a common space, it becomes easier to extend it to the dynamic OT framework. Formally, we minimize
subject to
An application of this dynamic RBTI-OT is the spatiotemporal Video Cassette Recorder (stVCR) framework89, which reconstructs cell differentiation, migration, and proliferation/apoptosis from time-series ST data that jointly measure gene expression \({\{{{\bf{X}}}_{i}\}}_{i = 0}^{K}\) and spatial coordinates \({\{{{\bf{Z}}}_{i}\}}_{i = 0}^{K}\). In stVCR, the authors solve a minimization problem with the cost
subject to
where b = (bx, bz) characterizes the migration velocity in coordinate space and gene space, respectively. In addition, stVCR incorporates optional biological priors, including known cell-type transitions and spatial structure-preserving priors. These priors are introduced as constraints on the reconstruction dynamics, effectively guiding the model based on established biological knowledge. In scenarios with sparsely sampled time-point data, the inclusion of such biological priors is indispensable and significantly enhances the accuracy of the dynamical reconstruction.
stVCR has been applied to reconstruct the continuous spatiotemporal dynamics of brain regeneration in the Mexican axolotl following injury91. It has also been used to model the 3D development of Drosophila embryos and organs, particularly the central nervous system and midgut, capturing the spatiotemporal dynamics from 7 to 10 hours post-fertilization92,93, especailly at unseen time points. These applications demonstrate stVCR’s ability to recover complex biological processes with high temporal and spatial resolution, providing insights into tissue regeneration and organogenesis.
Algorithms implementation
In this section, we summarize the existing trajectory inference algorithms that are implemented based on the mathematical theories described above (Fig. 2B, Table 2). Often, these methods aim to learn the “optimal" mapping from one distribution to another, which can be induced by point-to-point correspondences or continuous flows within the data space, and is determined by the form of the “action" defined for the cell state-transition process. We categorize these trajectory inference methods based on three characteristics: Data Assumption, Modeling Strategy, and Training Methods.
Data assumption
Different trajectory inference methods rely on distinct assumptions about the input data, reflecting diverse types of biological priors. For example, several methods propose considering the effects of cell division and death, which lead to an unnormalized total mass in the distribution (Unbalanced Data). In such cases, one assumes that the distribution follows an unbalanced Fokker-Planck equation and uses the static unbalanced OT distance or the WFR distance as the action (Eq. (3), Eq. (8)). Typical methods that adopt the Unbalanced Data Assumption include TIGON67, DeepRUOT51, and stVCR89.
In scRNA-seq data, the gene expression count numbers are inherently discrete, typically following count distributions such as the Poisson or negative binomial (Count Data). As a result, data points cannot evolve continuously within the data space. To nevertheless capture the continuous evolution of the continuous cell fates, one approach is to model the dynamics of a parameterized probability measure and employ the geodesic distance, e.g., defined via the Fisher information metric on a finite-dimensional statistical manifold as the action functional, and then adopt the least action method to calculate the transitional paths. For instance, Euclidean VAE94 adopts the idea by assuming that the VAE’s decoder is a smooth mapping from the latent space to the probability measure manifold, and directly considering the evolution trajectory in the latent space.
Most computational methods assume that data reside in a Euclidean space; however, scRNA-seq data are often governed by intrinsic biological structures and thus are better represented as lying on a low-dimensional manifold (Low-Dim Manifold). For example, although gene expression measurements are collected in a G-dimensional gene space, cellular states typically occupy only a restricted region determined by regulatory programs, developmental lineages, or other biological constraints95. Consequently, the effective dimensionality of the data is substantially much lower than G. Methods like MIOFlow96 and Metric FM97 exploit the low-dimensional manifold structure underlying the data and perform geodesic interpolation in this manifold rather than in Euclidean space. Wasserstein Lane-Riesenfeld (WLR) algorithm98 approximates B-spline curves in the Wasserstein space through iterative averaging of geodesics. In the meantime, methods such as Topological SB99 treat each dimension of the data vector as a feature on the vertices of an undirected graph, thus designating the diffusion on the graph as the reference process in the SB problem.
At times, the data points analyzed are sampled from different modalities (e.g., transcriptomic, proteomic, or morphological measurements) or from distinct biological systems (e.g., samples collected across individuals, tissues, or developmental time points) with different metric spaces (Cross-Domain Mapping), making the optimal transport problem directly on these data points not well defined. A typical theory used for analyzing and processing such cross-space data is GWOT (Eq. (13)), which computes a transport plan using the geodesic distance between data points on each manifold, therefore assessing the similarity of the two manifolds’ geometric structures. Building on this framework, methods such as MOSCOT36, GENOT100, and SCOT+101 apply GWOT theory to address multi-modal and cross-system integration challenges in trajectory inference tasks.
In single-cell omics, a special type of data is ST data, which includes not only gene expression counts within cells but also the physical spatial location information of each cell (Spatial Data). Methods like stVCR89, Dest-OT102 and STORIES103 are specifically designed to handle such data. Both DeST-OT and STORIES adopt Fused Gromov-Wasserstein OT (FGWOT) to model ST across time. More specifically, DeST-OT incorporates cell proliferation by employing semi-unbalanced OT within the static OT framework, while STORIES directly uses FGWOT as the loss function to reconstruct gene expression dynamics. Instead of using GWOT-related theory, stVCR applies RBTI-OT to model spatial coordinates, which makes it possible to simultaneously reconstruct cell differentiation, migration, and proliferation dynamics in a continuous setting. Besides, other related applications for spatiotemporal data have also been developed. For example, PASTE104 and PASTE2105 align adjacent tissue slices by employing the Gromov-Wasserstein OT framework. CODA106 uses an image registration-based approach to align histological images and reconstruct 3D tissues from serial sections. Furthermore, other methods focus on inferring spatial locations for scRNA-seq data; for example, STALocator107 uses a supervised auto-encoder to localize single cells onto ST data, while iSORT108 maps gene expressions to spatial locations via transfer learning.
Lastly, in perturbation studies, the gene expression matrices are often accompanied by categorical labels or other experimental conditions (e.g., treatment type, dosage, or time point), which needs to be explicitly incorporated during training or inference (Conditional Modeling). Methods such as CFGen109 and CellFlow110 enable the generation of cell states under specified perturbations, while MMFM111 extends trajectory inference frameworks to account for conditional information.
Modeling strategy
Trajectory inference methods also differ in the dynamical models they adopt, which could be formulated in either discrete or continuous time and space. The choice of dynamical model specifies the underlying structure of the governing equations, while the inference procedure estimates the unknown components (typically time-dependent scalar or vector fields). When the continuous temporal evolution of the distribution is not of primary interest, one may instead employ discrete-time dynamics, focusing on the mapping of cell states observed at an initial time point to those at a subsequent one (Eq. (1)). Methods such as Waddington OT35, MOSCOT36 and Multistage OT112 adopt this setting. In particular, optimal transport between Gaussian mixtures admits an analytical solution, which scEGOT113 takes advantage of. Moreover, OTVelo114 attempts to estimate RNA Velocity using the solution of discrete OT. Discrete OT also has several variants. For example, HM-OT115 can handle partially observed data by learning a latent representation for each data point and determining the transition matrix in the latent representation. By harmonizing discrete and continuous-time modeling, the CT-OT Flow method116 estimates finer-grained time labels from the data, and then proceeds to solve the OT problem and reconstruct the continuous ODE/SDE dynamics.
In practice, cellular processes are subject to numerous unobserved perturbations and intrinsic variability, and coarse-graining of underlying deterministic dynamics naturally gives rise to stochastic dynamics. A principled framework for inferring such stochastic dynamics is provided by the SB (Eq. (4), Eq. (5), Eq. (6)). This formulation augments single-particle dynamics with a Brownian motion term and introduces a diffusion term into the corresponding Fokker-Planck equation, while retaining an action functional equivalent to that of Dynamical Optimal Transport. Methods like SB between Gaussian117, SF2M77, PISDE118, FBSDE Model119, Probability Flow Inference120 and Likelihood Training SB121 discuss various solutions for the SB problem. Among them, SB between Gaussian provides an analytical solution for cases with Gaussian Mixture marginal distributions; Likelihood Training SB mimics the Likelihood Training in score matching to offer a framework for solving the SB problem. Several methods are proposed solve more generalized SB problems. For example, Lagrangian SB122 allows for solving the evolution of particle distributions in any given potential field; mvOU-OTFM123 sets the reference process of the SB to an OU process; and Smooth SB124 adopts a smooth Gaussian process as the reference process. In order to handle branching data for improved downstream tasks such as cell fate prediction, Branched SB125 matches a single initial distribution to multiple terminal distributions with unequal weights. Moreover, to simultaneously address the previously mentioned Unbalanced Distribution and Stochastic Dynamics, Pseudo Dynamics126 uses the Fokker-Planck equation with diffusion and non-equilibrium terms, and employs maximum likelihood estimation to determine the parameters; Unbalanced Diffusion SB127 proposes a SB that incorporates growth and death; ARTEMIS128 solves such a SB in the latent space of a VAE, further enhancing the model’s expressive capacity ; DeepRUOT51 adopts the RUOT framework (Eq. (7), Eq. (8)), where the Fokker-Planck equation includes both diffusion and unbalanced terms, using the WFR Distance as the action.
Compared to the commonly used first-order dynamical frameworks, incorporating momentum dynamics allows modeling of more complex cellular processes, where the history or “inertia” of transcriptional changes influences future cell states. Methods such as 3MSBM129 explicitly account for this effect. Moreover, many existing approaches assume that cells evolve independently; however, in biological systems this assumption is often violated. Cell-cell interaction dynamics arising from processes such as ligand-receptor signaling or cell-cell contact can play a central role in shaping cell-fate trajectories. To address this, methods including MetaFM130, scIMF55, GraphFP131, and CytoBridge54 incorporate intercellular interactions into trajectory inference.
Training methods
Trajectory inference methods also differ in their training paradigms. A foundational class of approaches builds on the Neural ODE framework132,133. For instance, TrajectoryNet65 and scNODE134 approximate population dynamics by evolving an empirical particle system, where velocity fields are parameterized by neural networks to capture the underlying transcriptional dynamics. The associated action functional and distribution-matching error can be computed from this neural ODE formulation and incorporated into the loss function for backpropagation-based training. A recent alternative, Cell-MNN135, learns a locally linearized ODE representation of dynamics by predicting the system’s linear operator. To further address the challenge of highly unbalanced cell state distributions, TIGON67 employs a weighted particle system by additionally parameterizing growth rate to approximate the evolution of both cellular mass and densities. To solve stochastic dynamics, PISDE118 and Var-RUOT48 also adopt the neural SDE methods136.
However, neural ODE or SDE-based methods require iterative numerical integration of continuous dynamics during training, which leads to computational overhead. As a result, their scalability is limited when applied to high-dimensional gene expression spaces or large-scale single-cell datasets. In response, a series of simulation-free training methods exemplified by Conditional Flow Matching39,40,137 have emerged. These methods are typically designed based on analytical solutions for simple cases (for instance, mapping a Dirac distribution to another Dirac distribution), allowing for the direct estimation of the target scalar or vector fields without simulating ODEs. SF2M77 employs the Flow Matching method to solve the SB problem; Score-Based NF138 uses the Flow Matching method to solve the velocity field of the PF-ODE in Score Matching; Unbalanced Monge Map139 and VGFM41 combine Unbalanced Optimal Transport with Flow Matching. In particular, VGFM can simultaneously learn v and g in the Unbalanced Dynamical Optimal Transport framework to address Unbalanced Distribution. Curly FM140 is capable of learning non-gradient velocity fields, while Metric FM97 first estimates geodesics on a low-dimensional manifold and then performs geodesic interpolation. Furthermore, Wasserstein FM141 performs interpolation directly in the space of probability measures and has proven to be effective in generating high-dimensional distributions; MMSFM142 allows the connection of data between time points via multi-marginal SB.
Furthermore, the first-order optimality conditions for optimal transport and its variants can be derived via variational principles (Eq. (9) and Eq. (10)), providing the foundation for designing efficient computational algorithms. PRESCIENT143,Action Matching144 and PISDE118 constrain the dynamic search space to the set of gradients of a scalar field, where the HJB equation is enforced as a loss term in PISDE. Wasserstein Lagrangian Flow145 solves optimal transport and its variants by fitting covariant vectors on the probability measure manifold along with parameterized probability measures. GraphFP131 designs a gradient descent method based on the Pontryagin Maximum Principle for solving optimal control laws. HJ-Sampler146 employs the Cole-Hopf transformation to convert the nonlinear problem into a tractable linear or semi-linear form, then derives the control law by solving the HJB equation, ultimately obtaining the posterior distribution of the data. Recently, Var-RUOT48 further demonstrated that it is sufficient to solve the RUOT problem by merely parametrizing a single scalar function based on the HJB framework.
Practical guidelines
To help researchers utilize the proposed Spatiotemporal Dynamical Generative Model (stDGM) framework, here we outline the guidelines for applying dynamical generative modeling tools, covering the entire workflow from data input to biological discovery, as summarized in Fig. 3. Specifically, to put these principles into practice, we are actively developing CytoBridge, a Python package that integrates this entire workflow, and we invite contributions from the community to help shape its future. Below we describe the design philosophy and the workflow of applying the CytoBridge into spatiotemporal omics data analysis. We also provide a case study in Box 1 to demonstrate the stDGM workflow using CytoBridge.

The analysis pipeline starts with data preprocessing. Following preprocessing, a suitable model is configured by selecting from four primary components offered by the CytoBridge package: velocity for cell differentiation, growth for cell proliferation, score for stochasticity, and interaction for cell-cell communication. Different combinations of these components correspond to distinct theoretical frameworks and enable specific downstream analyses.
Data preprocessing
stDGM is primarily applied to temporally-resolved scRNA-seq data or spatial-transcriptomic data. The required input is a gene expression matrix accompanied by metadata for each cell, crucially specifying its sampling time point, and spatial coordinates if available. The initial step, data preprocessing, is essential for minimizing technical noise. This process involves normalizing the gene expression data to correct for library size variations, and aligning spatial coordinates across different time points. Then, feature selection of highly variable genes is utilized to isolate signals driving cellular change147. Subsequently, the gene expression data is projected into a low-dimensional space. Methods like PCA and AutoEncoders are recommended because they are reversible, they allow vectors to be projected from the reduced space back to the original gene expression space. This property is vital for enabling the downstream analysis of specific genes and pathways. Generally, this projected space should be kept below 100 dimensions, as higher dimensionality can obscure the key factors driving cell differentiation. These preprocessing steps can be carried out using CytoBridge, as shown in Step 1 of Box 1.
Tools selection
With a clean and properly structured dataset in hand, the core analysis begins: applying and configuring the dynamical models. These methods reconstruct trajectories from discrete time-point snapshots by using neural networks to model the driving factors of cellular state changes. The CytoBridge package supports four primary modeling components: velocity network, growth network, score network, and interaction network. Each of these components corresponds to a specific stDGM-based framework. Then, a crucial step is selecting the appropriate dynamical model, a choice guided by the biological assumptions one makes about the system.
The first consideration is the cell growth term. If changes in the number of cells across time points are not significant, or due to technical sampling artifacts, or not of biological interest, a standard dynamical OT formulation focused only on matching probability distributions can be applied. Representative methods include MioFlow96 or OT-CFM40. However, users still need to be cautious of the false-positive transitions incurred by unbalanced sample sizes35,51,67 and certain resampling strategies could be considered. Indeed, if population size changes are significant, or reflect genuine biological processes like development, including the growth term is recommended to gain deeper insights and more accurate inference. This places the analysis within the unbalanced optimal transport framework, often requiring an additional neural network to model growth, as implemented in tools like TIGON67 and the recently proposed simulation-free method VGFM41. In such methods, velocity and growth networks are used to simultaneously match both the distribution and the number of cells across different time points. An example of leveraging the dynamical unbalanced OT framework can be found in Step 2 of Box 1.
A second consideration is stochasticity. To capture the inherent randomness of biological processes, the problem can be framed as a SB Problem if the growth term is not considered. This can be addressed by directly simulating neural SDE with methods like PI-SDE118, or by augmenting a deterministic velocity field with a score-matching network to model probability densities, as seen in SF2M77. For systems exhibiting both unbalanced growth and stochasticity, Regularized Unbalanced Optimal Transport (RUOT) frameworks, implemented in methods like DeepRUOT51 and Var-RUOT48, are appropriate.
Most recently, the scope of modeling has expanded to include cell-cell interactions through the newly proposed Unbalanced Mean Field Schrödinger Bridge (UMFSB) problem54. The UMFSB framework can simultaneously infer interactions, growth, and stochastic effects. Built upon this theory, the CytoBridge package is aimed at serving as a unified toolkit. It enables users to selectively deactivate the interaction, growth, or stochastic terms, thereby tailoring the analysis precisely to their specific dataset and biological questions.
Downstream analysis
The next stage of the workflow is the downstream analysis and interpretation. Visualization is often the first step, where the inferred velocity can be projected onto a low-dimensional embedding like a UMAP148. This provides an intuitive view of the developmental flow and the major predicted lineage paths (Fig. 1C (a)). Moreover, the growth network, if available, can reveal the specific cell types that exhibit higher growth rates (Fig. 1C (c)). The score network identifies high-density regions corresponding to stable cell fates, which are analogous to the valleys in the Waddington epigenetic landscape4,51,149,150,151,152,153,154,155,156,157,158 (Fig. 1C (d)). The usage of basic visualization using CytoBridge can be found in Step 3 of Box 1. Beyond visualization, the trained neural networks are interpretable models that enable powerful quantitative analysis. The learned velocity field, for instance, can be used to infer Gene Regulatory Networks (GRNs) by computing its Jacobian67 (Fig. 1C (f)). Similarly, the gradient of the growth network can identify key genes driving cell proliferation. This principle extends to models that include cell-cell interactions. Methods modeling cellular interactions can distinguish a cell’s intrinsic differentiation drive from the influence of intercellular communication. By analyzing the properties of the interaction forces, it’s possible to identify which genes are most responsive to neighborly signaling and to characterize the nature of the interactions themselves by calculating the spatial autocorrelation of the similarity of interacting forces to the intrinsic drift54. For these analyses, the results from the low-dimensional space can be projected back to the original gene space to ensure biological interpretability. The lists of high-impact genes generated from these different analyses, whether they are GRN hubs, proliferation drivers, or interaction targets, can be subjected to Gene Set Enrichment Analysis159. This step connects the individual genes to the broader biological pathways and functions they collectively represent, completing the bridge from data to mechanistic insights. Thus, in-silico perturbations to specific driver genes can be applied in a straightforward way. It is also noteworthy that the overall drift, which integrates velocity, score, and interaction terms if available, provides a comprehensive representation of cellular dynamics. This drift is compatible with other downstream analysis tools such as scVelo34, and thus can be used to compute a cell-to-cell transition matrix and velocity graph. The constructed graph can be subsequently applied to CellRank160 to infer fate probabilities or driver genes (Fig. 1C (e)). The seamless integration of these stDGM methods with the broader ecosystem of downstream analysis tools opens the door to a wider array of analytical possibilities.
Trajectory generation
A fundamental advantage of the stDGM methods discussed here, distinguishing them from static methods, is their formulation as generative models. This generative capability allows one to simulate entire cellular trajectories forward in time from an initial population distribution. Consequently, these models can not only reconstruct the observed cell distributions at discrete time points but also interpolate to predict cell states at previously unobserved times (Fig. 1C (b)). Therefore, a final step in the workflow focuses on this trajectory reconstruction. Once trajectories are built, they provide an explicit mapping of an individual cell’s fate, revealing which cell state evolves into another. This enables the direct analysis of how cell types transition along a lineage. To achieve this, a cell annotation step is typically required, where a classifier is trained to predict a cell’s type from its gene expression vector. By applying these labels to the simulated trajectories, a complete lineage fate map can be constructed, offering interpretability into the underlying biological mechanisms of development and differentiation. For example, TrajectoryNet65 used the generated trajectory on an embryoid body dataset161 to identify how early the gene expression profiles of cells destined for different fates began to diverge. On the same dataset, MIOFlow96 generated and decoded trajectories back to the full gene space to accurately reconstruct complex, non-monotonic expression dynamics for individual genes, which align with known biology. TIGON67 interpolated data at unmeasured time points in an epithelial-to-mesenchymal transition (EMT) dataset162, revealing the changes of cell-cell communication patterns over time. We show CytoBridge’s function of generating trajectories in Step 4 of Box 1, and also visualize the generated trajectory of the mouse hematopoiesis dataset using CytoBridge in Fig. 4.

In the mouse hematopoiesis dataset that includes samples from Day 2, Day 4, and Day 6, CytoBridge reconstructs continuous dynamic trajectories to elucidate cellular dynamics. Specifically, based on the initial sampling at Day 2, it generates 100 time-step hypothetical cell trajectories from Day 2 to Day 6, depicted as red lines, with the states for Days 3, 4, 5, and 6 individually highlighted. At every time point, cells that arise from division during the generation interval are shown in orange. These data and figures are produced by calling Cytobrdige fit funciton with the unbalanced mode.
However, there remain several key limitations inherent to current generative models. A primary challenge is temporal generalization. While models may excel at interpolation within the training time ranges, their accuracy often degrades when forecasting far beyond it, as the underlying biological regulatory dynamics may shift. Another potential issue is the sensitivity to initial conditions and sampling noise; errors or biases in the early time point data can be amplified throughout the simulation, leading to divergent and biologically implausible trajectories. Furthermore, generalization across different cell types can be limited; a model trained on specific differentiation pathways may be unable to predict the emergence of a rare or previously unseen cell lineage. Rigorously validating generated trajectories remains a challenge. Experimental lineage tracing, when available, can serve as a standard for confirmation.
Conclusion and future directions
In this review, we have discussed recent progress toward the \(\underline{{\bf{s}}}{\rm{patio}}\underline{t}{\rm{emporal}}\) Dynamical Generative Model (stDGM) for single-cell and ST, with a particular emphasis on dissecting cell-fate trajectories from time-series scRNA-seq and spatiotemporal data. We first introduced the mathematical foundations of dynamical systems and generative modeling, including optimal transport and SB foundation formulations, highlighting how these concepts provide a framework for reconstructing and generating cellular dynamics. We then reviewed algorithmic advances that implement these frameworks in practice. Finally, we offered practical guidelines to help researchers select and apply methods to different types of application scenarios. By integrating mathematical principles, computational methods, and biological applications, our aim is to provide a systematic and accessible perspective for the study of cellular dynamics.
Looking forward, an important direction is the integration of richer data modalities, where transcriptomic time series are combined with epigenomic, proteomic, and imaging measurements to provide more comprehensive views of regulatory dynamics. Another promising direction is the incorporation of lineage tracing and clonal recording technologies163,164,165,166,167,168, which will allow computationally inferred trajectories to be validated and refined with experimentally observed ancestry, thus strengthening the biological interpretation of fate decisions. Advances in spatial and temporal resolution will also make it possible to explicitly couple intracellular dynamics with cell-cell interactions169,170,171,172,173,174,175 and tissue-level organization. Importantly, integrating studies of cell mechanics will help model cellular morphogenesis in physical space176,177,178, offering multiscale perspectives on development and disease. Finally, the continued refinement of computational packages into user-friendly and accessible tools will be critical for enabling a broader community of biologists to apply these methods in practice. Together, these developments point toward a future in which dynamical modeling is not only a theoretical framework but also a practical component of experimental biology, deepening our understanding of cellular dynamics in development, regeneration, and pathology.
Data availability
No datasets were generated during the current study. The single-cell mouse hematopoiesis lineage tracing dataset analysed was downloaded from https://github.com/AllonKleinLab/paper-data/tree/master/Lineage_tracing_on_transcriptional_landscapes_links_state_to_fate_during_differentiation (ref. 163).
Code availability
The source code and tutorials to reproduce the analysis in Figure 1 are publicly available at https://github.com/zhenyiizhang/DeepRUOTv2. The CytoBridge package is publicly available at https://github.com/zhenyiizhang/CytoBridge and is continuously updated.
References
Lei, J. Mathematical modeling of heterogeneous stem cell regeneration: from cell division to waddington’s epigenetic landscape. In Dynamics of Physiological Control: Contributions in Honor of Michael C. Mackey, 37–82 (Springer, 2025).
Hong, T. & Xing, J. Data-and theory-driven approaches for understanding paths of epithelial–mesenchymal transition. Genesis 62, e23591 (2024).
Xing, J. Reconstructing data-driven governing equations for cell phenotypic transitions: integration of data science and systems biology. Phys. Biol. 19, 061001 (2022).
Schiebinger, G. Reconstructing developmental landscapes and trajectories from single-cell data. Curr. Opin. Syst. Biol. 27, 100351 (2021).
Heitz, M., Ma, Y., Kubal, S. & Schiebinger, G. Spatial transcriptomics brings new challenges and opportunities for trajectory inference. Ann. Re. Biomed. Data Sci. 8, 1–19 (2024).
Waddington, C. H.The strategy of the genes (Routledge, 2014).
Moris, N., Pina, C. & Arias, A. M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 17, 693–703 (2016).
MacLean, A. L., Hong, T. & Nie, Q. Exploring intermediate cell states through the lens of single cells. Curr. Opin. Syst. Biol. 9, 32–41 (2018).
Zhu, L. & Wang, J. Quantifying landscape and flux from single-cell omics: unraveling the physical mechanisms of cell function. JACS Au 5, 3738–3757 (2025).
Ziegenhain, C. et al. Comparative analysis of single-cell RNA sequencing methods. Mol. cell 65, 631–643 (2017).
Tang, F. et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656 (2019).
Ding, J., Sharon, N. & Bar-Joseph, Z. Temporal modelling using single-cell transcriptomics. Nat. Rev. Genet. 23, 355–368 (2022).
Bunne, C., Schiebinger, G., Krause, A., Regev, A. & Cuturi, M. Optimal transport for single-cell and spatial omics. Nat. Rev. Methods Prim. 4, 58 (2024).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with slide-seqv2. Nat. Biotechnol. 39, 313–319 (2021).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792 (2022).
Oliveira, M. F. d., Romero, J. P. & Chung, M. High-definition spatial transcriptomic profiling of immune cell populations in colorectal cancer. Nat. Genet. 57, 1512–1523 (2025).
Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018).
Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by rna seqfish+. Nature 568, 235–239 (2019).
Wang, X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018).
Liu, L. et al. Spatiotemporal omics for biology and medicine. Cell 187, 4488–4519 (2024).
Bunne, C. et al. How to build the virtual cell with artificial intelligence: priorities and opportunities. Cell 187, 7045–7063 (2024).
Qian, L., Dong, Z. & Guo, T. Grow AI virtual cells: three data pillars and closed-loop learning. Cell Res. 35, 319–321 (2025).
Roohani, Y. H. et al. Virtual cell challenge: toward a turing test for the virtual cell. Cell 188, 3370–3374 (2025).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 1–16 (2018).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Soldatov, R. A., Kharchenko, P. V. & Theis, F. J. RNA velocity—current challenges and future perspectives. Mol. Syst. Biol. 17, e10282 (2021).
Wang, K. et al. Phylovelo enhances transcriptomic velocity field mapping using monotonically expressed genes. Nat. Biotechnol. 42, 778–789 (2024).
Liu, Y., Huang, K. & Chen, W. Resolving cellular dynamics using single-cell temporal transcriptomics. Curr. Opin. Biotechnol. 85, 103060 (2024).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Schiebinger, G. et al. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 928–943 (2019).
Klein, D. et al. Mapping cells through time and space with Moscot. Nature 638, 1065–1075 (2025).
Zhang, Z., Sun, Y., Peng, Q., Li, T. & Zhou, P. Integrating dynamical systems modeling with spatiotemporal scRNA-seq data analysis. Entropy 27, 453 (2025).
Lavenant, H. & Zhang, S. et al. Toward a mathematical theory of trajectory inference. Ann. Appl. Probab. 34, 428–500 (2024).
Lipman, Y., Chen, R. T. Q., Ben-Hamu, H., Nickel, M. & Le, M. Flow matching for generative modeling. In The Eleventh International Conference on Learning Representations (OpenReview.net, 2023).
Tong, A. et al. Improving and generalizing flow-based generative models with minibatch optimal transport. Trans. Mach. Learn. Res. (OpenReview.net, 2024).
Wang, D. et al. Joint velocity-growth flow matching for single-cell dynamics modeling. Adv. Neural Inform. Process. Syst. (Curran Associates, Inc., 2025).
Zhang, Y. & Levin, M. Equilibrium flow: from snapshots to dynamics. Preprint at https://doi.org/10.48550/arXiv.2509.17990 (2025).
Morehead, A. et al. How to go with the flow: flow matching in bioinformatics and computational biology. Preprint at https://doi.org/10.22541/au.175382408.89466370/v3 (2025).
Li, Z. et al. Flow matching meets biology and life science: a survey. Preprint at https://doi.org/10.48550/arXiv.2507.17731 (2025).
Chizat, L., Peyré, G., Schmitzer, B. & Vialard, F.-X. An interpolating distance between optimal transport and fisher–rao metrics. Found. Comput. Math. 18, 1–44 (2018).
Chizat, L., Peyré, G., Schmitzer, B. & Vialard, F.-X. Unbalanced optimal transport: dynamic and kantorovich formulations. J. Funct. Anal. 274, 3090–3123 (2018).
Gangbo, W., Li, W., Osher, S. & Puthawala, M. Unnormalized optimal transport. J. Comput. Phys. 399, 108940 (2019).
Sun, Y., Zhang, Z., Wang, Z., Li, T. & Zhou, P. Variational regularized unbalanced optimal transport: single network, least action. Adv. Neural Inform. Process. Syst. (Curran Associates, Inc., 2025).
Léonard, C. A survey of the schrödinger problem and some of its connections with optimal transport. Discret. Contin. Dyn. Syst. Ser. A 34, 1533–1574 (2014).
Gentil, I., Léonard, C. & Ripani, L. About the analogy between optimal transport and minimal entropy. Ann. Fac. Sci. de Toulouse Math. 26, 569–600 (2017).
Zhang, Z., Li, T. & Zhou, P. Learning stochastic dynamics from snapshots through regularized unbalanced optimal transport. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Yang, L., Daskalakis, C. & Karniadakis, G. E. Generative ensemble regression: learning particle dynamics from observations of ensembles with physics-informed deep generative models. SIAM J. Sci. Comput. 44, B80–B99 (2022).
Ruthotto, L., Osher, S. J., Li, W., Nurbekyan, L. & Fung, S. W. A machine learning framework for solving high-dimensional mean field game and mean field control problems. Proc. Natl. Acad. Sci. USA 117, 9183–9193 (2020).
Zhang, Z., Wang, Z., Sun, Y., Li, T. & Zhou, P. Modeling cell dynamics and interactions with unbalanced mean field schrödinger bridge. Adv. Neural Inform. Process. Syst. (Curran Associates, Inc., 2025).
Jiang, Q., Zhang, L., Li, L. & Wan, L. Learning collective multi-cellular dynamics from temporal scRNA-seq via a transformer-enhanced neural SDE. Preprint at https://doi.org/10.48550/arXiv.2505.16492 (2025).
Wang, L. et al. Current progress and potential opportunities to infer single-cell developmental trajectory and cell fate. Curr. Opin. Syst. Biol. 26, 1–11 (2021).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Li, T., Shi, J., Wu, Y. & Zhou, P. On the mathematics of RNA velocity I: theoretical analysis. CSIAM-AM. 2, 1–55 (2021).
Li, T., Wang, Y., Yang, G. & Zhou, P. On the mathematics of RNA velocity ii: algorithmic aspects. CSIAM-AM. 5, 182-220 (2024).
Jiang, Q. & Wan, L. Dynamic modeling, optimization, and deep learning for high-dimensional complex biological data. SCI. SIN. Math. 55, 1–14 (2025).
Monge, G. Mémoire sur la théorie des déblais et des remblais. Mem. Math. Phys. Acad. Royale Sci., 666−704 (1781).
Kantorovich, L. V. On the translocation of masses. Dokl. Akademii Nauk SSSR 37, 227–229 (1942).
Cheng, Y.-C. et al. Reconstruction of single-cell lineage trajectories and identification of diversity in fates during the epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 121, e2406842121 (2024).
Benamou, J.-D. & Brenier, Y. A computational fluid mechanics solution to the Monge-Kantorovich mass transfer problem. Numer. Math. 84, 375–393 (2000).
Tong, A., Huang, J., Wolf, G., Van Dijk, D. & Krishnaswamy, S. Trajectorynet: A dynamic optimal transport network for modeling cellular dynamics. In International conference on machine learning, 9526–9536 (PMLR, 2020).
Liero, M., Mielke, A. & Savaré, G. Optimal entropy-transport problems and a new hellinger–kantorovich distance between positive measures. Invent. Math. 211, 969–1117 (2018).
Sha, Y., Qiu, Y., Zhou, P. & Nie, Q. Reconstructing growth and dynamic trajectories from single-cell transcriptomics data. Nat. Mach. Intell. 6, 25–39 (2024).
Nie, Q. Stem cells: a window of opportunity in low-dimensional emt space. Oncotarget 9, 31790 (2018).
Jolly, M. K. et al. Coupling the modules of EMT and stemness: a tunable ‘stemness window’model. Oncotarget 6, 25161 (2015).
Bocci, F., Zhou, P. & Nie, Q. Single-cell RNA-seq analysis reveals the acquisition of cancer stem cell traits and increase of cell–cell signaling during EMT progression. Cancers 13, 5726 (2021).
Sha, Y., Wang, S., Zhou, P. & Nie, Q. Inference and multiscale model of epithelial-to-mesenchymal transition via single-cell transcriptomic data. Nucleic Acids Res. 48, 9505–9520 (2020).
Sha, Y., Wang, S., Bocci, F., Zhou, P. & Nie, Q. Inference of intercellular communications and multilayer gene-regulations of epithelial–mesenchymal transition from single-cell transcriptomic data. Front. Genet. 11, 604585 (2021).
Zhou, P. et al. Stochasticity triggers activation of the s-phase checkpoint pathway in budding yeast. Phys. Rev. X 11, 011004 (2021).
Schrödinger, E. Sur la théorie relativiste de l’électron et l’interprétation de la mécanique quantique. Ann. de. l’Inst. Henri Poincaré 2, 269–310 (1932).
Dai Pra, P. A stochastic control approach to reciprocal diffusion processes. Appl. Math. Optim. 23, 313–329 (1991).
Chen, Y., Georgiou, T. T. & Pavon, M. On the relation between optimal transport and schrödinger bridges: a stochastic control viewpoint. J. Optim. Theory Appl. 169, 671–691 (2016).
Tong, A. et al. Simulation-free schrödinger bridges via score and flow matching. In International Conference on Artificial Intelligence and Statistics, 1279–1287 (PMLR, 2024).
Chen, Y., Georgiou, T. T. & Pavon, M. The most likely evolution of diffusing and vanishing particles: Schrodinger bridges with unbalanced marginals. SIAM J. Control Optim. 60, 2016–2039 (2022).
Baradat, A. & Lavenant, H. Regularized unbalanced optimal transport as entropy minimization with respect to branching Brownian motion. Preprint at https://doi.org/10.48550/arXiv.2111.01666 (2021).
Bertucci, C. Stochastic optimal transport and Hamiltoni-Jacobi-Bellman equations on the set of probability measures. Ann. Inst. H. Poincaré‚ Anal. Non Linéaire 42, 1543–1600 (2025).
Chen, Y., Georgiou, T. T. & Pavon, M. The most likely evolution of diffusing and vanishing particles: Schrödinger bridges with unbalanced marginals. SIAM J. Control Optim. 60, 2016–2039 (2022).
Backhoff, J., Conforti, G., Gentil, I. & Léonard, C. The mean field schrödinger problem: ergodic behavior, entropy estimates and functional inequalities. Probab. Theory Relat. Fields 178, 475–530 (2020).
Hernández, C. & Tangpi, L. Propagation of chaos for mean field schrödinger problems. SIAM J. Control Optim. 63, 112–150 (2025).
McKean, H. P. Propagation of chaos for a class of non-linear parabolic equations. In Stochastic Differential Equations, Lecture Series in Differential Equations, Session 7, Catholic Univ., 41–57 (Air Force Office of Scientific Research, 1967).
Mémoli, F. Spectral gromov–wasserstein distances for shape matching. In Proc. 2009 IEEE 12th International Conference on Computer Vision Workshops (ICCV Workshops), 256–263 (IEEE, 2009).
Mémoli, F. Gromov–wasserstein distances and the metric approach to object matching. Found. Comput. Math. 11, 17–83 (2011).
Zhang, Z., Goldfeld, Z., Greenewald, K., Mroueh, Y. & Sriperumbudur, B. K. Gradient flows and Riemannian structure in the Gromov-Wasserstein geometry. Found. Comput. Math. (2025).
Cohen, S. & Guibasm, L. The earth mover’s distance under transformation sets. Proc. Seventh IEEE Int. Conf. Computer Vis. 2, 1076–1083 (1999).
Peng, Q., Zhou, P. & Li, T. stVCR: Reconstructing spatio-temporal dynamics of cell development using optimal transport. Preprint at https://doi.org/10.1101/2024.06.02.596937 (2024).
Schönemann, P. H. A generalized solution of the orthogonal procrustes problem. Psychometrika 31, 1–10 (1966).
Wei, X. et al. Single-cell stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science 377, eabp9444 (2022).
Wang, M. et al. High-resolution 3d spatiotemporal transcriptomic maps of developing Drosophila embryos and larvae. Dev. Cell 57, 1271–1283 (2022).
Wang, M. et al. A Drosophila single-cell 3d spatiotemporal multi-omics atlas unveils panoramic key regulators of cell-type differentiation. Cell 188, 4734–4753 (2025).
Palma, A., Rybakov, S., Hetzel, L., Günnemann, S. & Theis, F. J. Enforcing latent euclidean geometry in single-cell VAEs for manifold interpolation. In Forty-second International Conference on Machine Learning (PMLR, 2025).
Ling, Y., Zhang, P., Zhang, Z. & Zhou, P. CellStream: Dynamical Optimal Transport Informed Embeddings for Reconstructing Cellular Trajectories from Snapshots Data. The Fortieth AAAI Conference on Artificial Intelligence (AAAI Press, 2025).
Huguet, G. et al. Manifold interpolating optimal-transport flows for trajectory inference. Adv. Neural Inf. Process. Syst. 35, 29705–29718 (2022).
Kapusniak, K. et al. Metric flow matching for smooth interpolations on the data manifold. Adv. Neural Inf. Process. Syst. 37, 135011–135042 (2024).
Banerjee, A., Lee, H., Sharon, N. & Moosmüller, C. Efficient Trajectory Inference in Wasserstein Space Using Consecutive Averaging. Proceedings of The 28th International Conference on Artificial Intelligence and Statistics, in Proceedings of Machine Learning Research 258, 2260−2268 (2025).
Yang, M. Topological schrödinger bridge matching. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Klein, D., Uscidda, T., Theis, F. & Cuturi, M. GENOT: Entropic (Gromov) Wasserstein Flow Matching with Applications to Single-Cell Genomics. Adv. Neural Inf. Process. Syst. (Curran Associates, Inc., 2024).
Baker, C. et al. Scot+: A comprehensive software suite for single-cell alignment using optimal transport. bioRxiv 2025–05 (2025).
Halmos, P. et al. Dest-ot: Alignment of spatiotemporal transcriptomics data. Cell Systems 16 (2025).
Huizing, G.J. et al. STORIES: learning cell fate landscapes from spatial transcriptomics using optimal transport. Nat. Methods (2025).
Zeira, R., Land, M., Strzalkowski, A. & Raphael, B. J. Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022).
Liu, X., Zeira, R. & Raphael, B. J. Partial alignment of multislice spatially resolved transcriptomics data. Genome Res. 33, 1124–1132 (2023).
Kiemen, A. L. et al. Coda: quantitative 3d reconstruction of large tissues at cellular resolution. Nat. Methods 19, 1490–1499 (2022).
Li, S., Shen, Q. & Zhang, S. Spatial transcriptomics-aided localization for single-cell transcriptomics with stalocator. Cell Systems 16 (2025).
Tan, Y. et al. Transfer learning of multicellular organization via single-cell and spatial transcriptomics. PLOS Comput. Biol. 21, e1012991 (2025).
Palma, A. et al. Multi-modal and multi-attribute generation of single cells with cfgen. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Klein, D. et al. Cellflow enables generative single-cell phenotype modeling with flow matching. bioRxiv 2025–04 (2025).
Rohbeck, M. et al. Modeling complex system dynamics with flow matching across time and conditions. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Tronstad, M., Karlsson, J. & Dahlin, J. S. MultistageOT: Multistage optimal transport infers trajectories from a snapshot of single-cell data. Preprint at https://doi.org/10.48550/arXiv.2502.05241 (2025).
Yachimura, T. et al. scEGOT: single-cell trajectory inference framework based on entropic gaussian mixture optimal transport. BMC Bioinform. 25, 388 (2024).
Zhao, W., Larschan, E., Sandstede, B. & Singh, R. Optimal transport reveals dynamic gene regulatory networks via gene velocity estimation. PLOS Comput. Biol. 21, e1012476 (2025).
Halmos, P., Gold, J., Liu, X. & Raphael, B. J. Learning latent trajectories in developmental time series with hidden-markov optimal transport. In International Conference on Research in Computational Molecular Biology, 367–370 (Springer, 2025).
Kawano, K., Kutsuna, T., Hayashi, N., Esaki, Y. & Tanaka, H. CT-OT Flow: Estimating continuous-time dynamics from discrete temporal snapshots. Preprint at https://doi.org/10.48550/arXiv.2505.17354 (2025).
Bunne, C., Hsieh, Y.-P., Cuturi, M. & Krause, A. The schrödinger bridge between Gaussian measures has a closed form. In International Conference on Artificial Intelligence and Statistics, 5802–5833 (PMLR, 2023).
Jiang, Q. & Wan, L. A physics-informed neural SDE network for learning cellular dynamics from time-series scRNA-seq data. Bioinform. 40, ii120–ii127 (2024).
Zhang, K., Zhu, J., Kong, D. & Zhang, Z. Modeling single cell trajectory using forward-backward stochastic differential equations. PLOS Comput. Biol. 20, e1012015 (2024).
Maddu, S., Chardès, V. & Shelley, M. J. Learning stochastic processes with intrinsic noise from cross-sectional biological data. Proc. Natl. Acad. Sci. USA 122, e2420621122 (2025).
Chen, T., Liu, G.-H. & Theodorou, E. Likelihood training of schrödinger bridge using forward-backward SDEs theory. In International Conference on Learning Representations (OpenReview.net, 2022).
Koshizuka, T. & Sato, I. Neural lagrangian schrödinger bridge: Diffusion modeling for population dynamics. In The Eleventh International Conference on Learning Representations (OpenReview.net, 2023).
Zhang, S. Y. & Stumpf, M. P. Learning non-equilibrium diffusions with schrödinger bridges: from exactly solvable to simulation-free. Adv. Neural Inform. Process. Syst. (Curran Associates, Inc., 2025).
Hong, W., Shi, Y. & Niles-Weed, J. Trajectory inference with smooth schrödinger bridges. In Forty-second International Conference on Machine Learning (PMLR, 2025).
Tang, S., Zhang, Y., Tong, A. & Chatterjee, P. Branched schrödinger bridge matching. Preprint at https://doi.org/10.48550/arXiv.2506.09007 (2025).
Fischer, D. S. et al. Inferring population dynamics from single-cell rna-sequencing time series data. Nat. Biotechnol. 37, 461–468 (2019).
Pariset, M., Hsieh, Y.-P., Bunne, C., Krause, A. & Bortoli, V. D. Unbalanced diffusion schrödinger bridge. In ICML Workshop on New Frontiers in Learning, Control, and Dynamical Systems (OpenReview.net, 2023).
Alatkar, S. A. & Wang, D. Artemis integrates autoencoders and schrödinger bridges to predict continuous dynamics of gene expression, cell population, and perturbation from time-series single-cell data. Bioinform. 41, i189–i197 (2025).
Theodoropoulos, P., Saravanos, A. D., Theodorou, E. A. & Liu, G.-H. Momentum multi-marginal schrödinger bridge matching. Adv. Neural Inform. Process. Syst. (Curran Associates, Inc., 2025).
Atanackovic, L. et al. Meta flow matching: Integrating vector fields on the wasserstein manifold. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Jiang, Q., Zhang, S. & Wan, L. Dynamic inference of cell developmental complex energy landscape from time series single-cell transcriptomic data. PLoS Comput. Biol. 18, e1009821 (2022).
E, W. A proposal on machine learning via dynamical systems. Commun. Math. Stat. 5, 1–11 (2017).
Chen, R. T., Rubanova, Y., Bettencourt, J. & Duvenaud, D. K. Neural ordinary differential equations. Adv. Neural Inform. Process. Syst. 31 (2018).
Zhang, J., Larschan, E., Bigness, J. & Singh, R. Scnode: generative model for temporal single cell transcriptomic data prediction. Bioinform. 40, ii146–ii154 (2024).
von Bassewitz, J.-P. et al. Learning explicit single-cell dynamics using ODE representations. Preprint at https://doi.org/10.48550/arXiv.2510.02903 (2025).
Li, X., Wong, T.-K. L., Chen, R. T. & Duvenaud, D. Scalable gradients for stochastic differential equations. In International Conference on Artificial Intelligence and Statistics, 3870–3882 (PMLR, 2020).
Holderrieth, P. et al. Generator matching: generative modeling with arbitrary Markov processes. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Zhou, M., Osher, S. & Li, W. Simulating Fokker-Planck equations via mean field control of score-based normalizing flows. Preprint at https://doi.org/10.48550/arXiv.2506.05723 (2025).
Eyring, L. et al. Unbalancedness in neural monge maps improves unpaired domain translation. In The Twelfth International Conference on Learning Representations (OpenReview.net, 2024).
Petrović, K. et al. Curly flow matching for learning non-gradient field dynamics. In Learning Meaningful Representations of Life (LMRL) Workshop at ICLR 2025 (ICLR, 2025).
Haviv, D., Pooladian, A.-A., Pe’er, D. & Amos, B. Wasserstein flow matching: Generative modeling over families of distributions. In Forty-second International Conference on Machine Learning (PMLR, 2025).
Lee, J., Moradijamei, B. & Shakeri, H. Multi-marginal stochastic flow matching for high-dimensional snapshot data at irregular time points. In Forty-second International Conference on Machine Learning (PMLR, 2025).
Yeo, G. H. T., Saksena, S. D. & Gifford, D. K. Generative modeling of single-cell time series with prescient enables prediction of cell trajectories with interventions. Nat. Commun. 12, 3222 (2021).
Neklyudov, K., Brekelmans, R., Severo, D. & Makhzani, A. Action matching: Learning stochastic dynamics from samples. In International conference on machine learning, 25858–25889 (PMLR, 2023).
Neklyudov, K. et al. A computational framework for solving Wasserstein Lagrangian flows. In Forty-first International Conference on Machine Learning (PMLR, 2024).
Meng, T., Zou, Z., Darbon, J. & Karniadakis, G. E. Hj-sampler: a Bayesian sampler for inverse problems of a stochastic process by leveraging Hamilton-Jacobi PDEs and score-based generative models. Preprint at https://doi.org/10.48550/arXiv.2409.09614 (2024).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
McInnes, L., Healy, J. & Melville, J. Umap: uniform manifold approximation and projection for dimension reduction. Preprint at https://doi.org/10.48550/arXiv.1802.03426 (2018).
Zhou, P. & Li, T. Construction of the landscape for multi-stable systems: Potential landscape, quasi-potential, a-type integral and beyond. J. Chem. Phys. 144, 094109 (2016).
Shi, J., Aihara, K., Li, T. & Chen, L. Energy landscape decomposition for cell differentiation with proliferation effect. Natl. Sci. Rev. 9, nwac116 (2022).
Zhao, Y., Zhang, W. & Li, T. Epr-net: constructing a non-equilibrium potential landscape via a variational force projection formulation. Natl. Sci. Rev. 11, nwae052 (2024).
Li, C. & Wang, J. Quantifying cell fate decisions for differentiation and reprogramming of a human stem cell network: landscape and biological paths. PLoS Comput. Biol. 9, e1003165 (2013).
Wang, J., Li, C. & Wang, E. Potential and flux landscapes quantify the stability and robustness of budding yeast cell cycle network. Proc. Natl Acad. Sci. 107, 8195–8200 (2010).
Li, C. & Wang, J. Landscape and flux reveal a new global view and physical quantification of mammalian cell cycle. Proc. Natl. Acad. Sci. USA 111, 14130–14135 (2014).
Zhou, P., Bocci, F., Li, T. & Nie, Q. Spatial transition tensor of single cells. Nat. Methods 21, 1053–1062 (2024).
Torregrosa, G. & Garcia-Ojalvo, J. Mechanistic models of cell-fate transitions from single-cell data. Curr. Opin. Syst. Biol. 26, 79–86 (2021).
Zhu, L. & Wang, J. Quantifying landscape-flux via single-cell transcriptomics uncovers the underlying mechanism of cell cycle. Adv. Sci. 11, 2308879 (2024).
Zhu, L. et al. Uncovering underlying physical principles and driving forces of cell differentiation and reprogramming from single-cell transcriptomics. Proc. Natl. Acad. Sci. USA 121, e2401540121 (2024).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Lange, M. et al. Cellrank for directed single-cell fate mapping. Nat. Methods 19, 159–170 (2022).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
Cook, D. P. & Vanderhyden, B. C. Context specificity of the EMT transcriptional response. Nat. Commun. 11, 2142 (2020).
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381 (2020).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427 (2020).
Ventre, E. et al. Trajectory inference for a branching SDE model of cell differentiation. Preprint at https://doi.org/10.48550/arXiv.2307.07687 (2023).
Forrow, A. & Schiebinger, G. Lineageot is a unified framework for lineage tracing and trajectory inference. Nat. Commun. 12, 4940 (2021).
Lange, M. et al. Mapping lineage-traced cells across time points with Moslin. Genome Biol. 25, 277 (2024).
Mao, S. et al. Cell lineage tracing: Methods, applications, and challenges. Quant. Biol. 13, e70006 (2025).
Almet, A. A., Cang, Z., Jin, S. & Nie, Q. The landscape of cell–cell communication through single-cell transcriptomics. Curr. Opin. Syst. Biol. 26, 12–23 (2021).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Jin, S., Plikus, M. V. & Nie, Q. Cellchat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat. Protoc. 20, 180–219 (2025).
Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Almet, A. A., Tsai, Y.-C., Watanabe, M. & Nie, Q. Inferring pattern-driving intercellular flows from single-cell and spatial transcriptomics. Nat. Methods 21, 1806–1817 (2024).
Wada, T., Hironaka, K.-I. & Kuroda, S. Cell-to-cell variability serves as information not noise. Curr. Opin. Syst. Biol. 27, 100339 (2021).
Topolewski, P. & Komorowski, M. Information-theoretic analyses of cellular strategies for achieving high signaling capacity—dynamics, cross-wiring, and heterogeneity of cellular states. Curr. Opin. Syst. Biol. 27, 100352 (2021).
Ichbiah, S., Delbary, F., Mcdougall, A., Dumollard, R. & Turlier, H. Embryo mechanics cartography: inference of 3d force atlases from fluorescence microscopy. Nat. Methods 20, 1989–1999 (2023).
Hallou, A., He, R., Simons, B. D. & Dumitrascu, B. A computational pipeline for spatial mechano-transcriptomics. Nat. Methods 1–14 (2025).
Lange, M. et al. A multimodal zebrafish developmental atlas reveals the state-transition dynamics of late-vertebrate pluripotent axial progenitors. Cell 187, 6742–6759 (2024).
Acknowledgements
The authors are grateful for the helpful discussions with Professor Weinan E. This work was supported by the National Key R&D Program of China (No. 2021YFA1003301 to T.L.) and the National Natural Science Foundation of China (NSFC No. 12288101 to T.L. & P.Z., and 8206100646, T2321001 to P.Z.). We acknowledge the support from the High-performance Computing Platform of Peking University for computation.
Author information
Authors and Affiliations
Contributions
Z.Z., P.Z., and T.L. conceived the study. All authors conducted the investigation. Z.Z., Z.W., Y.S., J.S., and Q.P. prepared the original draft. All authors reviewed and edited the manuscript. Z.Z., Z.W., Y.S., and J.S. prepared the visualizations. P.Z. and T.L. supervised the work. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Wang, Z., Sun, Y. et al. Deciphering cell-fate trajectories using spatiotemporal single-cell transcriptomic data. npj Syst Biol Appl 12, 2 (2026). https://doi.org/10.1038/s41540-025-00624-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41540-025-00624-9
